Back to Journals » Journal of Pain Research » Volume 11
Sonic hedgehog signaling in spinal cord contributes to morphine-induced hyperalgesia and tolerance through upregulating brain-derived neurotrophic factor expression
Authors Liu S, Yao J ![]() , Wan X, Song Z, Miao S
, Wan X, Song Z, Miao S ![]() , Zhao Y, Wang X
, Zhao Y, Wang X ![]() , Liu Y
, Liu Y
Received 9 October 2017
Accepted for publication 23 December 2017
Published 3 April 2018 Volume 2018:11 Pages 649—659
DOI https://doi.org/10.2147/JPR.S153544
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Su Liu,1,2,* Jun-Li Yao,1,3,* Xin-Xin Wan,1,* Zhi-Jing Song,1 Shuai Miao,1,2 Ye Zhao,1,2 Xiu-Li Wang,1,2 Yue-Peng Liu4
1Jiangsu Province Key Laboratory of Anesthesiology, Xuzhou Medical University, Xuzhou, Jiangsu, China; 2Department of Anesthesiology, Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China; 3Department of Anesthesiology, Xuzhou Children’s Hospital, Xuzhou, Jiangsu, China; 4Center of Clinical Research and Translational Medicine, Lianyungang Oriental Hospital, Lianyungang, Jiangsu, China
*These authors contributed equally to this work
Purpose: Preventing opioid-induced hyperalgesia and tolerance continues to be a major clinical challenge, and the underlying mechanisms of hyperalgesia and tolerance remain elusive. Here, we investigated the role of sonic hedgehog (Shh) signaling in opioid-induced hyperalgesia and tolerance.
Methods: Shh signaling expression, behavioral changes, and neurochemical alterations induced by morphine were analyzed in male adult CD-1 mice with repeated administration of morphine. To investigate the contribution of Shh to morphine-induced hyperalgesia (MIH) and tolerance, Shh signaling inhibitor cyclopamine and Shh small interfering RNA (siRNA) were used. To explore the mechanisms of Shh signaling in MIH and tolerance, brain-derived neurotrophic factor (BDNF) inhibitor K252 and anti-BDNF antibody were used.
Results: Repeated administration of morphine produced obvious hyperalgesia and tolerance. The behavioral changes were correlated with the upregulation and activation of morphine treatment-induced Shh signaling. Pharmacologic and genetic inhibition of Shh signaling significantly delayed the generation of MIH and tolerance and associated neurochemical changes. Chronic morphine administration also induced upregulation of BDNF. Inhibiting BDNF effectively delayed the generation of MIH and tolerance. The upregulation of BDNF induced by morphine was significantly suppressed by inhibiting Shh signaling. In naïve mice, exogenous activation of Shh signaling caused a rapid increase of BDNF expression, as well as thermal hyperalgesia. Inhibiting BDNF significantly suppressed smoothened agonist-induced hyperalgesia.
Conclusion: These findings suggest that Shh signaling may be a critical mediator for MIH and tolerance by regulating BDNF expression. Inhibiting Shh signaling, especially during the early phase, may effectively delay or suppress MIH and tolerance.
Keywords: sonic hedgehog, tolerance, hyperalgesia, brain-derived neurotrophic factor, spinal cord
Introduction
Opioid, such as morphine, plays an indispensable role in pain relief. However, long-term and repeated use of morphine no doubt leads to serious side effects, such as morphine-induced hyperalgesia (MIH) and tolerance.1 MIH is characterized as a paradoxical increase of pain after long-term morphine use,2 and tolerance is defined as a gradual loss of drug potency and reduced duration of action.3 The two side effects usually emerged together. This phenomenon limits the beneficial therapeutic use of opioids in clinic. Thus, preventing and reversing MIH and tolerance is a clinical challenge. The mechanisms of MIH and tolerance are complex and involve many factors at different levels, such as the receptors, the cells, the ion channels, and the neural networks.4–7 However, despite decades of investigation, the specific cellular and molecular mechanisms underlying MIH and tolerance remain elusive.
Sonic hedgehog (Shh) is a secreted glycoprotein that plays a causal role in controlling the patterning of neural progenitor cells during development.8 The components of Shh signaling in vertebrates mainly include Shh ligand, patched (Ptch) and smoothened (Smo) receptor, and Gli transcription factors.8,9 In the canonical pathway, activation of Shh signaling by binding of Shh to Ptc results in the activation of Smo and nuclear translocation of Gli.10–12 Emerging findings suggest that Shh plays important roles in the formation of neuronal circuits and synaptic plasticity.8,9 Shh could increase the size of presynaptic terminals and the frequency of miniature excitatory postsynaptic currents at hippocampal neuron synapses.13,14 Although the role of Shh during neurodevelopment is well addressed, the effect of Shh in adults remains unclear. It was reported that activation of Shh signaling was induced by acute brain injury15 and mediated brain plasticity.16 Recently, it was reported that Shh signaling was involved in nociceptive regulation.17–19 Shh mutation resulted in lack of nociceptive sensitization in drosophila.18 Besides, inhibited Shh signaling significantly suppressed complete Freund’s adjuvant-induced thermal analgesia and sciatic nerve ligation-induced mechanical allodynia.18 More recently, it was found that Shh signaling contributed to pancreatic cancer pain.19 Accumulating evidence suggests an involvement of a similar molecular mechanism underlying the nociceptive pain in MIH and tolerance,20,21 but whether Shh signaling is involved in MIH and tolerance remains unclear.
In the present study, we provide integrated evidence that activation of Shh signaling contributed to the generation of MIH and tolerance by upregulating brain-derived neurotrophic factor (BDNF) expression. Inhibiting Shh signaling could effectively prevent MIH and tolerance.
Materials and methods
Animals
Adult male CD-1 mice (24–28 g) were purchased from Shanghai Experimental Animal Center of Chinese Academy of Science. The mice were housed in a temperature-controlled environment with 12-hour light–dark cycles and were fed standard laboratory diet and water ad libitum. All mice were handled daily for 5 days before the start of the experiment to minimize the stress reaction to manipulation. The experimental protocols were approved by the Animal Research Committee of Xuzhou Medical University and were in accordance with the Declaration of the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23.)
Drugs and anesthesia
Mice were anesthetized with pentobarbital (50 mg/kg, intraperitoneally [i.p.]) for preparing the spinal cord tissue for Western blotting and immunobiochemistry. Shh signaling inhibitor cyclopamine and activator smoothened agonist (SAG) were purchased from Selleck (Houston, TX, USA). Tyrosine kinase inhibitor K252, BDNF inhibitor, was purchased from Biomol (Plymouth Meeting, PA, USA). Each of these drugs was dissolved in PBS ordimethyl sulfoxide (DMSO) and diluted in PBS (final concentration of DMSO was 1%). Cyclopamine (10 mg/kg), SAG (5 mg/kg), and the vehicle controls were injected i.p. (1 mL each). K252 (80 μg/kg) and anti-BDNF antibody (2 μg/kg) were injected intrathecally (i.t.). To confirm the role of Shh in MIH, Shh siRNA (Santa Cruz Biotechnology Inc., Dallas, TX, USA) was injected (1 μg/10 μL, i.t.) by means of lumbar puncture at the intervertebral space of L4–L5.
MIH model and behavior test
To construct the animal model of MIH, mice were administered morphine repeatedly (10 mg/kg, i.p.) twice a day for 7 consecutive days. Mice in the sham group were administered the same dose of saline (1 mL, i.p.) at the same time points. The anti-nociceptive effect was measured 30 minutes after each injection of morphine. Morphine tolerance was tested by hot plate. Mice were placed on a 55°C hot plate apparatus, and the latency to lick a paw was measured. In order to avoid empyrosis, the maximal latency was set as 60 seconds. Data were calculated as percentage maximal possible effect (MPE%), which was calculated by the following formula: MPE% = [(drug response time – basal response time)]/(30 seconds – basal response time)]×100. Thermal hyperalgesia was assessed by an analgesia meter (IITC Model 336 Analgesia Meter, Series 8; IITC Life Science Inc.; Woodland Hills, CA, USA). In brief, animals were placed in a box with a temperature-controlled glass floor. The heat source was focused on a portion of the hindpaw, which was flush against the glass, and a radiant thermal stimulus was delivered to that site. The stimulus shut off when the hindpaw moved (or after 20 seconds to prevent tissue damage). Thermal stimuli were delivered three times to each hindpaw at 5–6 minute intervals. To explore the role of Shh in MIH generation, cyclopamine was injected 15 minutes before morphine treatment twice a day from day 1 to day 3. To investigate the effect of Shh on MIH maintenance, cyclopamine was injected for 3 consecutive days after the MIH model was established, from day 5 to day 7, twice a day.
Western blotting
To identify temporal expression of Shh signaling (Shh, Ptch1, Smo, Gli1), whole-cell protein extract lysates were used. To identify the activation of Shh signaling, nuclear extracts were prepared using an NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer’s instruction. L1–L6 spinal cord segments and L4–L6 dorsal root ganglions (DRGs) were quickly removed from deeply anesthetized mice and homogenized in ice-cold radio-immunoprecipitation assay lysis buffer containing a cocktail of protease inhibitors. Total proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to 0.2 µm polyvinylidenefluoride membrane. The following primary antibodies were used: anti-Shh (1:200; Santa Cruz Biotechnology Inc.), anti-Ptch1 (1:1000; Sigma , St. Louis, MO, USA), anti-Smo (1:1000; Abcam , Cambridge, UK), anti-Gli1 (1:200; Santa Cruz Biotechnology Inc.), anti-BDNF (1:1000; Abcam), anti-Histone H3 (1:1000; Abcam), anti-c-fos (1:500; Cell Signaling Tech, Danvers, MA, USA), anti-CGRP (1:1000; Abcam), and anti-GAPDH (1:10000; Sigma). The filters were developed using ECL reagents (PerkinElmer Inc., Waltham, MA, USA) with secondary antibodies from Millipore Bioscience Research Reagents (EMD Millipore, Billerica, MA, USA). Data were analyzed with a Molecular Imager (Gel DocTM XR, 170-8170; Bio-Rad Laboratories Inc., Hercules, CA, USA) and the associated software Quantity One-4.6.5 (Bio-Rad Laboratories Inc.).
Immunohistochemistry
Under deep anesthesia, mice were transcardially perfused with PBS followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 M PBS (pH 7.2–7.4), and then the L4–L6 lumbar segment and L4–L6 DRGs were dissected out and postfixed in the same fixative overnight. The embedded blocks were sectioned as 30 µm thick and processed for immunofluorescence. Sections from each group (five mice in each group) were incubated with the following primary antibodies: anti-Shh (1:100, Santa Cruz Biotechnology Inc.), anti-c-Fos (1:100, Santa Cruz Biotechnology Inc.), and anti-CGRP (1:1000, EMD Millipore), respectively. Then the sections were washed in 50 mM Tris-HCl (pH 7.4) PBS for 5 minutes three times and incubated in the secondary antibody for 2 hours at room temperature. After washing three times in PBS, the sections were observed by a confocal microscope (Leica TCS SPEII; Leica Microsystems, Wetzlar, Germany) for morphologic details. Images were analyzed using MicroSuite image analysis software. For double staining, the same procedure was performed to the second primary and secondary antibodies. Sections were mounted on slides and covered with 90% glycerin for observation under a confocal microscope. The dilution of antibodies used included anti-Shh (1:100; Santa Cruz Biotechnology Inc.), anti-Gli1 (1:100; Santa Cruz Biotechnology Inc.), and anti-BDNF (1:200; Abcam). To obtain the quantitative measurements of CGRP immunofluorescence, 15–20 fields covering the entire dorsal horn in each group were evaluated and photographed at the same exposure time to generate the raw data. The average green fluorescence intensity of each pixel was normalized to the background intensity in the same image. Tissues were collected at days 4 and 7 after the first injection of morphine.
Enzyme-linked immunosorbent assay (ELISA)
Under deep anesthesia, the L1–L6 spinal cord segments and L4–L6 DRGs of mice were rapidly removed and homogenized in ice-cold 0.01 mol/L PBS. Protein concentrations were determined by BCA protein assay. The level of Shh concentration was measured by ELISA using Sonic Hedgehog N-Terminus Quantikine ELISA Kit (R&D Systems, Inc., Minneapolis, MN, USA), according to the manufacturer’s instruction.
Statistical analysis
SPSS 16 (SPSS Inc., Chicago, IL, USA) was used to conduct all the statistical analyses. Alteration of expression of the proteins detected and the behavioral responses to MIH were analyzed with one-way analysis of variance (ANOVA), and the differences in withdrawal threshold over time among groups were tested with two-way ANOVA with repeated measures, followed by Bonferroni post hoc test. All data are presented as mean ± standard error of the mean (SEM). Statistical results were considered significant if P<0.05.
Results
Inhibiting Shh signaling delayed and suppressed tolerance and MIH generation, but showed no effect on the maintenance of tolerance and MIH
To address whether Shh signaling was involved in the regulation of morphine tolerance, we first conducted behavioral tests by using Shh signaling inhibitor cyclopamine. As shown in Figure 1, mice that received chronic morphine treatment showed significant decrease of MPE% (Figure 1A and B), which refers to morphine tolerance, from day 3 after morphine administration. Besides, with morphine treatment, mice showed obvious thermal hyperalgesia from day 5 after morphine injection, lasting at least to day 11 (Figure 1C and D). Repetitive treatment with cyclopamine 15 minutes before morphine injection (10 mg/kg, i.p., twice a day) at days 1, 2, and 3 significantly delayed and suppressed morphine-induced tolerance (Figure 1A) and thermal hyperalgesia (Figure 1C) (P<0.05). On the contrary, the same dose of cyclopamine injected at days 5, 6, and 7 failed to reverse or suppress morphine-induced tolerance (Figure 1B) and thermal hyperalgesia (Figure 1D) (P>0.05). The normal pain sensation was not altered in the sham group. Taken together, these results demonstrated that Shh signaling may be involved in the regulation of tolerance and MIH generation, but not the maintenance of tolerance and MIH.
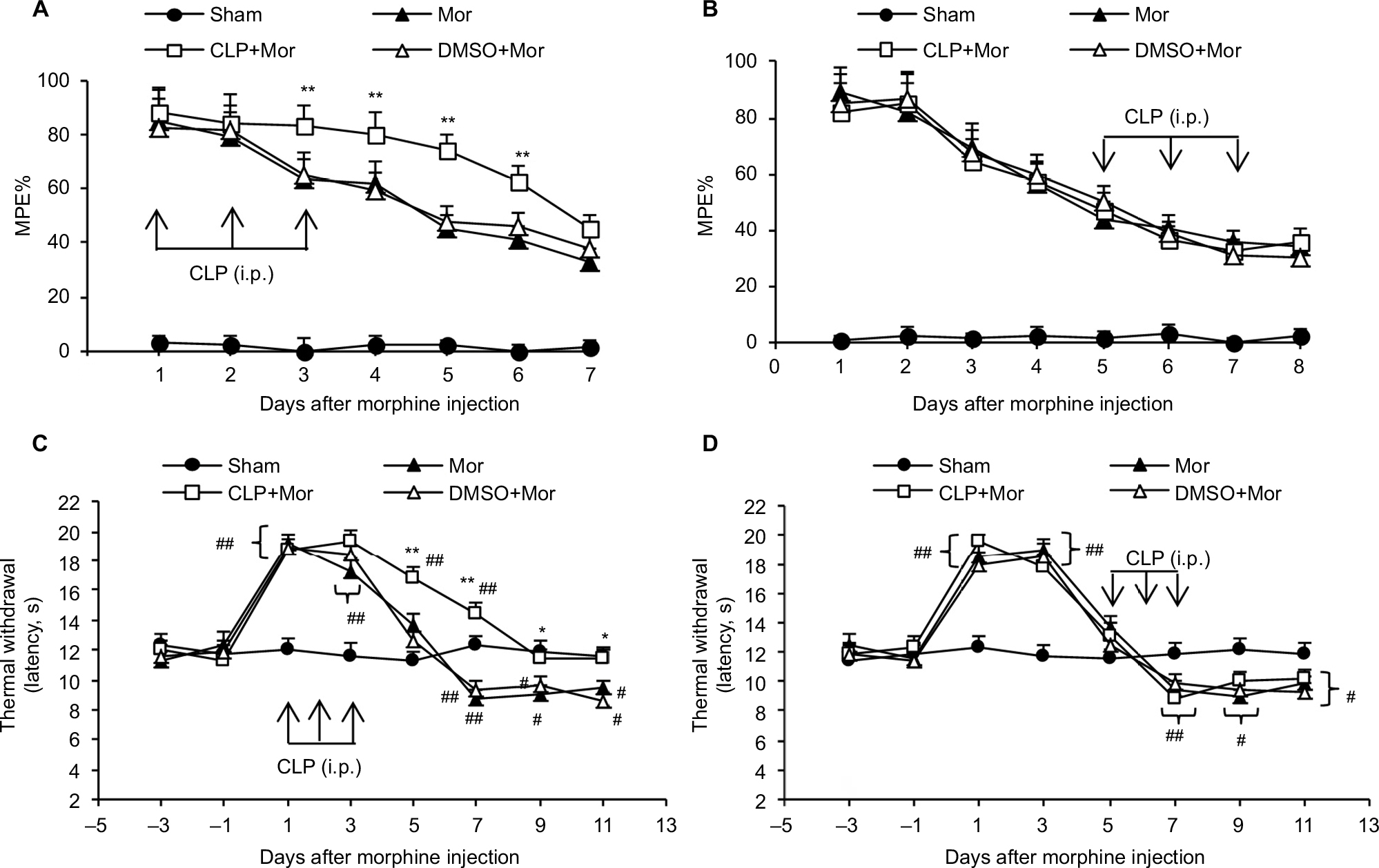 | Figure 1 Effects of cyclopamine on morphine-induced hyperalgesia and tolerance. Notes: The MPE% (refers to tolerance) decreased after repeated morphine injection (A and B). Coadministration of morphine with cyclopamine (10 mg/kg) on days 1, 2, and 3 significantly delayed MPE% decrease (A). However, coadministration of morphine with cyclopamine on days 5, 6, and 7 showed no effect on the reduction of MPE% (B). The thermal withdrawal latency (refers to hyperalgesia) of mice was significantly decreased after chronic morphine treatment (C and D). Coadministration of morphine with cyclopamine on days 1, 2, and 3 significantly delayed or attenuated hyperalgesia (C), but coadministration of morphine with cyclopamine on days 5, 6, and 7 showed no effect on hyperalgesia induced by chronic morphine treatment. *P<0.05, **P<0.01 versus sham group; #P<0.05, ##P<0.01 versus Mor group. Ten mice were included in each group. Abbreviations: i.p., intraperitoneally; MPE%, percentage of maximal possible effect; Mor, morphine; CLP, cyclopamine; DMSO, dimethyl sulfoxide. |
To confirm the role of Shh signaling in MIH and tolerance, Shh siRNA was used. Shh siRNA (1 μg/10 μL) was injected i.t. 3 consecutive days before morphine administration. Western blotting showed that spinal administration of Shh siRNA for 3 consecutive days effectively decreased the expression of Shh in spinal cord (Figure 2A). Furthermore, behavioral tests showed that with Shh gene knockdown, the reduction of morphine-induced MPE% (Figure 2B) was significantly delayed and suppressed. The normal pain sensation showed no obvious changes in the siRNA + sham group, and control siRNA (con-siRNA) showed no effect on tolerance (Figure 2B). In addition, siRNA administration significantly inhibited morphine treatment-induced Shh protein upregulation in spinal cord, while con-siRNA showed no effect on Shh expression (Figure 2C).
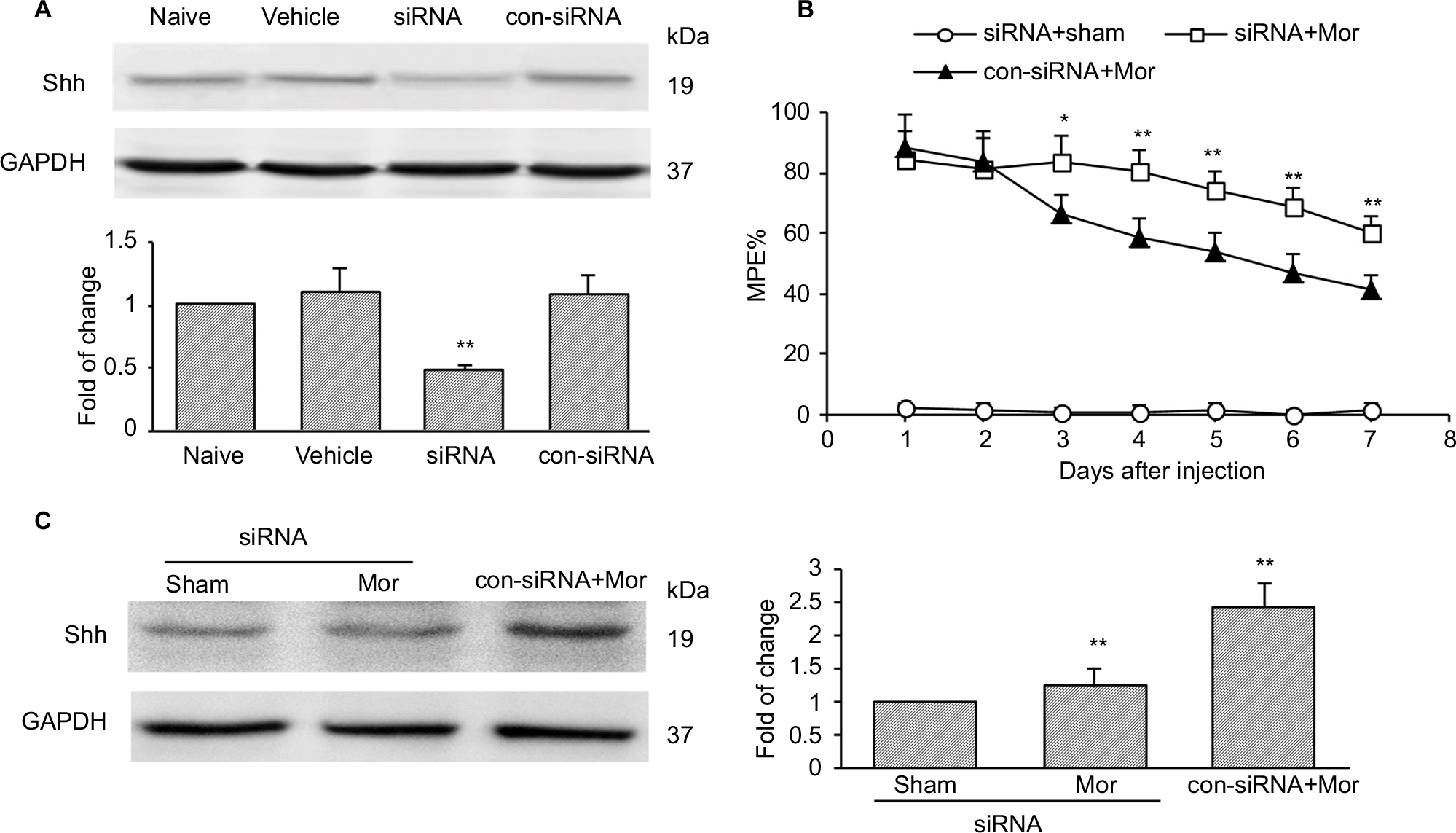 | Figure 2 Knockdown of Shh in spinal cord prevents the reduction of chronic morphine treatment-induced MPE%. Notes: (A) Effects of Shh targeting siRNA (1 μg i.t., daily for 3 consecutive days) on the expression of Shh in naïve mice (n=6 in each group). Whole spinal cord tissues were collected 2 hours after the last injection. (B) Knockdown of Shh significantly prevented the reduction of chronic morphine treatment-induced MPE% (n=10 in each group). (C) Effects of Shh targeting siRNA (1 μg i.t., daily for 3 consecutive days) on the expression of Shh in morphine-treated mice (n=6 in each group). Whole spinal cord tissues were collected at day 4 after the first morphine injection. *P<0.05, **P<0.01 versus con-siRNA+Mor group. Abbreviations: MPE%, percentage of maximal possible effect; siRNA, small interfering RNA; i.t., intrathecally; Mor, morphine; con-siRNA, control-siRNA. |
Inhibiting Shh signaling suppressed chronic morphine treatment-induced neurochemical signs in spinal cord
It was reported that chronic morphine exposure could cause neurochemical alterations in spinal cord, including c-fos and CGRP.22–25 Consistent with previous studies, both immunofluorescence staining (Figure 3A) and Western blot test (Figure 3B) showed that with repetitive administration of morphine, the expressions of c-fos and CGRP significantly increased in the spinal cord of the mice. Pretreatment with cyclopamine to inhibit Shh signaling significantly delayed or suppressed the upregulation of c-fos and CGRP (Figure 3).
 | Figure 3 Shh signaling inhibition with cyclopamine suppressed morphine treatment-induced upregulation of c-fos and CGRP in the spinal cord of mice. Notes: (A) Examples of immunofluorescence images and data summary (n=6 in each group) showing the expression of c-fos and CGRP in the dorsal horn of the spinal cord. Magnification: 200×. (B) Examples of Western blot analysis and data summary (n=6 in each group) showing the expression of c-fos and CGRP in the spinal cord. Samples were collected at day 4 after the first morphine injection. Cyclopamine (10 mg/kg, i.p.) was coadministrated with morphine on days 1, 2, and 3. *P<0.05, **P<0.01 versus sham group. ##P<0.01 versus Mor group. Abbreviations: i.p., intraperitoneally; Mor, morphine; CLP, cyclopamine. |
Shh signaling upregulated and activated following repeated morphine treatment
Given that inhibiting Shh signaling can delay or suppress morphine exposure-induced behavioral and neurochemical changes, we examined the expression and activity of Shh signaling in spinal cord under the same conditions. Western blotting showed that repetitive administration of morphine induced a time-dependent and long-lasting increase in the expressions of Shh and its receptor Ptch1 and Smo in spinal cord, which began on postinjection day 3. The upregulation of Shh signaling remained until day 7 (the last examination) (Figure 4A). Gli1 is a nuclear transcription factor and will transfer to the nucleus after the activation of Shh signaling.8,9 In order to prove the activation of Shh signaling, the expression of Gli1 in nuclear extract was tested. It was shown that the expression of Gli1 in the nucleus was also increased significantly after repetitive administration of morphine, from day 3 after morphine injection (Figure 4B). These results suggested that repeated morphine treatment could increase and activate Shh signaling in spinal cord. The immunofluorescence staining also showed that after morphine treatment, the expression of Shh was significantly increased in the dorsal horn of the spinal cord (Figure 4C). Furthermore, we also investigated the expression and activation of Shh signaling in DRG. Both the Western blot analysis (Figure 4D) and immunofluorescence staining (Figure 4E) showed that, after chronic morphine treatment, the expression of Shh protein and the nuclear translocation of Gli1 were significantly increased in a time-dependent pattern. Besides, immunofluorescence staining indicated that the increased Shh proteins were primarily located in small and medium neurons (Figure 4E). ELISA showed that the concentration and release of Shh protein was significantly increased in DRG and spinal cord after morphine treatment (Figure 4F). Taken together, these results suggested that Shh signaling was significantly upregulated and activated in DRG and spinal cord during morphine tolerance.
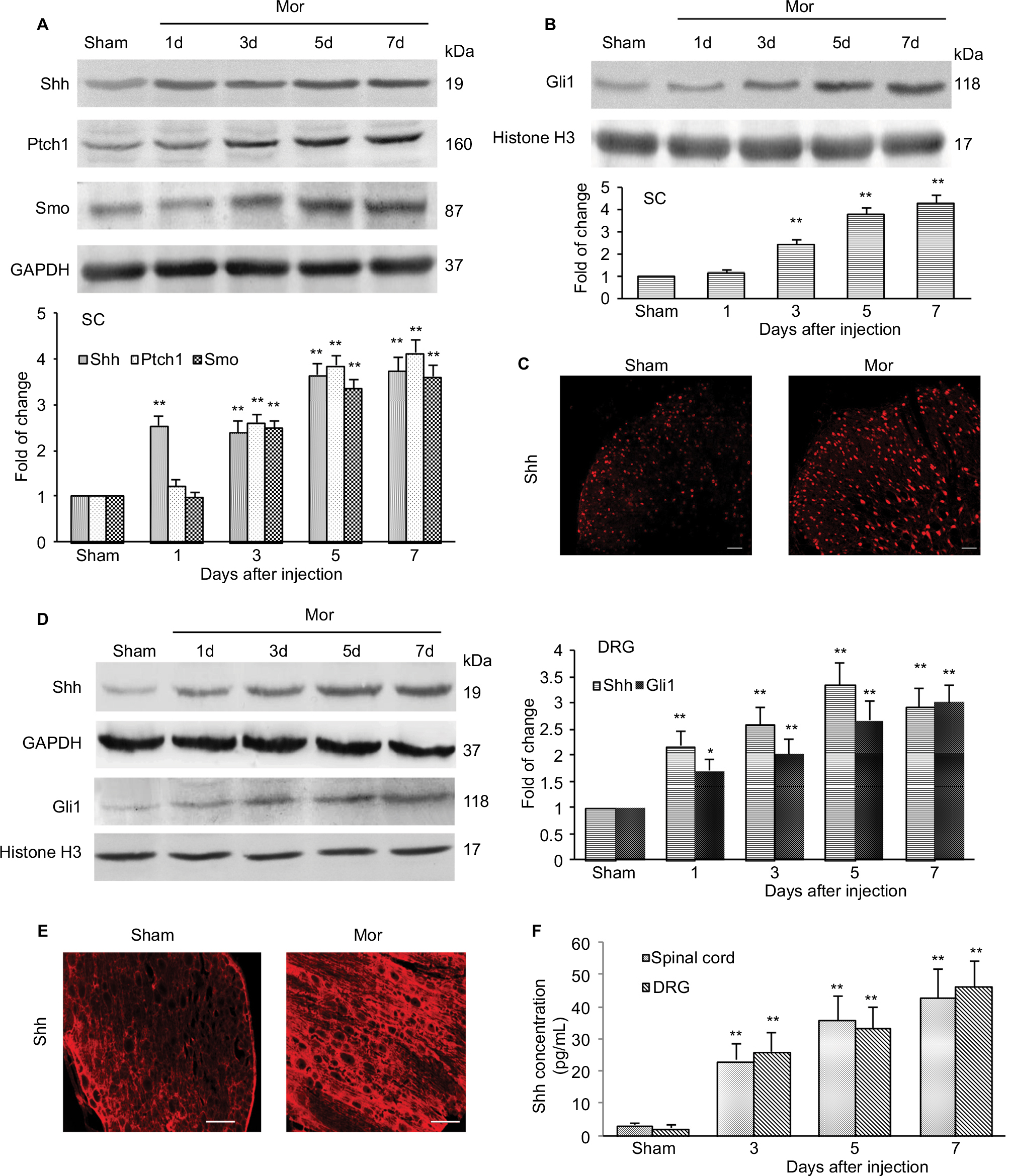 | Figure 4 Expression and activation of Shh signaling after chronic morphine treatment. Notes: (A) Examples of Western blot analysis and data summary (n=6 in each group) showing a time-dependent increased expression of Shh, Ptch1, and Smo in the SC of the mice. (B) Examples of Western blot analysis and data summary (n=6 in each group) showing a time-dependent increased expression of Gli1 in nuclear extract of the SC of the mice. (C) Immunofluorescence staining shows the expression and location of Shh in the dorsal horn of the SC (n=4 in each group) (bar: 50 μm). (D) Examples of Western blot analysis and data summary showing a time-dependent increased expression of Shh and Gli1 in DRG (n=6 in each group). (E) Immunofluorescence staining shows the expression of Shh in DRG (n=6 in each group) (bar: 250 μm). (F) ELISA showing the concentration of Shh in DRG and SC (n=6 in each group). For immunofluorescence staining, samples were collected on day 7 after the first morphine injection. *P<0.05, **P<0.01 versus sham group. Abbreviations: SC, spinal cord; DRG, dorsal root ganglion; i.t., intrathecally; Mor, morphine; ELISA, enzyme linked immunosorbent assay. |
Activation of Shh signaling regulating the expression of BDNF in spinal cord during MIH and tolerance
To explore the mechanisms by which Shh signaling induces MIH and tolerance, we first examined the expression of BDNF, which is reported as the important mediator for MIH and tolerance.26,27 Consistent with previous studies, we found that the expression of BDNF increased significantly after chronic morphine treatment. It increased from day 3 after morphine administration and lasted at least 5 days until final detection (Figure 5A). Repetitive administration of cyclopamine (10 mg/kg, i.p., at days 1, 2, and 3) significantly inhibited morphine-induced upregulation of BDNF (Figure 5B). Similarly, genetic knockdown of Shh with Shh siRNA also significantly inhibited increase in morphine-induced BDNF (Figure 5C). In naïve mice, intraperitoneal injection of SAG (5 mg/kg), Shh signaling activator, caused a rapidly increased (within 1 hours) expression of BDNF protein (Figure 5D), and this effect of SAG on BDNF expression was prevented by cyclopamine pre-administration (Figure 5E). SAG injection can also induce thermal hyperalgesia in naïve mice (Figure 5F). Pretreatment with K252 (80 μg/kg, i.t.), BDNF inhibitor, or anti-BDNF antibody (2 μg/kg, i.t.), significantly prevented and suppressed SAG-induced hyperalgesia (Figure 5F). Besides, repetitive injection of K252 (80 μg/kg, i.t.) also effectively prevented and delayed the generation of tolerance (Figure 5G) and MIH (Figure 5H). Furthermore, to confirm BDNF was under regulated by Shh signaling during morphine tolerance, immunofluorescence double staining was used to investigate the location of BDNF. It was shown that, in the dorsal horn of spinal cord, increased BDNF was colocalized with Shh (Figure 5I, left) and Gli1 (Figure 5I, right). These results confirm that it was indeed possible that BDNF was regulated by Shh signaling. Taken together, these results indicated that the upregulation of BDNF in the spinal cord after chronic morphine administration may be induced by Shh signaling activation and that Shh signaling contributes to MIH and tolerance by regulating BDNF expression.
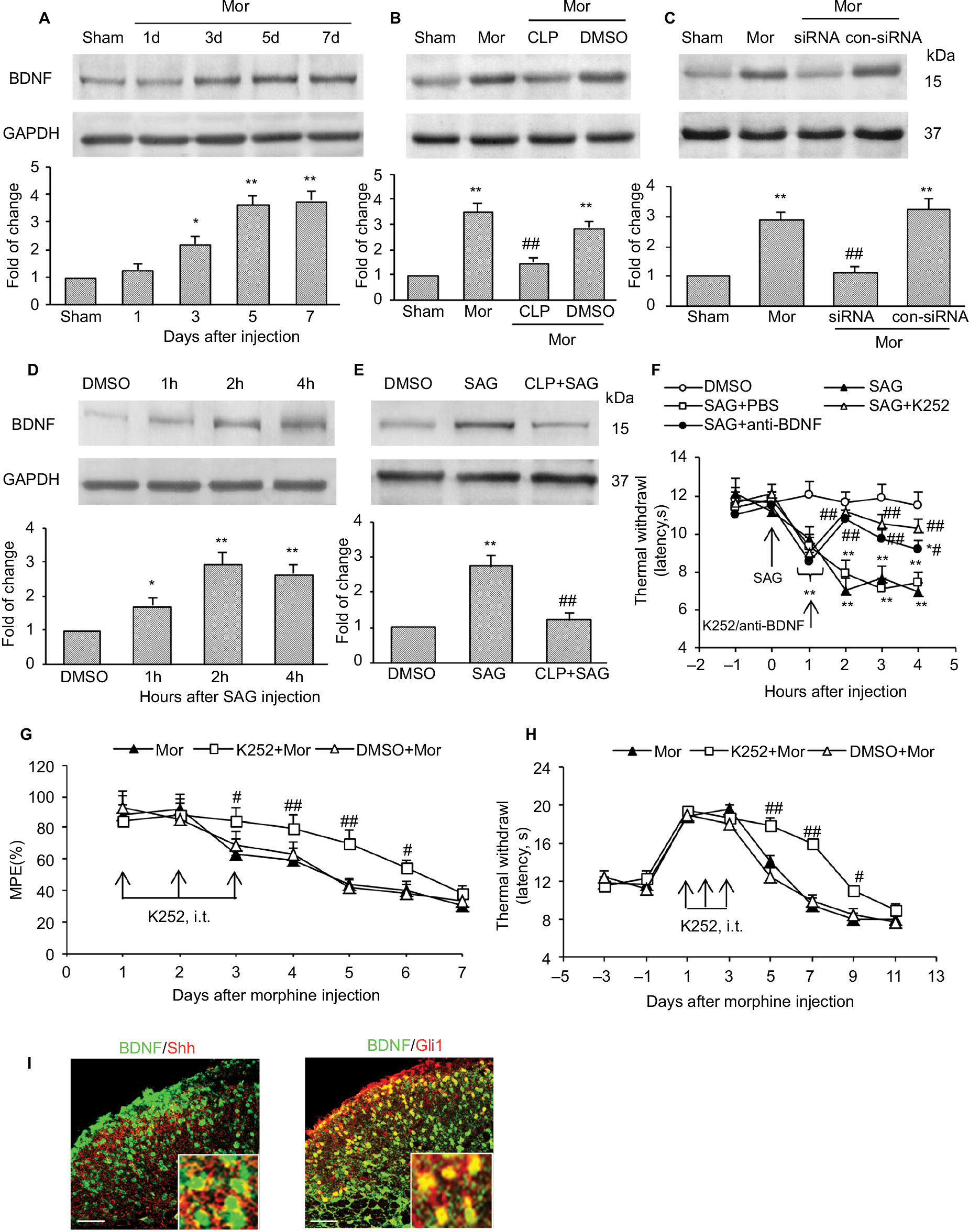 | Figure 5 Shh signaling contributed to tolerance and MIH through regulating BDNF expression. Notes: (A) Expression of BDNF increased in a time-dependent manner after chronic morphine treatment. Inhibiting Shh signaling by cyclopamine (B) and Shh-target siRNA (C) significantly suppressed upregulation of BDNF. Tissues were collected on day 4 after morphine injection (B and C). (D) Activating Shh signaling by SAG injection (i.p.) in naïve mice significantly increased BDNF expression, and pretreatment with cyclopamine effectively prevented the upregulation of BDNF induced by SAG (E). Tissues were collected at 2 hours after SAG injection (D and E). Six mice were included in each group (A–E). BDNF inhibitor K252 and anti-BDNF antibody significantly reversed SAG-induced thermal hyperalgesia in naïve mice (F). BDNF inhibitor K252 effectively suppressed and delayed chronic morphine treatment-induced MPE decrease (G) and thermal hyperalgesia (H). BDNF (green) were colocalized with Shh (I, left) and Gli1 (I, right) in the spinal cord (n=4, bar: 50 μm). Insets are magnified areas. Yellow represents colocalization. Tissues were collected on day 7 after the first morphine injection. Ten mice were included in each group (F–H). *P<0.05, **P<0.01 versus sham group (A–C) or DMSO group (D–F). #P<0.05, ##P<0.01 versus Mor group (B, C, G, and H) or SAG group (E and F). Abbreviations: MIH, morphine-induced hyperalgesia; BDNF, brain-derived neurotrophic factor; siRNA, small interfering RNA; i.p., intraperitoneally; i.t., intrathecally; Mor, morphine; CLP, cyclopamine; DMSO, dimethyl sulfoxide; SAG, smoothened agonist; MPE, maximal possible effect. |
Discussion
In our present study, Shh was identified as an important molecular signal for the regulation of MIH and tolerance. Activation of Shh signaling is involved in the development of MIH and tolerance, and inhibition or downregulation of Shh signaling significantly delayed and alleviated MIH and tolerance. The possible mechanism of Shh on MIH may be mediated by BDNF. To our knowledge, this is the first demonstration of a regulatory role of Shh signaling in MIH and tolerance through BDNF.
MIH and tolerance to morphine is one of the major side effects associated with its long-term administration. This phenomenon limits the beneficial therapeutic use of opioids. Many attempts have been made to find agents that can prevent this adverse effect. Although MIH and tolerance are not fully understood, many potential mechanisms have been identified for this process, including sensitization of the nerve system and synaptic plasticity.21 The withdrawal from opioids, such as morphine, fentanyl, and remifentanyl, induced a potentiation of the synapse between nociceptive C fibers and neurons in the superficial dorsal horn of the spinal cord.28,29
Shh is a secreted protein that controls the patterning of neural progenitor cells, and their neuronal and glial progeny, during development. Emerging findings suggest that Shh also has important roles in the formation and plasticity of neuronal circuits in hippocampus.8 It was reported that Shh is present in both pre- and postsynaptic terminals, where it may be associated with synaptic vesicles and endosomes.14 Besides, activation of Shh signaling can also increase the size of presynaptic terminals at hippocampal neuron synapses.13 The ability of the Shh signaling pathway to regulate neuronal connectivity and synaptic plasticity both during and after the development has been observed in many parts of the nervous system.30–32 Recently, Shh signaling was reported to be required for modulating nociception and analgesia.18,19,33 The expression of Shh significantly increased after sciatic nerve injury and lasted at least 4 weeks,17 which is consistent with pain-related behavioral changes. Inhibiting Shh signaling by cyclopamine effectively attenuated neuropathic pain18 and decreased the concentration of cytokines, such as tumor necrosis factor-α and interleukin-1β.34 In our present study, we also found that, after repetitive administration of morphine, the expression of Shh and its related proteins significantly increased in both spinal cord and DRG in a time-dependent manner. And, the expression and activation of Shh signaling were consistent with MIH and tolerance in the time course. The increased Shh was located not only in laminae I and II of the dorsal horn but also in deeper dorsal horn (laminae VI-V), which receive Aδ nociceptor project. There are two major classes of nociceptive afferents; Aδ fibers and C fibers. Aδ nociceptor projects to lamina I as well as to deeper dorsal horn (lamina VI-V). C nociceptors project more superficially to laminae I and II. Pharmacological and genetic blocking of Shh signaling successfully rescued the analgesic effect of morphine and delayed the generation of MIH and tolerance. Meanwhile, the increased levels of both c-fos and CGRP following repetitive administration of morphine were also inhibited by blocking Shh signaling. These data strongly suggested that Shh signaling is involved in MIH and tolerance. Here we chose to focus on chronic morphine tolerance, rather than acute morphine tolerance, because it resembles more closely clinical practice.
Although the present data indicated that Shh may be involved in the regulation of MIH and tolerance, it will also be important to determine how Shh signaling regulates them. Previous studies performed in the cavernous nerve35 and in the sciatic nerve17 suggest that BDNF could be a target of Shh.36 Activation of Shh pathway upregulates BDNF to protect the cortical neurons against oxidative stress.37 BDNF is a neurotrophic factor. It is implicated in neuronal plasticity and synaptic function.38,39 Recently, BDNF was found to be involved in MIH. Upregulation of BDNF expression by DNA methylation inhibitor augmented MIH.40 In our present study, we found that after repeated morphine administration, the expression of BDNF in spinal cord increased significantly. Pharmacological and genetic blocking of Shh signaling effectively suppressed the increase of BDNF induced by MIH. Furthermore, we also found that in naïve mice, after SAG (Shh signaling activator) administration, the expression of BDNF increased obviously. Besides, the hyperalgesia induced by SAG injection could be significantly suppressed and reversed by BDNF inhibitor and anti-BDNF antibody. Also, immunofluorescence double staining showed that increased BDNF was colocalized with Shh and Gli1 in the dorsal horn of spinal cord. Taken together, these data suggested that BDNF may be downstream of Shh signaling during MIH. Nevertheless, the possible mechanism of how the Shh signaling induced the upregulation of BDNF needs to be further studied. After chronic morphine treatment, Shh signaling was upregulated and activated in DRG and spinal cord. Activated Shh signaling induced nociceptive neuron hyperexcitability and nociceptive inputs increased by upregulating BDNF expression, and then induced morphine tolerance and MIH.
Interestingly, there was an unexpected finding that Shh signaling inhibitor showed no effect on the maintenance of MIH and tolerance. Administration of cyclopamine at days 5, 6, and 7 after morphine injection, when MIH and tolerance had already emerged, showed no effect on MPE% and thermal withdrawal latency decrease. This is an interesting finding. This result demonstrated that the mechanisms of MIH and tolerance generation may be different from those of maintenance, and Shh signaling activation may be the trigger of MIH and tolerance generation.
Conclusion
The present study suggested a new clinical strategy for treating and preventing MIH and tolerance. Shh signaling in spinal cord may play a key role in chronic morphine tolerance and MIH by regulating BDNF expression. Inhibiting Shh signaling, especially during the early phase of MIH and tolerance, may effectively delay or suppress MIH and tolerance.
Acknowledgments
The authors thank Xue-jun Song, MD, PhD, and Zhi-jiang Huang, PhD, (Department of Neurobiology, Parker University, Dallas, TX, 75229, USA) for their help in this study. This work was supported by a grant from the National Natural Science Foundation of China (NSFC-81371242, NSFC-81671084), Qing Lan Project of Jiangsu province, Nature Science Foundation of Jiangsu province (BK20161175), and “Six One” Project of Jiangsu province (LGY2016039).
Author contributions
SL, J-LY, and X-XW designed the studies. J-LY performed the molecular biological test. X-XW and SM performed the animal behavioral tests. Z-JS, YZ, and X-LW performed data collection and statistical analysis. SL and Y-PL oversaw the execution of the project. SL, J-LY, and X-XW interpreted the results and drafting the manuscript. All the authors discussed the results and revised the manuscript critically for important intellectual content.
Disclosure
The authors report no conflicts of interest in this work.
References
Mcquay HJ. Opioids in pain management. Lancet. 1999;353:2229–2232. | ||
Hua Z, Liu L, Shen J, et al. Mesenchymal stem cells reversed morphine tolerance and opioid-induced hyperalgesia. Sci Rep. 2016;6:32096. | ||
Wang J, Xu W, Zhong T, et al. miR-365 targets β-arrestin 2 to reverse morphine tolerance in rats. Sci Rep. 2016;6:38285. | ||
Hayashi Y, Morinaga S, Zhang J, et al. BK channels in microglia are required for morphine-induced hyperalgesia. Nat Commun. 2016;7:11697. | ||
Cheppudira BP, Trevino AV, Petz LN, et al. Anti-nerve growth factor antibody attenuates chronic morphine treatment-induced tolerance in the rat. BMC Anesthesiol. 2016;16:73. | ||
Sanchezblazquez P, Rodriguezmunoz M, Berrocoso E, et al. The plasticity of the association between mu-opioid receptor and glutamate ionotropic receptor N in opioid analgesic tolerance and neuropathic pain. Eur J Pharmacol. 2013;716:94–105. | ||
Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol. 2007;17:556–564. | ||
Yao PJ, Petralia RS, Mattson MP. Sonic hedgehog signaling and hippocampal neuroplasticity. Trends Neurosci. 2016;39:840–850. | ||
Luca AD, Cerrato V, Fucà E, et al. Sonic hedgehog patterning during cerebellar development. Cell Mol Life Sci. 2016;73:291–303. | ||
Ingham PW, Mcmahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. | ||
Ruel L, Rodriguez R, Gallet A, et al. Stability and association of Smoothened, Costal2 and Fused with Cubitus interruptus are regulated by hedgehog. Nat Cell Biol. 2003;5:907–913. | ||
Briscoe J Small S. Morphogen rules: design principles of gradient-mediated embryo patterning. Development. 2015;142:3996–4009. | ||
Mitchell N, Petralia RS, Currier DG, et al. Sonic hedgehog regulates presynaptic terminal size, ultrastructure and function in hippocampal neurons. J Cell Sci. 2012;125:4207–4213. | ||
Petralia RS, Wang Y, Mattson MP, Yao PJ. Sonic hedgehog distribution within mature hippocampal neurons. Commun Integr Biol. 2011;4:775–777. | ||
Amankulor N, Hambardzumyan D, Pyonteck SM, Becher OJ, Joyce JA, Holland EC. Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J Neurosci. 2009;29:10299–10308. | ||
Ding X, Li Y, Liu Z, et al. The sonic hedgehog pathway mediates brain plasticity and subsequent functional recovery after bone marrow stromal cell treatment of stroke in mice. J Cereb Blood Flow Metab. 2013;33:1015–1024. | ||
Hashimoto M, Ishii K, Nakamura Y, et al. Neuroprotective effect of sonic hedgehog up-regulated in Schwann cells following sciatic nerve injury. J Neurochem. 2008;107:918–927. | ||
Babcock DT, Shi S, Jo J, Shaw M, Gutstein HB, Galko MJ. Hedgehog signaling regulates nociceptive sensitization. Curr Biol. 2011;21:1525–1533. | ||
Han L, Ma J, Duan W, et al. Pancreatic stellate cells contribute pancreatic cancer pain via activation of sHH signaling pathway. Oncotarget. 2016;7:18146–18158. | ||
Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci U S A. 1999;96:7731–7736. | ||
Roeckel LA, Le Coz GM, Gavériaux-Ruff C, Simonin F. Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience. 2016;338:160–182. | ||
Yan H, Yu L. Expression of calcitonin gene-related peptide receptor subunits in cultured neurons following morphine treatment. Neurosci Lett. 2013;544:52–55. | ||
Wang Z, Chabot J, Quirion R. On the possible role of ERK, p38 and CaMKII in the regulation of CGRP expression in morphine-tolerant rats. Mol Pain. 2011;7:68. | ||
Hao S, Mata M, Goins WF, Glorioso JC, Fink DJ. Transgene-mediated enkephalin release enhances the effect of morphine and evades tolerance to produce a sustained antiallodynic effect in neuropathic pain. Pain. 2003;102:135–142. | ||
Liu S, Liu W, Liu Y, et al. Blocking EphB1 receptor forward signaling in spinal cord relieves bone cancer pain and rescues analgesic effect of morphine treatment in rodents. Cancer Res. 2011;71:4392–4402. | ||
Wen YR, Tan PH, Cheng JK, Liu YC, Ji RR. Microglia: a promising target for treating neuropathic and postoperative pain, and morphine tolerance. J Formos Med Assoc. 2011;110:487–494. | ||
Hu XM, Cao SB, Zhang HL, et al. Downregulation of miR-219 enhances brain-derived neurotrophic factor production in mouse dorsal root ganglia to mediate morphine analgesic tolerance by upregulating CaMKIIγ. Mol Pain. 2016;12:14670–14683. | ||
Drdla R, Gassner M, Gingl E, Sandkühler J, et al. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210. | ||
Drdla-Schutting R, Benrath J, Wunderbaldinger G, Sandkühler J, et al. Erasure of a spinal memory trace of pain by a brief, high-dose opioid administration. Science. 2012;335:235–238. | ||
Chou Y, Zheng X, Beachy PA, Luo L. Patterning axon targeting of olfactory receptor neurons by coupled hedgehog signaling at two distinct steps. Cell. 2010;142:954–966. | ||
Yam PT, Charron F. Signaling mechanisms of non-conventional axon guidance cues: the Shh, BMP and Wnt morphogens. Curr Opin Neurobiol. 2013;23:965–973. | ||
Yao PJ, Petralia RS, Ott C, Wang YX, Lippincott-Schwartz J, Mattson MP. Dendrosomatic sonic hedgehog signaling in hippocampal neurons regulates axon elongation. J Neurosci. 2015;35:16126–16141. | ||
Moreau N, Mauborgne A, Bourgoin S, et al. Early alterations of Hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation, and neuropathic pain development. Pain. 2016;157:827–839. | ||
Li R, Cai L, Ding J, Hu CM, Wu TN, Hu XY. Inhibition of hedgehog signal pathway by cyclopamine attenuates inflammation and articular cartilage damage in rats with adjuvant-induced arthritis. J Pharm Pharmacol. 2015;67:963–971. | ||
Bond CW, Angeloni N, Harrington DA, Stupp S, Podlasek CA. Sonic hedgehog regulates brain-derived neurotrophic factor in normal and regenerating cavernous nerves. J Sex Med. 2013;10:730–737. | ||
He W, Cui L, Zhang C, Zhang X, He J, Xie Y. Sonic hedgehog promotes neurite outgrowth of primary cortical neurons through up-regulating BDNF expression. Neurochem Res. 2015;41:687–695. | ||
Dai R, Zhu S, Xia Y, et al. Sonic hedgehog protects cortical neurons against oxidative stress. Neurochem Res. 2010;36:67–75. | ||
Antal A, Chaieb L, Moliadze V, et al. Brain-derived neurotrophic factor (BDNF) gene polymorphisms shape cortical plasticity in humans. Brain Stimul. 2010;3:230–237. | ||
Castillo DV, Escobar ML. A role for MAPK and PI-3K signaling pathways in brain-derived neurotrophic factor modification of conditioned taste aversion retention. Behav Brain Res. 2011;217:248–252. | ||
Chao Y, Xie F, Li X, et al. Demethylation regulation of BDNF gene expression in dorsal root ganglion neurons is implicated in opioid-induced pain hypersensitivity in rats. Neurochem Int. 2016;97:91–98. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.