Back to Journals » Journal of Pain Research » Volume 19
SMR Analysis Integrating GWAS and eQTL Data Reveals UHRF1BP1 and SNRPC as Potential Drug Targets for Low Back Pain
Authors Xie XY, Lin JW, Li PC, Fang JY, Chen ZC, Huang XY, Cao MH, Lin LL ![]() , Guo MY
, Guo MY
Received 2 January 2026
Accepted for publication 30 June 2026
Published 10 July 2026 Volume 2026:19 593304
DOI https://doi.org/10.2147/JPR.S593304
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Alaa Abd-Elsayed
Xiao-Yan Xie,1,2,* Jian-Wei Lin,3,* Peng-Cheng Li,1,* Jia-Yu Fang,1 Zhuo-Chen Chen,1 Xiao-Yan Huang,1 Ming-Hui Cao,1,2 Li-Ling Lin,1 Ming-Yan Guo1,2
1Department of Anesthesiology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, People’s Republic of China; 2Shenshan Medical Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Shanwei, 516621, People’s Republic of China; 3Big Data Laboratory, Joint Shantou International Eye Center of Shantou University and the Chinese University of Hong Kong, Shantou, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Li-Ling Lin, Department of Anesthesiology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, No. 107 Yan Jiang West Road, Guangzhou, 510120, People’s Republic of China, Email [email protected] Ming-Yan Guo, Department of Anesthesiology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, No. 107 Yan Jiang West Road, Guangzhou, 510120, People’s Republic of China, Email [email protected]
Purpose: Low back pain (LBP) is a leading cause of disability worldwide with limited effective pharmacotherapies. We aimed to identify novel therapeutic candidate targets for LBP through integrative genomics.
Methods: We employed summary-data-based Mendelian randomization (SMR) with GWAS data from FinnGen (13,178 cases/164,682 controls) and tissue-specific expression quantitative trait loci (eQTLs) from peripheral blood (Westra cohort: n = 5,311; 15,636 genes) and brain tissue (UKBEC: n = 134; 16,309 genes). Heterogeneity in dependent instruments (HEIDI) analysis validated causal associations. Candidate targets were further assessed by pathway enrichment, drug prediction, and phenome-wide association studies (PheWAS).
Results: Peripheral blood eQTLs identified four genes associated with LBP (PSMR < 3.2× 10−, HEIDI P > 0.05): BTN2A3P, GFPT1, UHRF1BP1, SNRPC; brain eQTLs identified four genes associated with LBP (PSMR < 3.07× 10−, HEIDI P > 0.05): CHST3, DCC, UHRF1BP1, SNRPC. Cross-tissue integration prioritized UHRF1BP1 and SNRPC as consensus candidates. Drug prediction suggested levamisole and taxifolin as potential UHRF1BP1-modulating compounds. PheWAS indicated low pleiotropic risk, with associations mainly with hypertension and celiac disease.
Conclusion: This multi-omics framework prioritizes UHRF1BP1 (involved in epigenetic regulation) and SNRPC (RNA splicing modulator) as mechanistically novel, genetically supported candidate targets for LBP, providing a foundation for future experimental validation and therapeutic development.
Keywords: LBP, therapeutic targets, SMR, HEIDI, UHRF1BP1, SNRPC
Background
As a common clinical complaint, low back pain (LBP) can be clinically classified into three main types: overt injury pain (eg., myofascial LBP), neuropathic pain (eg., lumbar radiculopathy), and centrally sensitized pain (nonspecific LBP),1,2 each potentially involving distinct molecular pathways and thus requiring different management strategies. The World Health Organization’s Global Burden of Disease Study shows that LBP has become the leading cause of disability in disability-adjusted life years (DALYs) in 126 countries,3 with a one-month prevalence rate of 23.3%,4 lifetime prevalence is as high as 47%,5 resulting in a loss of social productivity that exceeds that of most chronic diseases. Notably, the development of chronic LBP involves a multidimensional interaction of biomechanical abnormalities, altered neuroplasticity, and psychosocial factors,6 a complexity that often makes it difficult to achieve sustained relief with existing treatment options.
Current clinical guidelines recommend a stepwise therapeutic strategy: non-steroidal anti-inflammatory drugs (NSAIDs) are preferred for mild-to-moderate pain in the acute phase, and weak opioids can be used in combination with NSAIDs for moderate-to-severe pain; for chronic skeletal and musculoskeletal pain, central neuromodulators such as duloxetine can inhibit pain signaling by modulating 5-hydroxytryptamine/norepinephrine reuptake.7–9 However, the limitations of insufficient response to conventional medications in partial patients, the potential for gastrointestinal damage10 and cardiovascular disease risk11 with long-term use of NSAIDs, the risk of addiction with long-term opioid use highlight the urgent need for the development of novel targeted therapeutic regimens.
The traditional drug discovery model relies on phenotypic screening and randomized controlled trials, which is characterized by low target discovery efficiency and long development cycle. Genomics-based integration strategy provides a new direction to break through this dilemma: by integrating genome-wide association studies (GWAS) and expression quantitative trait loci (eQTL) data, the causal chain between genetic variation, gene expression and disease phenotype can be systematically analyzed.12,13 Mendelian randomization (MR) uses genetic variants as instrumental variables to infer causality, reducing bias from confounding and reverse causation.14,15 The crucial step in MR is selecting instrumental variables that satisfy three assumptions: (i) the instrumental variable should be closely related to the exposure of interest; (ii) the instrumental variable should be independent of the outcome of interest; (iii) the instrumental variable should be independent of all confounding factors related to the relationship between exposure and outcome.
Among MR approaches, summary-data-based Mendelian randomization (SMR) is particularly effective in detecting gene–trait associations, as it integrates GWAS and eQTL summary data and incorporates a heterogeneity in dependent instruments (HEIDI) test to distinguish pleiotropy from linkage.16
Therefore, inspired by the successful experience of SMR in Parkinson’s disease,17–19 Alzheimer’s disease,20–22 as well as in other pain conditions such as migraine,23,24 we applied cross-tissue SMR (MR based on summarized data) integrating GWAS with blood and brain eQTL data, combined with pathway, PheWAS, and pharmacogenomic resources, to identify and prioritize druggable gene targets for LBP.
Methods
Study Design
Figure 1 illustrates the analytical framework of this study based on cross-omics Mendelian randomization, and the specific process includes the following key steps:
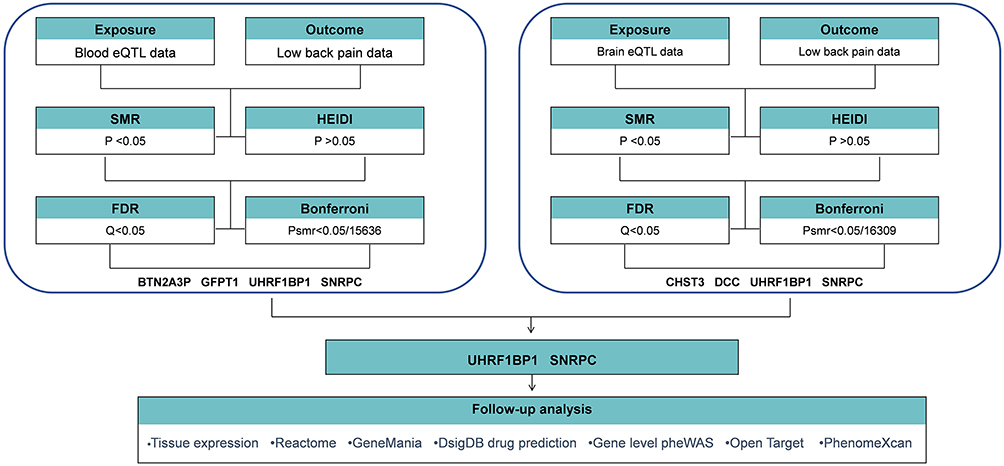 |
Figure 1 Overview of the study design. Abbreviations: eQTL, expression quantitative trait loci; SMR, Summary-data-based Mendelian randomization; HEIDI, heterogeneity in dependent instruments; FDR, False Discovery Rates. |
(i) multi-tissue eQTL data integration: the peripheral blood eQTL dataset25 constructed by Westra et al (sample size = 5,311) and the brain tissue eQTL26 atlas of the United Kingdom Brain Expression Consortium (UKBEC) (covering 10 brain regions) were selected as the sources of instrumental variables, which systematically covered the peripheral and central gene regulatory networks related to pain signaling.
(ii) causal effect inference: based on the summarized GWAS data for LBP released by the FinnGen Consortium (version R11), gene-phenotype association analysis was performed using the Mendelian randomization method based on the summarized data, and multiple tests were rigorously corrected using the Bonferroni correction and FDR (False Discovery Rates) correction.
(iii) verification of genetic homogeneity: in order to exclude the interference of pleiotropy on the results, gene loci with a single causal pathway with the target phenotypes were screened by heterogeneity in dependent instruments (HEIDI) analysis (HEIDI P > 0.05) to ensure the genetic association specificity of the candidate target genes.
(iv) molecular pathway mining: based on the Reactome and GeneMania platforms, the pathways and interactions of the screened target genes will be analyzed.
(v) clinical translational assessment: Phenome-wide association scanning (PheWAS) was performed on validated targets to assess potential off-target effects, and pharmacological matching analysis was carried out based on the Drug Signatures Database (DsigDB) to predict repositionable drugs and their mechanisms of action, and finally, based on the Open targets and PhenomeXcan platforms, a further comprehensive assessment of the association of the target genes for LBP with other human traits or diseases.
Exposure Data
Table S1 details the multi-tissue eQTL data sources used in this study and their quality control processes:
Peripheral Blood eQTL Dataset
Based on the cross-cohort meta-analysis published by Westra’s team (5,311 whole blood samples from healthy individuals were included), a total of 19,725 protein-coding genes were examined for their expression profiles using a unified RNA sequencing process and a standardized genotyping platform. The screening criteria in this study included (i) cis-acting eQTL (cis-eQTL, defined within ±1 Mb from the gene transcription start site); (ii) genome-wide significant association signal (P < 5×10−8); (iii) independent locus screening (r2 < 0.001 by PLINK clumping algorithm, window size 10 Mb). A final set of 15,636 gene probes meeting the above criteria was included. The raw data are publicly available through the European Bioinformatics Institute (EBI) database (accession number: EGAS00001000910).
Brain Tissue eQTL Dataset
Raw data derives from the UK Brain Expression Consortium (UKBEC) multi-brain region exome study, covering 134 individuals of European descent without a history of neurological disease (postmortem autopsy samples) and covering 10 pain-related brain regions such as prefrontal cortex and hippocampus. Gene expression quantification was performed using Illumina HumanHT-12v4 microarrays, and confounders such as age, gender and postmortem interval were corrected by mixed linear modeling. Strict screening conditions included (i) cis-acting eQTL (defined as above); (ii) genome-wide significance threshold (P < 5×10−8); (iii) probe-specific filtering (removal of cross-hybridization risk probes). 16,309 high-confidence probes were retained after QC, and data access is available on the consortium website (http://www.ukbec.org).
Outcome Data
Low back pain
The outcome dataset is from FinnGen version 11 and is available at https://r11.finngen.fi. LBP was defined using FinnGen endpoint codes M13_LOWBACKPAIN (inclusion of ICD-10 M54.4-M54.5 and corresponding ICD-8/9 codes). In this dataset, the total number of LBP was 13,178, while 164,682 cases were categorized as healthy.
Statistical Analysis
SMR Analysis and HEIDI Testing
In contrast to most other methods used for comprehensive analysis of GWAS and eQTL data, the SMR and HEIDI methods were able to distinguish between multiple and chained models.16 SMR analysis was performed using SMR software as a means of confirming causal relationships between LBP and gene expression.27 Analyses were performed using SMR v1.3.1 with default parameters, using the European 1000 Genomes reference panel for LD clumping. R v4.2.0 was used for secondary analyses.
The HEIDI test was used to demonstrate that proteins associated with LBP were not caused by genetic linkage. A significant SMR association was defined as P < 0.05, whereas a HEIDI P > 0.05 indicated that the association was caused by shared genetic variation. Sensitivity analyses were then performed using both Bonferroni correction and FDR correction (Q-value) for significance thresholds. Peripheral blood P < 3.2×10−6 and brain P < 3.07×10−6 was considered significant in the Bonferroni correction. Q < 0.05 was considered significant in the FDR correction. The study was conducted according to current guidelines.
Tissue Expression Analysis
Human Protein Atlas (www.proteinatlas.org) was used to examine mRNA and protein expression across 44 normal human tissues cover 16,975 genes.28 Exploring the expression of target genes in different tissues and different brain regions could suggest possible mechanisms for their use as therapeutic targets for LBP.
Pathway Analysis
Reactome (https://reactome.org) and GeneMANIA29 (https://genemania.org) were used for functional enrichment and network analysis.
Drug Prediction
DSigDB30(v1.0, http://dsigdb.tanlab.org) via Enrichr31 (https://maayanlab.cloud/modEnrichr/) was used to identify potential drug candidates.
PheWAS
AstraZeneca PheWAS Portal32(https://azphewas.com) and PheWeb were used to assess pleiotropic effects and potential off-target risks.33
Cross-Trait Analysis
Open Targets (https://www.targetvalidation.org) and PhenomeXcan34 were used to evaluate associations with other human traits and diseases.
Results
SMR and HEIDI Test for the Association Between Peripheral Blood Gene Expression and LBP
Using FDR correction (Q < 0.05), SMR analysis of peripheral blood eQTL data identified numerous genes significantly associated with LBP risk (Figure 2A and Table S2). Under the more stringent Bonferroni correction (PSMR < 3.2×10−6), only four genes remained significant: BTN2A3P (OR (95% CI) = 1.733 (1.425–2.109); P = 2.70×10−8), GFPT1 (OR (95% CI) = 1.094 (1.052–1.138); P = 1.34×10−7), UHRF1BP1 (OR (95% CI)= 1.051 (1.031–1.072); P = 6.56×10−7), SNRPC (OR (95% CI)= 0.869 (0.820–0.922); P = 8.01×10−7). All four passed HEIDI test (P > 0.05), supporting a causal role independent of linkage (Figure 2C and Table S2).
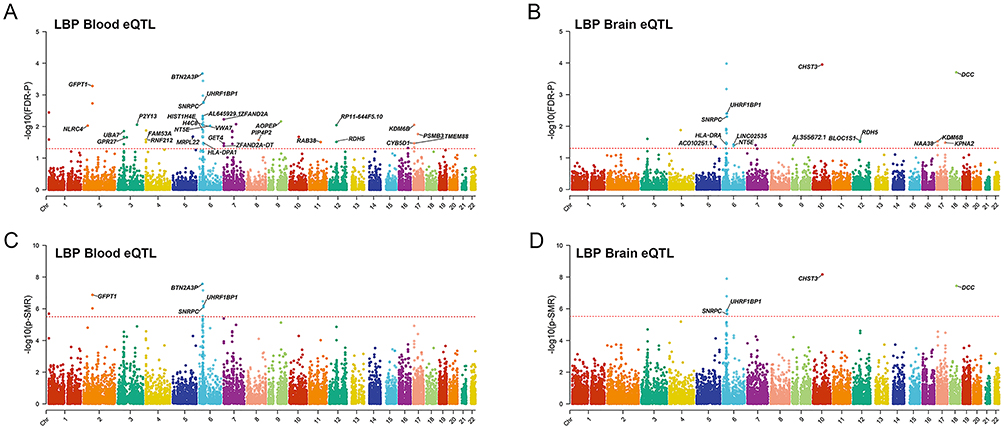 |
Figure 2 Manhattan plot for correlation of target genes with LBP in SMR and HEIDI test The y-axis shows –log10(P) or –log10(Q) values from SMR tests. The red horizontal line indicates the genome‑wide significance threshold for each panel. (A) Causal associations with LBP identified in peripheral blood eQTLs using FDR correction (FDR-P =0.05). (B) Causal associations with LBP identified in peripheral blood eQTLs using Bonferroni correction (PSMR = 3.2×10−6 = 0.05/15,636). (C) Causal associations with LBP identified in brain eQTLs using FDR correction (FDR-P = 0.05). (D) Causal associations with LBP identified in brain eQTLs using Bonferroni correction (PSMR =3.07×10−6 = 0.05/16,309). Genes passing the respective threshold are labeled. |
SMR and HEIDI Test for the Association Between Brain Gene Expression and LBP
Similarly, in brain eQTL data, FDR correction identified a large set of genes (Figure 2B). After Bonferroni correction (PSMR < 3.07×10−6), only four genes remained significant: CHST3 (OR (95% CI) = 0.923 (0.905–0.941); P = 6.87×10−9), DCC (OR (95% CI) = 1.174 (1.106–1.245); P = 3.63×10−8), UHRF1BP1(OR (95% CI) = 1.051 (1.031–1.072); P = 1.21×10−6), SNRPC (OR (95% CI) = 0.942 (0.923–0.960); P = 2. 13×10−6). All four passed HEIDI test (P > 0.05) (Figure 2D and Table S3).
UHRF1BP1 and SNRPC are the Concordant Genes Across Peripheral Blood and Brain eQTL Analyses
Notably, when comparing the Bonferroni-significant genes from the two tissues, UHRF1BP1 and SNRPC were the only genes that appeared in both lists (Figure 3C). Although other genes (eg., BTN2A3P, GFPT1 in blood; CHST3, DCC in brain) passed Bonferroni correction in a single tissue, their effects were tissue-specific. In contrast, UHRF1BP1 and SNRPC showed consistent causal associations with LBP in both peripheral blood and brain, and they also passed the stringent Bonferroni correction in each tissue. This cross-tissue consistency strongly suggests that these two genes are the most robustly prioritized candidate targets. The two genes that were highly concordant between the SMR results in peripheral blood eQTL data (Figure 3A) and brain eQTL data (Figure 3B) were UHRF1BP1 and SNRPC. This indicates the existence of a single potentially causal variant with pleiotropic effects on the three “phenotypes”: gene expression levels in blood, expression levels in brain, and LBP risk.
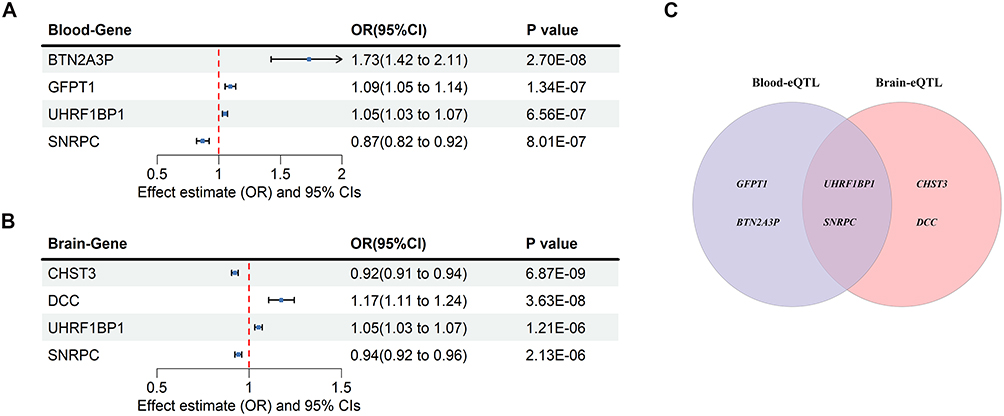 |
Figure 3 The causal relationship between peripheral blood genes and brain genes with the risk of LBP (A) The forest plot shows significant causal associations between four peripheral blood genes and the risk of LBP; (B) The forest plot shows significant causal associations between four brain genes and the risk of LBP; The effect estimates represent the odds ratio per SD increase of target genes on LBP, and the error bars represent 95% CIs. A P value of <0.05 was considered significant; (C)Venn diagram of target genes from two datasets. |
Taken together, these findings highlight the clinical relevance of UHRF1BP1 and SNRPC as genetically supported candidate targets for LBP, warranting further investigation in therapeutic development.
Expression of UHRF1BP1 and SNRPC in Different Tissues
The differences in the expression of UHRF1BP1 and SNRPC in human tissues and brain regions by Human Protein Atlas were determined. The results showed that UHRF1BP1 was mainly expressed in skeletal muscle, bone marrow and cerebellum, cerebral cortex, and choroid plexus (Figure 4A and B). SNRPC was mainly expressed in cardiac muscle, bone marrow, skeletal muscle and spinal cord, white matter, and choroid plexus (Figure 4C and D). This may provide an explanation for the target tissues and pathways of UHRF1BP1 and SNRPC.
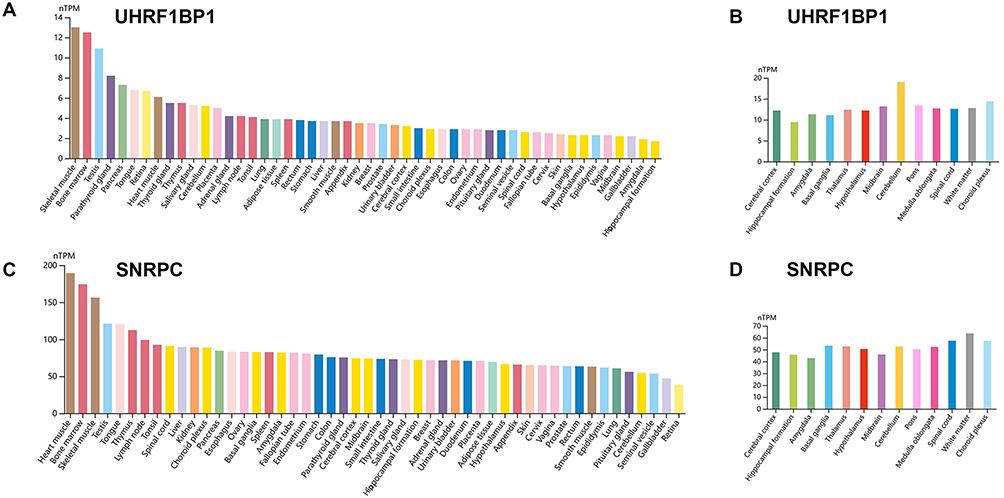 |
Figure 4 The expression of UHRF1BP1 and SNRPC in human tissues and brain regions (A) Expression of UHRF1BP1 in human tissues. (B) Expression of UHRF1BP1 in brain regions. (C) Expression of SNRPC in human tissues. (D) Expression of SNRPC in brain regions. |
Reactome
Figure 5 shows the results of Reactome pathway analysis, indicating that the up-regulated gene UHRF1BP1 is involved in the CREB3 signaling pathway and pathways such as stress response and stimulus response. The expression of the down-regulated gene SNRPC was mainly enriched through the apoptotic signaling pathway by activation of cysteinyl asparaginase, and the detailed data is shown in Table S4.
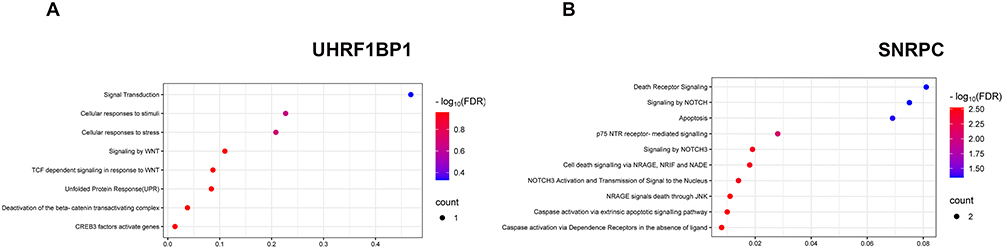 |
Figure 5 Pathways involved in the alteration of UHRF1BP1 and SNRPC for LBP based on Reactome database (A) Pathways associated with UHRF1BP1 (upregulated in relation to LBP risk). (B) Pathways associated with SNRPC (downregulated in relation to LBP risk). The x‑axis represents the gene ratio (or enrichment fraction), and the y‑axis lists the pathways. Color intensity indicates –log10(FDR), and dot size represents the number of genes enriched in each pathway. |
GeneMANIA
The network processing data tool GeneMANIA was used to identify new genes that may co-express or physically interact with our targets. The inner circle in Figure 6 includes target genes that we have entered into the engine, while the outer circle indicates new targets that show co-expression or physical interaction with our targets. This analysis was able to identify the most important biological pathways (all FDR P values < 0.05), with the top-ranked pathway being involvement in the small nuclear ribonucleoprotein complex. SNRPA, SNRNP70 were identified in the biological pathway analysis as target genes interacting with target genes related to LBP (eg. SNRPC). With this strategy, SNRPA, SNRNP70 might also be hypothesized to serve as a candidate target for LBP treatment.
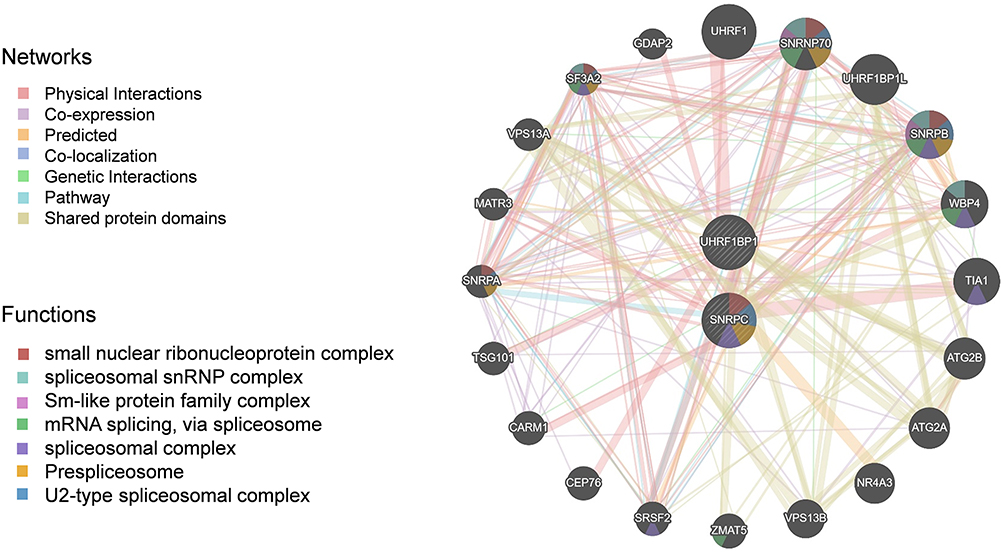 |
Figure 6 Pathways and genetic interactions mainly related to the UHRF1BP1 and SNRPC based on GeneMANIA database. |
DsigDB
Evaluation of UHRF1BP1 and SNRPC as possible drug targets required assessment of protein-drug interactions. To find possible drugs, UHRF1BP1 and SNRPC were examined using the DSigDB drug database on Enrichr. Based on the results, the top four drugs associated with UHRF1BP1 were levamisole (CTD00006205), dihydroquercetin (CTD00000115), nickel (CTD00006389), and hexestrol (CTD 00005818) (Table 1). The first four drugs associated with SNRPC queried in the DsigDB database all downregulate SNRPC expression, which increases the risk of developing LBP, and therefore these four drugs were not considered.
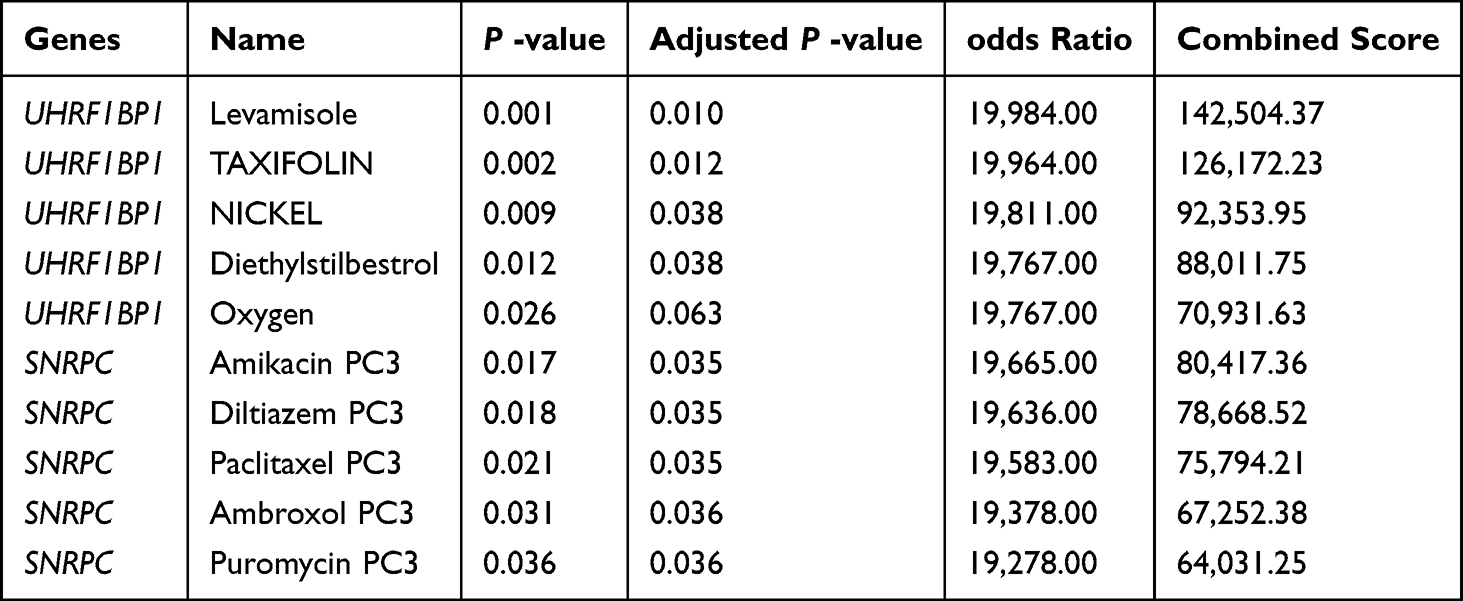 |
Table 1 Candidate Drug Predicted Using DSigDB |
PheWAS
By Using the PheWAS Portal and PheWeb databases, phenotypic group-wide MR was performed to identify possible side effects of targeting UHRF1BP1 and SNRPC. The results revealed evidence of significant associations between UHRF1BP1, SNRPC and other phenotypes, ie., a strong association with hypertension, celiac disease, in either the PheWeb database or the PheWAS portal at the genome-wide significance level (P < 5×10−8) (Figure 7 and Table S5). These results reinforce the comprehensiveness of our findings, suggesting that if targeting UHRF1BP1, SNRPC is therapeutic, there is a low risk of adverse drug reactions or unexpected horizontal pleiotropic effects, but with attention to hypertension and celiac disease.
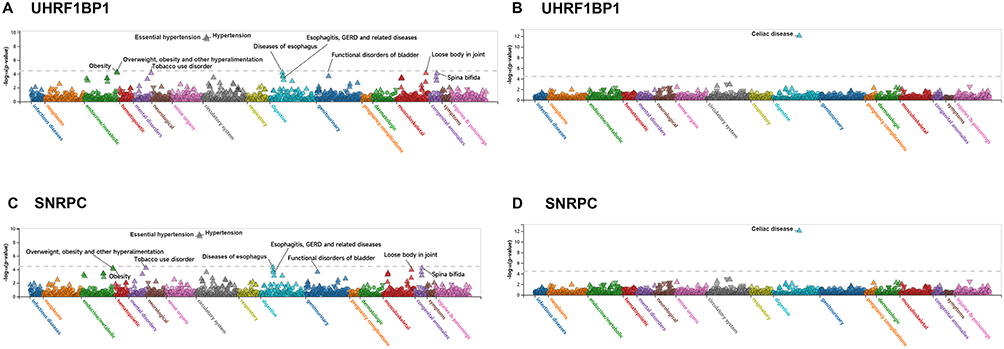 |
Figure 7 PheWAS analysis of off‑target risk for UHRF1BP1 and SNRPC based on the PheWeb database (A) Strong association between UHRF1BP1 and hypertension. (B) Strong association between UHRF1BP1 and celiac disease. (C) Strong association between SNRPC and hypertension. (D) Strong association between SNRPC and celiac disease. The dashed horizontal line indicates the genome‑wide significance threshold (P = 5×10−8). |
Open Targets
Because many target genes are associated with other phenotypes, we aimed to determine in a more comprehensive manner which disease characteristics are affected by these exposures. A systematic analysis of potential diseases or phenotypes associated with target genes in the Open Target Platform was performed. Table 2 shows the phenotypes closely associated with UHRF1BP1 (also known as BLTP3A) and SNRPC, in which globalScore is the total association score and otGeneticsPortal (Open Target Genetics Portal) is the genetic association score. Phenotypes strongly associated with UHRF1BP1 were height, HDL cholesterol measurements, and body mass index. Phenotypes closely associated with SNRPC were LDL cholesterol assay, body mass index and platelet index. More phenotypes associated with UHRF1BP1 and SNRPC is detailed in Table S6.
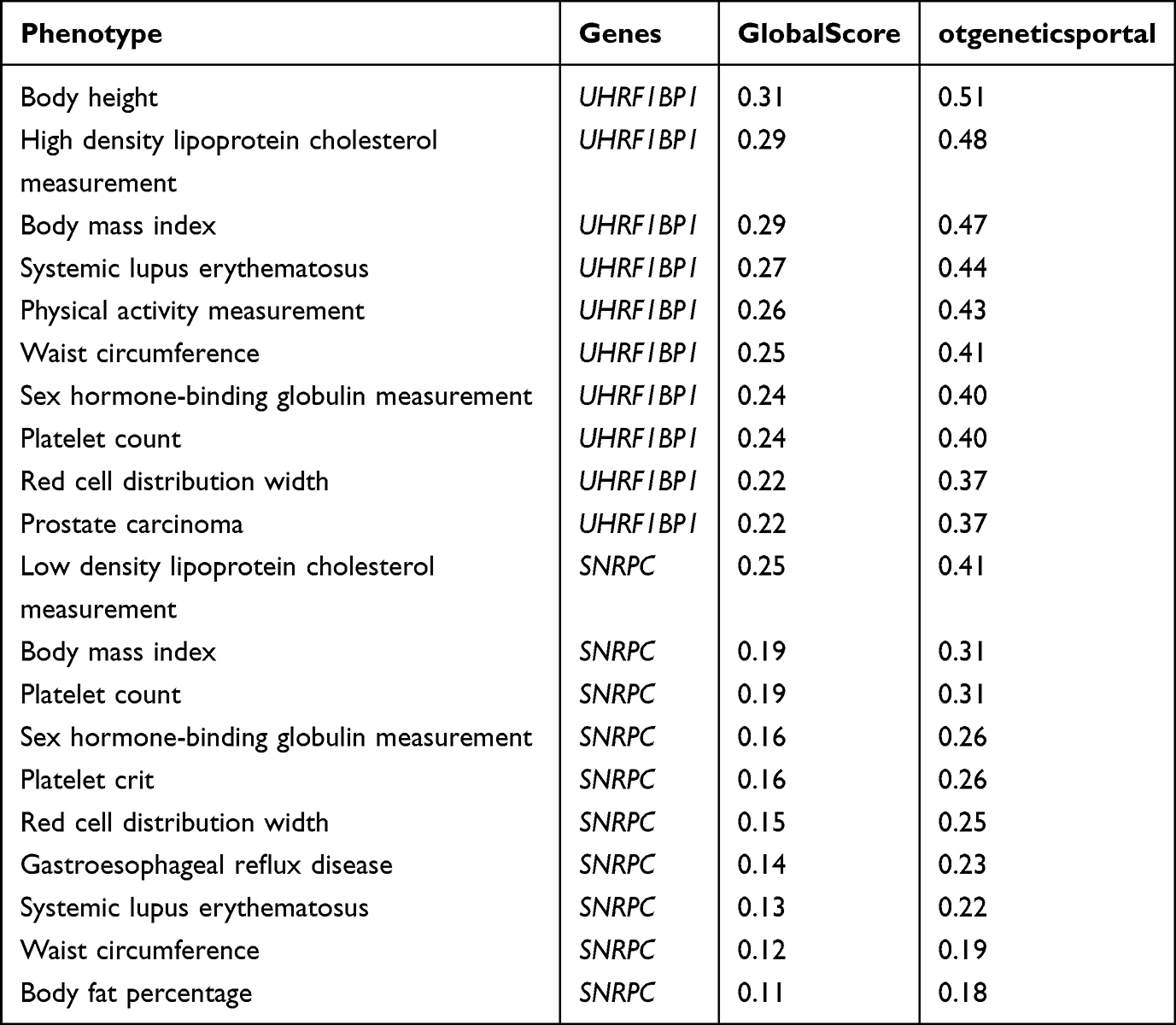 |
Table 2 Phenotypes Closely Related to UHRF1BP1 and SNRPC Based on Open Targets |
PhenomeXcan
In order to more comprehensively assess the association of LBP target genes with other phenotypes, PhenomeXcan was also applied to further analyze the unearthed target genes. Among them, RCP > 0.1 was defined as the causative gene. Table 3 shows that UHRF1BP1 is a risk gene for 18 phenotypes, of which the most closely related phenotypes are trunk fat mass, body fat percentage and waist circumference, and the detailed associations are shown in Table S7.
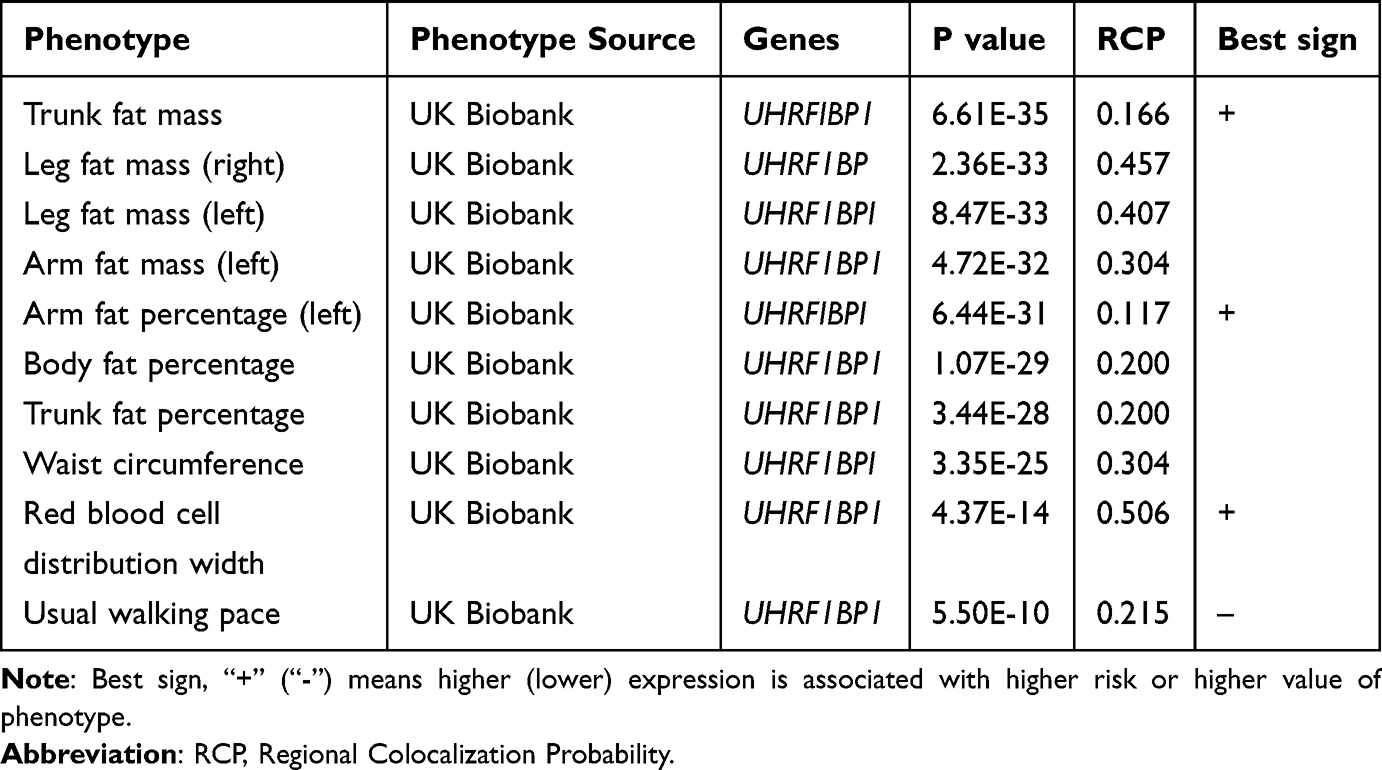 |
Table 3 Phenotypes Closely Related to UHRF1BP1 Based on PhenomeXcan |
Discussion
As a global public health challenge, LBP is causing increasing physical and mental health damage and socioeconomic burden.35 We identified UHRF1BP1 and SNRPC as genetically supported, mechanistically plausible candidate targets for LBP, through SMR-HEIDI integration of GWAS and multi-tissue eQTL data. Notably, the cross-tissue convergence of these two genes represents a more robust prioritization strategy than single-tissue or locus-based approaches.
The pathogenesis of LBP involves multi-system interactions, including local inflammatory cascades, dorsal root ganglion sensitization, and central nervous system remodeling. Among them, intervertebral disc degeneration (IVDD), as a key pathological basis,36 has a central mechanism focusing on the imbalance of cellular homeostasis in the nucleus pulposus.37 As the biomechanical core of the intervertebral disc, the nucleus pulposus (NP) has an extracellular matrix (ECM) composed mainly of type II collagen and proteoglycan. Human studies have shown that apoptosis in the nucleus pulposus not only leads to a decrease in ECM synthesis, but also activates neuroimmune interactions through the release of damage-associated molecular patterns (DAMPs),38 a finding that echoes Yang et al’s observation that apoptosis inhibition ameliorates intervertebral disc degeneration in a rat model.39 Therefore, targeted modulation of nucleus pulposus cell survival and inflammatory microenvironment may become an important strategy for IVDD intervention.
SNRPC encodes a core component of the U1 small nuclear ribonucleoprotein (snRNP) complex, which plays a fundamental role in pre-mRNA splicing and, more recently, has been implicated in neuronal gene regulation. Although direct evidence linking U1 snRNP or its core components to neuropathic pain or central sensitization is currently lacking, converging lines of evidence support the plausibility of such a connection. First, U1 snRNP dysfunction has been shown to cause RNA splicing defects that lead to neuronal hyperexcitability in neurodegenerative models,40 establishing a precedent that splicing dysregulation can directly affect neuronal function. Second, the U1 snRNP core protein SNRNP70 (U1-70K) has been detected in axonal RNA-associated granules, suggesting that snRNP components can function locally in neuronal processes to regulate the axonal transcriptome and influence neural connectivity.41 Third, alternative splicing regulation has been directly implicated in neuropathic pain pathogenesis through other splicing factors,42 highlighting that the splicing machinery as a whole is relevant to pain processing. Given the high expression of SNRPC in the spinal cord and white matter observed in this study, we hypothesize that SNRPC may influence pain signaling in LBP through alternative splicing of genes involved in neuronal excitability, neuroinflammation, or apoptotic cascades—a hypothesis that requires experimental validation in future studies.
UHRF1BP1 (also known as BLTP3A) encodes a bridge-like lipid transfer protein involved in lipid transport and metabolism regulation.43 Emerging evidence has established a strong link between lipid metabolism dysregulation and intervertebral disc degeneration (IDD), the primary pathological basis of LBP. Case-control and Mendelian randomization studies have demonstrated that dyslipidemia—particularly elevated triglycerides and apolipoprotein ratios—is significantly associated with increased risk of IDD and LBP.44,45 Mechanistically, oxidized low-density lipoprotein (ox-LDL) drives endplate chondrocyte senescence and calcification via the ROS/P38-MAPK/NF-κB axis,46 while excessive palmitic acid accumulation in nucleus pulposus cells induces endoplasmic reticulum stress-dependent senescence.47 These findings directly link lipid dysregulation to key pathological processes in the degenerating disc, supporting the hypothesis that UHRF1BP1 may contribute to LBP through its role in lipid metabolism. Notably, this study found that UHRF1BP1 is highly expressed in the choroid plexus and cerebral cortex, suggesting potential involvement in central pain processing. Mechanistically, UHRF1BP1 binds to UHRF1, which has been shown to recruit histone deacetylase 1 (HDAC1) via its SRA domain to mediate histone deacetylation,48 providing a molecular basis for UHRF1BP1 to potentially influence epigenetic regulation through the UHRF1-HDAC axis. HDACs have been well-established as key regulators of central sensitization and neuropathic pain: spinal HDAC upregulation induces nociceptive sensitization by inhibiting the GABAergic system;49 HDAC6 inhibition alleviates neuropathic pain by modulating spinal STAT3 acetylation and downstream chemokine release;50 and HDACs in the spinal cord and dorsal root ganglia regulate neuronal excitability and neuroinflammation through histone and non-histone deacetylation.51 Given the abundant HDAC evidence in CNS pain modulation, together with the cerebral expression of UHRF1BP1 and its indirect link to HDACs, we hypothesize that UHRF1BP1 may also participate in LBP through HDAC-mediated epigenetic regulation of central sensitization pathways. Together, we propose a dual-mechanism hypothesis: UHRF1BP1 may influence LBP pathogenesis through peripheral lipid-mediated disc degeneration and, simultaneously, through central HDAC-related epigenetic modulation of pain sensitization—both of which require direct functional validation in future studies.
Notably, PheWAS analysis showed that UHRF1BP1/SNRPC had a low risk of pleiotropy, and the DsigDB database suggested that natural compounds such as TAXIFOLIN might exert neuroprotective effects by modulating UHRF1BP1, which provided a basis for lead compound selection for translational medicine research. Multi-tissue eQTL integration analysis based on the SMR framework further strengthened the biological rationality of target selection: the strong expression signal of SNRPC in the spinal cord was highly consistent with its potential function of regulating peripheral sensitization, while the specific expression of UHRF1BP1 in the choroid plexus might be involved in the process of central sensitization by influencing the cerebrospinal fluid metabolism.
Several limitations should be acknowledged. First, the GWAS data are from European populations only, limiting generalizability. Second, the brain eQTL sample size (n=134) is modest, which may affect statistical power. Third, our analyses focus on transcript levels; proteomic and single-cell data are needed to resolve cell-type specific mechanisms. Fourth, the mechanistic interpretations for SNRPC and UHRF1BP1 are hypotheses generated from expression patterns and literature, not direct experimental evidence. Finally, LBP is clinically heterogeneous (radicular vs. mechanical), and stratified analyses were not possible.
Several translational challenges should be noted. First, the modest effect size (OR ≈ 1.05) implies that targeting a single gene may have limited impact, supporting the need for combination approaches—including psychological,6 physical, and muscle-strengthening interventions52—as important adjuncts to pharmacotherapy. Second, the safety associations with hypertension and celiac disease, although not strong, warrant monitoring in future trials. Third, genetic findings reflect lifelong effects, whereas drugs act over shorter periods; dose-effect and duration studies are needed.
In conclusion, this integrative genomics study prioritizes UHRF1BP1 and SNRPC as genetically supported candidate therapeutic targets for LBP. These findings are hypothesis-generating and require experimental validation in cellular and animal models, as well as dose-effect studies to bridge the gap between genetic association and clinical translation.
Data Sharing Statement
All data generated or analyzed during this study are included in this article and its Supplementary Materials. Additional materials can be obtained from the corresponding author (Liling Lin, [email protected]) upon reasonable request.
Institutional Review Board Statement
This study used only publicly available summary-level data from GWAS (FinnGen), eQTL (Westra et al, UKBEC) and other public resources. No individual-level human data were collected, and no direct interaction with human subjects occurred. According to Article 32, items 1 and 2 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects (jointly issued by the National Health Commission, the Ministry of Science and Technology, and other Chinese government authorities on February 18, 2023), research that uses lawfully obtained public data without involving personal privacy or causing harm to human subjects may be exempt from institutional ethical review. Therefore, this study was exempt from additional approval by our Institutional Review Board (IRB). The original GWAS and eQTL studies from which the summary data were derived had obtained their own ethical approvals and informed consent from.
Acknowledgments
Xiao-Yan Xie, Jian-Wei Lin and Peng-Cheng Li are co-first authors in this study. We would like to thank the FinnGen database for the GWAS summary data of LBP.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Guangdong Basic and Applied Basic Research Foundation (No.2023A1515012939) and supported by the “Three million for Three Years” Project of the Academic Backbone of Shenshan Medical Center, Sun Yat-Sen Memorial Hospital (No.1320900002).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Nijs J, Apeldoorn A, Hallegraeff H, et al. Low back pain: guidelines for the clinical classification of predominant neuropathic, nociceptive, or central sensitization pain. Pain Physician. 2015;18(3):E333–13.
2. Knezevic NN, Candido KD, Vlaeyen JWS, Van Zundert J, Cohen SP. Low back pain. Lancet. 2021;398(10294):78–92. doi:10.1016/S0140-6736(21)00733-9
3. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1789–1858. doi:10.1016/S0140-6736(18)32279-7
4. Hoy D, Bain C, Williams G, et al. A systematic review of the global prevalence of low back pain. Arthritis Rheum. 2012;64(6):2028–2037. doi:10.1002/art.34347
5. Morris LD, Daniels KJ, Ganguli B, Louw QA. An update on the prevalence of low back pain in Africa: a systematic review and meta-analyses. BMC Musculoskelet Disord. 2018;19(1):196. doi:10.1186/s12891-018-2075-x
6. Canli İ, Özüdoğru A. Biopsychosocial factors affecting disability in individuals with chronic nonspecific low back pain: a cross-sectional study. J Pain Res. 2026;19:590191. doi:10.2147/JPR.S590191
7. Chou R, Deyo R, Friedly J, et al. Systemic pharmacologic therapies for low back pain: a systematic review for an American college of physicians clinical practice guideline. Ann Intern Med. 2017;166(7):480–492. doi:10.7326/M16-2458
8. Bernstein IA, Malik Q, Carville S, Ward S. Low back pain and sciatica: summary of NICE guidance. BMJ. 2017;
9. Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478–491.
10. Hernández-Díaz S, Rodríguez LA. Association between nonsteroidal anti-inflammatory drugs and upper gastrointestinal tract bleeding/perforation: an overview of epidemiologic studies published in the 1990s. Arch Intern Med. 2000;160(14):2093–2099.
11. Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332(7553):1302–1308.
12. Namba S, Konuma T, Wu K-H, Zhou W, Okada Y. A practical guideline of genomics-driven drug discovery in the era of global biobank meta-analysis. Cell Genom. 2022;2(10):100190. doi:10.1016/j.xgen.2022.100190
13. Kreitmaier P, Katsoula G, Zeggini E. Insights from multi-omics integration in complex disease primary tissues. Trends Genet. 2023;39(1):46–58. doi:10.1016/j.tig.2022.08.005
14. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–R98. doi:10.1093/hmg/ddu328
15. Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: a review. Res Synth Methods. 2019;10(4):486–496. doi:10.1002/jrsm.1346
16. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi:10.1038/ng.3538
17. Storm CS, Kia DA, Almramhi MM, et al. Finding genetically-supported drug targets for Parkinson’s disease using Mendelian randomization of the druggable genome. Nat Commun. 2021;12(1):7342. doi:10.1038/s41467-021-26280-1
18. Lyu Q, Chen R, Qiu Z, Wang C, Liu R. Druggable targets for Parkinson’s disease: transcriptomics based Mendelian randomization study. Sci Rep. 2024;14(1):25763. doi:10.1038/s41598-024-77401-x
19. Li X, Zhang L, Xia J, Zheng M, Zhou Z, Cai J. Multi-omics Mendelian randomization and machine learning identify candidate therapeutic targets for Alzheimer’s and Parkinson’s diseases. Mamm Genome. 2026;37(1):24. doi:10.1007/s00335-025-10191-3
20. Ou Y-N, Yang Y-X, Deng Y-T, et al. Identification of novel drug targets for Alzheimer’s disease by integrating genetics and proteomes from brain and blood. Mol Psychiatry. 2021;26(10):6065–6073. doi:10.1038/s41380-021-01251-6
21. Zhai Y, Li N, Zhang Y, et al. Identification of JAZF1, KNOP1, and PLEKHA1 as causally associated genes and drug targets for Alzheimer’s disease: a summary data-based Mendelian randomization study. Inflammopharmacology. 2024;32(6):3913–3923. doi:10.1007/s10787-024-01583-z
22. Gao J, Bi X, Jiang W, Wang Y. Integration of multi-omics quantitative trait loci evidence reveals novel susceptibility genes for Alzheimer’s disease. Sci Rep. 2025;15(1):30158. doi:10.1038/s41598-025-12290-2
23. Xiong Z, Zhao L, Mei Y, et al. Proteome-wide Mendelian randomization identified potential drug targets for migraine. J Headache Pain. 2024;25(1):148. doi:10.1186/s10194-024-01853-9
24. Gao R, Wang R, Xiong Z. Causal proteomic insights into drug target discovery for tension-type headache. J Headache Pain. 2025;27(1):7. doi:10.1186/s10194-025-02235-5
25. Westra H-J, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238–1243. doi:10.1038/ng.2756
26. Ramasamy A, Trabzuni D, Guelfi S, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17(10):1418–1428. doi:10.1038/nn.3801
27. Wu Y, Zeng J, Zhang F, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun. 2018;9(1):918. doi:10.1038/s41467-018-03371-0
28. Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi:10.1126/science.1260419
29. Franz M, Rodriguez H, Lopes C, et al. GeneMANIA update 2018. Nucleic Acids Res. 2018;46(W1):W60–W64. doi:10.1093/nar/gky311
30. Yoo M, Shin J, Kim J, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31(18):3069–3071. doi:10.1093/bioinformatics/btv313
31. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi:10.1093/nar/gkw377
32. Gagliano Taliun SA, VandeHaar P, Boughton AP, et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat Genet. 2020;52(6):550–552. doi:10.1038/s41588-020-0622-5
33. Wang Q, Dhindsa RS, Carss K, et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature. 2021;597(7877):527–532. doi:10.1038/s41586-021-03855-y
34. Pividori M, Rajagopal PS, Barbeira A, et al. PhenomeXcan: mapping the genome to the phenome through the transcriptome. Sci Adv. 2020;6(37). doi:10.1126/sciadv.aba2083
35. Katz JN. Lumbar disc disorders and low-back pain: socioeconomic factors and consequences. J Bone Joint Surg Am. 2006;88(2):21–24.
36. Vergroesen PPA, Kingma I, Emanuel KS, et al. Mechanics and biology in intervertebral disc degeneration: a vicious circle. Osteoarthritis Cartilage. 2015;23(7):1057–1070. doi:10.1016/j.joca.2015.03.028
37. Wang J, Markova D, Anderson DG, Zheng Z, Shapiro IM, Risbud MV. TNF-α and IL-1β promote a disintegrin-like and metalloprotease with thrombospondin type I motif-5-mediated aggrecan degradation through syndecan-4 in intervertebral disc. J Biol Chem. 2011;286(46):39738–39749. doi:10.1074/jbc.M111.264549
38. Hunter CJ, Matyas JR, Duncan NA. Cytomorphology of notochordal and chondrocytic cells from the nucleus pulposus: a species comparison. J Anat. 2004;205(5):357–362.
39. Yang Y-H, Gu X-P, Hu H, et al. Ginsenoside Rg1 inhibits nucleus pulposus cell apoptosis, inflammation and extracellular matrix degradation via the YAP1/TAZ pathway in rats with intervertebral disc degeneration. J Orthop Surg Res. 2022;17(1):555. doi:10.1186/s13018-022-03443-4
40. Chen P-C, Han X, Shaw TI, et al. Alzheimer’s disease-associated U1 snRNP splicing dysfunction causes neuronal hyperexcitability and cognitive impairment. Nat Aging. 2022;2(10):923–940. doi:10.1038/s43587-022-00290-0
41. Nikolaou N, Gordon PM, Hamid F, et al. Cytoplasmic pool of U1 spliceosome protein SNRNP70 shapes the axonal transcriptome and regulates motor connectivity. Curr Biol. 2022;32(23). doi:10.1016/j.cub.2022.10.048
42. He L, Guo H, Wang H, et al. Rbfox1 regulates alternative splicing of Nrcam in primary sensory neurons to mediate peripheral nerve injury-induced neuropathic pain. Neurotherapeutics. 2023;21(1):e00309. doi:10.1016/j.neurot.2023.e00309
43. McEwan DG, Ryan KM. ATG2 and VPS13 proteins: molecular highways transporting lipids to drive membrane expansion and organelle communication. FEBS J. 2022;289(22):7113–7127. doi:10.1111/febs.16280
44. Tan B, Xiang S, Zheng Y, Ouyang J, Zhou N. Association of dyslipidemia with intervertebral disc degeneration: a case-control study. Eur J Med Res. 2025;30(1):194. doi:10.1186/s40001-025-02455-0
45. Pang Z-Y, Zhu Y-B, Hu J-X, et al. The impact of lipidome on intervertebral disk degeneration, low back pain, and sciatica: a Mendelian randomization study. Sci Rep. 2025;15(1):18045. doi:10.1038/s41598-025-99914-9
46. Bing T, Shanlin X, Jisheng W, et al. Dysregulated lipid metabolism and intervertebral disc degeneration: the important role of ox-LDL/LOX-1 in endplate chondrocyte senescence and calcification. Mol Med. 2024;30(1):117. doi:10.1186/s10020-024-00887-8
47. Chen X, Chen K, Hu J, et al. Palmitic acid induces lipid droplet accumulation and senescence in nucleus pulposus cells via ER-stress pathway. Commun Biol. 2024;7(1):539. doi:10.1038/s42003-024-06248-9
48. Unoki M, Nishidate T, Nakamura Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. 2004;23(46):7601–7610.
49. Wen Z-H, Chen N-F, Cheng H-J, et al. Upregulated spinal histone deacetylases induce nociceptive sensitization by inhibiting the GABA system in chronic constriction injury-induced neuropathy in rats. Pain Rep. 2024;9(6):e1209. doi:10.1097/PR9.0000000000001209
50. Chi Z, Lu B, Liu R, et al. Inhibition of histone deacetylase 6 alleviates neuropathic pain via direct regulating post-translation of spinal STAT3 and decreasing downstream C-C Motif Chemokine Ligand 7 synthesis. J Neuroinflammation. 2025;22(1):74. doi:10.1186/s12974-025-03400-y
51. Zhang W, Jiao B, Yu S, et al. Histone deacetylase as emerging pharmacological therapeutic target for neuropathic pain: from epigenetic to selective drugs. CNS Neurosci Ther. 2024;30(5):e14745. doi:10.1111/cns.14745
52. Sönmez E, Tekin A, Oğuzhanasiltürk DÜ, et al. The effect of paraspinal muscle morphology on the development of osteoporotic lumbar vertebral fractures. J Back Musculoskelet Rehabil. 2025;38(6):1468–1476. doi:10.1177/10538127251340350
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.