Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Small Airways Disease, Biomarkers and COPD: Where are We?
Authors Chukowry PS ![]() , Spittle DA
, Spittle DA ![]() , Turner AM
, Turner AM ![]()
Received 4 September 2020
Accepted for publication 11 December 2020
Published 18 February 2021 Volume 2021:16 Pages 351—365
DOI https://doi.org/10.2147/COPD.S280157
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Priyamvada S Chukowry,1 Daniella A Spittle,2 Alice M Turner3
1Respiratory Research Department, University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK; 2Institute of Inflammation and Ageing, University of Birmingham, Birmingham, B15 2TT, UK; 3Institute for Applied Health Research, University of Birmingham, Birmingham, B15 2TT, UK
Correspondence: Alice M Turner
Institute for Applied Health Research, University of Birmingham, Birmingham, B15 2TT, UK
Tel +44 0121 3713885
; +44 07825683519
Email [email protected]
Abstract: The response to treatment and progression of Chronic Obstructive Pulmonary Disease (COPD) varies significantly. Small airways disease (SAD) is being increasingly recognized as a key pathological feature of COPD. Studies have brought forward pathological evidence of small airway damage preceding the development of emphysema and the detection of obstruction using traditional spirometry. In recent years, there has been a renewed interest in the early detection of SAD and this has brought along an increased demand for physiological tests able to identify and quantify SAD. Early detection of SAD allows early targeted therapy and this suggests the potential for altering the course of disease. The aim of this article is to review the evidence available on the physiological testing of small airways. The first half will focus on the role of lung function tests such as maximum mid-expiratory flow, impulse oscillometry and lung clearance index in detecting and quantifying SAD. The role of Computed Tomography (CT) as a radiological biomarker will be discussed as well as the potential of recent CT analysis software to differentiate normal aging of the lungs to pathology. The evidence behind SAD biomarkers sourced from blood as well as biomarkers sourced from sputum and broncho-alveolar lavage (BAL) will be reviewed. This paper focuses on CC-16, sRAGE, PAI-1, MMP-9 and MMP-12.
Keywords: spirometry, targeted treatment, normal aging, pathological process
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous disorder, characterized by irreversible airway obstruction.1 It is widely recognized that disease of the small airways contributes hugely to this obstruction.2,3 Colloquially referred to as the “silent zone” of the lungs, it is estimated that 75% of the small airways must first be destroyed before changes in standard lung function are observed.4 The number of small airways is reportedly lower in mild COPD patients, compared to controls, with the number depleting with increasing GOLD stage.5
Understanding of early pathological changes is particularly important, given the suggestion that disease of the small airways precedes emphysema in COPD patients; micro-CT performed in ex vivo lungs revealed loss of terminal bronchioles occurred before emphysematous changes.5,6 In a recent longitudinal study, 70% of a COPD cohort followed a disease progression trajectory, whereby small airway and emphysematous changes preceded large airway abnormalities.7
Despite the advent of several physiological tests of the small airways, none have yet been validated for use in the clinical assessment of COPD. The aim of this review is to discuss current physiological testing of the small airways, and how the underlying mechanisms may contribute to our understanding. We will allude to the use of potential biological markers that, following further validation, may serve as predictors of early disease and disease progression.
Methods
Using Ovid, the following databases – Medline (1946 to April 20) and Embase (1974 to April 2020) including the Cochrane Library were searched. The search was limited to papers written in English and that had been published in the last 20 years. To ensure a thorough review we conducted literature searches using the following terms: “small airways disease”, “COPD”, “spirometry”, “oscillometry”, “lung clearance index/LCI”, “maximum mid-expiratory flow/MMEF”, “Computed Tomography/CT”, “Parametric Response Mapping/PRM”, “biomarker”, “sputum”, “bronchoalveolar lavage/BAL”, “plasma”, “serum”. Boolean Operators AND and OR were used to find the most relevant papers. Titles and abstracts were reviewed within the study team for inclusion and relevant findings summarized within sections that focus on each pathophysiological mechanism or biomarker in turn. Papers older than 20 years were used as reference when there was no other suitable latest alternative.
Lung Function Testing to Detect SAD
As mentioned earlier, COPD is a heterogenous disorder.8,9 Its clinical presentation, progression, clinical response to treatment, imaging and physiological changes vary significantly. The main pathological processes driving COPD include emphysema, large airway inflammation and small airway disease, each varying in extent and in severity in patients.10,11 These pathological processes are triggered by the complex interaction between cigarette smoke, environmental toxicants, genetics and infectious agents.12,13 Bacterial and viral insults have been shown to disrupt host immune defense systems in patients with COPD; tuberculosis is well known for causing bronchiectasis and COPD patients with a history of tuberculosis are well known to have worse outcomes.14–19
The small size of small airways renders imaging difficult and their inaccessibility makes obtaining biopsies challenging. Hence, they have proven difficult to study and this explains the paucity of data about small airways disease in the literature. However, over the last years, there has been a renewed interest in small airways disease. This renewed interest has led to the emergence of an increased demand for methods to identify and quantify small airway disease accurately. Currently, research efforts are being aimed at early detection and targeting of therapies even before symptoms occur and traditional spirometry results are abnormal. In this section of the review, we will explore the different clinical tests available to measure small airways disease.
Expiratory Flow
Several tests are available to assess small airways. A recent study conducted by Stockley et al20 showed that reduced Maximal Mid-Expiratory (MMEF) Flow is an early indicator of lung damage in patients with alpha-one antitrypsin deficiency (AATD). Not only did the reduction in MMEF precede spirometry abnormality but it also preceded detecting of emphysema using CT. As not all patients with AATD develop COPD, MMEF would be useful in predicting who are at higher risk of developing clinically significant disease.
Whilst other work has validated the use of MMEF to detect small airway disease,21 MMEF is not routinely used in clinical practice for several reasons. Firstly, it is highly dependent on FVC hence a false picture of airflow obstruction can be obtained if the patient has a smaller body habitus than the average for his/her height, ethnicity, sex and age; whilst use of lower limit of normal could ameliorate this error interpretation it may not eliminate it completely. Secondly, MMEF is derived from spirometry results which is effort dependent. Hence older patients, patients with poor physical health and those with cognitive impairment will struggle to carry out this test.22,23
Additionally, a study published by Bhatt et al showed that the use of all data points on expiratory spirometry curves to assess their shape is more sensitive to the detection of mild airway obstruction. The novel indices they used included Parameter D (derived from the shape of the volume–time curve) as well as Transition Point and Transition Distance (both derived from flow–volume curves). These new and easily obtainable metrics were found to pick up small airway obstruction which was previously missed using traditional methods.24 Similarly, the Peak Index, the number of peaks in the descending part of the expiratory flow–volume curve (adjusted for lung volume), is another novel spirometry metric with potential to help in detection of airflow obstruction and prognostication.25 Unfortunately, detailing how these novel indices are calculated is beyond the scope of this paper.
Impulse Oscillometry
Impulse Oscillometry (IOS) is a non-invasive pulmonary function test which involves superimposing soundwaves onto tidal breathing to measure respiratory mechanics. The sound waves emitted vary in frequency (usually between 5 and 25 Hz); higher frequencies penetrate proximal airways whereas lower frequencies penetrate the more distal airways.22 The resistance and reactance readings for different frequencies can be translated into the mechanical properties of different regions of the lungs. R20 is the resistance measured at 20Hz and represents the resistance of the large airways. R5 is the resistance measured at 5Hz and represents the total airway resistance. The difference between these two values (R5-R20) represents the resistance of small airways. Hence, one key advantage of IOS is its ability to differentiate between small and large airway obstruction. IOS also provides information about elastic recoil of the small airways through the measurement of X5, the reactance at 5Hz. A decrease in elasticity is reflected by a more negative X5.26 Wei et al27 have shown that IOS had good correlation with the GOLD staging and several other studies have shown IOS to be more sensitive than spirometry in detecting small airways disease. For example, results obtained from 488 male firefighters found that IOS identified airway abnormalities even though spirometry results were within range.28 Mondal et al29 found that IOS was more sensitive than spirometry in detecting bronchodilator response in children with sickle-cell disease. Saadeh et al30 also showed that IOS had greater sensitivity than spirometry for monitoring bronchodilator response in patients with COPD. Data from the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) trial showed that smokers without COPD had an elevated AX (integrated area of low-frequency reactance which is a marker of small airway abnormality) but normal CT-scan densitometry. Moreover, although their FEV1 was within normal range, it was lower when compared to non-smoking controls.31 This adds to the body of evidence that IOS plays a valuable role in detecting small airways disease.
IOS has several advantages. It requires minimal cooperation from the patient and is effort independent. Hence, unlike MMEF, the elderly, those with poor physical reserve and cognitive impairment find it less challenging to perform IOS. It can also be used in patients post-surgery and acute-coronary syndromes.
Limitations of IOS include bulky and expensive equipment; unlike spirometry IOS needs a laboratory. Moreover, there are no set reference values for different respiratory conditions and there is high intra-subject variability.26,32
Lung Clearance Index (LCI)
LCI is a marker of deranged ventilation and is considered to reflect small airway abnormalities. It is more sensitive than traditional spirometry in detecting small airway disease.33–35 LCI is obtained by measurement of the elimination of an inert tracer gas such as nitrogen or helium through tidal breathing. Small airway disease causes unevenness in ventilation and this results in the inert gas taking longer to be washed out from the lungs and hence require a greater number of breaths. From these washout tracings, the LCI can be calculated.36
Studies have shown that LCI can be used to predict exacerbations in cystic fibrosis (CF) patients.37 LCI is currently used as an endpoint in clinical trials but there is currently limited data about its utility in routine clinical care of CF patients.38,39
LCI offers various advantages. It is effort independent as it involves normal tidal breathing and hence can be done by elderly patients. Moreover, unlike spirometry, it is independent of gender, age and height33,36 and hence the interpretation of LCI is more straightforward. It has been shown to have good reproducibility in patients with stable COPD.40
There are however several barriers that prevent LCI from being used as a biomarker for COPD. Fähndrich et al41 found that measurement of LCI in patients who were on oxygen led to invalid results. In addition, they found that LCI did not differentiate GOLD II–IV and did not correlate with 6-MWT. However, the high drop-out rate in this study could have contributed to these results. Furthermore, Horsley36 has argued that in patients with severe obstruction, therapeutic interventions targeting infection and inflammation may increase the heterogeneity between adequately and poorly ventilated parts of the lung hence worsening LCI.
Computed Tomography
Computed Tomography (CT) is the most commonly used imaging tool in clinical practice to assess the extent and distribution of emphysema. As high-resolution CT (HRCT) can only detect airways more than 2mm, microCT has been used in studies assessing the degree of small airway disease. One such landmark study by McDonough et al found that in patients with various stages of COPD as defined by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) scale and in lungs removed from COPD patients who underwent transplantation, small airway narrowing as measured by microCT preceded the development of emphysema and could explain the increase in airway resistance present in COPD patients. Small airway narrowing was defined as the reduced number of small airways measuring between 2.0mm and 2.5mm in diameter detected by microCT when compared to controls.42 A very recent study conducted by Young et al11 was able to demonstrate, for the first time, 2 trajectories in the progression of COPD: a “Tissue to Airway” subtype and an “Airway to Tissue” subtype. This study used a technique called Subtype and stage Inference (SuStain) to analyze CT images of GOLD 1–4 patients and controls from the COPDGene study. SuStain is a novel computational technique with the ability to separate temporal and phenotypical heterogeneity in order to identify subtypes and hence patterns of disease progression. They found that in the “Tissue→Airway” subtype (n = 2354, 70.4%), small airway dysfunction and emphysema occurred before large airway wall abnormalities whereas in the “Airway→Tissue” subtype (n = 988, 29.6%), large airway abnormalities preceded small airway dysfunction and emphysema. Interestingly, they were able not only to verify that disease subtype remained the same at 5-year follow-up but they were also successful in demonstrating that patients in GOLD 1–2 had a steeper stage progression compared to GOLD 3–4 patients and described a steeper initial decline in FEV1 in patients belonging to the “Airway→ Tissue” subtype. This was particularly important since previous studies had reported opposite results when assessing disease activity in early disease.9,43,44 This study also identified patients with normal spirometry at risk of developing COPD. Therefore, CT images analyzed using SuStain could serve as a biomarker of early COPD and subtype hence allowing for targeted therapy. Moreover, it could be used in clinics to stratify patients, monitor progression and assess treatment effect.
Biomarkers to Measure SAD
Biomarkers give indication to both normal physiological processes and pathological processes and are a useful method in observing clinical progression of disease, monitoring response to therapeutic intervention and for understanding the underlying mechanisms of pathogenesis in a research setting.45 Exposure to toxicants, such as prolonged cigarette smoke inhalation, may influence biomarkers by having direct effects on the small airway epithelium (SAE).46
Pulmonary biomarkers can be investigated using various biofluids; sputum, bronchoalveolar lavage (BAL) and blood. Biomarkers detectable in induced or spontaneous sputum, and BAL, reflect the local environment within the lung where blood biomarkers represent the periphery and are therefore more indicative of a systemic process. Several studies have evaluated the source of biomarkers in lung disease, assessing the efficacy of each biofluid in giving an accurate representation of clinical parameters such as severity and prognosis.47,48 Such studies have concluded that sputum, either induced or spontaneous, appears to be the superior source of biomarkers in COPD cohorts. More research is required, however, to investigate longitudinal progression of sputum-derived biomarkers in larger cohorts as well as identifying those derived from the small airways. Whilst BAL also gives a clear representation of the small airways, the procedure for obtaining the fluid is invasive, thus impairing its utility and potentially its reproducibility. Despite not directly reflecting processes within the airways, blood biomarkers have been vastly studied. Although highly reproducible, blood biomarkers are highly confounded by other co-morbidities, particularly those that are associated with systemic inflammation. Further, biomarkers must first be in adequate concentrations within the lung for them to diffuse into the blood, thereby limiting detection to those biomarkers that are in abundance within the lung.
Ability to observe changes in the small airways is important for understanding pathological changes at an epithelial level. The narrow diameter (<2mm) of the small airways renders them particularly susceptible to particle deposition and subsequent inflammation49 such that at an epithelial level several alterations are observed in a diseased airway compared to healthy. Due to their position at the luminal interface, airway epithelial cells (AECs) are exposed to inhaled gases and particles.50 Particularly in response to smoke exposure, a significant risk factor for COPD development, cells of the SAE undergo various morphological and functional alterations (Figure 1).51 A diseased SAE undergoes excessive tissue repair, eventually resulting in airway remodeling52 with pseudostratified and ciliated epithelium transforming into a simple cuboidal type, and the cilia that remain becoming markedly shorter. Further metaplastic changes of ciliated cells occur, such that they differentiate to become squamous cells,53 and morphological and functional changes are observed, notably hyper- and metaplasia of secretory goblet cells. The combination of depleted ciliary function and goblet cell hyperplasia contributes to mucous hypersecretion, with impaired ability for mucus to be removed. This, in turn, culminates in lumen obstruction.54
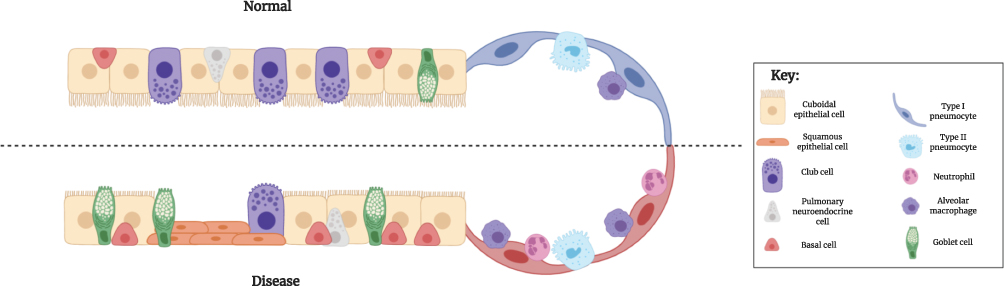 |
Figure 1 Epithelial differences between normal and diseased small airways. Ciliated, cuboidal epithelial cells constitute the majority of a normal bronchiolar epithelium. In a diseased SAE, however, these cell types undergo metaplasia and become a more squamous phenotype. Secretory goblet cells are found in small numbers within a normal SAE, but under diseased conditions, increase in prevalence. By contrast, the number of secretory club cells are reduced in a diseased SAE, compared to normal, with a subsequent reduction in the secretion of the protective protein, CC-16. Their proposed progenitor cell, the basal cell, is however present in greater abundance in a diseased epithelium, compared to normal. Abbreviations: SAE, small airway epithelium; CC-16, club cell protein 16. |
Increased immune cell infiltration within the alveoli, particularly alveolar macrophages and neutrophils, is characteristic of a diseased lung. Presence of these innate immune cells is driven by inhaled insults, namely chronic smoke exposure and microbial pathogens, which exacerbate airway remodeling through release of various proteolytic enzymes and inflammatory mediators.50
Fluctuations of biomarkers in SAD may also be influenced at a genetic level. Gene expression profiling of the SAE has revealed a distinct signature in a COPD cohort; increased expression of the keratin genes KRT5, −6a and −14 has been shown in COPD patients, compared to a healthy control cohort, and complements histological observations of squamous cell metaplasia. Further, cigarette smoke exposure appears to promote the expression of MUC5AC, and differentiation of goblet cells, as well as reduced expression of SCGB1A1 in club cells.55
The following proteins have been postulated as biological markers of SAD, based on their derivation in the small airways, links to epithelial processes or association with alveolar macrophages.
Club Cell Protein 16
Secretoglobin, family 1A, member 1 (SCGB1A1) is expressed almost exclusively by Club cells in the small airways.56 This SAE-enriched gene encodes uteroglobin, a secretory multifunctional protein that mediates both anti-inflammatory and immunomodulatory actions.57 Uteroglobin, also referred to as Club cell protein 16 (CC16), readily diffuses along a concentration gradient from the respiratory tract into the bloodstream. Although found in greater abundance in BAL fluid, CC16 can be detected in serum, urine and sputum.58 Hence, serum concentration of CC16 has been suggested as a biomarker of lung epithelium permeability and integrity.59 In fact, several studies have demonstrated an association with serum concentration of CC16 and lung function decline. In the ECLIPSE study cohort, serum CC16 levels were lower in COPD patients compared to smokers without COPD60 and over a 3 year period, lower CC16 levels were associated with lung function decline. In a separate cohort, a longitudinal study showed an association between low serum CC16 levels and accelerated lung function decline.61 Using a cohort grouped into sustained quitters, intermittent quitters and continued smokers, the group demonstrated the lowest serum CC16 levels in the latter group.62
Whilst these studies have described a clear association with CC16 levels and lung function, it may be worthy to investigate the role of CC16 in “earlier” stage COPD, perhaps measuring levels in asymptomatic smokers. Given the high specificity of club cells to the small airways, coupled with their abundance, this may be a suitable biomarker of SAD.
Receptor for Advanced Glycation Endproducts (RAGE) and Its Ligands
Receptor for Advanced Glycation Endproducts (RAGE) is a well-studied biomarker of inflammatory disease, with increasing studies suggesting its role in small airways disease.63–65
With constitutive expression in type I pneumocytes, RAGE is expressed at uniquely high levels within the lungs.66–69 Common ligands of RAGE include advanced glycation endproducts (AGEs), S100/calgranulin proteins and high mobility box group 1 protein (HMGB1).70 RAGE recognizes the binding of these ligands to the transmembrane isoform (mRAGE), based on their 3D structure, to initiate downstream signaling, culminating in the NF-kB-transcription of various pro-inflammatory cytokines.71 No known negative feedback system has been reported between RAGE and its ligands and rather, it would appear that RAGE ligands accumulate, thereby perpetuating an inflammatory response.72
A soluble isoform of RAGE (sRAGE) acts as a decoy receptor by binding and clearing ligands of RAGE within extracellular fluid.72–74 Binding of ligands to sRAGE, rather than mRAGE, prevents the downstream inflammatory cascade and for such reason, it is believed the presence of sRAGE serves as a protective mechanism (Figure 2).75 Using a murine model, it was found that treatment with recombinant sRAGE significantly reduced neutrophil infiltration and NF-kB activation.76 In addition, a marked reduction of pro-inflammatory cytokine production was observed, with less TNF-ɑ and macrophage inflammatory proteins (MIP-1) present in the sRAGE treated cohort compared to the vehicle.
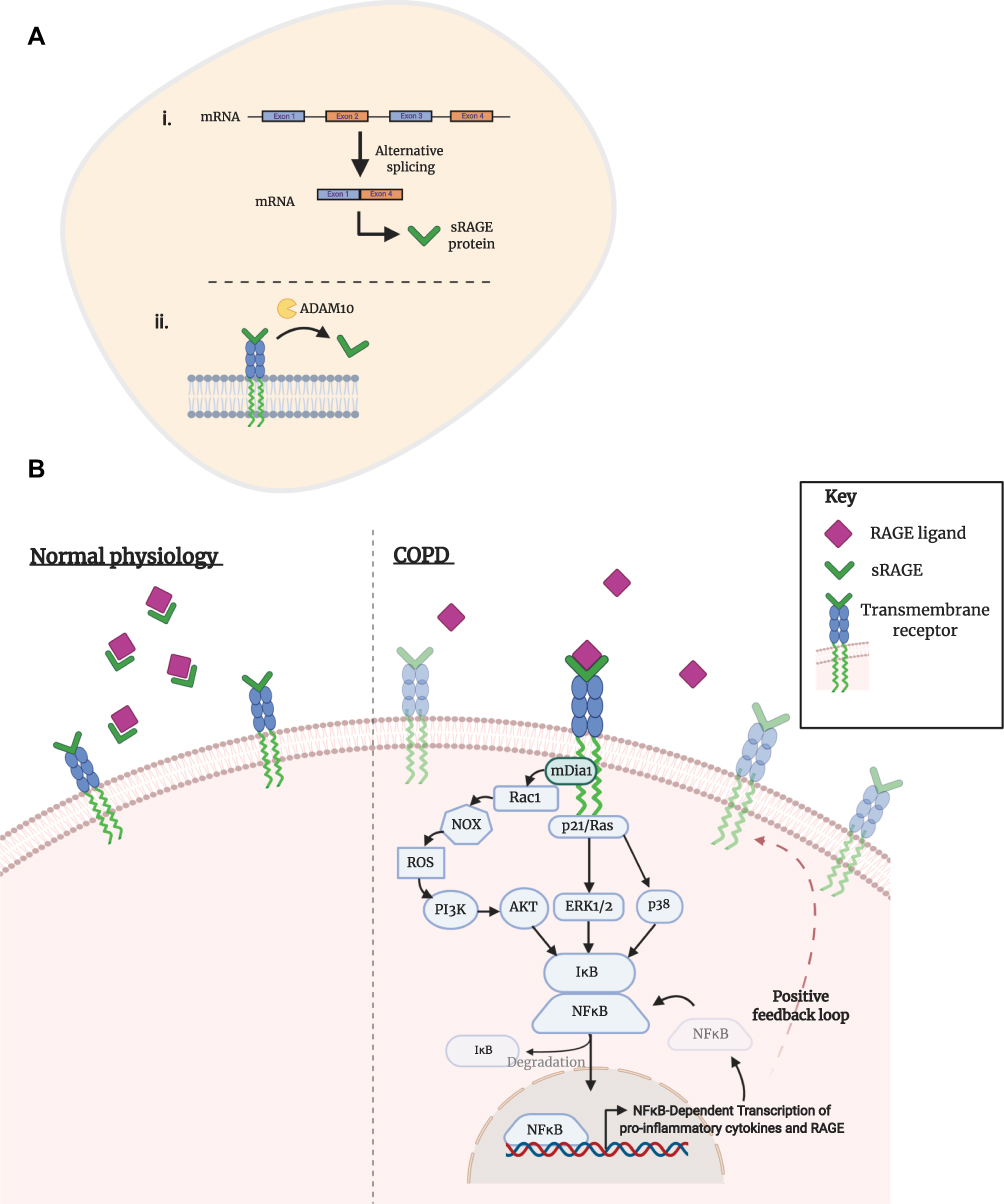 |
Figure 2 Activation of transmembrane RAGE in COPD. (A) The soluble isoform of receptor for advanced glycation endproducts (sRAGE) can arise from (i) alternative mRNA splicing or (ii) through ADAM10-mediated proteolytic cleavage of the transmembrane isoform. (B) Under normal physiological conditions, sRAGE performs as a decoy receptor; ligands of RAGE preferentially bind to the soluble isoform, creating a complex that is then degraded. Studies in the context of COPD have shown an enhanced expression of transmembrane RAGE and its ligands, as well as a decreased concentration of sRAGE. Consequently, RAGE ligands are able to bind to their cognate receptor, initiating a downstream pathway that eventuates in an inflammatory response. Dia-1, a Rho effector protein, is bound to the C-terminal of RAGE and is responsible for activation of a Rho-like small GTPase: Rac1. NADPH oxidases (NOXes) signal through Rac1 to generate downstream reactive oxygen species (ROS). ROS are an intracellular signaling intermediate, which increase tyrosine kinase activity and initiate the PI3K/Akt pathway. Activation of Akt promotes the nuclear translocation of NFkB and subsequent transcription of several genes, including pro-inflammatory cytokines TNF-ɑ and interleukin- (IL-) 1, 2, 6, 8 and 12. Alternative pathways of NFkB initiation are achieved through p21/Ras-ERK1/2 or p38 activation. A positive feedback loop is initiated by the NFkB transcription of RAGE. |
Several studies have described involvement of RAGE in the presence of COPD, showing a particular association with emphysema.73,77 Studies have investigated the levels of sRAGE in both systemic biofluids, such as serum and plasma, as well as localized fluids such as BAL and sputum.63,64,75,78
Using the COPDgene cohort, Carolan et al79 discovered that lower plasma concentrations of sRAGE were associated with increased emphysema.79 Similarly, using the ECLIPSE cohort, higher serum levels of sRAGE were associated with less emphysema.80 This trend appears to extend to other SAD-related pathologies; reduced levels of sRAGE have been detected in the BAL of both COPD patients and persistent asthmatics, compared to a healthy control cohort.64
Whilst the RAGE-signaling pathway may initially perform as a protective mechanism against lung insults, including cigarette smoke, sustained and constitutive expression of RAGE is likely to contribute to the pathogenesis of chronic lung disease.
It is likely that the observed increase in RAGE signaling in COPD is both by result of increased RAGE expression and decreased levels of local sRAGE within the lung. Assessment of RAGE and/or sRAGE may be a useful indicator of alveolar damage, particularly type I pneumocytes. Given its detectability within serum, RAGE may be a potential biomarker of SAD, though whether it is sufficiently specific to this phenotype remains to be determined.
Plasminogen Activator Inhibitor-1 (PAI-1)
Plasminogen activator inhibitor (PAI-1) is a serine protease inhibitor which has been associated with a number of inflammatory and metabolic conditions such as diabetes and cardiovascular disease.81–83 Several studies have reported an association between PAI-1 levels in the serum and sputum of COPD patients.84,85 Given the suggested inhibition of alveolar epithelial repair by PAI-1,86 elevated levels of the protease inhibitor may contribute to the pathology of SAD. As with markers centered on RAGE, whether PAI-1 is specific for SAD is not yet clear, though association with pathophysiological processes in the airway epithelium suggests potential for this.
In a cohort of subjects with cystic fibrosis or COPD, PAI-1 levels were elevated in the induced sputum, when compared to healthy controls.87 Similarly, sputum PAI-1 levels were increased in COPD patients, compared to smokers without COPD and healthy, non-smokers.88 Authors of the latter study suggested PAI-1 levels were closely associated with oxidative stress-induced NF-kB activation in sputum macrophages. Further suggestion of the association of airway macrophages with PAI-1 levels was described by Wang et al.85 Comparison of a cohort of COPD patients with smokers without COPD and healthy non-smokers demonstrated elevated serum levels of PAI-1 in the former. Following further division of the COPD cohort, based upon their smoking status, it was shown that PAI-1 was highest in the serum of COPD smokers. Moreover, serum PAI-1 levels appeared to positively associate with matrix metalloproteinase-9 (MMP-9) and its inhibitor, tissue inhibitor of matrix metalloproteinase-1 (TIMP-1).
Assuming both the association of PAI-1 with metabolic and cardiovascular diseases, and the prevalence of these morbidities in COPD patients, it was suspected that this may be the cause of PAI-1 elevation. This hypothesis was assessed in a cohort of moderate-to-severe COPD patients (GOLD stage II and III), on the premise that these groups display the highest prevalence of co-morbidities.89,90 As with previous studies, serum PAI-1 levels were elevated in the COPD cohort, with levels highest in GOLD stage II and III patients.84 Importantly, COPD was an independent predictor for higher PAI-1 levels following adjustment for age, sex, BMI and the presence of several comorbidities (such as diabetes, atherosclerosis and history of coronary artery disease). Smoking status and statin use did not appear to confound the level of PAI-1.
Immunohistochemistry (IHC) staining of lung tissue revealed alveolar macrophages and epithelia as a major source of PAI-1 in the airways,88 consistent with studies that showed PAI-1 expression in alveolar macrophages and epithelia in murine models of pulmonary fibrosis.91,92 The upregulation of PAI-1 in COPD is potentially attributed to the prevalence of oxidative stress within the lungs of this population.88 Oxidant-induced nuclear translocation of NF-kB triggers the expression of PAI-1 and several other inflammatory genes, notably in alveolar macrophages.93
Matrix Metalloproteinases (MMPs)
Matrix metalloproteinases (MMPs) are a family of enzymes that are involved in the degradation of extracellular matrix (ECM) proteins and are essential for tissue repair under normal physiological conditions.94 Within the airways, MMPs are principally produced and secreted by alveolar macrophages.95 Categorized based upon the type of ECM they degrade, a number of MMPs have been implicated in the pathogenesis and protease-antiprotease imbalance, characteristic of COPD (Figure 3). Tissue inhibitor of metalloproteinases (TIMPs) are the principle inhibitors of MMPs; measurement of TIMPs, alongside MMPs, is crucial for the understanding of the protease-antiprotease imbalance.96–99 Strong associations between MMP levels and presence of SAD, as confirmed by FEF25%-75% measurement (analogous to MMEF), have been reported.100 In spite of this, the exact role of MMPs in disease pathogenesis remains elusive and several conflicting studies suggest a complex mechanism (Table 1).
 |
Figure 3 Prolonged smoke exposure initiates various pathways, resulting in disease of the small airways and eventuating in the onset of emphysema. Upon exposure to smoke, both alveolar macrophages and epithelial cells within the small airways release various chemoattractants, including interleukin (IL)-8, monocyte chemoattractant protein (MCP)-1, IL-1B, tumor necrosis factor (TNF)-α and macrophage inflammatory protein (MIP)-1β. These, in turn, recruit neutrophils, monocytes and lymphocytes and promote their infiltration of the small airways. Alongside alveolar macrophages, neutrophils are a source of matrix metalloproteinases (MMPs) within the lungs. MMPs have been shown to contribute to several of the phenotypic changes in SAD, namely remodeling, inflammation, mucus hypersecretion and airflow obstruction. Recruited monocytes differentiate into macrophages, contributing almost to a positive feedback loop. Lymphocytes represent a major source of cytokines, particularly Th1 cytokines such as IFN-γ, which contribute greatly to local inflammation. Reactive oxygen species (ROS) generation by neutrophils inhibits the action of antiprotease, alpha-1 antitrypsin (AAT), thus causing an imbalance with its cognate protease, neutrophil elastase. Similarly, the antiprotease activity of TIMPs is hindered by ROS. By result, a large imbalance is observed, in favor of protease activity. Cumulatively, these prior pathways culminate in tissue remodeling and elastin degradation within the alveoli, leading to emphysema. Elastin fibers, as liberated by elastin degradation, activate alveolar macrophages. |
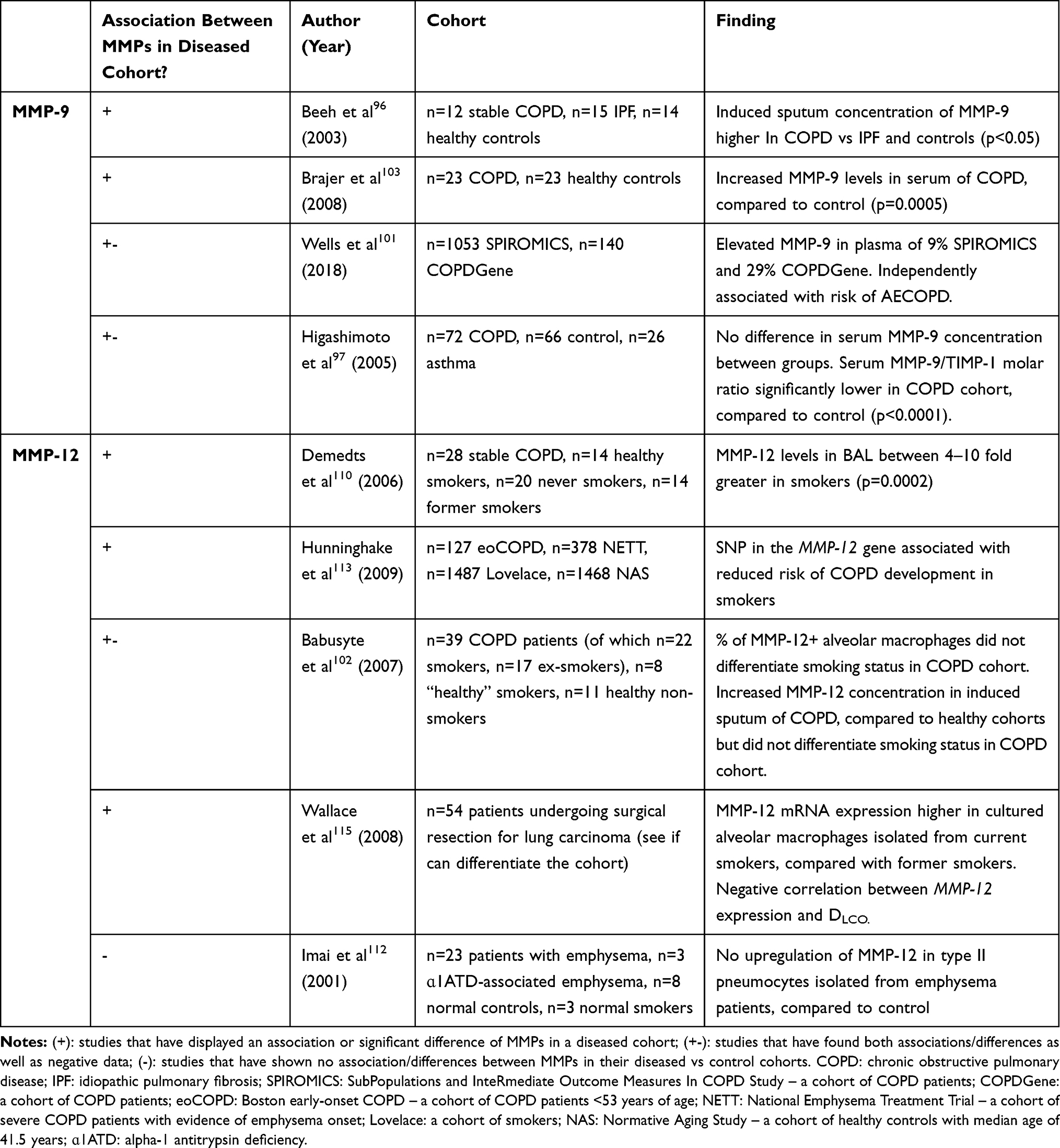 |
Table 1 Summary of Evidence Surrounding the Role of MMP-9 and -12 in COPD and Emphysema |
MMP-9
MMP-9, a gelatinase, has especially attracted much attention for its role in COPD. Due to its ability to degrade extracellular matrix proteins, promote neutrophil chemotaxis and mediate inflammation, MMP-9 is believed to contribute to the development of emphysema.101,102 Minimal evidence exists for the role of MMP-9 in SAD, specifically; however given the abundance of alveolar macrophages within the small airways, it is likely that MMP-9 is a contributor to the pathogenesis of SAD as well as emphysema, or when airways disease is a precursor to emphysema.
Multiple cross-sectional studies have reported elevated levels of MMP-9 in COPD cohorts and inverse association with FEV1.96,97,103,104 Within 2 established COPD cohorts, SPIROMICS and COPDGene, elevated plasma MMP-9 concentrations appeared to predict the risk of acute exacerbation in COPD.101 Across both cohorts, elevated MMP-9 levels occurred more frequently in the current smokers but did not associate with lung function, smoking history (in pack years) or presence of comorbidities. In another cohort, levels of circulating MMP-9 appeared to sequentially increase with progression of GOLD stage, with the highest levels observed in emphysematous patients.105
Whilst protein levels of MMP-9 may be altered in COPD, the proteolytic activity does not appear to significantly differ from non-COPD subjects.106,107 In fact, reduced MMP-9 activity was associated with increased airflow obstruction in a small cohort of COPD patients.106 A possible explanation for this may be due to the observed increased levels of TIMP-1 in COPD subjects, which would inhibit the proteolytic capacity of the MMPs.108
MMP-12
The involvement of MMP-12, an elastase, has also been discussed as playing a role in the underlying disease mechanism of COPD. Early studies showed that MMP-12 knockout mice did not develop emphysema following cigarette smoke exposure109 and in humans, MMP-12 levels in BAL were shown to be up to 10-fold greater in smokers.110
Several conflicting reports have questioned the importance of MMP-12 in the context of COPD. No differences in the level of MMP-12 in the induced sputum between COPD patients and a control cohort were found.111 At a transcriptomic level, MMP-12 did not appear to be upregulated in smokers,112 contrasting a landmark study that showed an association between a SNP in the MMP-12 gene and reduced risk of COPD development in smokers.113
In further support of the latter, MMP-12 expression was found to be higher in the alveolar macrophages of a current smoker COPD group, compared to ex-smoker COPD.114,115
In spite of conflicting evidence, it remains likely that MMP-12 is involved within the network of protease-driven mechanism of disease and rather the inconsistencies within the literature reflect the complexity. It may be the case that MMP-12 is not directly involved in the degradation of elastin within the lung,116 but instead contributes to alveolar damage through local macrophage recruitment, thus having a similar relationship to phenotype of COPD to MMP-9.109
MMP-8
The collagenase MMP-8 has also been associated with COPD, although to a lesser extent than MMP-9 and -12. In a small cohort of COPD patients, MMP-8 appeared to be upregulated in lung tissue, with expression being enriched in neutrophils located in the alveoli.117 At a systemic level, both MMP-8 mRNA levels in PBMCs, and plasma MMP-8 concentrations, were significantly higher in a COPD cohort, compared to the control group.118
To explore a possible association between smoking and changes in MMP expression, Ilumets et al used a cohort comprised of symptomatic smokers (stage 0, GOLD criteria) and healthy, asymptomatic smokers. Concomitant analysis of MMP-8, −9, and −12 identified MMP-8 as the most sensitive marker of COPD.107 The induced sputum of stage 0 COPD subjects contained higher MMP-8 levels, which significantly associated with small airway flow parameters (MEF50 and MEF25).
In order to minimize spurious correlations being suggested, it is important that the activity of proteases and antiproteases is measured. Measurement of protease and antiprotease concentrations alone may be not be sufficient for confirming their role in COPD; detection of zymogens, inactive enzymes or those bound with their cognate substrate may give a false representation of underlying pathophysiology.
Conflicting literature surrounding the involvement of MMPs in chronic lung disease may imply a complex network constitutes the underlying pathophysiology, though some studies could have been affected by small sample sizes.
Consolidation of data reporting alterations in MMP protein levels, and their localization within alveolar space, highlights them as a potential biomarker of small airway dysfunction. On the assumption that alveolar macrophages are a major source of MMPs within the airways, detection and measurement of these enzymes may be indicative of damage within the small airways.
Discussion
Small airway dysfunction contributes immensely to the pathogenesis of COPD.5,7 Current clinical detection of SAD involves the measurement of various physiological parameters, such as gas transfer, or the use of (high resolution) CT imaging.119
Such assessments are not always specific to small airway abnormalities and often require the use of specialized equipment, which limits their capacity in research. Hence, biological markers are required to improve the understanding of underlying mechanisms, which may in turn guide clinical management. Careful consideration must be taken in assessing and potentiating biomarkers. Firstly, the biomarker must display specificity to the small airways, with strong evidence supporting its role in the pathophysiology of disease. Secondly, and important in a clinical context, is the ability of the biomarker to be readily detectable in biofluids. Blood and induced sputum are non-invasive fluids that give representation of the systemic and local environments, respectively. Reproducibility is especially important for the monitoring of disease progression in individuals with COPD and may influence clinical management.
Whilst our review has delineated some plausible biomarkers of SAD it was also apparent from the results that a significant area of unmet need is the differentiation between SAD and emphysema in COPD patients. Currently, only limited studies have shown an association between biomarker levels and other physiological measures of small airway function. It remains unclear whether the biological markers are associated with SAD alone, or both SAD and emphysema. Further research must be conducted that investigates the presence of biological markers with already-established physiological measures of SAD, with consideration for possible overlap between the two disease phenotypes.
In order to better target therapy, it is crucial to differentiate age-related decline in function of tissues to disease. Physiological “normal” aging of the lung involves loss of elastic recoil,120 increased air trapping,121 hyperinflation,122 decline in FEV1.123 Due to the similarities between emphysema and age-related gas trapping, particularly on CT, controversies exist over whether emphysema is a feature of aging in healthy elderly people.124,125 Small airway abnormalities also occur as a normal part of the aging process. However, when these small airway abnormalities develop earlier and to a greater extent, a disease develops – small airway disease. COPD has been described as “accelerated aging of the lung”.126,127 Specialized CT analysis software with the ability to better visualize small airways, whilst also quantifying emphysema may be one way to aid differentiation of aging and disease.
Parametric Response Mapping is one such recent CT analysis software with the ability to differentiate emphysema from gas trapping associated with functional small airway abnormality (PRMFSA). Martinez et al used PRM to analyze inspiratory/expiratory CT imaging of patients without airflow obstruction or mild to moderate airflow obstruction involved in SPIROMICS (Subpopulations and Intermediate Outcome Measures in COPD Study) and made several important observations. Firstly, they found a strong link between PRMFSA and increased FVC. Secondly, in patients with the same degree of airway obstruction, those who were older were subjected to a greater extent of PRMFSA. Moreover, in those without airflow obstruction PRMFSA increased with age, irrespective of respiratory symptoms and smoking status. These findings help to shed more light on normal aging of the lungs and what can be classified as disease. Small airway abnormality associated with normal aging have minimal effect of FEV1 decline but contribute to increase FVC (by gas trapping). Hence healthy, elderly patients are at risk of being wrongly diagnosed with COPD due to their FEV1/FVC ratio being less than <0.7.128 Furthermore, they found a low burden of emphysema in elderly patients without airflow obstruction.129 This study provides evidence that the development of emphysema does not form part of the normal physiological process of aging.
The identification of the key role played by small airways in the pathogenesis of asthma and COPD has resulted in a need for more effective drug delivery mechanisms to the small airways. The ability of extrafine drug particles to be better distributed in the small airways has been associated with better asthma control.130,131 However, a study investigating the effectiveness of beclomethasone dipropionate/formoterol fumarate (BDP/FF) extrafine combination versus fluticasone propionate/salmeterol (FP/S) combination in COPD patients (NCT01245569) found that both drugs improved shortness of breath to the same extent although BDP/FF had faster onset of bronchodilation.132 As this study used changes in FEV1 as outcome measure, it can be argued that FEV1 is not a sensitive measure of the functional status of the small airways and results might have been different if pulmonary function tests more sensitive at measuring the function of small airways were used.
Conclusion
It is crucial not only to detect small airways disease early so that treatment can be targeted to slow down the trajectory of disease but also to be able to separate normal aging from a pathological process. Whilst specialized radiological and physiological tests in early disease show promise, addition of biomarkers of pathological processes is in its infancy such that further research in highly phenotyped patients is warranted.
Acknowledgment
All figures have been created using Biorender.com.
Disclosure
AMT has current research grants from AstraZeneca and Chiesi, both of whom manufacture products which could be used in SAD and/or COPD.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
1. Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(4):347–365.
2. Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Eng J Med. 1968;278(25):1355–1360.
3. Koo H-K, Vasilescu DM, Booth S, et al. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir Med. 2018;6(8):591–602.
4. Cosio M, Ghezzo H, Hogg JC, et al. The relations between structural changes in small airways and pulmonary-function tests. N Eng J Med. 1978;298(23):1277–1281.
5. McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Eng J Med. 2011;365(17):1567–1575.
6. Hogg JC, McDonough JE, Suzuki M. Small airway obstruction in COPD: new insights based on micro-CT imaging and MRI imaging. Chest. 2013;143(5):1436–1443.
7. Young AL, Bragman FJS, Rangelov B, et al. Disease progression modeling in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2019;201(3):294–302.
8. Agusti A, Calverley PM, Celli B, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res. 2010;11:122.
9. Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365(13):1184–1192.
10. Timmins SC, Diba C, Farrow CE, et al. The relationship between airflow obstruction, emphysema extent, and small airways function in COPD. Chest. 2012;142(2):312–319.
11. Young AL, Bragman FJS, Rangelov B, et al. Disease progression modeling in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2020;201(3):294–302.
12. Molfino NA, Coyle AJ. Gene-environment interactions in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3(3):491–497.
13. Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122(8):2749–2755.
14. Yakar HI, Gunen H, Pehlivan E, Aydogan S. The role of tuberculosis in COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:323–329.
15. Sarkar M, Madabhavi I, Kumar K. Tuberculosis associated chronic obstructive pulmonary disease. Clin Respir J. 2017;11(3):285–295.
16. van Kampen SC, Wanner A, Edwards M, et al. International research and guidelines on post-tuberculosis chronic lung disorders: a systematic scoping review. BMJ Glob Health. 2018;3(4):e000745–e000745.
17. Guo-Parke H, Linden D, Weldon S, Kidney JC, Taggart CC. Mechanisms of virus-induced airway immunity dysfunction in the pathogenesis of COPD disease, progression, and exacerbation. Front Immunol. 2020;11.
18. Chow DP, Chung W Role of viral infections in asthma and chronic obstructive pulmonary disease. http://dxdoiorg/101165/rcmb2006-0199TR. 2012.
19. Singanayagam A, Joshi PV, Mallia P, Johnston SL. Viruses exacerbating chronic pulmonary disease: the role of immune modulation. BMC Med. 2012;10(1):27.
20. Stockley JA, Ismail AM, Hughes SM, Edgar R, Stockley RA, Sapey E. Maximal mid-expiratory flow detects early lung disease in α. Eur Respir J. 2017;49:3.
21. McFadden ER, Linden DA. A reduction in maximum mid-expiratory flow rate: a spirographic manifestation of small airway disease. Am J Med. 1972;52(6):725–737.
22. Stockley JA, Cooper BG, Stockley RA, Sapey E. Small airways disease: time for a revisit? Int J Chron Obstruct Pulmon Dis. 2017;12:2343–2353.
23. Quanjer PH, Weiner DJ, Pretto JJ, Brazzale DJ, Boros PW. Measurement of FEF25-75% and FEF75% does not contribute to clinical decision making. Eur Respir J. 2014;43(4):1051–1058.
24. Bhatt SP, Bhakta NR, Wilson CG, et al. New spirometry indices for detecting mild airflow obstruction. Sci Rep. 2018;8(1):17484.
25. Bhatt SP, Bodduluri S, Raghav V, et al. The peak index: spirometry metric for airflow obstruction severity and heterogeneity. Ann Am Thorac Soc. 2019;16(8):982–989.
26. Bickel S. Impulse Oscillometry. Adv Respir Med. 2019;87(4).
27. Wei X, Shi Z, Cui Y, et al. Impulse oscillometry system as an alternative diagnostic method for chronic obstructive pulmonary disease. Medicine. 2017;96:46.
28. Schermer T, Malbon W, Newbury W. Spirometry and Impulse Oscillometry (IOS) for Detection of Respiratory Abnormalities in Metropolitan Firefighters. Respirology. 2010;15:6.
29. Mondal P, Yirinec A, Midya V. Diagnostic value of spirometry vs impulse oscillometry: a comparative study in children with sickle cell disease. Pediatr Pulmonol. 2019;54:9.
30. Midya V. Advantage of impulse oscillometry over spirometry to diagnose chronic obstructive pulmonary disease and monitor pulmonary responses to bronchodilators: an observational study. SAGE Open Med. 2015;3.
31. Crim C, Celli B, Edwards LD, et al. Respiratory system impedance with impulse oscillometry in healthy and COPD subjects: ECLIPSE baseline results. Respir Med. 2011;105(7):1069–1078.
32. Desiraju K, Agrawal A. Impulse oscillometry: the state-of-art for lung function testing. Lung India. 2016;33(4):410–416.
33. Horsley AR, Gustafsson PM, Macleod KA, et al. Lung clearance index is a sensitive, repeatable and practical measure of airways disease in adults with cystic fibrosis. Thorax. 2008;63:135.
34. Aurora P, Bush A, Gustafsson P, et al. Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. Am J Respir Crit Care Med. 2005;171(3):249–256.
35. Gustafsson PM, Aurora P, Lindblad A. Evaluation of ventilation maldistribution as an early indicator of lung disease in children with cystic fibrosis. Eur Respir J. 2003;22(6):972–979.
36. Horsley A. Lung clearance index in the assessment of airways disease. Respir Med. 2009;103(6):793–799.
37. Vermeulen F, Proesmans M, Boon M, Havermans T, De Boeck K. Lung clearance index predicts pulmonary exacerbations in young patients with cystic fibrosis. Thorax. 2014;69(1):39–45.
38. Poncin W, Lebecque P. [Lung clearance index in cystic fibrosis]. Rev Mal Respir. 2019;36(3):377–395.
39. Perrem L, Rayment JH, Ratjen F. The lung clearance index as a monitoring tool in cystic fibrosis: ready for the clinic? Curr Opin Pulm Med. 2018;24(6):579–585.
40. Daynes E, Soares M, Greening N, Owers-Bradley J, Singh S, Siddiqui S. P231 The feasibility and repeatability of the lung clearance index via multiple breath washout measurements in stable chronic obstructive pulmonary disease. 2018.
41. Fähndrich S, Lepper PM, Trudzinski F, Seibert M, Wagenpfeil S, Bals R. Lung Clearance Index is Increased in Patients with COPD – LCI Measurements in the Daily Routine | OMICS International. J Pulmonary Respir Med. 2016;6(3):1.
42. McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–1575.
43. Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1(6077):1645–1648.
44. Bridevaux PO, Gerbase MW, Probst-Hensch NM, Schindler C, Gaspoz JM, Rochat T. Long-term decline in lung function, utilisation of care and quality of life in modified GOLD stage 1 COPD. Thorax. 2008;63(9):768–774.
45. Doyle TJ, Pinto-Plata V, Morse D, Celli BR, Rosas IO. The expanding role of biomarkers in the assessment of smoking-related parenchymal lung diseases. Chest. 2012;142(4):1027–1034.
46. Seiler CL, JuM S, Kotandeniya D, et al. Inhalation exposure to cigarette smoke and inflammatory agents induces epigenetic changes in the lung. Sci Rep. 2020;10(1):11290.
47. Koutsokera A, Kostikas K, Nicod LP, Fitting J-W. Pulmonary biomarkers in COPD exacerbations: a systematic review. Respir Res. 2013;14(1):111.
48. Holz O, Waschki B, Roepcke S, et al. Potential prognostic value of biomarkers in lavage, sputum and serum in a five year clinical follow-up of smokers with and without COPD. BMC Pulm Med. 2014;14:30.
49. Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364(9435):709–721.
50. Hiemstra PS, McCray PB, Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J. 2015;45(4):1150.
51. Gohy ST, Hupin C, Pilette C, Ladjemi MZ. Chronic inflammatory airway diseases: the central role of the epithelium revisited. Clin Exp Allergy. 2016;46(4):529–542.
52. Hogg JC, Timens W. The Pathology of Chronic Obstructive Pulmonary Disease. Ann Rev Pathol. 2009;4(1):435–459.
53. Araya J, Cambier S, Markovics JA, et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest. 2007;117(11):3551–3562.
54. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–2247.
55. Gindele JA, Kiechle T, Benediktus K, et al. Intermittent exposure to whole cigarette smoke alters the differentiation of primary small airway epithelial cells in the air-liquid interface culture. Sci Rep. 2020;10(1):6257.
56. Lakind JS, Holgate ST, Ownby DR, et al. A critical review of the use of Clara cell secretory protein (CC16) as a biomarker of acute or chronic pulmonary effects. Biomarkers. 2007;12(5):445–467.
57. Mukherjee AB, Zhang Z, Chilton BS. Uteroglobin: a Steroid-Inducible Immunomodulatory Protein That Founded the Secretoglobin Superfamily. Endocr Rev. 2007;28(7):707–725.
58. Stenberg H, Wadelius E, Moitra S, et al. Club cell protein (CC16) in plasma, bronchial brushes, BAL and urine following an inhaled allergen challenge in allergic asthmatics. Biomarkers. 2018;23(1):51–60.
59. Van Miert E, Dumont X, Bernard A. CC16 as a marker of lung epithelial hyperpermeability in an acute model of rats exposed to mainstream cigarette smoke. Toxicol Lett. 2005;159(2):115–123.
60. Faner R, Tal-Singer R, Riley JH, et al. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax. 2014;69(7):666.
61. Guerra S, Halonen M, Vasquez MM, et al. Relation between circulating CC16 concentrations, lung function, and development of chronic obstructive pulmonary disease across the lifespan: a prospective study. Lancet Respir Med. 2015;3(8):613–620.
62. Park HY, Churg A, Wright JL, et al. Club Cell Protein 16 and Disease Progression in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2013;188(12):1413–1419.
63. Mulrennan S, Baltic S, Aggarwal S, et al. The role of receptor for advanced glycation end products in airway inflammation in CF and CF related diabetes. Sci Rep. 2015;5:8931.
64. Sukkar MB, Wood LG, Tooze M, et al. Soluble RAGE is deficient in neutrophilic asthma and COPD. Eur Respir J. 2012;39(3):721.
65. Wu L, Ma L, Nicholson LFB, Black PN. Advanced glycation end products and its receptor (RAGE) are increased in patients with COPD. Respir Med. 2011;105(3):329–336.
66. Fehrenbach H, Kasper M, Tschernig T, Shearman MS, Schuh D, Müller M. Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cell Mol Biol. 1998;44(7):1147–1157.
67. Dahlin K, Mager EM, Allen L, et al. Identification of Genes Differentially Expressed in Rat Alveolar Type I Cells. Am J Respir Cell Mol Biol. 2004;31(3):309–316.
68. Shirasawa M, Fujiwara N, Hirabayashi S, et al. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes to Cells. 2004;9(2):165–174.
69. Uchida T, Shirasawa M, Ware LB, et al. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med. 2006;173(9):1008–1015.
70. Buckley ST, Ehrhardt C. The receptor for advanced glycation end products (RAGE) and the lung. J Biomed Biotechnol. 2010;2010:917108.
71. Oczypok EA, Perkins TN, Oury TD. All the “RAGE” in lung disease: the receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr Respir Rev. 2017;23:40–49.
72. Hoonhorst SJM. Lo Tam Loi AT, Pouwels SD, et al. Advanced glycation endproducts and their receptor in different body compartments in COPD. Respir Res. 2016;17(1):46.
73. Miniati M, Monti S, Basta G, Cocci F, Fornai E, Bottai M. Soluble receptor for advanced glycation end products in COPD: relationship with emphysema and chronic cor pulmonale: a case-control study. Respir Res. 2011;12(1):37.
74. Haider SH, Oskuei A, Crowley G, et al. Receptor for advanced glycation end-products and environmental exposure related obstructive airways disease: a systematic review. Eur Respir Rev. 2019;28(151):180096.
75. Smith DJ, Yerkovich ST, Towers MA, Carroll ML, Thomas R, Upham JW. Reduced soluble receptor for advanced glycation end-products in COPD. Eur Respir J. 2011;37(3):516.
76. Zhang H, Tasaka S, Shiraishi Y, et al. Role of Soluble Receptor for Advanced Glycation End Products on Endotoxin-induced Lung Injury. Am J Respir Crit Care Med. 2008;178(4):356–362.
77. Sanders KA, Delker DA, Huecksteadt T, et al. RAGE is a Critical Mediator of Pulmonary Oxidative Stress, Alveolar Macrophage Activation and Emphysema in Response to Cigarette Smoke. Sci Rep. 2019;9(1):231.
78. Robinson AB, Johnson KD, Bennion BG, Reynolds PR. RAGE signaling by alveolar macrophages influences tobacco smoke-induced inflammation. Am J Physiol Lung Cell Mol Physiol. 2012;302(11):L1192–L1199.
79. Carolan BJ, Hughes G, Morrow J, et al. The association of plasma biomarkers with computed tomography-assessed emphysema phenotypes. Respir Res. 2014;15(1):127.
80. Cheng DT, Kim DK, Cockayne DA, et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(8):948–957.
81. Cesari M, Pahor M, Incalzi RA. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc Ther. 2010;28(5):e72–e91.
82. Alessi M-C J-VI. PAI-1 and the Metabolic Syndrome. Arterioscler Thromb Vasc Biol. 2006;26(10):2200–2207.
83. Yamamoto K, Takeshita K, Kojima T, Takamatsu J, Saito H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. 2005;66(2):276–285.
84. Waschki B, Watz H, Holz O, et al. Plasminogen activator inhibitor-1 is elevated in patients with COPD independent of metabolic and cardiovascular function. Int J Chron Obstruct Pulmon Dis. 2017;12:981–987.
85. Wang H, Yang T, Li D, et al. Elevated circulating PAI-1 levels are related to lung function decline, systemic inflammation, and small airway obstruction in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2016;11:2369–2376.
86. Lazar MH, Christensen PJ, Du M, et al. Plasminogen activator inhibitor–1 impairs alveolar epithelial repair by binding to vitronectin. Am J Respir Cell Mol Biol. 2004;31(6):672–678.
87. Xiao W, Hsu Y-P, Ishizaka A, Kirikae T, Moss RB, Cathelicidin S. Urokinase plasminogen activation system components, and cytokines discriminate cystic fibrosis, COPD, and Asthma Inflammation. Chest. 2005;128(4):2316–2326.
88. To M, Takagi D, Akashi K, et al. Sputum plasminogen activator inhibitor-1 elevation by oxidative stress-dependent nuclear factor-κB activation in COPD. Chest. 2013;144(2):515–521.
89. Cavaillès A, Brinchault-Rabin G, Dixmier A, et al. Comorbidities of COPD. Eur Respir Rev. 2013;22(130):454.
90. Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135(1):173–180.
91. Wygrecka M, Markart P, Ruppert C, et al. Cellular origin of pro-coagulant and (anti)-fibrinolytic factors in bleomycin-injured lungs. Eur Respir J. 2007;29(6):1105.
92. Ueno M, Maeno T, Nomura M, et al. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;300(5):L740–L752.
93. Caramori G, Romagnoli M, Casolari P, et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax. 2003;58(4):348–351.
94. Loffek S, Schilling O, Franzke CW. Biological role of matrix metalloproteinases: a critical balance. Eur Respir J. 2011;38(1):191.
95. Ishii T, Abboud RT, Wallace AM, et al. Alveolar macrophage proteinase/antiproteinase expression in lung function and emphysema. Eur Respir J. 2014;43(1):82.
96. Beeh KM, Beier J, Kornmann O, Buhl R. Sputum matrix metalloproteinase-9, tissue inhibitor of metalloprotinease-1, and their molar ratio in patients with chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and healthy subjects. Respir Med. 2003;97(6):634–639.
97. Higashimoto Y, Yamagata Y, Iwata T, et al. Increased serum concentrations of tissue inhibitor of metalloproteinase-1 in COPD patients. Eur Respir J. 2005;25(5):885.
98. Owen CA, Hu Z, Barrick B, Shapiro SD. Inducible expression of tissue inhibitor of metalloproteinases–resistant matrix metalloproteinase-9 on the cell surface of neutrophils. Am J Respir Cell Mol Biol. 2003;29(3):283–294.
99. Culpitt SV, Rogers DF, Traves SL, Barnes PJ, Donnelly LE. Sputum matrix metalloproteases: comparison between chronic obstructive pulmonary disease and asthma. Respir Med. 2005;99(6):703–710.
100. Ostridge K, Williams N, Kim V, et al. Relationship between pulmonary matrix metalloproteinases and quantitative CT markers of small airways disease and emphysema in COPD. Thorax. 2016;71(2):126.
101. Wells JM, Parker MM, Oster RA, et al. Elevated circulating MMP-9 is linked to increased COPD exacerbation risk in SPIROMICS and COPDGene. JCI Insight. 2018;3(22):e123614.
102. Xu X, Jackson PL, Tanner S, et al. A self-propagating matrix metalloprotease-9 (MMP-9) dependent cycle of chronic neutrophilic inflammation. PLoS One. 2011;6(1):e15781–e15781.
103. B B-gh B, Nowicka A, Kuznar-Kaminska B, Szczepanik A. Concentration of matrix metalloproteinase-9 in serum of patients with chronic obstructive pulmonary disease and a degree of airway obstruction and disease progression. J Physiol Pharmacol. 2008;59:145–152.
104. Linder R, Rönmark E, Pourazar J, Behndig A, Blomberg A, Lindberg A. Serum metalloproteinase-9 is related to COPD severity and symptoms - cross-sectional data from a population based cohort-study. Respir Res. 2015;16(1):28.
105. Uysal P, Uzun H. Relationship between circulating serpina3g, matrix metalloproteinase-9, and tissue inhibitor of metalloproteinase-1 and −2 with chronic obstructive pulmonary disease severity. Biomolecules. 2019;9(2):62.
106. Lowrey GE, Henderson N, Blakey JD, Corne JM, Johnson SRMMP-9. protein level does not reflect overall MMP activity in the airways of patients with COPD. Respir Med. 2008;102(6):845–851.
107. H RP I, Demedts I, Brusselle GG, et al. Matrix metalloproteinases −8, −9 and −12 in smokers and patients with Stage 0 COPD. Int J Chron Obstruct Pulmon Dis. 2007;2(3):369–370.
108. Vernooy JHJ, Lindeman JHN, Jacobs JA, Hanemaaijer R, Wouters EFM. Increased activity of matrix metalloproteinase-8 and matrix metalloproteinase-9 in induced sputum from patients with COPD. Chest. 2004;126(6):1802–1810.
109. Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002.
110. Demedts IK, Morel-Montero A, Lebecque S, et al. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax. 2006;61(3):196.
111. LaPan P, Brady J, Grierson C, et al. Optimization of total protein and activity assays for the detection of MMP-12 in induced human sputum. BMC Pulm Med. 2010;10:40.
112. Imai K, Dalal SS, Chen ES, et al. Human Collagenase (Matrix Metalloproteinase-1) Expression in the Lungs of Patients with Emphysema. Am J Respir Crit Care Med. 2001;163(3):786–791.
113. Hunninghake GM, Cho MH, Tesfaigzi Y, et al. MMP12, lung function, and COPD in high-risk populations. N Engl J Med. 2009;361(27):2599–2608.
114. Babusyte A, Stravinskaite K, Jeroch J, Lötvall J, Sakalauskas R, Sitkauskiene B. Patterns of airway inflammation and MMP-12 expression in smokers and ex-smokers with COPD. Respir Res. 2007;8(1):81.
115. Wallace AM, Sandford AJ, English JC, et al. Matrix metalloproteinase expression by human alveolar macrophages in relation to emphysema. COPD. 2008;5(1):13–23.
116. Filippov S, Caras I, Murray R, et al. Matrilysin-dependent elastolysis by human macrophages. J Exp Med. 2003;198(6):925–935.
117. Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M. Upregulation of gelatinases a and b, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117(3):684–694.
118. Sng JJ, Prazakova S, Thomas PS, Herbert CMMP-8. MMP-9 and neutrophil elastase in peripheral blood and exhaled breath condensate in COPD. COPD. 2017;14(2):238–244.
119. McNulty W, Usmani OS. Techniques of assessing small airways dysfunction. Eur Clin Respir J. 2014;1. doi:10.3402/ecrj.v3401.25898
120. Subramaniam K, Kumar H, Tawhai MH. Evidence for age-dependent air-space enlargement contributing to loss of lung tissue elastic recoil pressure and increased shear modulus in older age. J Appl Physiol. 2017;123(1):79–87.
121. Lee KW, Chung SY, Yang I, Lee Y, Ko EY, Park MJ. Correlation of aging and smoking with air trapping at thin-section CT of the lung in asymptomatic subjects. Radiology. 2000;214(3):831–836.
122. Fukuchi Y. The aging lung and chronic obstructive pulmonary disease: similarity and difference. Proc Am Thorac Soc. 2009;6(7):570–572.
123. Thomas ET, Guppy M, Straus SE, Bell KJL, Glasziou P. Rate of normal lung function decline in ageing adults: a systematic review of prospective cohort studies. 2019.
124. Verbeken EK, Cauberghs M, Mertens I, Clement J. The senile lung. Comparison with normal and emphysematous lungs. 1. Structural aspects. Chest. 1992;101(3):793–799.
125. Teramoto S, Ishii M Aging, the Aging Lung, and Senile Emphysema Are Different. http://dxdoiorg/101164/ajrccm1752197. 2012.
126. Mercado N, Ito K, Barnes PJ. Accelerated ageing of the lung in COPD: new concepts. 2015.
127. Walters MS, De BP, Salit J, et al. Smoking accelerates aging of the small airway epithelium. Respir Res. 2014;15:94.
128. Swanney MP, Ruppel G, Enright PL, et al. Using the lower limit of normal for the FEV1/FVC ratio reduces the misclassification of airway obstruction. Thorax. 2008;63(12):1046–1051.
129. Martinez CH, Diaz AA, Meldrum C, et al. Age and small airway imaging abnormalities in subjects with and without airflow obstruction in SPIROMICS. Am J Respir Crit Care Med. 2017;195(4):464–472.
130. van der Molen T, Postma DS, Martin RJ, et al. Effectiveness of initiating extrafine-particle versus fine-particle inhaled corticosteroids as asthma therapy in the Netherlands. BMC Pulm Med. 2016;16(1):80.
131. Scichilone N, Battaglia S, Sorino C, et al. Effects of extra-fine inhaled beclomethasone/formoterol on both large and small airways in asthma. Allergy. 2010;65(7):897–902.
132. Singh D, Nicolini G, Bindi E, et al. Extrafine beclomethasone/formoterol compared to fluticasone/salmeterol combination therapy in COPD. BMC Pulm Med. 2014;14:43.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.