Back to Journals » Drug Design, Development and Therapy » Volume 13
Small-Molecule Intervention At The Dimerization Interface Of Survivin By Novel Rigidized Scaffolds
Authors Ibrahim TM ![]() , Ernst C, Lange A
, Ernst C, Lange A ![]() , Hennig S, Boeckler FM
, Hennig S, Boeckler FM ![]()
Received 24 July 2019
Accepted for publication 12 November 2019
Published 18 December 2019 Volume 2019:13 Pages 4247—4263
DOI https://doi.org/10.2147/DDDT.S224561
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Manfred Ogris
Tamer M Ibrahim,1,2 Christoph Ernst,1 Andreas Lange,1 Susanne Hennig,1 Frank M Boeckler1
1Laboratory for Molecular Design and Pharmaceutical Biophysics, Department of Pharmaceutical and Medicinal Chemistry, Institute of Pharmacy, Eberhard Karls University Tübingen, Tübingen, Germany; 2Pharmaceutical Chemistry Department, Faculty of Pharmacy, Kafrelsheikh University, Kafr El-Sheikh, Egypt
Correspondence: Tamer M Ibrahim
Pharmaceutical Chemistry Department, Faculty of Pharmacy, Kafrelsheikh University, El-Geish Street, Kafr El-Sheikh 33516, Egypt
Email [email protected]
Introduction: Survivin is a nodal protein involved in several cellular pathways. It is a member of the IAP family and an integral component of the chromosomal passenger complex, where it binds to borealin and INCENP through its dimerization interface. By targeting survivin with a small molecule at its dimerization interface, inhibition of the proliferation of cancer cells has been suggested. With Abbott 8, a small-molecule dimerization inhibitor has been recently reported. The structure–activity relationship of this series of inhibitors implied that the middle pyridin-2(1H)-one ring did not tolerate modifications of any kind.
Methods: Based on the synthetic strategy of Abbott 8 using multicomponent reactions, we synthesized a series of small molecules bearing a novel rigidized core scaffold. This rigidization strategy was accomplished by integrating the pyridin-2(1H)-one and its 6-phenyl substituent into a tricyclic structure, linking position 5 of pyridin-2(1H)-one to the phenyl substituent by rings of different sizes. The new scaffolds were designed based on in silico molecular dynamics of survivin.
Results: Binding of these rigidized scaffolds to the recombinant L54M mutant of survivin was evaluated, revealing affinities in the low micromolar range.
Conclusion: This easily accessible, new class of survivin-dimerization modulators is an interesting starting point for further lead optimization.
Keywords: pyridin-2(1H)-one derivatives, one-pot synthesis, molecular dynamics and design, survivin-dimerization modulators
Introduction
Survivin is a nodal protein involved in several cellular pathways. By targeting survivin, it has been suggested that multiple tumor-signaling pathways may be simultaneously disabled.1 Survivin is a member of the IAP family and highly overexpressed in many common types of human cancer,1–7 but its levels are low in normal tissue and undetectable in nondividing cells.8 Survivin inhibits apoptosis by binding to caspases through its baculovirus IAP repeat (BIR) domain, which is strongly attributable to mediating therapeutic resistance in cancer cells.9–12
In addition, survivin plays an important role in modulating the formation of the mitotic spindle, which modulates accurate chromosome segregation during mitosis.13,14 Survivin is an integral component of the chromosomal passenger complex (CPC), where the survivin monomer binds to borealin and INCENP in a way that mimics its dimerization to form a survivin homodimer through its dimerization interface.15,16 Survivin is required for localization of the CPC to the centromere.15,17 The CPC ensures the proper localization of Aurora B kinase for successful phosphorylation of potential targets for mitotic progression.16 Therefore, by targeting survivin with a small molecule at its dimerization interface, inhibition of the proliferation of the cancer cells is expected.18
The survivin homodimer has two distinct binding hot spots: the conventional BIR domain and the dimerization interface. However, only the dimerization interface has been reported to accommodate small-molecule binders (eg, Abbott 8),16,19 as well as an allosteric site in the near proximity of the dimerization interface.20 Abbott 8 is the prototype of pyridone-based small molecules that binds at the dimerization interface of survivin with a KD value of 75 µM (Figure 1A).19 Further and extensive modifications on rings A and C have been proposed by Abbott Laboratories to improve binding affinity.19 In addition, Chettiar et al discovered recently potent small molecules that prolong mitotic progression with relatively large substituents on ring A (LLP3 in Figure 1A).16 However, Abbott Laboratories reported that ring B (the 3-cyano-pyridin-2(1H)-one moiety) did not tolerate further modifications of any kind.19
 |
Figure 1 (A) Abbott 8 and LLP3 are examples of the reported survivin binders at the dimerization interface. (B) This study proposes new binders with rigidized-pyridin-2(1H)-one scaffolds (n=1, 2, or 3), as discussed in the “Design approach and proof of concept” section. R1 is an open-chain alkyl group, while R2 represents certain substituents. |
In this study, we report the design, synthesis, and biological evaluation of novel survivin binders with a pyridin-2(1H)-one core structure. Moreover, we provide a proof of concept demonstrating that pyridin-2(1H)-one (ring B) and its 6-phenyl substituent (ring C) can be integrated into a tricyclic structure using differently sized spacers to link both rings while retaining binding affinity to survivin (Figure 1).
Methods
Molecular Dynamics
To establish the manually docked survivin–Abbott 8 complex, the phenyl side chain of Phe93 was rotated to make the binding site of the dimerization interface accessible. This rotation of the Phe93 side chain was also proposed from the REMD study of the Apo structure of survivin (wild type).21 To equilibrate and further study the complex, molecular dynamic (MD) simulations were performed using Amber (version 11).22–24 The X-ray apo-structure of the L54M mutant of survivin (homodimer) was retrieved from the PDB (code 1F3H).25 Residues Δ118–142 from the long amphipathic C-terminal helix of each monomer were truncated to reduce the size of the solvated water shell and hence reduce the computational expense of simulation time. This C-terminal truncation strategy has been reported in experimental studies, where it did not affect dimerization,26 and for REMD simulations for the wild-type apo structure of survivin.21 To model the Zn-ion atom type, the tetrahedral dummy-atom model was implemented.27
GAFF force-field28 parameters were used for the ligand (Abbott 8), and missing parameters were determined using the Parmchk29 module in Amber. The correct GAFF atom types of the ligand were determined with the Antechamber program.29 FF99SB30 force-field parameters were used for the protein atoms. Missing protons were added to the protein using the program Leap. To neutralize the system, sodium ions were added to the complex structure. With Leap, the complex was then solvated with the TIP3P31 octahedron solvent model with borders of 10 Å away from the protein, which resulted in the addition of about 60,000 water atoms to the system. Then, the starting coordinate and topology files for the simulations were constructed. Ions and solvent atoms were minimized with the Sander module over 500 steps using the steepest descent algorithm, with the subsequent 500 steps using the conjugate gradient, while the complex structure (Survivin — Abbott 8) was held fixed during this initial minimization. A subsequent minimization for the whole system was performed (1,000 steps steepest descent followed by 1,500 steps conjugate gradient) with no restraints.
The system was then heated to 300 K in 200 ps under constant-volume periodic boundary conditions with weak restraints (force constant of 10 Å) for complex atoms’ positions. Consequently, the whole system was equilibrated for 1 ns with no restraints. In the subsequent production phase (~16 ns), the trajectory was calculated under constant-pressure periodic boundary conditions, using an NPT ensemble (isothermal–isobaric ensemble, ie, constant number of particles n, constant pressure P, and constant temperature T at 1 atm pressure, and a time step of 2 fs. Initial MD velocities were applied randomly according to the Boltzmann distribution. The temperature (300 K) was maintained using the Langevin dynamics algorithm32 to guarantee an equal distribution of temperature. The Shake33 algorithm was applied to all bonds involving hydrogen atoms. The particle-mesh Ewald method34 was employed to treat long-range electrostatic interactions, and a 10 Å cutoff for van der Waals interactions was applied. Coordinates were saved every 0.2 ps and energy data every 100th time step. The generated trajectories were all centered to the protein, projected back to their initial solvent octahedron, and RMS fitted to the backbone atom positions of the first frame using the program PTRAJ. The clustering of the trajectory was performed with the average-linkage algorithm using PTRAJ at the tenth frame interval.35 Cluster analysis was performed according to the metrics reported in the literature and implemented in the Amber package.35 Metric values per cluster count were produced by PTRAJ. 2D-RMSD values for the 2D-RMSD plots were also calculated using PTRAJ, prepared by in-house Python and Java scripts and visualized by GNUPlot (http://www.gnuplot.info) and Origin (http://www.originlab.com). Clustering data were prepared by Newick Perl script provided by the Amber community to produce the Newick format.36 Then, the phenogram was plotted using the web service https://www.bioinformatics.nl/tools/plottree.html
Survivin Expression And Purification
The gene coding for residues 1–142 (full length) of the human survivin L54 mutant was synthesized (Eurofins) and finally cloned into a pET24a+ vector,37 which encodes an N-terminal hexahistidine tag. Expression was carried out in BL21 (DE3) Escherichia coli. Purification from E. coli lysates was accomplished using Ni2+ chelation chromatography with standard procedures.25,38 The histidine tag was removed using TEV protease digestion during dialysis in 50 mM Tris (pH 80) 150 mM NaCl, 5 mM β-mercaptoethanol, and 10% (v:v) glycerol at 4°C for 24 hours. Peak fractions were collected from the reverse Ni2+ chelation chromatography, dialyzed against 5 mM HEPES-Na+ (pH 7.5) and 3 mM β-mercaptoethanol, concentrated, and stored at −80°C.
Fluorescence-Based Assay
A 1:3 series dilution of the survivin (L54M) protein was incubated with 1 µM testing compound for 30 minutes in 5 mM HEPES-Na+ (pH 7.5) buffer, 3% (v:v) DMSO, and 3 mM β-mercaptoethanol. Fluorescence intensity was measured by ClarioStar (BMG Labtech) with excitation wavelength of 380–440 nm and emission wavelength 440–550 nm. KD values were derived assuming a two-component binding equilibrium. Data were fitted to a four-parameter sigmoidal curve.
Docking And Benchmarking
Representative Structure Preparation
MOE39 was used to prepare the three MD representative structures for benchmarking and docking. The structures were prepared with restrained relaxing by applying Tether energy minimization (MMFF94x force field) to the side chains with a strength of 0.5 Å SD from the original coordinates, while the backbone atoms were kept fixed. The rest of the parameters were kept at the default setting.
Preparation Of DEKOIS 2.0 Benchmark Set
The DEKOIS 2.040 protocol was applied on 29 survivin bioactives extracted from BindingDB, to generate 870 challenging decoys (1:30 ratio). Then, all molecules were prepared with the LigPrep module (version 2.4)41 of Maestro (version 9.1).42 Molecules were minimized using the OPLS 2005 force field. Only one conformer was retrieved, and one protonation state was generated at pH 7 for each molecule. The specified stereoconfiguration of all bioactives and decoys of the data sets was retained. All prepared molecules were saved as SD files for GOLD docking.
Benchmarking
GOLD (version 5.1)43–46 was used for benchmarking on the three representative structures of the MD clustering. Residues of the binding site were defined by specifying the Abbott 8 coordinates and using a cutoff radius of 10 Å, with the “detect cavity” option enabled. The scoring function used for GOLD docking experiments was ChemPLP. The search efficiency of the genetic algorithm was set at 200%. Docking was terminated early when the top three solutions were within 1.5 Å RMSD. The docking score of best pose was then retrieved for calculating the pROC-AUC utilizing R-Snippet components of KNIME.47 The pROC-Chemotype plots were generated by the pROC-Chemotype plot tool, available from http://www.dekois.com.48,49
Conformational Search (Synthesized Compounds)
Synthesized compounds were built using MOE. Then, the molecule-wash function was employed at default settings. A conformational search was conducted at the default settings using LowModeMD and an RMS gradient of 0.005, and at most 10,000 iteration-limit attempts were made to generate conformations. A total of 500 iterations of energy minimization were performed for each conformer using the MMFF94x force field. Partial charges were calculated according to the standard parameters of the force field. This protocol produced one, two, and four ring conformations per compound of the five-, six-, and seven-member rigidized systems, respectively.
Docking For Synthesized Compounds
All generated conformations were subjected to docking experiments on the third MD-cluster representative. Docking was performed with GOLD (version 5.2) using settings similar to the previously mentioned benchmarking. The total number of generated poses was 12, 27, and 52 for compounds of five, six, and seven-member rigidized systems, respectively.
Chemistry
All reagents and solvents were of commercial quality and utilized without further purification. Reactions were carried out in standard laboratory glassware under air, except when noted otherwise. Reaction temperatures refer to the temperature of the oil/sand/water bath used if not denoted differently. The purity of the compounds, which were tested in the survivin (L54M) assays, was >95%, determined via HPLC on an HP 1090 series II LC equipped with a UV diode-array detector (detection at 230 nm and 254 nm). Chromatographic separation was performed on a Phenomenex Luna 5u C8 column (150×4.6 mm, 5 µm) at 35°C oven temperature. The injection volume was 5 µL and flow 1.5 mL/min using the following gradient: 0.01 M KH2PO4, pH 2.30 (solvent A), methanol (solvent B), 40% B to 85% B in 8 minutes; 85% B for 5 minutes; 85% to 40% B for 1 minute; 40% B for 2 minutes; stop time 16 minutes. TLC reaction controls were performed for all reactions using fluorescent silica gel 60 F254 plates (Merck) and visualized under natural light and UV illumination at 254 and 365 nm. Column chromatography was performed on Davisil LC60A 20-45 µm silica from Grace Davison and Geduran Si60 63-200 µm silica from Merck for the precolumn using an Interchim PuriFlash 430 automated flash chromatography system. 1H NMR/13C NMR spectra were measured on a Bruker Avance 400 NMR spectrometer at 400/100 MHz. Chemical shifts are reported in parts per million relative to TMS. All spectra were calibrated against the residual proton peak of the deuterated solvent used. Mass spectra (EI-MS or FAB-MS) were obtained from the Mass Spectrometry Department, Institute of Organic Chemistry, Eberhard Karls University of Tübingen.
Synthesis Of Reported Compound: 6-(5-Bromo-2-Hydroxyphenyl)-4-(2-Chloro-5-(Trifluoromethyl)Phenyl)-2-Oxo-1,2-Dihydropyridine-3-Carbonitrile19 (Abbott 16)
A mixture of 1-(5-bromo-2-hydroxyphenyl)ethan-1-one (1 mmol, 0.215 g) and ammonium acetate (8 mmol, 0.617 g) in ethanol was stirred at room temperature for 10 minutes. Then, 2-chloro-5-(trifluoromethyl)benzaldehyde (1 mmol, 0.208 g) and ethyl cyanoacetate (1 mmol, 0.113 g) were added to the mixture and heated under reflux for 12 hours. The reaction mixture was cooled and the resulting solution dried in vacuo to give a yellow residue that was then purified by column chromatography using an n-hexane:ethyl acetate 6.5:3.5 solvent system. The product was canary yellow and solid: yield (0.165 g, 35%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 6.92 (d, J=8.59 Hz, 2 H, Ar-H), 7.40 (d, J=8.34 Hz, 1 H, Ar-H), 7.76 (br s, 1 H, Ar-H), 7.82 (m, 4 H, Ar-H + Ar-OH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 108.71, 110.28, 114.74, 119.01, 120.28, 121.82, 126.80, 127.57, 128.29, 128.61, 130.74, 131.36, 134.57, 135.41, 136.09, 150.98, 155.32, 155.83, 161.05; HPLC: tret= 9.29 min (98.7% at 254 nm); EI-MS: m/z = 470.1 [M·]+.
Synthesis Of 4-Aryl-6-(5-Bromo-2-Hydroxyphenyl)-5-Methyl-2-Oxo-1,2-Dihydropyridine-3-Carbonitriles (8–10)
A mixture of 1-(5-bromo-2-hydroxyphenyl)propan-1-one (4, 1 mmol, 0.229 g), aldehyde (1–3), ethyl cyanoacetate (1 mmol, 0.113 g), and ammonium acetate (8 mmol, 0.617 g) in ethanol was heated under reflux for 12–24 hours. The reaction mixture was cooled and the resulting solution dried in vacuo to give a yellow residue that was then purified by column chromatography. Solvent mixtures for column chromatography are stated for individual compounds.
6-(5-Bromo-2-Hydroxyphenyl)-4-(2-Chloro-5-(Trifluoromethyl)phenyl)-5-Methyl-2-Oxo-1,2-Dihydropyridine-3-Carbonitrile (8)
2-Chloro-5-(trifluoromethyl)benzaldehyde (1, 1 mmol, 0.209 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The compound was purified by column chromatography using dichloromethane as a solvent. The product was a yellow solid: yield (0.1 g, 21%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.56 (s, 3 H, -CH3), 7.00 (d, J=8.84 Hz, 1 H, Ar-H), 7.50 (s, 1 H, Ar-H), 7.55 (s, 1 H, Ar-H), 8.01 (s, 3 H, Ar-H), 10.50 (br s, 1 H, Ar-OH), 12.86 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 14.32, 102.35, 109.72, 112.82, 115.29, 118.24, 122.17, 129.08, 129.45, 130.97, 131.50, 132.30, 132.45, 133.92, 136.63, 147.77, 154.19, 157.94, 159.57; HPLC: tret= 8.28 min (98.6% at 254 nm); EI-MS: m/z = 483.1 [M·]+.
6-(5-Bromo-2-Hydroxyphenyl)-4-(2,5-Dibromophenyl)-5-Methyl-2-Oxo-1,2-Dihydropyridine-3-Carbonitrile (9)
2,5-Dibromobenzaldehyde (2, 1 mmol, 0.263 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The compound was purified by column chromatography using dichloromethane:methanol 9.5:0.5 as solvent. The product was a yellow solid: yield (0.095 g, 18%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.52 (s, 3 H, -CH3) 6.93 (br s, 1 H, Ar-H) 7.22–8.03 (m, 5H, Ar-H), 10.37 (s, 1H, Ar-OH), 12.66 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 14.34, 102.36, 109.65, 112.47, 115.14, 118.19, 119.65, 121.18, 122.12, 131.56, 132.17, 132.90, 133.81, 134.71, 139.00, 147.52, 154.15, 159.53, 162.14; HPLC: tret= 8.45 min (98.1% at 254 nm); EI-MS: m/z = 539 [M·]+.
6-(5-Bromo-2-Hydroxyphenyl)-4-(2,5-Dichlorophenyl)-5-Methyl-2-Oxo-1,2-Dihydropyridine-3-Carbonitrile (10)
2,5-Dichlorobenzaldehyde (3, 1 mmol, 0.175 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The compound was purified by column chromatography using dichloromethane:methanol 9.5:0.5 as solvent. The product was a yellow solid: yield (0.09 g, 20%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.52 (s, 3 H, -CH3), 6.94 (d, J=8.84 Hz, 1 H, Ar-H), 7.43 (br s, 1 H, Ar-H), 7.50 (d, J=8.84 Hz, 1 H, Ar-H), 7.58–7.81 (m, 3 H, Ar-H); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 14.32, 102.35, 109.72, 112.82, 115.29, 118.24, 122.17, 129.08, 129.45, 130.97, 131.50, 132.30, 132.45, 133.92, 136.63, 147.77, 154.19, 157.94, 159.57; HPLC: tret= 7.99 min (97% at 254 nm); EI-MS: m/z = 450 [M·]+.
Synthesis Of 8-Bromo-4-Aryl-2-Oxo-2,5-Dihydro-1H-indeno[1,2-b]Pyridine-3-Carbonitriles (11–13)
A mixture of 6-bromo-2,3-dihydro-1H-inden-1-one (5, 1 mmol, 0.211 g), aldehyde (1–3), ethyl cyanoacetate (1 mmol, 0.113 g), and ammonium acetate (8 mmol, 0.617 g) in ethanol was heated under reflux for 12–24 hours. The reaction mixture was cooled and the precipitate formed filtered, washed with ethanol, and then excessively with water, dried in vacuo and purified with column chromatography. Solvent mixtures for column chromatography are stated for individual compounds.
8-Bromo-4-(2-Chloro-5-(Trifluoromethyl)Phenyl)-2-Oxo-2,5-Dihydro-1H-Indeno[1,2-b]Pyridine-3-Carbonitrile (11)
2-Chloro-5-(trifluoromethyl)benzaldehyde (1, 1 mmol, 0.209 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The compound was firstly crystallized from DMF:ethanol 1:5, further purified by column chromatography using an n-hexane–ethyl acetate gradient, and then eluted with ethyl acetate. The product was a pale-yellow solid: yield (0.09 g, 20%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 3.42–3.62 (m, 2 H, -CH2-), 7.58 (d, J=7.83 Hz, 1 H, Ar-H), 7.73 (d, J=7.58 Hz, 1 H, Ar-H), 8.00 (s, 2 H, Ar-H), 8.10 (s, 1 H, Ar-H), 8.40 (br s, 1 H, Ar-H), 13.80 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 33.13, 115.73, 119.43, 120.67, 122.14, 124.79, 124.85, 127.01, 127.67, 128.35, 128.74, 131.19, 133.03, 134.63, 135.31, 136.28, 144.92, 150.09, 153.18, 161.22; HPLC: tret= 8.38 min (99% at 254 nm); FAB-MS: 467.1 [M+H]+.
8-Bromo-4-(2,5-dibromophenyl)-2-Oxo-2,5-Dihydro-1H-indeno[1,2-b]Pyridine-3-Carbonitrile (12)
2,5-Dibromobenzaldehyde (2, 1 mmol, 0.263 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The compound was purified by column chromatography using a solvent gradient starting with n-hexane 100% to ethyl acetate 100%, and then the product was eluted with ethyl acetate. The product was a dark-yellow solid: yield (0.111 g, 22%); 1H NMR (DMSO-d6) δ [ppm]: 3.43–3.63 (m, 2 H, -CH2-), 7.58 (d, J=7.58 Hz, 1 H, Ar-H), 7.72 (d, J=7.33 Hz, 2 H, Ar-H), 7.77–8.03 (m, 2 H, Ar-H), 8.38 (br s, 1 H, Ar-H), 13.69 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 33.16, 115.71, 119.59, 120.64, 121.18, 123.84, 124.73, 126.74, 127.66, 132.01, 132.94, 134.28, 134.84, 136.36, 137.68, 144.85, 150.04, 154.65, 161.34; HPLC: tret= 8.40 min (99% at 254 nm); EI-MS: m/z = 521 [M·]+.
8-Bromo-4-(2,5-Dichlorophenyl)-2-Oxo-2,5-Dihydro-1H-Indeno[1,2-b]Pyridine-3-Carbonitrile (13)
2,5-Dichlorobenzaldehyde (3, 1 mmol, 0.175 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. Dried raw residue was washed with DCM and the purity was sufficient. The product was a yellow solid: yield (0.146 g, 34%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 3.41–3.62 (m, 2 H, -CH2-), 7.57 (d, J=8.08 Hz, 1 H, Ar-H), 7.64–7.84 (m, 4 H, Ar-H), 8.37 (br s, 1 H, Ar-H), 13.75 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 33.11, 115.74, 119.63, 120.64, 121.82, 124.78, 127.64, 129.54, 131.34, 131.62, 132.37, 132.97, 135.21, 136.17, 137.64, 144.88, 149.57, 153.44, 161.09; HPLC: tret= 8.15 min (96.3% at 254 nm); EI-MS: m/z = 432.0 [M·]+.
Synthesis Of 9-Bromo-4-Aryl-2-Oxo-1,2,5,6-Tetrahydrobenzo[h]Quinoline-3-Carbonitriles (14–16)
A mixture of 7-bromo-3,4-dihydronaphthalen-1(2H)-one (6, 1 mmol, 0.225 g), aldehyde (1–3), ethyl cyanoacetate (1mmol, 0.113 g), and ammonium acetate (8 mmol, 0.617 g) in ethanol was heated under reflux for 18 hours. The reaction mixture was cooled and the precipitate formed filtered, washed with ethanol, dried in vacuo, and crystallized from DMF:ethanol 1:10.
9-Bromo-4-(2-Chloro-5-(Trifluoromethyl)Phenyl)-2-Oxo-1,2,5,6-Tetrahydrobenzo[h]Quinoline-3-Carbonitrile (14)
2-Chloro-5-(trifluoromethyl)benzaldehyde (1, 1 mmol, 0.209 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The product was a canary-yellow solid: yield (0.148 g, 31%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 2.31 (br s, 2 H, -CH2-CH2-), 2.64–2.89 (m, 2 H, -CH2-CH2-), 7.32 (d, J=8.08 Hz, 1 H, Ar-H), 7.65 (d, J=7.83 Hz, 1 H, Ar-H), 7.86–8.10 (m, 3 H, Ar-H), 8.34 (s, 1 H, Ar-H), 12.88 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 22.80, 26.31, 115.02, 119.98, 121.09, 122.08, 124.80, 125.93, 127.13, 127.87, 128.10, 128.41, 128.74, 130.38, 130.98, 133.59, 135.00, 135.62, 138.36, 153.37, 161.59; HPLC: tret= 8.83 min (100% at 254 nm); EI-MS: m/z = 480.0 [M·]+.
9-Bromo-4-(2,5-Dibromophenyl)-2-Oxo-1,2,5,6 Tetrahydrobenzo[h]Quinoline-3-Carbonitrile (15)
2,5-Dibromobenzaldehyde (2, 1 mmol, 0.263 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The product was a canary-yellow solid: yield (0.174 g, 33%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 2.16–2.38 (m, 2 H, -CH2-CH2-), 2.62–2.90 (m, 2 H, -CH2-CH2-), 7.32 (d, J=8.08 Hz, 1 H, Ar-H), 7.56–7.91 (m, 4 H, Ar-H), 8.32 (s, 1 H, Ar-H), 12.85 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 22.90, 26.41, 115.12, 120.05, 120.21, 121.30, 127.89, 128.20, 130.44, 131.21, 131.59, 132.20, 132.62, 133.63, 134.13, 134.72, 138.09, 138.37, 151.64, 161.42; HPLC: tret= 8.84 min (100% at 254 nm); EI-MS: m/z = 535 [M·]+.
9-Bromo-4-(2,5-Dichlorophenyl)-2-Oxo-1,2,5,6-Tetrahydrobenzo[h]Quinoline-3-Carbonitrile (16)
2,5-Dichlorobenzaldehyde (3, 1 mmol, 0.175 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The product was a canary-yellow solid: yield (0.076 g, 17%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 2.18–2.46 (m, 2 H, -CH2-CH2-), 2.71–2.91 (m, 2 H, -CH2-CH2-), 7.31 (d, J=7.83 Hz, 1 H, Ar-H), 7.58–7.88 (m, 4 H, Ar-H), 8.32 (s, 1 H, Ar-H), 12.87 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 22.83, 26.39, 115.11, 120.03, 121.03, 125.93, 127.91, 129.62, 129.89, 130.42, 130.70, 131.16, 131.46, 131.72, 132.42, 133.62, 135.64, 138.41, 152.07, 161.30; HPLC: tret= 8.69 min (100% at 254 nm); EI-MS: m/z = 446.0 [M·]+.
Synthesis Of 3-Bromo-6,7,8,9-Tetrahydro-5H-Benzo[7]Annulen-5-One (7)50,51 and Methyl 5-(4-Bromophenyl)-5-Oxopentanoate (20)51
Anhydrous AlCl3 (4.8 g, 36 mmol, 2.2 eq) was added portion-wise to vigorously stirred bromobenzene (20 mL) on ice. After stirring for 30 minutes, methyl 5-chloro-5-oxopentanoate (2.7 g, 16 mmol) was added dropwise, followed by stirring for 3.5 hours at room temperature. Then, crushed ice and concentrated HCl (15 mL) were added and the mixture warmed until the suspension formed had dissolved. The aqueous layer was extracted with CH2Cl2 (3×20 mL). The combined organic layers were washed with water (40 mL) and brine (30 mL), dried over Na2SO4 and concentrated in vacuo to give 20 as an orange oil.
5-(4-Bromophenyl)-5-Oxo-Pentanoic Acid (21)51
The oil (20) was dissolved in a mixture of MeOH (20 mL) and 2N NaOH (20 mL) and the mixture heated under reflux for 3 hours. The mixture was concentrated in vacuo and the residue acidified with concentrated HCl and extracted with CH2Cl2 (4×20 mL). The combined organic layers were washed with water (30 mL) and brine (30 mL), dried over Na2SO4 and evaporated in vacuo to give a crude product of 5-(4-bromophenyl)-5-oxo-pentanoic acid (21) as a slightly yellow solid (4.2 g).
5-(4-Bromophenyl)Pentanoic Acid (22)51
To the previous crude solid 21, KOH (13.9 g, 4.4 eq) and hydrazine monohydrate (4.4 g, 5.5 eq) were added and the mixture heated in triethylene glycol to reflux (200°C) in a Dean–Stark apparatus for overnight. The mixture was poured into cold water and washed with Et2O. The aqueous layer was acidified with concentrated HCl and extracted with CH2Cl2 (4×20 mL). The combined organic layers were washed with water (2×20 mL) and brine (30 mL), dried over Na2SO4, and concentrated in vacuo to give a crude product of 5-(4-bromophenyl)pentanoic acid (22) as a yellow solid (3.5 g).
3-Bromo-6,7,8,9-Tetrahydro-5H-Benzo[7]Annulen-5-One (7)50,51
Then, the crude product 22 was added portion-wise to polyphosphoric acid (30 g) at 90°C. The mixture was stirred overnight and turned brown during the first hour. Then, it was poured into crushed ice and warmed until the suspension formed had dissolved. The mixture was extracted with CH2Cl2 (4×20 mL). The combined organic layers were washed with water (2×30 mL) and brine (2×30 mL), dried over Na2SO4, and concentrated in vacuo to give the crude product 7 as a dark-brown oil, which was further purified by column chromatography using n-hexane:ethyl acetate 9:1 to give a dark-orange oil: yield (0.8 g, 25%); 1H NMR (400 MHz, chloroform-d) δ [ppm]: 1.84 (m, 4 H, -CH2-CH2-CH2-CH2-), 2.73 (m, 2 H, -CH2-CH2-CH2-CH2-), 2.88 (m, 2 H, -CH2-CH2-CH2-CH2-), 7.09 (m, 1 H, Ar-H), 7.52 (d, J=8.08 Hz, 1 H, Ar-H), 7.84 (br s, 1 H, Ar-H); 13C NMR (101 MHz, chloroform-d) δ [ppm]: 20.81, 25.05, 32.03, 40.70, 120.57, 131.38, 131.48, 134.82, 140.07, 140.42, 204.30; HPLC: tret= 7.33 min (100% at 254 nm); EI-MS: m/z = 239 [M·]+.
Synthesis Of 10-Bromo-4-Aryl-2-Oxo-2,5,6,7-Tetrahydro-1H-benzo[6,7]Cyclohepta[1,2-b]Pyridine-3-Carbonitriles (17–19)
A mixture of the prepared 3-bromo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (7, 0.5 mmol, 0.119 g) aldehyde (1–3), ethyl cyanoacetate (0.5 mmol, 0.057 g), and ammonium acetate (4 mmol, 0.308 g) in ethanol was heated under reflux for 18 hours. The reaction mixture was cooled and the precipitate formed filtered, washed excessively with ethanol, washed again with DCM, and dried in vacuo. No further purification steps were required, since the HPLC purity grade was sufficient.
10-Bromo-4-(2-Chloro-5-(Trifluoromethyl)Phenyl)-2-Oxo-2,5,6,7-Tetrahydro-1H-benzo[6,7]Cyclohepta[1,2-b]Pyridine-3-Carbonitrile (17)
2-Chloro-5-(trifluoromethyl)benzaldehyde (1, 0.5 mmol, 0.104 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The product was a pale-yellow solid: yield (0.06 g, 25%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.80–2.06 (m, 4 H, -CH2-CH2-CH2-), 2.60–2.69 (m, 2 H, -CH2-CH2-CH2-), 7.34 (d, J=8.08 Hz, 1 H, Ar-H), 7.68 (d, J=7.83 Hz, 1 H, Ar-H), 7.82 (br s, 1 H, Ar-H), 7.97 (s, 2 H, Ar-H), 8.06 (br s, 1 H, Ar-H), 13.05 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 24.56, 29.46, 32.07, 113.13, 115.19, 119.28, 126.85, 128.00, 128.28, 128.61, 129.00, 129.64, 130.14, 130.93, 131.10, 131.32, 133.31, 133.56, 135.49, 139.65, 156.65, 158.87; HPLC: tret= 8.97 min (100% at 254 nm); EI-MS: m/z = 494.0 [M·]+.
10-Bromo-4-(2,5-Dibromophenyl)-2-oxo-2,5,6,7-Tetrahydro-1H-Benzo[6,7]Cyclohepta[1,2-b]Pyridine-3-Carbonitrile (18)
2,5-Dibromobenzaldehyde (2, 0.5 mmol, 0.132 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The product was a pale-yellow solid: yield (0.084 g, 31%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.81–1.95 (m, 4 H, -CH2-CH2-CH2-), 2.53–2.68 (m, 2 H, -CH2-CH2-CH2-), 7.29–7.40 (m, 1 H, Ar-H), 7.61–7.85 (m, 5 H, Ar-H); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 24.53, 29.54, 32.07, 115.32, 119.30, 120.15, 121.12, 131.11, 131.28, 131.93, 133.24, 133.61, 133.99, 134.23, 134.65, 135.13, 138.54, 139.61, 140.49, 157.65, 160.53; HPLC: tret= 8.84 min (100% at 254 nm); EI-MS: m/z = 549 [M·]+.
10-Bromo-4-(2,5-Dichlorophenyl)-2-Oxo-2,5,6,7-Tetrahydro-1H-Benzo[6,7]Cyclohepta[1,2-b]Pyridine-3-Carbonitrile (19)
2,5-Dichlorobenzaldehyde (3, 1 mmol, 0.087 g) was used as the aldehyde for the previously described reaction mixture in the general procedure. The product was a pale-yellow solid: yield (0.2 g, 87%); 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.60–1.97 (m, 4 H, -CH2-CH2-CH2-), 2.47–2.64 (m, 2 H, -CH2-CH2-CH2-), 7.27 (d, J=7.58 Hz, 1 H, Ar-H), 7.50–7.85 (m, 5 H, Ar-H), 12.95 (br s, 1 H, -NH); 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 24.50, 29.51, 32.09, 115.19, 116.72, 116.97, 119.27, 129.27, 129.78, 131.03, 131.08, 131.31, 131.37, 131.59, 132.31, 132.85, 133.27, 136.06, 139.62, 156.25, 160.26; HPLC: tret= 8.61 min (100% at 254 nm); EI-MS: m/z = 459.9 [M·]+.
Results And Discussion
Design Approach And Proof Of Concept
The SAR suggested by Abbott Laboratories' study on survivin proposed that the middle pyridin-2(1H)-one ring did not tolerate any modification in position 5.19 The NMR interaction pattern of the survivin–Abbott 8 complex showed that both rings B and C should be coplanar.19 The hydroxyl group of ring C helps to keep this coplanarity by forming an intramolecular hydrogen bond with the nitrogen atom of the pyridin-2(1H)-one ring.
Since there is no crystal structure available in the PDB to represent small-molecule binding with survivin, we modeled this binding scenario for Abbott 8 by manually docking Abbott 8 into the dimerization interface of the apo survivin mutant (L54M) dimer (PDB: 1F3H). Starting from this modeled complex, we figured that any substitution in position 5 of the pyridin-2(1H)-one ring would lead to a clash with Phe101 residue of the binding site, as well as a loss of the coplanarity between rings B and C of Abbott 8. Therefore, a successful introduction of any substituent in position 5 of the pyridin-2(1H)-one ring should be accommodated by an induced fit of the binding site backbone and/or side chain rotation of the Phe101. However, to retain the relative coplanarity between the two rings (B and C) while introducing a substituent on ring B, we considered a rigidization strategy to link ring B and C with differently sized spacers, producing five-, six-, and seven-member cyclic moieties fused to the core pyridin-2(1H)-one ring (Figure 1B). Since we focused on modifying the middle pyridin-2(1H)-one ring, we kept the bromo group in position 3 of ring C in our scaffolds, because it facilitates further synthetic expansion of the system by adding lengthy peptide-like substituents, as suggested in the literature.19 Based on considerations reflecting the dynamics of the binding site, we aimed to demonstrate that our intended modification strategy leads to novel fused cyclic chemotypes that can be accommodated by the flexibility of the binding site.
Molecular Dynamic Simulation
In this section, we aim at extracting a rational model of a survivin–ligand complex that can be used for further modeling and docking experiments for the proposed rigidized scaffolds. Starting from the manually docked complex of survivin (L54M) dimer with Abbott 8, we conducted a medium time–course and explicit-solvent MD simulation for 16 ns to assess the flexibility of the dimerization interface, as shown in Figure S1. After finishing the MD-trajectory simulation, we assessed binding-site flexibility by visualizing the pairwise RMSD of the backbone MD frames of the binding site (Figure 2). The first 200 ps (ten frames of the contour map) shows an obvious fluctuation of the binding-site backbone, with many yellow and red areas (2.25–3 Å RMSD), representing the heating phase of the system. Continuing to ~0.6 ns, the binding-site backbone exhibits fewer changes, indicated by the appearance of green areas with smaller RMSD values. Between ~0.6 ns and ~16 ns, roughly one major batch of binding-site conformations (green ~ RMSD 1.5 A) can be observed, with minor yellow (2.25 Å RMSD) and blue (0.75 Å RMSD) regions.
 |
Figure 2 2D-RMSD of the backbone of the binding site is represented as a contour map with 20 ps–resolution intervals. One major batch of close frames (RMSD ~ 1.5 A) can be observed. |
For further investigation, we performed a cluster analysis of the MD frames. This suggested that three clusters would be an optimum choice (Figure S2). We found that the occurrence of the first two clusters did not exceed approximately 5% of the trajectory, while the third cluster accounted for about 95% of the trajectory and extended toward the end of the simulation. This highlights that the third cluster was the biggest and most dominant cluster, which agrees with the conclusion drawn from the 2D-RMSD contour map in Figure 2.
Benchmarking The Representative Structures Of The MD Clusters
To evaluate the in silico druggability of the binding sites, we generated a challenging decoy set by our DEKOIS 2.040,52,53 protocol from the available bioactives of survivin (retrieved from BindingDB). Then, we conducted a benchmarking study using GOLD as the docking program and the three representative structures of the clusters. The idea was to identify the appropriate representative structure that is able efficiently to separate bioactive ligands from generated challenging decoys. Based on the benchmarking results, the second and third cluster representatives showed similarly good performance, with pROC (AUC) values of 0.91 and 0.93, respectively (Figure 3, B and C), compared to the performance of the first cluster representative, with a pROC (AUC) value of 0.71 (Figure 3A). It should be noted that this performance was much better than the statistical average (pROC [AUC] of 0.43). As such, we concluded that both binding-site conformations were able to identify and accommodate survivin-dimerization modulators.
 |
Figure 3 An overview of the benchmarking results on the three representative structures of the clustering. The structures were docked with the DEKOIS 2.0 benchmark set by GOLD and the docking scores normalized by n2/3, where n is the number of heavy atoms. The x-axis represents the false-positive rate (FPR), while the y-axis represents the true-positive rate (TPR). |
Visualizing Chemotype Behavior Via pROC-Chemotype Plots
To decide which cluster should be selected, we visualized chemotype behavior by pROC-Chemotype48,49 plots (see Figure 4). Only 29 small-molecule binders to survivin have been recently introduced and collected by the BindingDB repository,54 comprising 25 pyridin-2(1H)-one–based molecules, while the other four are miscellaneous structures reported in the early stage of affinity screening by Abbott.19 Maximum common substructure (MCS) chemotype clustering demonstrated five main clusters, as seen in Figure 4. All the reported pyridin-2(1H)-one derivatives are clustered in cluster 1, while the rest (clusters 2–5) were clustered into singletons (ie, one compound per cluster). Such MCS clustering behavior reflects the diversity of the chemotypes. In a comparison of the second and third MD cluster representatives, the pROC-Chemotype plot demonstrated better diversity of the hits being enriched (at 1%) for the third MD cluster representative. This diversity is evident by the presence of MCS clusters 1 and 3, while only MCS cluster 1 is seen for the second MD representative. Since the performance in this case is presented by the normalized (by the N2/3) docking score, we converted the affinity values (KD values) into ligand efficiency (LE)55 values. This allows us to have a reasonable assessment when normalizing both the docking and bioactivity information by the molecular-size features. In this case, the third MD cluster representative enriched effectively ligands with better LE values compared to the second MD cluster representative (see Figure 4). Therefore, the third MD cluster representative satisfied the assumption to be used for further predictions as a main MD cluster representative.
 |
Figure 4 pROC-Chemotype plot49 of the three cluster representatives of the MD. Bioactivity information represented by rank of ligand efficiency (LE) values. Lower-rank numbers (red) demonstrate better ligand efficiency. The enrichment factor (EF) evaluates the ability of the docking program to find true positives throughout the score-order list compared to the random selection. The EF was calculated in our protocol thus:56 |
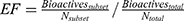
Elucidating the distance between position C5 of the middle pyridin-2(1H)-one ring and Phe101 side chain for both starting the survivin–Abbott 8 complex structure (ie, before the MD) and the main MD representative structure (ie, third cluster representative) showed that the latter possessed a better chance of accommodating a substituent at C5 of the pyridin-2(1H)-one ring (see Figure S3). This suggests that survivin, and particularly the residue Phe101, is possibly able to adopt by certain conformational changes the designed rigidized scaffolds forming reasonable interactions with them. Docking assessment of the designed compounds showed proper distances and hydrophobic contacts with Phe101, as shown in the Docking Assessment section.
Chemistry
The general synthesis of the five-, six, and seven-member rigidized derivatives fused with the middle pyridin-2(1H)-one ring, namely 4-aryl-8-bromo-2-oxo-2,5-dihydro-1H-indeno[1,2-b]pyridine-3-carbonitriles (11–13), 4-aryl-9-bromo-2-oxo-1,2,5,6-tetrahydrobenzo[h]quinoline-3-carbonitriles (14–16), and 4-aryl-10-bromo-2-oxo-2,5,6,7-tetrahydro-1H-benzo[6,7]cyclohepta[1,2-b]pyridine-3-carbonitriles (17–19), respectively, as well as the open methyl scaffolds of 4-aryl-6-(5-bromo-2-hydroxyphenyl)-5-methyl-2-oxo-1,2-dihydropyridine-3-carbonitriles (8–10), as illustrated in Scheme 1. For this, we adopted a one-pot reaction approach utilizing in-solution phase MCRs.57,58 Briefly, four reactants were mixed together and heated for reflux in ethanol. The four reactants were composed of: a suitable phenyl ketone derivative 4–7, namely 1-(5-bromo-2-hydroxyphenyl)propan-1-one (4), 6-bromo-2,3-dihydro-1H-inden-1-one (5), 7-bromo-3,4-dihydronaphthalen-1(2H)-one (6) and 3-bromo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (7); a substituted benzaldehyde 1–3, namely 2-chloro-5-(trifluoromethyl)benzaldehyde (1), 2,5-dibromobenzaldehyde (2), and 2,5-dichlorobenzaldehyde (3); ethyl cyanoacetate; and ammonium acetate.
 |
Scheme 1 Synthesis of 5-methyl-pyridin-2(1H)-ones 8–10, as well as rigidized scaffolds where the 6-phenylpyridin-2(1H)-one substructure is fused to a five, six, and seven-membered ring system 11–19. Rigidization changes are highlighted in red. Reagents and conditions (a) reflux in ethanol. |
For all 5-methyl-pyridin-2(1H)-one 8–10, their methyl protons are found as a shielded singlet peak at the upfield region δ≈1.5 ppm. The five-member fused-ring system 11–13 shows the methylene protons as a shielded multiplet peak (two strongly coupled geminal doublets with Δν/J=2.4) in the upfield regions δ≈3.4 and 3.5 ppm. For the six-member rigidized products 14–16, two shielded peaks (multiplets) in the upfield regions δ≈2.2 and 2.6 ppm can be observed, representing the two methylene groups. Likewise, the seven-member rigidized products 17–19 demonstrated two shielded peaks (multiplets) in the upfield regions δ≈1.9 and 2.6 ppm, representing the three methylene groups. All the previously mentioned 1H NMR observations confirmed the successful formation of the differently sized rigidized compounds. Further confirmation was conducted by 13C NMR, since the relevant peaks of the aliphatic carbons were observed in the upfield region. Mass spectrometry of all bromo- and chloro-containing products demonstrated peaks at M+, M++ 2, and M++ 4, due to the isotopic nature of the respective halogen. Representative spectral data can be found in the Supplementary material (Figures S4–S6).
To synthesize the seven-member rigidized compounds 17–19, the ketone 3-bromo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (7), which is not commercially available, was prepared starting from bromobenzene (Scheme 2).50,51 Friedel–Crafts acylation was carried out using methyl 5-chloro-5-oxopentanoate in the presence of anhydrous AlCl3 as a catalyst, followed by an ester hydrolysis of 20. Then, Wolff–Kishner reduction (Huang Minlon modification)59 was utilized for selective reduction of the carbonyl ketone 21. Lastly, a successful cyclization of 22 was performed by intramolecular Friedel–Crafts acylation using polyphosphoric acid as a Brønsted acid.
 |
Scheme 2 Reagents and conditions: (a) H3COCO(CH2)3COCl, AlCl3, rt; (b) 1.MeOH, NaOH, Δ, 2.HCl, H2O; (c) 1. KOH, NH2-NH2·H2O, triethylene glycol, 200°C, 2. HCl, H2O; (d) PPA, 90°C. |
The preparation of the compound Abbott 16,19 to be used as a reference for comparison, was not straightforward. Setting up the original one-pot reaction conditions, we ended up with a relatively low yield that was difficult separate. However, when performing the one-pot reaction condition in a multistep fashion, a better yield was afforded. For this, we mixed only two reactants (the ketone and ammonium acetate) for 10 minutes, and then added in situ the remaining two reactants (aldehyde and ethyl cyanoacetate) to complete the reaction successfully. This order of adding the reactants was inspired by a proposed mechanism of the synthesis of 2-amino-3-cyanopyridines.60
Biological Evaluation
In order to evaluate the binding affinity (KD) of the prepared rigidized systems to survivin (L54M), we cloned the (L54M) mutant gene of BIRC5 into our in-house vector. Then, recombinant survivin (L54M) expression and purification cycles were conducted. The choice of the point mutant (L54M) over survivin wild type was encouraged based on the preferred crystallographic properties, which may be needed in future for optimized ligands based on the proposed chemotypes in this study. Scanning the prepared compounds against purified survivin using differential scanning fluorimetry was not adequate, due to interference of the compounds’ intrinsic fluorescence with the dye used (Sypro orange). Isothermal titration calorimetry was problematic, due to the limited aqueous solubility of the compounds. Making use of the intrinsic fluorescence of the rigidized pyridin-2(1H)-one systems, we successfully monitored the increasing intensity of the fluorescence upon compound–survivin complex formation.19 All binding affinity data are summarized in Table 1.
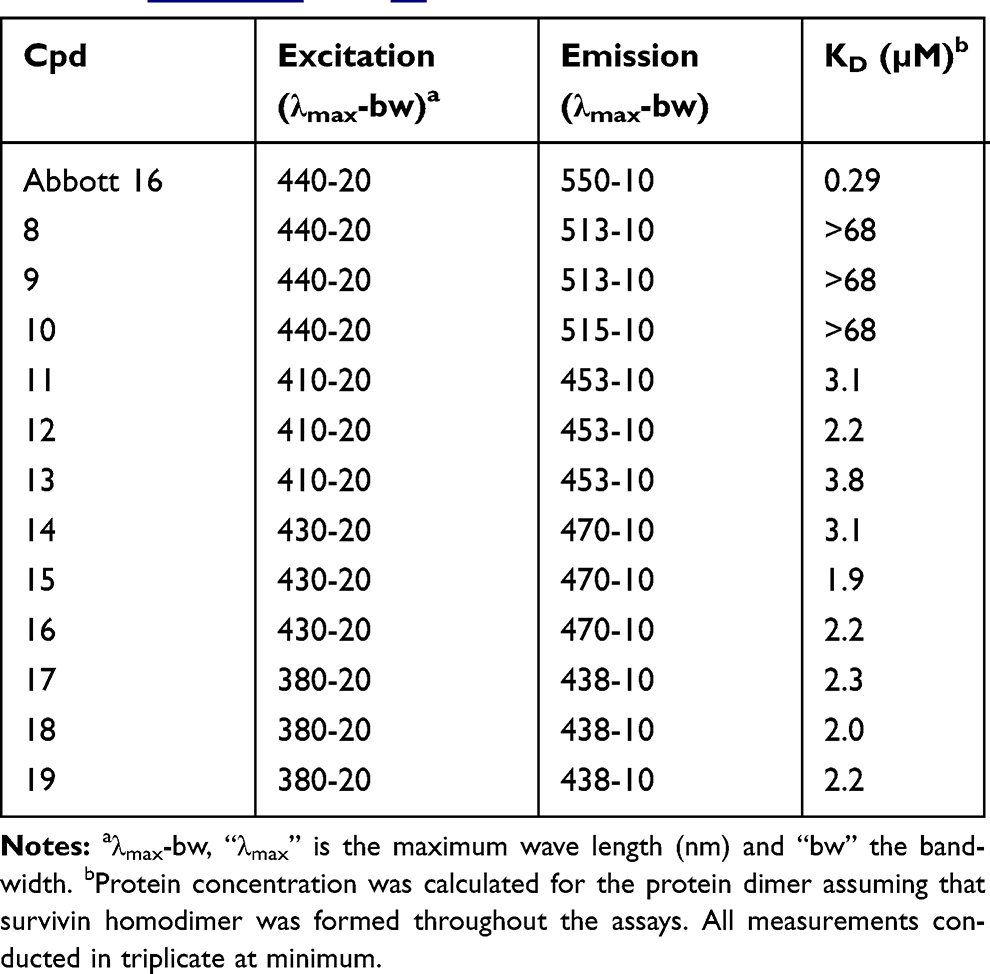 |
Table 1 Binding Affinity Of The Synthesized Compounds Upon Binding To Survivin (L54M) Mutant. Chemical Structures Of The Compounds And Representative Curves For The Assay Can Be Found In Figures S7 And S8, Respectively |
None of the compounds of the open methyl-scaffold 8–10 demonstrated binding affinity (Table 1). This is not surprising, since introduction of a 5-methyl group forces dominant orthogonal noncoplanar (rings B and C) conformations. Although the formation of coplanar conformations would be augmented by hydrogen-bonding formation between rings B and C, the orthogonal conformations should exert substantially higher energy to adopt the desired coplanarity, in order to pack optimally into the relatively narrow dimerization interface. Therefore, the desired coplanarity for binding is not tolerable in this case.
Interestingly, all the nine compounds of the rigidized systems 11–19 demonstrated evidence of binding at a low micromolar range of affinity. The conformational search of the five-, six-, and seven-membered systems showed one, two, and four main low-energy ring conformations in vacuum, respectively (Figure S9). This affected the relative positioning of rings B and C consequently. The dihedral angles of rings B and C did not show remarkable deviations within compound series. Although the seven-member rigidized compounds 17–19 showed relatively larger dihedral angles (larger deviations from the ideal coplanar conformations), still their biological affinity did not deviate remarkably from the average affinity of the other rigidized systems. The relative positioning of the substituted C ring allowed the bromo group to pack optimally against the shallow cleft of the binding site, as seen from the docking experiment in Figure S10C. However, the exact mechanism of how our proposed chemotypes intervene with survivin, eg, via inhibiting or stabilizing the dimerization form, is still unclear.
Docking Assessment
Assessment of the distance between Phe101 side chain and the best poses (best score) of the rigidized compounds showed acceptable proximities implying reasonable hydrophobic contacts (Figure 5). The general binding mode of these compounds did not deviate significantly from Abbott 8’s binding mode in the main MD representative structure. The main difference can be attributed to the relative positioning of the ring C due to relative deviations of the coplanarity. In Figures 5 and S10, one can observe that six and seven-member rigidized compounds showed optimum packing of the bromo group against the shallow cleft formed by residues Arg18 and Phe86, while the five-member rigidized scaffolds forced the bromo group to be slightly deviated, yet reasonably interacting with the Ph86 side chain.
 |
Figure 5 (A–C) Overlay of the best docking poses of the five, six, and seven-membered rigidized compounds, respectively, with Abbott 8 (yellow sticks) in the binding site of the MD representative structure. Such poses show one similar ring conformation per rigidized system. Apolar hydrogens (for compounds) and all hydrogens (for residues) are omitted for clarity. |
The best docking pose per ligand superposed optimally with Abbott 8 in the main MD representative structure (Figure 5). Interestingly, all the best docking poses showed one similar ring conformation per each rigidized series, as observed from Figure S10. In addition, such ring conformation of the best docking pose does not necessary resemble the respective energy minimum in vacuum. For example, unlike compounds 14 and 16, the best docking pose for 15 showed dissimilar ring conformation to its respective energy minimum. This observation is attributable to the small energy barrier (ΔE=0.07 kcal/mol) between their ring conformations (Figure S9B). Interestingly, none of the best docking poses for the seven-member rigidized compounds 17–19 showed similar ring conformation to their respective energy minima, although the energy barrier for selecting the ring conformation of the docking pose was relatively higher (ΔE≈3.5 kcal/mol, Figure S9C). This was likely due to the presence of a relatively narrow cleft formed by Phe93 and Phe13 in the binding site that favored only one ring conformation for optimum positioning and hydrophobic interactions (Figure S10D).
As such, when elucidating the distribution of the docking score (fitness) for the best poses of 11–19 compounds (Table 2), a relatively narrow SD around the mean can be perceived, while relatively lower means and wider SDs are observed when considering the other ring conformations. In addition, the narrow deviation of the distribution of all best poses (∑ in Table 2) also agreed with the narrow range of measured biological affinity values (1.9–3.8 µM).
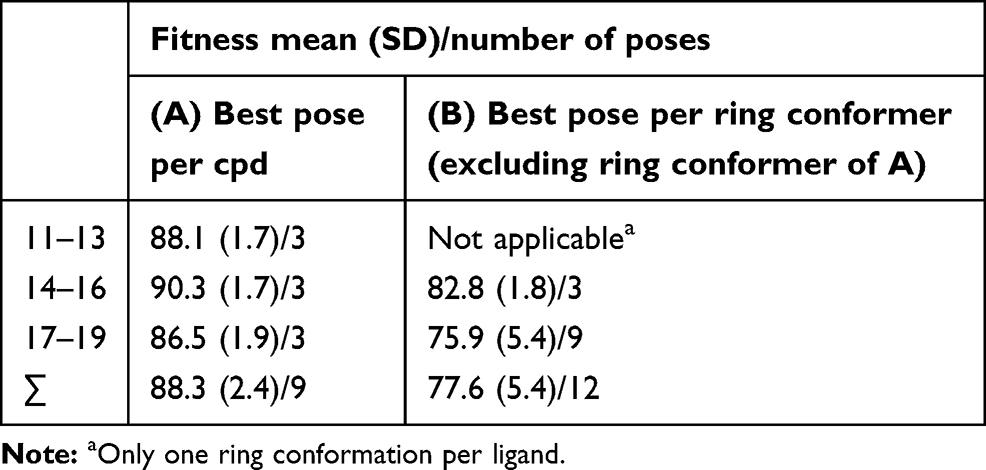 |
Table 2 Docking Score Distribution For Different Ring Conformations |
Conclusion
We conducted a medium time–course MD simulation for the survivin (L54M)–Abbott 8 complex to extract a rational representative model that highlights the possibility of addressing Phe101 by tolerable contact. Based on an in-solution phase MCR methodology, rigidized analogues of Abbott 8 were prepared by producing five-, six-, and seven-member cyclic moieties fused to the middle pyridin-2(1H)-one ring, namely 4-aryl-8-bromo-2-oxo-2,5-dihydro-1H-indeno[1,2-b]pyridine-3-carbonitriles (11–13), 4-aryl-9-bromo-2-oxo-1,2,5,6-tetrahydrobenzo[h]quinoline-3-carbonitriles (14–16), and 4-aryl-10-bromo-2-oxo-2,5,6,7-tetrahydro-1H-benzo[6,7]cyclohepta[1,2-b]pyridine-3-carbonitriles (17–19), respectively. Similarly, open methyl scaffolds of 4-aryl-6-(5-bromo-2-hydroxyphenyl)-5-methyl-2-oxo-1,2-dihydropyridine-3-carbonitriles (8–10) were also prepared for comparison purposes.
Applying biophysical methods in order to quantify binding affinity of the prepared compounds against survivin showed that differential scanning fluorimetry and isothermal titration calorimetry were not adequate, due to fluorescence interference and limited aqueous solubility of the compounds. Alternatively, monitoring the intrinsic fluorescence of the rigidized-pyridin-2(1H)-one scaffolds upon survivin–compound complex formation showed well-defined increasing intensity fitted to four-parameter curve models, and thus KD values were derived assuming a two-component binding equilibrium.
Biological evaluation of all 12 compounds against recombinant survivin (L54M) revealed that the five-, six-, and seven-member rigidized scaffolds 11–19 possessed binding affinity (KD) in the low micromolar range: 1.9–3.8 µM. Conformational search of five-, six-, and seven-member rigidized compounds showed one, two, and four main low-energy ring conformations in vacuum, respectively. Docking experiments demonstrated that only one ring conformation of the six- and seven-member rigidized compounds was favored upon binding at the dimerization interface of survivin. Such ring conformation of the best docking pose does not necessarily resemble the respective energy minimum in vacuum. This is likely due to the presence of a relatively narrow cleft formed by Phe93 and Phe13 in the binding site, which favored only one ring conformation for optimum hydrophobic interactions.
In conclusion, our study provides a solid proof of concept of the possibility of modifying the middle pyridin-2(1H)-one ring. In addition, it introduces novel rigidized chemotypes that can be a starting step for further lead-optimization cycles in the near future.
Acknowledgments
TMI is thankful to the GERLS (German–Egyptian Research Long-Term Scholarship) program of the German Academic Exchange Service (DAAD) for funding his PhD fellowship, and GERSS program (German–Egyptian Research Short-Term Scholarship/DAAD) for funding a research stay at Eberhard Karls University Tübingen. We thank Matthias R Bauer for generating the DEKOIS 2.0 benchmark set from survivin bioactives and Johannes Heidrich for preparing the raw 2D-RMSD matrix for plotting.
Author Contributions
TMI and FMB designed the experiments. TMI carried out all experiments. CE, AL, and SH helped in gene cloning and survivin expression and purification. TMI and FMB wrote the manuscript. FMB supervised the project and provided advice and expertise. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
FMB reports grants from the pharmaceutical industry outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Kelly RJ, Lopez-Chavez A, Citrin D, Janik JE, Morris JC. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol Cancer. 2011;10:35. doi:10.1186/1476-4598-10-35
2. Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998;58(22):5071–5074.
3. Adida C, Berrebi D, Peuchmaur M, Reyes-Mugica M, Altieri DC. Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet. 1998;351(9106):882–883. doi:10.1016/S0140-6736(05)70294-4
4. Monzo M, Rosell R, Felip E, et al. A novel anti-apoptosis gene: re-expression of survivin messenger RNA as a prognosis marker in non-small-cell lung cancers. J Clin Oncol. 1999;17(7):2100–2104. doi:10.1200/JCO.1999.17.7.2100
5. Tanaka K, Iwamoto S, Gon G, Nohara T, Iwamoto M, Tanigawa N. Expression of survivin and its relationship to loss of apoptosis in breast carcinomas. Clin Cancer Res. 2000;6(1):127–134.
6. Chakravarti A, Zhai GG, Zhang M, et al. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene. 2004;23(45):7494–7506. doi:10.1038/sj.onc.1208049
7. Liu BB, Wang WH. Survivin and pancreatic cancer. World J Clin Oncol. 2011;2(3):164–168. doi:10.5306/wjco.v2.i3.164
8. Wang Q, Greene MI. EGFR enhances Survivin expression through the phosphoinositide 3 (PI-3) kinase signaling pathway. Exp Mol Pathol. 2005;79(2):100–107. doi:10.1016/j.yexmp.2005.05.002
9. Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396(6711):580–584. doi:10.1038/25141
10. Muchmore SW, Chen J, Jakob C, et al. Crystal structure and mutagenic analysis of the inhibitor-of-apoptosis protein survivin. Mol Cell. 2000;6(1):173–182. doi:10.1016/S1097-2765(05)00019-5
11. Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 2003;22(53):8581–8589. doi:10.1038/sj.onc.1207113
12. Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 2004;64(20):7183–7190. doi:10.1158/0008-5472.CAN-04-1918
13. Skoufias DA, Mollinari C, Lacroix FB, Margolis RL. Human survivin is a kinetochore-associated passenger protein. J Cell Biol. 2000;151(7):1575–1582. doi:10.1083/jcb.151.7.1575
14. Uren AG, Wong L, Pakusch M, et al. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol. 2000;10(21):1319–1328. doi:10.1016/S0960-9822(00)00769-7
15. Jeyaprakash AA, Klein UR, Lindner D, Ebert J, Nigg EA, Conti E. Structure of a Survivin-Borealin-INCENP core complex reveals how chromosomal passengers travel together. Cell. 2007;131(2):271–285. doi:10.1016/j.cell.2007.07.045
16. Chettiar SN, Cooley JV, Park IH, et al. Design, synthesis and biological studies of survivin dimerization modulators that prolong mitotic cycle. Bioorg Med Chem Lett. 2013;23(19):5429–5433. doi:10.1016/j.bmcl.2013.07.034
17. Ainsztein AM, Kandels-Lewis SE, Mackay AM, Earnshaw WC. INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol. 1998;143(7):1763–1774. doi:10.1083/jcb.143.7.1763
18. Lens SM, Vader G, Medema RH. The case for Survivin as mitotic regulator. Curr Opin Cell Biol. 2006;18(6):616–622. doi:10.1016/j.ceb.2006.08.016
19. Wendt MD, Sun C, Kunzer A, et al. Discovery of a novel small molecule binding site of human survivin. Bioorg Med Chem Lett. 2007;17(11):3122–3129. doi:10.1016/j.bmcl.2007.03.042
20. Berezov A, Cai Z, Freudenberg JA, et al. Disabling the mitotic spindle and tumor growth by targeting a cavity-induced allosteric site of survivin. Oncogene. 2012;31(15):1938–1948. doi:10.1038/onc.2011.377
21. Park IH, Li C. Dynamic ligand-induced-fit simulation via enhanced conformational samplings and ensemble dockings: a survivin example. J Phys Chem B. 2010;114(15):5144–5153. doi:10.1021/jp911085d
22. AMBER [computer program]. Version 11: University of California, San Francisco; 2010.
23. Pearlman DA, Case DA, Caldwell JW, et al. Amber, a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free-energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91(1–3):1–41. doi:10.1016/0010-4655(95)00041-D
24. Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. doi:10.1002/(ISSN)1096-987X
25. Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat Struct Biol. 2000;7(7):602–608. doi:10.1038/77929
26. Sun C, Nettesheim D, Liu Z, Olejniczak ET. Solution structure of human survivin and its binding interface with Smac/Diablo. Biochemistry. 2005;44(1):11–17. doi:10.1021/bi0485171
27. Pang YP, Xu K, Yazal JE, Prendergas FG. Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. 2000;9(10):1857–1865.
28. Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi:10.1002/(ISSN)1096-987X
29. Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25(2):247–260. doi:10.1016/j.jmgm.2005.12.005
30. Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65(3):712–725. doi:10.1002/prot.v65:3
31. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi:10.1063/1.445869
32. Wu XW, Brooks BR. Self-guided Langevin dynamics simulation method. Chem Phys Lett. 2003;381(3–4):512–518. doi:10.1016/j.cplett.2003.10.013
33. Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–341. doi:10.1016/0021-9991(77)90098-5
34. Darden T, York D, Pedersen L. Particle mesh Ewald - an N.Log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. doi:10.1063/1.464397
35. Shao JY, Tanner SW, Thompson N, Cheatham TE. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J Chem Theory Comput. 2007;3(6):2312–2334. doi:10.1021/ct700119m
36. Shao J. Amber archive. Available from: http://archiveambermdorg/200808/0008html.
37. Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A. 2008;105(30):10360–10365. doi:10.1073/pnas.0805326105
38. Jez JM, Ferrer JL, Bowman ME, Dixon RA, Noel JP. Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction mechanism of a plant polyketide synthase. Biochemistry. 2000;39(5):890–902. doi:10.1021/bi991489f
39. Molecular Operating Environment (MOE) [computer program]. Version 2012: chemical Computing Group Inc.. 1010 Sherbooke St. West, Montreal, QC, Canada.
40. Bauer MR, Ibrahim TM, Vogel SM, Boeckler FM. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0–a public library of challenging docking benchmark sets. J Chem Inf Model. 2013;53(6):1447–1462. doi:10.1021/ci400115b
41. Ligprep [computer program]. Version 2.4: schrödinger, LLC. New York, NY; 2010.
42. Maestro [computer program]. Version 9.1: schrödinger, LLC. New York, NY; 2010.
43. Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267(3):727–748. doi:10.1006/jmbi.1996.0897
44. Jones G, Willett P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol. 1995;245(1):43–53. doi:10.1016/S0022-2836(95)80037-9
45. Jones G, Willett P, Glen RC. A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J Comput-Aided Mol Des. 1995;9(6):532–549. doi:10.1007/BF00124324
46. Hartshorn MJ, Verdonk ML, Chessari G, et al. Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem. 2007;50(4):726–741. doi:10.1021/jm061277y
47. Berthold MR, Cebron N, Dill F, et al. KNIME: the Konstanz information miner. In: Studies in Classification, Data Analysis, and Knowledge Organization (GFKL 2007). Heidelberg-Berlin: Springer-Verlag; 2007.
48. Ibrahim TM, Bauer MR, Boeckler FM. Probing the impact of protein and ligand preparation procedures on chemotype enrichment in structure-based virtual screening using DEKOIS 2.0 benchmark sets. J Cheminform. 2014;6(Suppl 1):p19. doi:10.1186/1758-2946-6-S1-P19
49. Ibrahim TM, Bauer MR, Dorr A, Veyisoglu E, Boeckler FM. pROC-chemotype plots enhance the interpretability of benchmarking results in structure-based virtual screening. J Chem Inf Model. 2015;55(11):2297–2307. doi:10.1021/acs.jcim.5b00475
50. Shiraishi M, Aramaki Y, Seto M, et al. Discovery of novel, potent, and selective small-molecule CCR5 antagonists as anti-HIV-1 agents: synthesis and biological evaluation of anilide derivatives with a quaternary ammonium moiety. J Med Chem. 2000;43(10):2049–2063. doi:10.1021/jm9906264
51. Junker A, Schepmann D, Yamaguchi J, et al. Diverse modifications of the 4-methylphenyl moiety of TAK-779 by late-stage Suzuki-Miyaura cross-coupling. Org Biomol Chem. 2014;12(1):177–186. doi:10.1039/C3OB41873A
52. Vogel SM, Bauer MR, Boeckler FM. DEKOIS: demanding evaluation kits for objective in silico screening–a versatile tool for benchmarking docking programs and scoring functions. J Chem Inf Model. 2011;51(10):2650–2665. doi:10.1021/ci2001549
53. Boeckler FM, Bauer MR, Ibrahim TM, Vogel SM. Use of DEKOIS 2.0 to gain insights for virtual screening. J Cheminform. 2014;6(Suppl 1):O24. doi:10.1186/1758-2946-6-S1-O24
54. Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35(Databaseissue):D198–D201. doi:10.1093/nar/gkl999
55. Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9(10):430–431. doi:10.1016/S1359-6446(04)03069-7
56. Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322(2):339–355. doi:10.1016/S0022-2836(02)00777-5
57. Abadi AH, Ibrahim TM, Abouzid KM, et al. Design, synthesis and biological evaluation of novel pyridine derivatives as anticancer agents and phosphodiesterase 3 inhibitors. Bioorg Med Chem. 2009;17(16):5974–5982. doi:10.1016/j.bmc.2009.06.063
58. Abadi A, Al-Deeb O, Al-Afify A, El-Kashef H. Synthesis of 4-alkyl (aryl)-6-aryl-3-cyano-2(1H)-pyridinones and their 2-imino isosteres as nonsteroidal cardiotonic agents. Farmaco. 1999;54(4):195–201. doi:10.1016/S0014-827X(99)00004-X
59. Minlon H. A simple modification of the wolff-kishner reduction. J Am Chem Soc. 1946;68(12):2487–2488. doi:10.1021/ja01216a013
60. Shi F, Tu S, Fang F, Li T. One-pot synthesis of 2-amino-3-cyanopyridine derivatives under microwave irradiation without solvent. ARKIVOC. 2005;i:137–142.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.