")
Back to Journals » International Journal of General Medicine » Volume 7
Some mechanisms of the protective effect of ischemic preconditioning on rat liver ischemia-reperfusion injury
Authors Adam ANI
Received 24 April 2014
Accepted for publication 1 July 2014
Published 31 October 2014 Volume 2014:7 Pages 483—489
DOI https://doi.org/10.2147/IJGM.S66766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Abdel Nasser Ismail Adam
Physiology Department, Medical Research Institute, Alexandria University, Alexandria, Egypt
Abstract: Ischemia-reperfusion (I/R) injury is a multifactorial process that affects graft function after liver transplantation. An understanding of the mechanisms involved in I/R injury is essential for the design of therapeutic strategies to improve the outcome of liver transplantation. The generation of reactive oxygen species subsequent to reoxygenation inflicts tissue damage and initiates a cascade of deleterious cellular responses, leading to inflammation, cell death, and ultimate organ failure. Increasing experimental evidence has suggested that Kupffer cells and T-cells mediate activation of neutrophil inflammatory responses. Activated neutrophils infiltrate the injured liver in parallel with increased expression of adhesion molecules on endothelial cells. The heme oxygenase system is among the most critical of the cytoprotective mechanisms activated during cellular stress, exerting antioxidant and anti-inflammatory functions, modulating the cell cycle, and maintaining the microcirculation. Finally, the activation of toll-like receptors on Kupffer cells may play a fundamental role in exploring new therapeutic strategies based on the concept that hepatic I/R injury represents a case for host “innate” immunity. In the present study, there was a significant decrease in hepatic activity of glycogen in the I/R group as compared with corresponding values in the control group. On the other hand, there was a significant increase in the hepatic activity of glycogen in the I/R-IP (ischemic preconditioning) group as compared with corresponding values in the I/R group.
Keywords: liver, ischemic preconditioning, reperfusion injury, oxidative stress, nitric oxide, caspase, glycogen
Introduction
Ischemia-reperfusion injury (IRI) is a phenomenon whereby cellular damage in a hypoxic organ is accentuated following the restoration of oxygen delivery. In the liver, this form of injury is recognized as a clinically important prolonged disorder.1
Liver IRI occurs as a result of some liver surgeries, liver transplantation, hemorrhagic shock, and prolonged portal triad clamping followed by reperfusion performed as an elective preplanned procedure or as an emergent maneuver to control excessive bleeding from the cut hepatic surface. Many other medical situations are also encountered. It may also be said that liver IRI is a complex process involving numerous cell types and molecular mediators in various pathophysiological and biochemical ways. The end result is cell death via a combination of apoptosis and necrosis involving a complex web of interactions between the various cellular and humoral contributors to the inflammatory response of Kupffer cells, also producing proinflammatory cytokines, tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, lymphocytes, neutrophils, and hepatocytes.1,2
Our knowledge regarding the mitochondria in generating reactive oxygen species (ROS), nitrogen species, and ionic disturbances, and in initiating mitochondrial permeability transition with subsequent cellular death in IRI is continuously growing. However, the most promising protective strategy against IRI explored during the last few years is ischemic preconditioning (IP), which appears capable of increasing the resistance of liver cells to ischemia and reperfusion events. IP refers to brief periods of ischemia-reperfusion (I/R) followed by a prolonged one.3,4 The aim of the present study was to detect some of these mechanisms by subjecting rats to IRI.
Materials and methods
Thirty male white albino Sprague Dawley rats weighing 200–250 g were used. All rats were treated in accordance with the Guide for Care and Use of Laboratory Animals. The rats were kept at a constant room temperature under air conditioning at 25°C for the duration of the study.
The rats were divided into three groups (n=10 each) as follows: a sham operated control group (group 1); an I/R group in which the liver was rendered ischemic by portal triad occlusion with a small bulldog vascular clamp for 60 minutes followed by reperfusion for 3 hours (group 2); and an IP-I/R group in which animals were subjected to three cycles of 10 minutes of ischemia, each followed by 10 minutes of reperfusion prior to prolonged I/R, as in group 2 (group 3).
At the end of the experiment, the animals were sacrificed by decapitation, and blood and liver samples were collected for analysis. The plasma was separated, and the liver was excised, de-encapsulated, washed with ice-cold saline, and then homogenized in phosphate-buffered saline. The following parameters were measured in plasma and/or liver homogenate:
- liver function tests, ie, alanine aminotransferase (ALT)5 and aspartate aminotransferase (AST)5
- oxidative stress parameters, ie, lipid peroxidation end products, including malondialdehyde (MDA) using thiobarbituric acid,6 reduced glutathione (GSH),7–9 oxidized glutathione (GSSG),7–9 and ratio of nitrite to nitrate in plasma (NOx)10,11
- antioxidant enzyme activity, ie, superoxide dismutase (SOD)12 and glutathione peroxidase (GPx)13
- protein concentration in liver homogenate14
- proinflammatory cytokines, ie, IL-1β and TNF-α, by enzyme-linked immunosorbent assays (Predicta®; Genzyme, San Diego, CA, USA)
- caspase-315
- liver glycogen concentration.16
Statistical analysis
The data are presented as the mean ± standard error and were analyzed using two-way analysis of variance followed by the least significant difference test using Statistical Package for the Social Sciences version 20 software (IBM Corporation, Armonk, NY, USA).
Results
Table 1 shows an increase in serum AST, ALT, plasma IL-1β and TNF-α, and tissue IL-1β and TNF-α in the I/R group as compared with the sham operated control group. However, the IP group showed a decrease in all these parameters compared with the control and I/R groups, but plasma TNF-α was increased compared with the control and I/R groups. Tissue IL-1β was increased when compared with the control group and tissue TNF-α was increased when compared with the control and I/R groups.
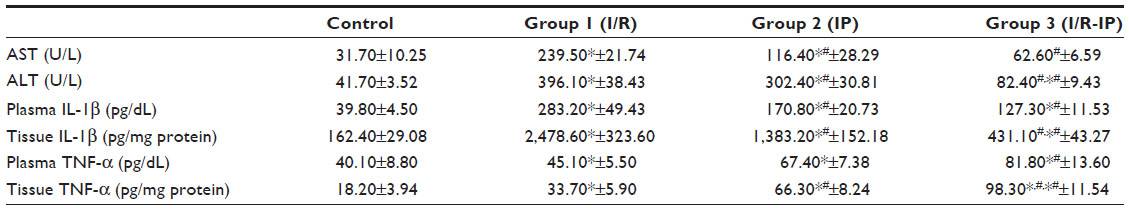 | Table 1 Comparison between study groups according to AST, ALT, plasma IL-1β and TNF-α, and tissue IL-1β and TNF-α levels |
The significant results shown in Table 1 show a decrease in AST in the I/R-IP group when compared with the I/R group. ALT and plasma IL-1β was decreased in the control and I/R groups whereas plasma TNF-α was decreased when compared with the I/R-IP group. Tissue IL-1β was increased significantly when compared with the control and I/R groups. Tissue TNF-α was increased compared with all the other groups, ie, control, T/R, and IP.
Table 2 shows the oxidative stress parameters, including MDA, GSH, GSSG, and NOx. MD and NOx were increased and GSH and GSSG were decreased in the I/R group when compared with controls. MDA was decreased in the IP group when compared with the control group and increased when compared with the I/R group. GSSG was increased in groups 1 and 2, and NOx was decreased compared with the I/R group and increased compared with the control group. In the IP-I/R group, MDA and NOx were decreased when compared with I/R, and GSH was increased as compared with the IP group and decreased when compared with the control group.
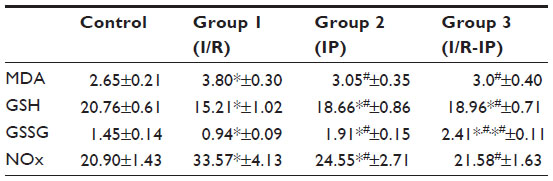 | Table 2 Comparison between study groups according to MDA, GSH, GSSG, and NOx levels |
Table 3 shows that SOD was increased significantly in the IP-I/R group when compared with the control, whereas GPx was decreased, but only GPx increased when compared with the I/R group, in which caspase-3 was decreased. IP (group 2) showed significantly increased caspase-3 compared with the control, but a decrease in GPx, While SOD showed a significant change in the IP group, the I/R group showed a significant decrease in GPx and an increase in caspase-3 compared to the control.
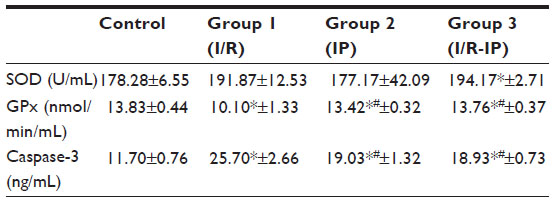 | Table 3 Comparison between study groups according to SOD, GPx, and caspase-3 levels |
Table 4 shows that there was a significant increase in glycogen phosphorylase in the I/R group compared with the control group. There was a significant decrease in glycogen synthase in the I/R group as compared with the control group. Glycogen synthase was increased in the I/R-IP group as compared with the I/R group, as shown in Table 5. There was a significant decrease in glycogen content in the I/R group compared with the control, and glycogen was significantly increased in the IR/Pr group when compared with the I/R group (Table 6).
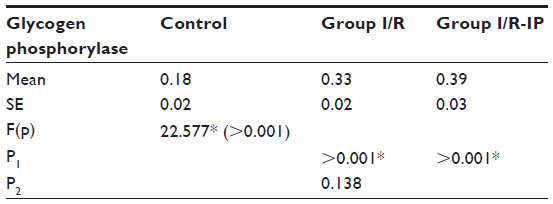 | Table 4 Effect of I/R and I/R-IP on rat liver glycogen phosphorylase level (μmol of inorganic phosphorylase) in study groups |
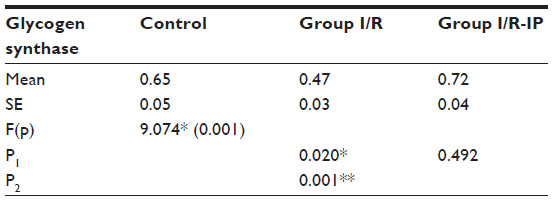 | Table 5 Effect of I/R and I/R-IP on rat liver glycogen synthase level (μM of substrate utilized/mg protein/hour) in study groups |
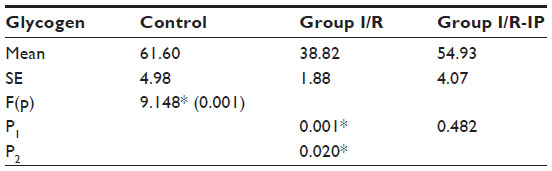 | Table 6 Effect of I/R and I/R-IP on rat liver glycogen level (mg of glycogen/mg of glycogen/100 mg tissue) in study groups |
Discussion
Although the precise mechanisms by which IP reduces IRI are not well understood, several factors have been reported to contribute to IP-mediated tissue protection.17 The protective effects of IP, a phenomenon by which a traumatic or stressful stimulus confers protection against subsequent injury, have been well documented in many organs, including the heart, brain, skeletal muscle, lung, intestine, kidney, retina, and endothelial cells.18
There is increasing evidence that cellular ischemic stressors activate protein kinase via G-protein-coupled receptor binding and membrane phospholipase activation.19 The signal transduction cascade for IP involves activation of protein kinase C, protein tyrosine kinase, and mitogen-activated protein kinase. However, it was found that protein kinase C inhibitors could attenuate the effects of IP induced by one cycle but not repetitive cycles in the heart. These data also suggest that repetitive IP may activate mechanisms other than the antioxidant system. For that reason, we performed three repetitive cycles of IP just before the main I/R in the present study. We also examined its effect on the inflammatory response of I/R as exemplified by IL-1β determination, in addition to its effects on the oxidant-antioxidant system.19–21
Increasing evidence has shown that proinflammatory cytokines and ROS are both key mediators of liver IRI.22 Shortly after hepatic IRI (1–6 hours), Kupffer cells are activated and release proinflammatory cytokines, such as TNF-α and IL-1β. These cytokines have a dual role: overexpression of TNF-α and IL-1β can induce more production of cytokines and granulocyte colony-stimulating factor, which enhances activation of Kupffer cells and promotes infiltration of neutrophils into the microcirculation of the liver,23 thereby aggravating sterile hepatic inflammation after ischemia and reperfusion. On the other hand, TNF-α and IL-1 are indispensable for liver regeneration.24 Measurement of MDA levels is used widely as an indicator of lipid peroxidation. Nitric oxide is also recognized as an important mediator of physiological and pathological processes in hepatic and renal IRI.25
Hepatic I/R can lead to damage and dysfunction (apoptosis) of liver parenchymal and sinusoidal cells. IP is extensively documented to reduce IRI in a variety of organs including the liver,26–28 and the caspase-3 enzyme is used as an indicator of apoptosis. This parameter was included in the present study to determine if IP can actually protect the liver from cell death. In this study, there was a significant increase in the serum activity of AST and ALT in the I/R group when compared with corresponding values in the control group. This result were in agreement with those of Wang et al28 and Jaeschke and Lemasters,29 who found that I/R resulted in a significant increase in AST and ALT levels when compared with controls.
AST and ALT concentrations are commonly used as indirect biochemical indices of liver injury.28,29 Studies have shown that increased AST and ALT levels in IRI probably result from cell membrane damage.30,31 On the other hand, the increase in AST and ALT activity observed in the I/R group by lipid peroxidation leads to cytolysis, which is caused by oxygen free radicals formed during the reperfusion phase.
There was a significant decrease in serum ALT activity in the I/R-IP group when compared with corresponding values in the I/R group. These results are in agreement with those of Romanque et al,32 who found that ALT was strongly decreased in the IP-I/R group, with net reductions of 57% in serum ALT level when compared with the I/R group. At the same time, Kupffer cells and hepatocytes also generate ROS, leading to direct damage of endothelial cells and hepatocytes. Due to their important role, ROS levels are tightly regulated through different pathways.33 The major regulators are ROS scavengers that include SOD, catalase, and glutathione peroxidase (GSH-Px). These ROS scavengers are responsible for reducing ROS inside tissues. Another type of oxidative stress regulation is mediated by nitric oxide (NO), which is created by endothelial and inducible NO synthases in the liver. NO can regulate endothelial function, and its levels can affect blood flow in an organ. Reduction of NO levels is always associated with IRI. ROS have been considered a major deleterious factor in reperfusion injury.34
In the present study, we found that liver I/R depleted glutathione stores in the liver and plasma. GSH is an important intracellular antioxidant that acts by directly scavenging ROS and also by being a cofactor for GPx-catalyzed reactions that degrade hydrogen peroxide.35 Depletion of GSH therefore renders cells susceptible to oxidative stress. In accordance with this, we found significant inhibition of GPx in the liver and plasma, resulting in excessive production of ROS. This results in local oxidative stress on the liver and also a systemic oxidative state as indicated by increased levels of lipid peroxidation end products (MDA) and decreased GSSG.
In this study, there was a significant increase in the hepatic activity of MDA in the I/R group when compared with corresponding values in the control group. This finding is in agreement with that of Wang et al,28 who found that the content of MDA, a marker of liver oxidative injury, was significantly increased in the model group when compared with the control group. The results reported by Yang et al36 show that MDA levels in liver tissue were elevated significantly when treated with vehicle after brain I/R in rats, indicating that the function of the liver, although a remote organ, was damaged. Lipid peroxidation is known to be responsible for cell membrane damage, and has been implicated in the pathogenesis of IRI.22
Accumulation of ROS may easily overcome endogenous antioxidative systems, such as SOD, catalase, GPx, and GSH, and because ROS exist in relatively low concentrations in the liver, they have been proposed to be a contributory factor to cellular mechanisms of inflammation and IRI.1,4 In the present study, it was found that the presented model of hepatic I/R was accompanied by a 254% increase in protein carbonyl content and a 37% decrease in hepatic GSH, with a net 481 enhanced protein carbonyl/GSH content ratio: sham-operated rats =0.26±0.03 (n=23) versus I/R animals =1.51±0.27 (n=6; P>0.0001). This pro-oxidant state was significantly attenuated by the IP maneuver, as evidenced by total recovery of the protein carbonyl content and an 89% net reduction in GSH depletion, when differences between the I/R and sham-operated groups were compared with those between IP-treated rats with and without I/R (P>0.05).
The role of NO in I/R remains controversial. On one hand, NO can induce cellular cytotoxicity and tissue injury via the peroxynitrite formation, protein tyrosine nitration, lipid peroxidation, DNA damage, and proapoptotic effects included in IRI.37 On the other hand, NO may have a protective effect in vasodilatation, antiapoptotic action, inhibition of platelet plug formation, and reduction of the inflammatory response.38 Thus, the cellular effects of NO may depend on its concentration, site of release, and duration of action. The discrepancy in the previously recorded results might be due to different levels of NO production associated with the degree or method of IRI.39 However, there is evidence that IP decreased NO levels significantly after three cycles of I/R.39
In contrast with this finding, group 2 (I/R) showed a slight decrease in this value, which may be attributed to different conditions of induction of I/R. SOD and GPx have been widely studied among the endogenous antioxidants. SOD catalyzes dysmutation of the superoxide anion to hydrogen peroxide and oxygen, but hydrogen peroxide still produces liver oxidative injury, and GPx further catalyzes the transformation of hydrogen peroxide to form water.40 I/R reduces liver antioxidant capacity, as evidenced by the downregulated activity of GPx. However, our study revealed that I/R treatment did not significantly change SOD, implying that I/R with its oxidant metabolites decreased the antioxidant capacity of SOD enzyme.
Previous research also showed that IP increased the expression and activity of antioxidant enzymes in the ischemic kidney and liver,41 which is in accordance with the present study, where IP decreased MDA and GSSG concentrations and avoided depletion of GSH, SOD and GSH-Px in the ischemic kidney. IP also decreased MDA and increased GSH in the heart, with no significant change in the other parameters in rats exposed to renal I/R. Increased GSH and decreased MDA and GSSG in the plasma of renal I/R rats reflects the actual effect of preconditioning.
Our data demonstrate that exposure of the rat liver to an I/P maneuver for 3 hours before the I/R protocol significantly diminishes hepatocellular injury, suggesting the development of a second or delayed phase of protection against IRI. This IP was characterized by development of transient oxidative stress of a considerably smaller magnitude than that elicited by I/R.
The IP mechanisms involved in hepatic IRI as investigated in our study suggest that the protective effects of IP against hepatic IRI are related to their role in reducing tissue oxidative stress levels. IP may regulate the activity of SOD, GSH-Px, NOS, and IP, and offer additive protection by increasing GSH-Px activity. It may also decrease cellular injury and promote cell survival by suppressing release of cytokines. As reported, IP has a similar physiological and cellular protection mechanism: they have the same catalytic substrate adenosine; they transport bioinformation through the phosphatidylinositol 3-kinase/Akt pathway; and they can decrease cytokines release.35 Another contributory factor to parenchymal inflammation in IRI is enhanced TNF-α generation by activated Kupffer cells.42,43 Accordingly, a 15.3-fold increase above that of sham operated rats was observed for serum TNF-α levels in I/R animals in the present study.
Our study also showed a high increase in plasma IL-1β and an increase in the liver of I/R rats was shown in other studies. Our studies also indicated that IP suppressed the TNF-α response in plasma and oxidative stress induced by I/R, as evidenced by normalization of I/R-induced GSH depletion and protein oxidation in the IP-I/R group. We also demonstrated liver protection by IP to be related to recovery of cellular signaling functions modified by the I/R protocol. These findings were found by other researchers to help upregulation of nuclear factor kappa beta and downregulation of activator protein 1 (AP-1) DNA binding, with significant reduction in the AP-1/nuclear factor kappa beta DNA binding ratio, which may afford liver protection by decreasing susceptibility to ROS and TNF-α-induced liver injury.
It was suggested that individual application of IP might reach the limit of decreasing cytokines release in rats. Recent studies have hypothesized a role for TNF-α and IL-1β in liver regeneration.44 Our data confirm that maintenance of TNF-α and IL-1β concentrations at a certain level might help recovery of hepatic function in the liver regeneration process.
Xanthine oxidase is capable of reducing molecular oxygen to both superoxide and hydrogen peroxide, and has been suggested to be the major source of ROS metabolites generated during I/R. Plasma xanthine oxidase activity was increased dramatically after liver and gut I/R.45
It has been shown that during I/R, sinusoidal cells die only via apoptosis while hepatocytes may die in both ways (necrosis and apoptosis). In sinusoidal cells, apoptosis occurs earlier and at a higher level than in hepatocytes, but the differences in extent of apoptosis between these cells tend to be improved after a prolonged reperfusion period.15 The present results also demonstrate improvement in caspase-3 levels by IP.
The protective effects of IP have been shown in various studies. Reduction of IRI has been associated with decreased serum aminotransferase, a finding which has also been confirmed in human models, especially in a younger population,27 and it has been shown that IP protects the liver by inhibiting apoptosis of sinusoidal endothelial cells.
Liver glycogen is the first and immediate source of glucose for the maintenance of blood glucose levels. In the liver, the glucose-6-phosphate generated from degradation of glycogen is hydrolyzed to glucose by glucose 6-phosphatase, an enzyme present only in the liver and kidneys. Glycogen degradation thus provides a readily mobilized source of blood glucose.46 The principal enzymes controlling glycogen metabolism, ie, glycogen phosphorylase and glycogen synthase, are regulated by allosteric mechanisms and covalent modifications due to reversible phosphorylation and dephosphorylation of proteins in response to hormone action.
In the present study, there was a significant decrease in hepatic glycogen activity in the I/R group as compared with corresponding values in the control group. On the other hand, there was a significant increase in hepatic glycogen activity in the I/R-IP group as compared with corresponding values in the I/R group. These results are in agreement with those of Peralta et al,35 who found that the glycogen concentration decreased as a function of duration of ischemia. However, glycogen levels were always higher in preconditioned livers.47 Glycogen concentration decreased as a function of duration of ischemia; however, glycogen levels were always higher in preconditioned livers.46
In summary, the present study shows that IP can protect sinusoidal endothelial cells as well as hepatocytes during liver IRI, and the mechanism partly involves modulation of the imbalance of the endogenous oxidant-antioxidant system in the organism. This may suggest a potential role for antioxidant enzymes in the management of IRI, but further studies are needed regarding their injury preventive effects.
Acknowledgment
Special thanks are due to the late Professor Nayer Mohammad Abourawash, Professor of Biochemistry, Biochemistry Department, Medical Research Institute, Alexandria University, Egypt, for his cooperation during the practical part of the research.
Disclosure
The author reports no conflicts of interest in this work.
References
Glantzounis GK, Salacinski HJ, Yang W, Davidson BR, Seifalian AM. The contemporary role of antioxidant therapy in attenuating liver ischemia-reperfusion injury: a review. Liver Transpl. 2005;11:1031–1047. | |
Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury: a fresh look. Exp Mol Pathol. 2003;74:86–93. | |
Chu MJ, Hickey AJ, Phillips AR, Bartlett AS. The impact of hepatic steatosis on hepatic ischemia-reperfusion injury in experimental studies: a systematic review. Bio Med Res Int. 2013;2013:192029. | |
Bruns H, Watanpour I, Gebhard MM, et al. Glycine and taurine equally prevent fatty livers from Kupffer cell-dependent injury: an in vivo microscopy study. Microcirculation. 2011;18:205–213. | |
Reitman S, Frankel S. A colorimetric method for the determination of SGPT and SGOT. Am J Clin Pathol. 1957;28:56–62. | |
Okhawa H, Ohishi N, Yagi K. Assay of lipid peroxides in normal tissue by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. | |
Ridnour LA, Winters RA, Ercal N, Spitz DR. Measurement of glutathione, glutathione disulfide in mammalian cell and tissue homogenates using high-performance liquid chromatography separation of N-(1-pyrenyl) maleimide derivatives. Methods Enzymol. 1999;299:258–267. | |
Winters RA, Zukowski J, Ercal N, Mathew RH, Spitz DR. Analysis of glutathione, glutathione disulfide, cysteine, homocysteine and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl) maleimide. Anal Biochem. 1995;227:14–21. | |
Leroy P, Nicolas A, Thioudellet C, Oster T, Wellman M, Siest G. Rapid liquid chromatographic assay of glutathione in cultured cells. Biomed Chromatogr. 1993;7:86–89. | |
Miranda KM, Espey MG, Wink DA. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide. 2001;5:62–71. | |
Manikandan R, Beulaja M, Thiagarajan R, Priyadarsini A, Saravanan R, Arumugam M. Ameliorative effects of curcumin against renal injuries mediated by inducible nitric oxide synthase and nuclear factor kappa B during gentamicin-induced toxicity in Wistar rats. Eur J Pharmacol. 2011;30:578–585. | |
Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247:3170–3175. | |
Kraus RJ, Ganther HE. Reaction of cyanide with glutathione peroxidase. Biochem Biophys Res Commun. 1980;16:1116–1122. | |
Shibko S, Koivistoinen P, Tratnyek CA, Newhall AR, Friedman L. A method for sequential quantitative separation and determination of protein, RNA, DNA, lipid, and glycogen from a single rat liver homogenate or from a subcellular fraction. Anal Biochem. 1967;19:514–528. | |
Liu X, Zou H, Slaughter C, Wang X. A heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. | |
Chan TM, Exton JH. A rapid method for the determination of glycogen content and radioactivity in small quantities of tissue or isolated hepatocytes. Anal Biochem. 1976;71:96–105. | |
Joo JD, Kim M, D’Agati VD, Lee HT. Ischemic preconditioning provides both acute and delayed protection against renal ischemia and reperfusion injury in mice. J Am Soc Nephrol. 2006;17:3115–3121. | |
Sahinkanat T, Ozkan KU, Tolun FI, Ciralik H, Imrek SS. The protective effect of ischemic preconditioning on rat testis. Reprod Biol Endocrinol. 2007;5:47. | |
Miura T, Goto M, Sakamoto J, et al. Effect of protein kinase C inhibitors on cardioprotection by ischemic preconditioning depends on the number of preconditioning episodes. Cardiovasc Res. 1998;37:700–709. | |
Yun N, Kim SH, Lee SM. Differential consequences of protein kinase C activation during early and late hepatic ischemic preconditioning. J Physiol Sci. 2012;62:199–209. | |
Yun N, Lee SM. Activation of protein kinase C delta reduces hepatocellular damage in ischemic preconditioned rat liver. J Surg Res. 2013;185:869–876. | |
Zhou G, Dada LA, Wu M, et al. Hypoxia induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1120–L1130. | |
Sínay L, Kürthy M, Horváth S, et al. Ischemic post-conditioning reduces peroxide formation, cytokine expression and leukocyte activation in reperfusion injury after abdominal aortic surgery in rat model. Clin Hemorheol Microcirc. 2008;40:133–142. | |
Jia C. Advances in the regulation of liver regeneration. Expert Rev Gastroenterol Hepatol. 2011;5:105–121. | |
Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–538. | |
Froh M, Zhong ZP, Walbrun P, et al. Dietary glycine blunts liver injury after bile duct ligation in rats. World J Gastroenterol. 2008;14:5996–6003. | |
Banga NR, Homer-Vanniasinkam S, Graham A, Al-Mukhtar A, White SA, Prasad KR. Ischaemic preconditioning in transplantation and major resection of the liver. Br J Surg. 2005;92:528–538. | |
Wang J, Kan Q, Li J, Zhang X, Qi Y. Effect of neferine on liver ischemia-reperfusion injury in rats. Transplant Proc. 2011;43:2536–2539. | |
Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2010;125:1246–1257. | |
Ambros JT, Herrero-Fresneda I, Borau OG, Boira JM. Ischemic preconditioning in solid organ transplantation: from experimental to clinics. Transpl Int. 2007;20:219–229. | |
Yildiz F, Coban S, Terzi A, et al. Nigella sativa relieves the deleterious effects of ischemia reperfusion injury on liver. World J Gastroenterol. 2008;14:5204–5209. | |
Romanque P, Díaz A, Tapia G, Uribe-Echevarría S, Videla LA, Fernandez V. Delayed ischemic preconditioning protects against liver ischemia-reperfusion injury in vivo. Transplant Proc. 2010;42:1569–1575. | |
Kuboki S, Sakai N, Tschöp J, Edwards MJ, Lentsch AB, Caldwell CC. Distinct contributions of CD4+ T cell subsets in hepatic ischemia/reperfusion injury. Am J Physiol. 2009;296:G1054–G1059. | |
Jaeschke H. Mechanisms of liver injury: mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1083–G1088. | |
Peralta C, Bartrons R, Riera L, et al. Hepatic preconditioning preserves energy metabolism during sustained ischemia. Am J Physiol Gastrointest Liver Physiol. 2000;279:G163–G171. | |
Yang XF, He W, Lu WH, Zeng FD. Effects of scutellarin on liver function after brain ischemia/reperfusion in rats. Acta Pharmacol Sin. 2003;24:1118–1124. | |
Aragno M, Cutrin JC, Mastrocola R, et al. Oxidative stress and kidney dysfunction due to ischemia/reperfusion in rat: attenuation by dehydroepiandrosterone. Kidney Int. 2003;64:836–843. | |
Phillips L, Toledo AH, Lopez-Neblina F, Anaya-Prado R, Toledo-Pereyra LH. Nitric oxide mechanism of protection in ischemia and reperfusion injury. J Invest Surg. 2009;22:46–55. | |
Siriussawakul A, Zaky A, Lang JD. Role of nitric oxide in hepatic ischemia-reperfusion injury. World J Gastroenterol. 2010;16:6079–6086. | |
Soncul H, Oz E, Kalaycioglu S. Role of ischemic preconditioning on ischemia-reperfusion injury of the lung. Chest. 1999;115:1672–1677. | |
Vaghasiya JD, Sheth NR, Bhalodia YS, Jivani NP. Exaggerated liver injury induced by renal ischemia reperfusion in diabetes: effect of exenatide. Saudi J Gastroenterol. 2010;16:174–180. | |
Perry BC, Soltys D, Toledo AH, Toledo-Pereyra LH. Tumor necrosis factor-α in liver ischemia/reperfusion injury. J Invest Surg. 2011;24:178–188. | |
Kumar R, Prakash S, Chhabra S, et al. Association of pro-inflammatory cytokines, adipokines and oxidative stress with insulin resistance and non-alcoholic fatty liver disease. Indian J Med Res. 2012;136:229–236. | |
Weigand K, Brost S, Steinebrunner N, Büchler M, Schemmer P, Müller M. Ischemia/reperfusion injury in liver surgery and transplantation: pathophysiology. HPB Surg. 2012;2012:176723. | |
Peglow S, Toledo AH, Anaya-Prado R, Lopez-Neblina F, Toledo-Pereyra LH. Allopurinol and xanthine oxidase inhibition in liver ischemia reperfusion. J Hepatobiliary Pancreat Sci. 2011;18:137–146. | |
Skurat AV, Roach PJ. Regulation of glycogen synthesis. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus: A Fundamental and Clinical Text. 2nd ed. New York, NY, USA: Lippincott, Williams & Wilkins; 2000. | |
Khandoga A, Kessler JS, Hanschen M, et al. Matrix metalloproteinase-9 promotes neutrophil and T cell recruitment and migration in the post ischemic liver. J Leukoc Biol. 2006;79:1295–1305. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.