Back to Journals » Nature and Science of Sleep » Volume 10
Sleep-related hypermotor epilepsy: prevalence, impact and management strategies
Authors Menghi V, Bisulli F ![]() , Tinuper P, Nobili L
, Tinuper P, Nobili L ![]()
Received 8 May 2018
Accepted for publication 26 June 2018
Published 10 October 2018 Volume 2018:10 Pages 317—326
DOI https://doi.org/10.2147/NSS.S152624
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Steven Shea
Veronica Menghi,1 Francesca Bisulli,2 Paolo Tinuper,2 Lino Nobili3,4
1Department of Biomedical and Neuromotor Sciences (DIBINEM), University of Bologna, Bologna, Italy; 2IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy; 3“Claudio Munari” Center for Epilepsy Surgery, Niguarda Hospital, Milan, Italy; 4Department of Neuroscience (DINOGMI), IRCCS, Giannina Gaslini Institute, University of Genoa, Genoa, Italy
Abstract: Sleep-related hypermotor epilepsy (SHE), previously called nocturnal frontal lobe epilepsy (NFLE), is a focal epilepsy characterized by asymmetric tonic/dystonic posturing and/or complex hyperkinetic seizures occurring mostly during sleep. SHE fulfills the definition of rare disease with an estimated minimum prevalence of 1.8/100,000 individuals, and it represents about 10% of drug-resistant surgical cases. Although SHE and autosomal-dominant SHE (ADSHE) have been considered benign epileptic conditions for a long time, emerging data have shed light on the severity of this disorder and some peculiar features can impact negatively on the quality of life of SHE patients. In fact, seizure frequency can be very high, resulting in nocturnal sleep fragmentation with possible diurnal consequences such as excessive sleepiness and fatigue. Moreover, recent studies, adopting a systematic neuropsychological assessment, have shown deficits in memory, executive functions and visuo-spatial abilities in almost half of SHE patients. Intellectual disabilities and psychiatric disorders have also been reported in some genetic forms. SHE may also exert a negative effect on health-related quality of life, especially in domains pertaining to a patient’s role in the family, social context and patient’s illness experience.
Despite a good response to pharmacological treatment, especially with carbamazepine, 30% of SHE patients suffer from drug-resistant seizures. Finally, recent studies suggest a poor prognosis in a high percentage of SHE patients with a 20.4% cumulative probability of achieving terminal remission at 10 years from onset. For selected drug-resistant SHE patients, epilepsy surgery is the only treatment offering high probability of recovery, both for seizures and for epilepsy-related sleep alterations.
Keywords: epilepsy, parasomnias, sleep-related hypermotor epilepsy, nocturnal frontal lobe epilepsy, epidemiology
Historical background
In 1977, Pedley and Guilleminault described an unusual type of sleepwalking in six patients; all experienced episodes characterized by screaming, vocalization, complex automatisms and ambulation. The episodes ceased after phenytoin or carbamazepine treatment. Four of these patients demonstrated epileptiform abnormalities in their electroencephalograms (EEGs). On the basis of the EEGs’ abnormalities and the favorable response to therapy, authors interpreted these episodes as epileptic manifestations, despite the fact that EEG recordings of two abortive attacks failed to correlate with any paroxysmal or other abnormal electrical activity.1 Some years later, further patients with complex motor attacks recurring every night during slow-wave sleep were reported; typically, the attacks consisted of twisting of the trunk and violent hyperkinetic movements of the limbs, and they were occasionally associated with tonic/dystonic posturing. Authors strongly debated the epileptic or nonepileptic origin of these episodes that were finally considered as rare motor disorders of sleep because of the complex motor clinical pattern that occurs only during sleep and the absence of EEG epileptiform abnormalities.2 The term “hypnogenic paroxysmal dystonia” was used first, changed later to “nocturnal paroxysmal dystonia” (NPD).3 At the same time, studies conducted in drug-resistant epileptic patients submitted to a presurgical evaluation have permitted a better understanding of the anatomo-electroclinical features of these paroxysmal manifestations, suggesting their epileptic origin.4–9 In particular, Tharp4 reported three children with seizures characterized by bizarre motor attacks recurring during wakefulness and resembling NPD episodes. One of them became seizure-free after the surgical resection of the orbital region. These data were successively confirmed by Wada et al, who demonstrated the epileptic nature and the frontal lobe origin of sleep-related seizures, similar to those classified as NPD.5,6 Finally, Tinuper et al recorded three patients with short-lasting motor attacks (similar to those described as NPD), showing clear-cut epileptic interictal and ictal abnormalities.9 All these observations led to the coining of the term “nocturnal frontal lobe epilepsy” (NFLE). In the following years, a large series of patients were described.10,11 A large video-polysomnographic study of 100 consecutive NFLE cases highlighted the different intensity and durations of these sleep-related manifestations (ranging from paroxysmal arousals to nocturnal wandering) that could occur in a single patient, during a single night. The hypothesis that these episodes could represent a continuum of the same seizure11,12 was demonstrated by further studies using intracerebral EEG recordings techniques during the presurgical evaluation of drug-resistant sleep-related hypermotor epilepsy (SHE) patients (video-stereo-electroencephalography, SEEG).13
In 2014, a Consensus Conference composed of a group of international experts in epilepsy, sleep and epidemiology was organized in order to better describe the electroclinical features of the disorder and to delineate more accurate diagnostic criteria.14 During the conference, a need for a new denomination emerged, and the term “sleep-related hypermotor epilepsy” (SHE) replaced NFLE. Indeed, the term nocturnal was judged not appropriate because seizures occur during sleep, both during the day and the night, independently of the daytime. Moreover, considering that sleep-related seizures with hyperkinetic automatisms and/or dystonic posturing have been shown to originate also from extrafrontal areas, such as temporal, insular and parietal regions,15–22 the term frontal was considered misleading. Finally, the term “hypermotor” was used because it was considered the best compromise able to represent both seizures with hyperkinetic automatisms or tonic–dystonic seizures. However, an update of the denomination might be required considering the last International League Against Epilepsy seizure classification.23 Experts agreed that SHE should be considered a unique syndrome, independently of the etiology (genetic, lesional or both) and the brain region involved.14
Clinical and diagnostic criteria for SHE
Seizures in SHE are usually brief (<2 minutes in duration), with an abrupt onset and offset and with stereotyped clinical features. Seizures are typically sleep-related (with several episodes per night) even if episodes occur rarely in wakefulness. The “hypermotor” semiology is the primary clinical pattern of the seizures, including asymmetric tonic or dystonic posturing and/or hyperkinetic automatisms. The level of awareness during and after the seizure is not a crucial clinical sign for a definitive diagnosis.
The clinical history and clinical semiology are the main criteria to establish the diagnosis as both interictal and ictal EEG may be uninformative. Three categories for the diagnosis with different levels of certainty have been identified: 1) Witnessed (possible), based on the description of the core clinical features; 2) Video Documented (clinical), based on the evaluation of a video recorded (preferably 2) hypermotor episode; and 3) Video-EEG Documented (confirmed), requiring the video-polygraphic recording of stereotyped events (one or two) and ictal or interictal epileptiform abnormalities.14
Epidemiological data
Studies indicate that about 12% of epileptic patients,24,25 mostly with a diagnosis of focal epilepsy,26,27 suffered from sleep-related seizures, defined as “seizures occurring exclusively or predominantly (>90 %) during sleep”. SHE represented the diagnosis in 13% of patients referred to a tertiary center for a video-polysomnographic evaluation of nocturnal motor disorders11 and in 9.4% of our population of focal drug-resistant epileptic patients (unpublished data). It is worth underlining the possible underestimation of SHE prevalence, as many patients may be misdiagnosed as parasomnias, particularly in the pediatric population.28,29
A prevalence study conducted in the northeast of Italy showed that SHE is a rare disease with a minimum prevalence of 1.8/100,000 individuals.30,31
SHE predominates in males (7:3); nonlesional forms are the most common; a family history of epilepsy is found in no more than 25% of cases; an autosomal-dominant inheritance pattern is rarely recognizable; onset of seizures peaks during childhood with the majority of cases presenting with seizures before the age of 20 years.10,11,32–34 Only 20% of drug-resistant SHE patients with hyperkinetic seizures present with this type of seizure before the age of 5 years (unpublished data). To date, the reason for this low percentage remains unknown and might reflect a later full development of behavioral neuronal networks.
Genetic background of SHE
In 1994, Sheffer et al reported a large Australian family with autosomal-dominant inherited NFLE/SHE (ADNFLE/ADSHE).35 A genetic mutation of the CHRNA4 gene coding for the alpha4 subunit of the neuronal nicotinic acetylcholine receptor (nAChR)36 was found. Successively, many studies described further mutations in genes (CHRNA2 and CHRNB2) coding for other nAChR subunits (alpha2 and beta2),37,38 the phenotype usually being indistinguishable.14
More recently, other mutations besides the ones coding for the nAChR subunits have been reported in both sporadic and ADSHE cases. Combi et al found two new nucleotide variations in the corticotropin-releasing hormone gene promoter in ADSHE pedigrees and two sporadic cases.39 Heron et al discovered missense mutations in the KCNT1 gene (coding for a sodium-gated potassium channel subunit) in patients with a severe phenotype, including frequent seizures, psychiatric symptoms and intellectual disabilities.40
Some ADSHE families and SHE sporadic cases have been found to have mutations in DEPDC5, and NPRL2 and 3,41–43 all encoding proteins of the mTORC1-regulating GATOR1 complex, a key regulator of cell growth.44,45 The mutations were first identified in patients with familial focal epilepsy with variable foci (FFEVF)46 and in epilepsy associated with a focal cortical dysplasia.47,48
Recently a novel missense mutation in the CABP4 gene encoding the neuronal Ca2+-binding protein 4 (CaBP4) has been found in a Chinese family, including 11 individuals diagnosed with ADSHE.49
Despite a great effort to study the genetic background of SHE, a genetic cause may be recognizable in a very low percentage of sporadic cases and in less than 30% of ADSHE families, with incomplete penetrance. Moreover, to date, there are no clear-cut correlations between disease severity, genetic findings and functional effects of the known genetic mutations,14 even if KCNT1 gene mutated SHE patients seem to present a more severe form.40 However, further studies focused on genotype–phenotype correlations in SHE are needed.
The genetic predisposition to present seizures especially during sleep is another interesting field of research. To date, a cholinergic pathway hyperactivation and an enhanced GABA ergic function were reported in in vitro and in vivo studies, suggesting that cortical–subcortical networks involved in the mechanism of arousal can contribute in the epileptogenesis of ADSHE.37 Very recently, studies performed in brain epileptogenic tissues have shown that defects in Circadian Locomotor Output Cycles Kaput (CLOCK) expression, a transcription factor that regulates the circadian rhythm and the mTOR pathway, could be responsible for the preferential occurrence of seizures during sleep.50
Impact on general life and prognostic features of SHE
Although SHE and ADSHE for a long time have been considered benign epileptic conditions due to the occurrence of seizures only during sleep and the good response to treatment, many studies reported some factors that might have a negative impact on the general life of these patients.
Sleep fragmentation
Frequency of seizures in SHE patients can be very high, ranging from 1 to 20 attacks per night,11 and minor motor events or paroxysmal arousals may be even more frequent. SHE patients may complain of nocturnal sleep discontinuity and consequently, excessive sleep inertia in the morning, daytime tiredness and excessive sleepiness,10,13,33,51–57 impacting negatively on their quality of life. Furthermore, sleep deprivation can facilitate seizure appearance, perpetuating an erroneous loop between seizures and triggering factors.
In a case–control study58 the occurrence of sleepiness-related symptoms and subjective sleep quality were examined using questionnaires (Epworth sleepiness scale and the Bologna Questionnaire on Sleepiness-related symptoms).59 SHE cases more commonly complained of “spontaneous midsleep awakenings”58 and “tiredness after awakening” compared with matched controls.58 Furthermore, a trend for higher values of excessive daytime somnolence was found in SHE patients, although not statistically different from the control group.58
ADSHE population studies, examining the macrostructure of sleep (ie, the “classical” sleep profile), reported that patients and controls were similar.10,53,60 However, SHE subjects with daytime complaints (ie, sleepiness, fatigue) had an increased sleep instability, as quantified by the analysis of the Cyclic Alternating Pattern (CAP).56,61 Moreover, seizures occurred mostly during unstable sleep,56 suggesting a probable connection between sleep-related motor manifestations, sleep disruption and daytime symptoms.62
The hypothesis that sleep-related epileptic manifestations in SHE patients could favor a substantial increase of sleep instability, and vice versa, was corroborated by SEEG recordings.63,64 Indeed, these studies suggested that interictal epileptic discharges (IEDs) may act as an internal trigger inducing an increase in the arousal level, which in turn favors the occurrence of motor events that can be epileptic or not65 (Figure 1). However, from a semiological point of view, minor motor events generated by an epileptic discharge or by a simple arousal are similar,63 thus a diagnosis of SHE based on the recording of only minor events is, to date, impossible. Finally, such a mutual relationship between arousal and epileptic discharges, together with genetic factors, may explain the high incidence of parasomnia and other sleep disorders in SHE patients.66,67
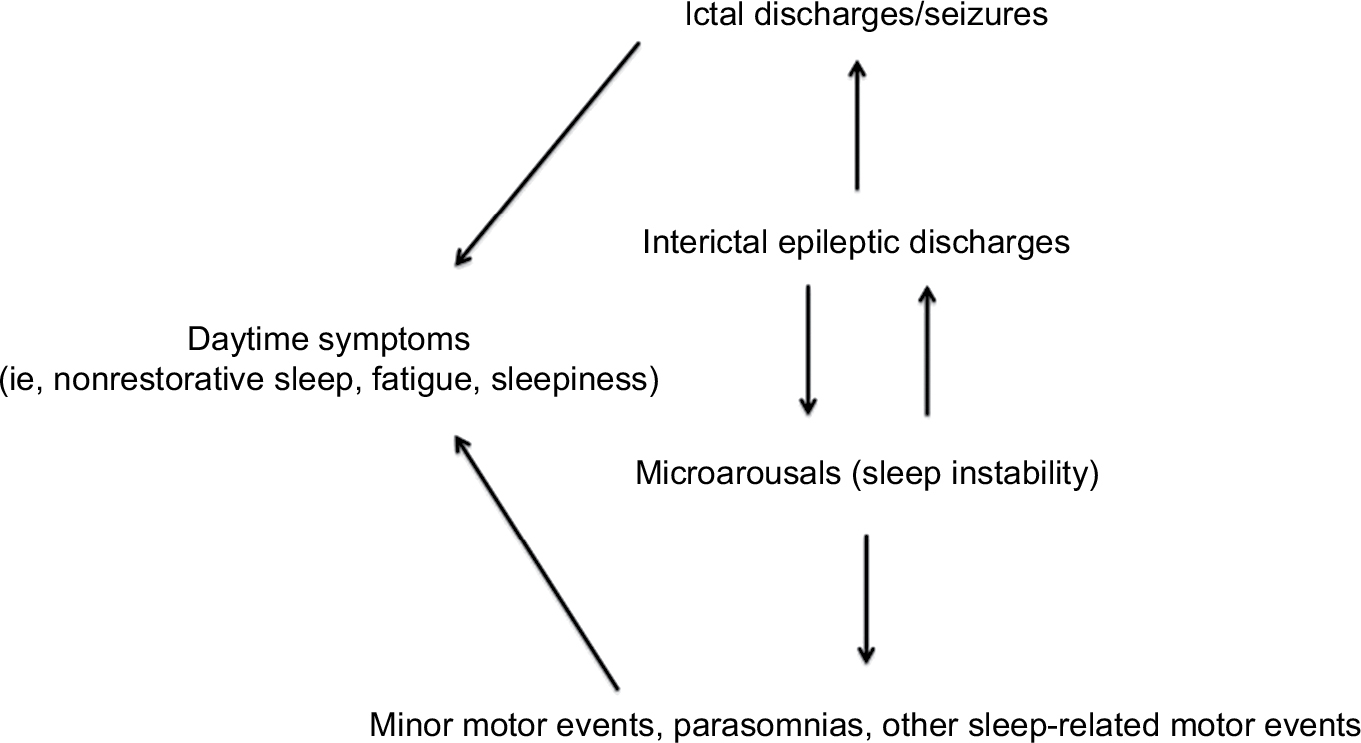 | Figure 1 The vicious loop of sleep-related epileptic discharges and microarousals. Notes: Interictal epileptic discharges may act as an internal trigger increasing the frequency of microarousals, which in turn favors the occurrence of motor events (epileptic or not). Sleep instability may have a negative impact on daytime functioning. Modified from Gibbs SA, Proserpio P, Terzaghi M, et al. Sleep-related epileptic behaviors and non-REM-related parasomnias: Insights from stereo-EEG. Sleep Med Rev. 2016;25:4–20. Copyright © 2015 The Authors. Published by Elsevier Ltd. http://creativecommons.org/licenses/by-nc-nd/4.0/.65 |
SHE and cognitive functions
Sleep deprivation exerts a negative impact on cognitive functions, including vigilance, memory retention, sensory perception and elements of executive function. Moreover, sleep deprivation may impair the functioning of cognitive systems connected to emotional networks.68
From a neuropsychological point of view, the largest series of sporadic and familial cases of SHE does not report gross cognitive deficits. Psychological and cognitive deficits associated with some forms of ADSHE originally reported were considered coincidental and nonsyndromic-specific.69,70 More recently, some studies have put in evidence intellectual disability and psychiatric problems in some ADSHE patients carrying nAChR subunits and KCNT1 genes mutations.71–77 In a large cohort of sporadic and familial SHE patients, Licchetta et al (2017) reported intellectual disabilities or a borderline IQ in about 12% of cases and psychiatric disorders in 24%.34 When present, the cognitive disorder in SHE seems to mainly affect memory functions as well as executive functions,75 irrespective of the treatment status. A recent study conducted in a large cohort of SHE patients, through a neuropsychological standardized assessment, revealed cognitive deficits in at least half of assessed patients and worse verbal IQ scores that might reflect deficits in working memory and phonemic fluency (executive functions).78 Deficits in selected executive, memory and visuospatial functions were found also in a high percentage of subjects without an apparent intellectual disability. Mutated SHE patients, compared to nonmutated ones, irrespective of the specific gene, presented a lower IQ score. Variables of clinical severity (ie, high seizure frequency, status epilepticus and bilateral convulsive seizures, poor response to antiepileptic treatment) were negatively correlated with memory and executive functions.78 The finding of memory deficit may be explained by the direct negative effect of interictal and ictal epileptic activity (originating from both frontal or extrafrontal regions) and by the alterations of sleep-related encoding memory processes induced by sleep fragmentation.79,80
Personal and social aspects
The injury potential of sleep-related seizures seems to be lower than that during wakefulness. This is probably related to the relative safe condition of being in bed.81 In SHE patients, injuries resulting from hyperkinetic seizures can be accidental.82 Nevertheless, epileptic attacks in SHE may be very violent, characterized by hypermotor patterns with bimanual/bipedal activity, ballistic, rocking axial and pelvic movements and sometimes agitated ambulation. Patients can fall from the bed and get injured, and sometimes countermeasures are necessary (for example, sleeping with a mattress on the floor or putting lateral pillows on the bed). In this scenario, it is not uncommon that patients adopt avoidance behaviors in their social and sentimental life.
Ictal aggression is exceptional83 and generally not intentional. It can take the form of pushing, grasping, hitting, shoving and spitting,84 and may happen if patients react to some environmental stimuli (such as the act of restraining them).85
A study evaluating health-related quality of life in SHE patients compared with the general population showed that SHE constitutes a restriction on health-related quality of life, especially in domains pertaining to the patient’s role in a family and social context and the patient’s illness experience.86
Seizures during wakefulness
To date, a study evaluating the risk of seizures during wake in typical SHE (100% sleep-related seizures) is lacking. Previous studies suggest that almost 30%–40% of SHE patients experience occasional seizures during wakefulness11,34 that may be similar to sleep-related manifestations or only characterized by isolated subjective symptoms. SHE patients may be exposed to driving license legal restrictions, especially in case of rare focal seizures during wake.87,88
The problem of misdiagnosis
The differential diagnosis between SHE and parasomnias may be challenging even for experts in epileptology and sleep medicine due to possible similarities between the two sleep-related manifestations. Indeed, misdiagnosis is frequent in SHE patients because the absence of typical convulsive seizures and the presence of behavioral patterns similar to those observed in non-rapid eye movement (NREM) parasomnias and rapid eye movement (REM) behavior disorders, at least at the anamnestic evaluation. Video-EEG and video-polysomnography, considered the gold standards for a confirmed diagnosis, may be unavailable or not definitively conclusive. The high prevalence of parasomnias in SHE probands and their healthy relatives11,66 may create a further challenge for the differential diagnosis. Moreover, the response to antiepileptic drugs is not discriminative for the diagnosis of SHE.89 A recent large cohort study reported a diagnostic delay of 12.8±10.1 years in 53.7% of SHE cases, parasomnias being the most frequent misdiagnosis (55.5%).34 The problem of a correct diagnosis is an important factor that may impact over the clinical course of the disease because of inappropriate treatment and management.
The difficulty to discriminate parasomnias from SHE led experts to develop a validated scale, named Frontal Lobe Epilepsy and Parasomnias (FLEP) scale.90 However, some criticism on the FLEP scale emerged, first of all its inability to distinguish nocturnal wandering from sleepwalking.91 The Structured Interview for NFLE/SHE (SINFLE) represents another tool that could improve the physician’s capability to distinguish parasomnias from SHE, with high specificity but unsatisfactory sensitivity.92 It is based on anamnestic information, including the presence or absence of the two major motor seizure patterns (ie, dystonic posturing or hyperkinetic automatisms) and four minor features regarding the duration of the episodes, the presence of a recognized aura, unstructured vocalization and a history of sleep-related convulsive seizures.
Prognostic features
Prognostic studies on SHE have originally included drug-resistant cases33,93 submitted to epilepsy surgery and showed a high percentage of patients (more than 70%) seizure-free after the intervention with a minimum follow-up of 24 months.93 Only a recent report34 focused on the analysis of the long-term evolution of SHE patients (lesional and nonlesional, sporadic and familial) showing a long-term poor prognosis. Indeed, in this study, the cumulative probability of remission ranged from 20.4% at 10 years to 28.4% at 40 years from onset. The most important favorable factors affecting remission appear to be 1) the absence of any direct or indirect brain disorder (pathologic neurological examination, intellectual disability, perinatal suffering and brain abnormalities, such as focal cortical dysplasia) and 2) typical SHE (ie, 100% sleep-related seizures).34
Drug resistance
Most patients show a good response to the pharmacological treatment, low doses of carbamazepine at bedtime being the first choice of therapy. However, about one-third of patients are drug-resistant.11,34,94 Although the surgical outcome seems to be relatively good in this population, especially in patients with positive brain MRI, the presurgical evaluation and the surgical approach (including risks related to the procedures) may have a negative impact on quality of life.
Sudden unexpected death in epilepsy (SUDEP) incidence in SHE
Most of SUDEP cases occur in bed while patients are probably sleeping. A possible relationship with sleep has been found in about 60% of SUDEP cases (after considering studies including more than ten subjects who died from SUDEP).95 Sleep-related settings (ie, lack of surveillance and sleeping position, especially prone position), generalized tonic–clonic seizures and autonomic changes could account for this relation, increasing SUDEP risk during sleep.95
Despite the fact that occurrence of sleep-related seizures is an important risk factor for SUDEP, a retrospective study showed an incidence of SUDEP in SHE patients similar to that observed in the general epilepsy population (0.36 per 1,000 person-years).96–98 A low occurrence of tonic–clonic seizures in SHE may be responsible for such a lower-than-expected risk of SUDEP.91 Although systematic studies are lacking, it is possible that SHE patients with an insular onset are at higher risk of SUDEP.96,99,100 Indeed, it has been shown that seizures originating from the insula may be accompanied by important autonomic alterations.95
Management strategies
Pharmacological treatment
Although controlled studies are lacking, carbamazepine seems to be the drug of choice in SHE patients.2,11,32 Given at low doses (200–400 mg at bedtime), carbamazepine abolishes or significantly reduces seizures in about 20% and 50% of patients.11 Seizures commonly recur after drug withdrawal.101 Interestingly, experimental observations have found an amplified sensitivity of mutated nAChR receptors to this drug.102
About one-third of SHE patients do not respond to therapy10,11,34 and maintain a very high seizure frequency. Indeed, about 70% of these subjects had more than 25 seizures per month and these numbers may be even higher, considering that many sleep-related seizures are probably underreported.11
Oxcarbazepine (at a mean dose of 30.4±11.7 mg/kg/day) has been shown to be effective in stopping seizures in a study including eight children diagnosed with SHE (ranging from 4 to 16 years of age) and followed up for a minimum of 12 months. Low doses of the drug allowed a rapid control of seizures (within 4 days from drug beginning) in six patients, whereas in two children, higher doses were required.103
In a small series of SHE patients, about 90% of cases benefited from topiramate use as single or add-on therapy (from 50 to 300 mg daily at bedtime); in particular, 25% of patients became seizure-free and 62% had a reduction in seizures of at least 50% during a follow-up period ranging from 6 months to 6 years.104
A study reported a good response to acetazolamide (500 mg at night) as add-on therapy to carbamazepine in three ADSHE family members.105
Recent reports conducted in small series of patients, including genetic cases, suggest lacosamide 200 or 400 mg/day, as a possible add-on treatment or in monotherapy.106,107
Seizure frequency improved in a single patient with refractory ADSHE after nicotine transdermal patches treatment.108 The favorable effect of nicotine on seizure frequency was also described in 9 of 22 patients from two European ADSHE families carrying CHRNA4 mutations.109 Considering the role of the cholinergic system in arousal regulatory processes, these observations suggested a possible link between nicotine defect, alteration of arousal regulation and seizures in SHE/ADSHE patients. However, despite the reported positive effect of nicotine in reducing seizure frequency, a case–control family study, did not find a higher tendency to smoke tobacco in SHE patients and their relatives compared with the control cases.110
Recently, Puligheddu et al have shown that fenofibrate, an agonist at peroxisome proliferator-activated receptor alpha (PPARa) acting as a negative modulator of nAChRs, may have beneficial effects both in mutated mouse models of SHE and pharmacoresistant SHE patients.111 Interestingly, good results with fenofibrate administration have been obtained both in the mutated (CHRNA2 and 4) and nonmutated SHE patients.
Finally, it has been shown that, despite a partial reduction of nocturnal seizures, standard antiepileptic treatment is not particularly effective in reducing sleep instability compared with the pretreatment condition.112
Very recently, a new therapeutic approach with quinidine was tested in a few drug-resistant epileptic patients carrying KCNT1 genetic mutations, none fitting SHE phenotype, with questionable results.113,114
Surgery
Epilepsy surgery provides excellent results in selected drug-resistant SHE cases both for seizures and for epilepsy-related sleep alterations (ie, frequent arousals, sleep deprivation and excessive daytime sleepiness).33,115
Some authors reported that more than two-thirds of patients with SHE were fully controlled after surgery (Engel’s Class Ia), and the remaining ones had a considerable decrease of both frequency and intensity of seizures (Engel’s Class II and III).33,93 Good surgical results are obtained in both frontal and extra-frontal SHE, although in this last group the number of operated-on patients is limited.17,20,93,99 A presurgical evaluation including SEEG recordings may be necessary, especially when brain MRI is uninformative.
An explanation for the excellent surgical results obtained in frontal and extrafrontal SHE seems to be dependent on the high incidence of cases with a histological diagnosis of type II focal cortical dysplasia (Taylor-type), a generally well-limited malformation of cortical development, known to increase the risk of sleep-related seizures independently of its location.21,93,116 Indeed, variables statistically associated with a favorable outcome are a positive MRI, a complete removal of the epileptogenic zone and the presence of focal cortical dysplasia type II.88 Patients submitted to SEEG investigation show a worse outcome as they represent more challenging cases, with an unremarkable MRI or discordant anatomo-electroclinical findings.
Other treatments
Finally, it is known that reducing factors that may promote seizures, such as sleep instability, could lead to more effective treatment strategies. Although specific studies in SHE population are lacking, disorders resulting in sleep disruption such as obstructive sleep apnea, insomnia and parasomnias may be associated with refractory focal epilepsy and have to be investigated and treated specifically.117,118 Hypothetically, considering that seizures in SHE are increased by sleep instability and mostly occur during N2 stage of sleep, a drug able to increase slow-wave sleep production could help in reducing seizure frequency; however, to date, there is no evidence that such a treatment may be effective.
Acknowledgments
We would like to acknowledge all of our collaborators from Niguarda Hospital (Milan) and Bellaria Hospital (Bologna).
Disclosure
The authors report no conflicts of interest in this work.
References
Pedley TA, Guilleminault C. Episodic nocturnal wanderings responsive to anticonvulsant drug therapy. Ann Neurol. 1977;2(1):30–35. | ||
Lugaresi E, Cirignotta F. Hypnogenic paroxysmal dystonia: epileptic seizure or a new syndrome? Sleep. 1981;4(2):129–138. | ||
Lugaresi E, Cirignotta F, Montagna P. Nocturnal paroxysmal dystonia. J Neurol Neurosurg Psychiatry. 1986;49(4):375–380. | ||
Tharp BR. Orbital frontial seizures. An unique electroencephalographic and clinical syndrome. Epilepsia. 1972;13(5):627–642. | ||
Wada JA, Purves SJ. Oral and bimanual-bipedal activity as ictal manifestation of frontal lobe epilepsy. Epilepsia. 1984;25:668. | ||
Wada JA. Nocturnal recurrence of brief, intensely affective vocal and facial expression with powerful bimanual, bipedal, axial, and pelvic activity with rapid recovery as manifestations of mesial frontal lobe seizure. Epilepsia. 1988;29:209. | ||
Williamson PD, Spencer DD, Spencer SS, Novelly RA, Mattson RH. Complex partial seizures of frontal lobe origin. Ann Neurol. 1985;18(4):497–504. | ||
Waterman K, Purves SJ, Kosaka B, Strauss E, Wada JA. An epileptic syndrome caused by mesial frontal lobe seizure foci. Neurology. 1987;37(4):577–582. | ||
Tinuper P, Cerullo A, Cirignotta F, Cortelli P, Lugaresi E, Montagna P. Nocturnal paroxysmal dystonia with short-lasting attacks: three cases with evidence for an epileptic frontal lobe origin of seizures. Epilepsia. 1990;31(5):549–556. | ||
Oldani A, Zucconi M, Asselta R, et al. Autosomal dominant nocturnal frontal lobe epilepsy. A video-polysomnographic and genetic appraisal of 40 patients and delineation of the epileptic syndrome. Brain. 1998;121(Pt 2):205–223. | ||
Provini F, Plazzi G, Tinuper P, Vandi S, Lugaresi E, Montagna P. Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain. 1999;122(Pt 6):1017–1031. | ||
Montagna P. Nocturnal paroxysmal dystonia and nocturnal wandering. Neurology. 1992;42(7 Suppl 6):61–67. | ||
Nobili L, Francione S, Mai R, et al. Nocturnal frontal lobe epilepsy: intracerebral recordings of paroxysmal motor attacks with increasing complexity. Sleep. 2003;26(7):883–886. | ||
Tinuper P, Bisulli F, Cross JH, et al. Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurology. 2016;86(19):1834–1842. | ||
Nobili L, Francione S, Cardinale F, Lo Russo G. Epileptic nocturnal wanderings with a temporal lobe origin: a stereo-electroencephalographic study. Sleep. 2002;25(6):669–671. | ||
Nobili L, Cossu M, Mai R, et al. Sleep-related hyperkinetic seizures of temporal lobe origin. Neurology. 2004;62(3):482–485. | ||
Mai R, Sartori I, Francione S, et al. Sleep-related hyperkinetic seizures: always a frontal onset? Neurol Sci. 2005;26 Suppl 3:s220–s224. | ||
Ryvlin P, Minotti L, Demarquay G, et al. Nocturnal hypermotor seizures, suggesting frontal lobe epilepsy, can originate in the insula. Epilepsia. 2006;47(4):755–765. | ||
Kaido T, Otsuki T, Nakama H, et al. Complex behavioral automatism arising from insular cortex. Epilepsy Behav. 2006;8(1):315–319. | ||
Dobesberger J, Ortler M, Unterberger I, et al. Successful surgical treatment of insular epilepsy with nocturnal hypermotor seizures. Epilepsia. 2008;49(1):159–162. | ||
Proserpio P, Cossu M, Francione S, et al. Epileptic motor behaviors during sleep: anatomo-electro-clinical features. Sleep Med. 2011;12 Suppl 2:S33–S38. | ||
Gibbs SA, Figorilli M, Casaceli G, Proserpio P, Nobili L. Sleep Related Hypermotor Seizures with a Right Parietal Onset. J Clin Sleep Med. 2015;11(8):953–955. | ||
Fisher RS, Acevedo C, Arzimanoglou A, et al. A practical clinical definition of epilepsy. Epilepsia. 2014;55:475–482. | ||
Thomas RH, King WH, Johnston JA, Smith PEM. Awake seizures after pure sleep-related epilepsy: a systematic review and implications for driving law. Journal of Neurology, Neurosurgery & Psychiatry. 2010;81(2):130–135. | ||
Derry CP, Duncan S. Sleep and epilepsy. Epilepsy Behav. 2013;26(3):394–404. | ||
Yaqub BA, Waheed G, Kabiraj MM. Nocturnal epilepsies in adults. Seizure. 1997;6(2):145–149. | ||
Fernández LB, Salas-Puig J. Pure sleep seizures: risk of seizures while awake. Epileptic Disord. 2007;9(1):65–70. | ||
Scheffer Iet al. Autosomal dominant frontal epilepsy misdiagnosed as sleep disorder. The Lancet. 1994;343(8896):515–517. | ||
Weinstock A, Giglio P, Kerr SL, Duffner PK, Cohen ME. Hyperkinetic seizures in children. J Child Neurol. 2003;18(8):517–524. | ||
Vignatelli L, Bisulli F, Giovannini G, et al. Prevalence of nocturnal frontal lobe epilepsy in the adult population of Bologna and Modena, Emilia-Romagna region, Italy. Sleep. 2015;38(3):479–485. | ||
Vignatelli L, Bisulli F, Giovannini G, et al. Prevalence of Nocturnal Frontal Lobe Epilepsy in the Adult Population of Bologna and Modena, Emilia-Romagna Region, Italy. Sleep. 2015;38(3):479–485. | ||
Scheffer IE, Bhatia KP, Lopes-Cendes I, et al. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain. 1995;118(Pt 1):61–73. | ||
Nobili L, Francione S, Mai R, et al. Surgical treatment of drug-resistant nocturnal frontal lobe epilepsy. Brain. 2007;130(Pt 2):561–573. | ||
Licchetta L, Bisulli F, Vignatelli L, et al. Sleep-related hypermotor epilepsy: Long-term outcome in a large cohort. Neurology. 2017;88(1):70–77. | ||
Scheffer IE, Bhatia KP, Lopes-Cendes I, et al. Autosomal dominant frontal epilepsy misdiagnosed as sleep disorder. Lancet. 1994;343(8896):515–517. | ||
Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11(2):201–203. | ||
Marini C, Guerrini R. The role of the nicotinic acetylcholine receptors in sleep-related epilepsy. Biochem Pharmacol. 2007;74(8):1308–1314. | ||
de Fusco M, Becchetti A, Patrignani A, et al. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet. 2000;26(3):275–276. | ||
Combi R, Ferini-Strambi L, Montruccoli A, et al. Two new putative susceptibility loci for ADNFLE. Brain Res Bull. 2005;67(4):257–263. | ||
Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44(11):1188–1190. | ||
Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet. 2013;45(5):552–555. | ||
Korenke GC, Eggert M, Thiele H, Nürnberg P, Sander T, Steinlein OK. Nocturnal frontal lobe epilepsy caused by a mutation in the GATOR1 complex gene NPRL3. Epilepsia. 2016;57(3):e60–e63. | ||
Ricos MG, Hodgson BL, Pippucci T, et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol. 2016;79(1):120–131. | ||
Bar-Peled L, Chantranupong L, Cherniack AD, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340(6136):1100–1106. | ||
Iffland PH, Baybis M, Barnes AE, Leventer RJ, Lockhart PJ, Crino PB. DEPDC5 and NPRL3 modulate cell size, filopodial outgrowth, and localization of mTOR in neural progenitor cells and neurons. Neurobiol Dis. 2018;114:184–193. | ||
Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013;45(5):546–551. | ||
Sim JC, Scerri T, Fanjul-Fernández M, et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol. 2016;79(1):132–137. | ||
Scheffer IE, Heron SE, Regan BM, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75(5):782–787. | ||
Chen ZH, Wang C, Zhuo MQ, et al. Exome sequencing identified a novel missense mutation c.464G>A (p.G155D) in Ca2+-binding protein 4 (CABP4) in a Chinese pedigree with autosomal dominant nocturnal frontal lobe epilepsy. Oncotarget. 2017;8(45):78940–78947. | ||
Li P, Fu X, Smith NA, et al. Loss of CLOCK Results in Dysfunction of Brain Circuits Underlying Focal Epilepsy. Neuron. 2017;96(2):387–401. | ||
Peled R, Lavie P. Paroxysmal awakenings from sleep associated with excessive daytime somnolence: a form of nocturnal epilepsy. Neurology. 1986;36(1):95–98. | ||
Maccario M, Lustman LI. Paroxysmal nocturnal dystonia presenting as excessive daytime somnolence. Arch Neurol. 1990;47(3):291–294. | ||
Oldani A, Zucconi M, Ferini-Strambi L, Bizzozero D, Smirne S. Autosomal dominant nocturnal frontal lobe epilepsy: electroclinical picture. Epilepsia. 1996;37(10):964–976. | ||
Oldani A, Zucconi M, Castronovo C, Ferini-Strambi L. Nocturnal frontal lobe epilepsy misdiagnosed as sleep apnea syndrome. Acta Neurol Scand. 1998;98(1):67–71. | ||
Schwalen S, Jörg J. Day time fatigue in frontal lobe epilepsy with primarily sleep-related seizures. A case report. Nervenarzt. 1998;69(2):166–170. | ||
Zucconi M, Oldani A, Smirne S, Ferini-Strambi L. The macrostructure and microstructure of sleep in patients with autosomal dominant nocturnal frontal lobe epilepsy. J Clin Neurophysiol. 2000;17(1):77–86. | ||
Alanis-Guevara I, Peña E, Corona T, López-Ayala T, López-Meza E, López-Gómez M. Sleep disturbances, socioeconomic status, and seizure control as main predictors of quality of life in epilepsy. Epilepsy Behav. 2005;7(3):481–485. | ||
Vignatelli L, Bisulli F, Naldi I, et al. Excessive daytime sleepiness and subjective sleep quality in patients with nocturnal frontal lobe epilepsy: a case-control study. Epilepsia. 2006;47 Suppl 5:73–77. | ||
Rinaldi R, Vignatelli L, D’Alessandro R, et al. Validation of symptoms related to excessive daytime sleepiness. Neuroepidemiology. 2001;20(4):248–256. | ||
Zucconi M, Oldani A, Ferini-Strambi L, Bizzozero D, Smirne S. Nocturnal Paroxysmal Arousals With Motor Behaviors During Sleep: Frontal Lobe Epilepsy or Parasomnia? Journal of Clinical Neurophysiology. 1997;14(6):513–522. | ||
Parrino L, Ferri R, Bruni O, Terzano MG. Cyclic alternating pattern (CAP): the marker of sleep instability. Sleep Med Rev. 2012;16:27–45. | ||
Parrino L, Halasz P, Tassinari CA, Terzano MG. CAP, epilepsy and motor events during sleep: the unifying role of arousal. Sleep Med Rev. 2006;10(4):267–285. | ||
Terzaghi M, Sartori I, Mai R, et al. Sleep-related minor motor events in nocturnal frontal lobe epilepsy. Epilepsia. 2007;48(2):335–341. | ||
Terzaghi M, Sartori I, Mai R, et al. Coupling of minor motor events and epileptiform discharges with arousal fluctuations in NFLE. Epilepsia. 2008;49(4):670–676. | ||
Gibbs SA, Proserpio P, Terzaghi M, et al. Sleep-related epileptic behaviors and non-REM-related parasomnias: Insights from stereo-EEG. Sleep Med Rev. 2016;25:4–20. | ||
Bisulli F, Vignatelli L, Naldi I, et al. Increased frequency of arousal parasomnias in families with nocturnal frontal lobe epilepsy: a common mechanism? Epilepsia. 2010;51(9):1852–1860. | ||
Nobili L. Nocturnal frontal lobe epilepsy and non-rapid eye movement sleep parasomnias: differences and similarities. Sleep Med Rev. 2007;11(4):251–254. | ||
Killgore WD. Effects of sleep deprivation on cognition. Prog Brain Res. 2010;185:105–129. | ||
Mclellan A, Phillips HA, Rittey C, et al. Phenotypic comparison of two Scottish families with mutations in different genes causing autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia. 2003;44(4):613–617. | ||
Khatami R, Neumann M, Schulz H, Kölmel HW. A family with autosomal dominant nocturnal frontal lobe epilepsy and mental retardation. J Neurol. 1998;245(12):809–810. | ||
Cho YW, Motamedi GK, Laufenberg I, et al. A Korean kindred with autosomal dominant nocturnal frontal lobe epilepsy and mental retardation. Arch Neurol. 2003;60(11):1625–1632. | ||
Cho YW, Yi SD, Lim JG, Kim DK, Motamedi GK. Autosomal dominant nocturnal frontal lobe epilepsy and mild memory impairment associated with CHRNB2 mutation I312M in the neuronal nicotinic acetylcholine receptor. Epilepsy Behav. 2008;13(2):361–365. | ||
Ito M, Kobayashi K, Fujii T, et al. Electroclinical picture of autosomal dominant nocturnal frontal lobe epilepsy in a Japanese family. Epilepsia. 2000;41(1):52–58. | ||
Magnusson A, Stordal E, Brodtkorb E, Steinlein O. Schizophrenia, psychotic illness and other psychiatric symptoms in families with autosomal dominant nocturnal frontal lobe epilepsy caused by different mutations. Psychiatr Genet. 2003;13(2):91–95. | ||
Wood AG, Saling MM, Fedi M, et al. Neuropsychological function in patients with a single gene mutation associated with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsy Behav. 2010;17(4):531–535. | ||
Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44(11):1188–1190. | ||
Picard F, Pegna AJ, Arntsberg V, et al. Neuropsychological disturbances in frontal lobe epilepsy due to mutated nicotinic receptors. Epilepsy Behav. 2009;14(2):354–359. | ||
Licchetta L, Poda R V, et al. Profile of neuropsychological impairment in Sleep-related Hypermotor Epilepsy. Sleep Med. 2018;48:8–15. | ||
Centeno M, Thompson PJ, Koepp MJ, Helmstaedter C, Duncan JS. Memory in frontal lobe epilepsy. Epilepsy Res. 2010;91(2-3):123–132. | ||
Kreutzmann JC, Havekes R, Abel T, Meerlo P. Sleep deprivation and hippocampal vulnerability: changes in neuronal plasticity, neurogenesis and cognitive function. Neuroscience. 2015;309:173–190. | ||
Wirrell EC. Epilepsy-related injuries. Epilepsia. 2006;47 Suppl 1:79–86. | ||
Schenck CH, Milner DM, Hurwitz TD, Bundlie SR, Mahowald MW. A polysomnographic and clinical report on sleep-related injury in 100 adult patients. Am J Psychiatry. 1989;146(9):1166–1173. | ||
Tassinari CA, Tassi L, Calandra-Buonaura G, et al. Biting behavior, aggression, and seizures. Epilepsia. 2005;46(5):654–663. | ||
Siclari F, Khatami R, Urbaniok F, et al. Violence in sleep. Brain. 2010;133(Pt 12):3494–3509. | ||
Delgado-Escueta AV, Mattson RH, King L, et al. Special report. The nature of aggression during epileptic seizures. N Engl J Med. 1981;305(12):711–716. | ||
Licchetta L, Vignatelli L, Bisulli F, et al. Health-related quality of life in patients with nocturnal focal epilepsy. Boll Lega It Epil. 2006 (133/134):299–300. | ||
Thomas RH, King WH, Johnston JA, Smith PE. Awake seizures after pure sleep-related epilepsy: a systematic review and implications for driving law. J Neurol Neurosurg Psychiatry. 2010;81(2):130–135. | ||
Beghi E, Sander JW. Epilepsy and driving. BMJ. 2005;331(7508):60–61. | ||
Tinuper P, Bisulli F. From nocturnal frontal lobe epilepsy to Sleep-Related Hypermotor Epilepsy: A 35-year diagnostic challenge. Seizure. 2017;44:87–92. | ||
Derry CP, Davey M, Johns M, et al. Distinguishing sleep disorders from seizures: diagnosing bumps in the night. Arch of Neurol. 2006;63:705–709. | ||
Manni R, Terzaghi M, Repetto A. The FLEP scale in diagnosing nocturnal frontal lobe epilepsy, NREM and REM parasomnias: data from a tertiary sleep and epilepsy unit. Epilepsia. 2008;49(9):1581–1585. | ||
Bisulli F, Vignatelli L, Naldi I, et al. Diagnostic accuracy of a structured interview for nocturnal frontal lobe epilepsy (SINFLE): a proposal for developing diagnostic criteria. Sleep Med. 2012;13(1):81–87. | ||
Losurdo A, Proserpio P, Cardinale F, et al. Drug-resistant focal sleep related epilepsy: results and predictors of surgical outcome. Epilepsy Res. 2014;108(5):953–962. | ||
Hirsch E, Sellal F, Maton B, Rumbach L, Marescaux C. Nocturnal paroxysmal dystonia: a clinical form of focal epilepsy. Neurophysiol Clin. 1994;24(3):207–217. | ||
Nobili L, Proserpio P, Rubboli G, Montano N, Didato G, Tassinari CA. Sudden unexpected death in epilepsy (SUDEP) and sleep. Sleep Med Rev. 2011;15(4):237–246. | ||
Mostacci B, Bisulli F, Vignatelli L, et al. Incidence of sudden unexpected death in nocturnal frontal lobe epilepsy: a cohort study. Sleep Med. 2015;16(2):232–236. | ||
Mostacci B, Bisulli F, Vignatelli L, et al. Incidence of sudden unexpected death in epilepsy in sleep-related hypermotor epilepsy, formerly named nocturnal frontal lobe epilepsy. Sleep Med. 2017;29:98. | ||
Lamberts RJ, Thijs RD, Laffan A, Langan Y, Sander JW. Sudden unexpected death in epilepsy: people with nocturnal seizures may be at highest risk. Epilepsia. 2012;53(2):253–257. | ||
Proserpio P, Cossu M, Francione S, et al. Insular-opercular seizures manifesting with sleep-related paroxysmal motor behaviors: a stereo-EEG study. Epilepsia. 2011;52(10):1781–1791. | ||
Ryvlin P. Avoid falling into the depths of the insular trap. Epileptic Disord. 2006;8 Suppl 2:S37–56. | ||
Provini F, Plazzi G, Lugaresi E. From nocturnal paroxysmal dystonia to nocturnal frontal lobe epilepsy. Clin Neurophysiol. 2000;111 Suppl 2:S2–S8. | ||
Picard F, Bertrand S, Steinlein OK, Bertrand D. Mutated nicotinic receptors responsible for autosomal dominant nocturnal frontal lobe epilepsy are more sensitive to carbamazepine. Epilepsia. 1999;40(9):1198–1209. | ||
Raju GP, Sarco DP, Poduri A, Riviello JJ, Bergin AM, Takeoka M. Oxcarbazepine in children with nocturnal frontal-lobe epilepsy. Pediatr Neurol. 2007;37(5):345–349. | ||
Oldani A, Manconi M, Zucconi M, Martinelli C, Ferini-Strambi L. Topiramate treatment for nocturnal frontal lobe epilepsy. Seizure. 2006;15(8):649–652. | ||
Varadkar S, Duncan JS, Cross JH. Acetazolamide and autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia. 2003;44(7):986–987. | ||
Liguori C, Romigi A, Placidi F, Sarpa MG, Mercuri NB, Izzi F. Effective treatment of nocturnal frontal lobe epilepsy with lacosamide: a report of two cases. Sleep Med. 2016;23:121–122. | ||
Samarasekera SR, Berkovic SF, Scheffer IE. A case series of lacosamide as adjunctive therapy in refractory sleep-related hypermotor epilepsy (previously nocturnal frontal lobe epilepsy). J Sleep Res. 2018:e12669–e12669. | ||
Willoughby JO, Pope KJ, Eaton V. Nicotine as an antiepileptic agent in ADNFLE: an N-of-one study. Epilepsia. 2003;44(9):1238–1240. | ||
Brodtkorb E, Picard F. Tobacco habits modulate autosomal dominant nocturnal frontal lobe epilepsy. Epilepsy Behav. 2006;9(3):515–520. | ||
Naldi I, Bisulli F, Vignatelli L, et al. Tobacco habits in nocturnal frontal lobe epilepsy. Epilepsy Behav. 2013;26(1):114–117. | ||
Puligheddu M, Melis M, Pillolla G, et al. Rationale for an adjunctive therapy with fenofibrate in pharmacoresistant nocturnal frontal lobe epilepsy. Epilepsia. 2017;58(10):1762–1770. | ||
de Paolis F, Colizzi E, Milioli G, et al. Effects of antiepileptic treatment on sleep and seizures in nocturnal frontal lobe epilepsy. Sleep Med. 2013;14(7):597–604. | ||
Mikati MA, Jiang YH, Carboni M, et al. Quinidine in the treatment of KCNT1-positive epilepsies. Ann Neurol. 2015;78(6):995–999. | ||
Abdelnour E, Gallentine W, Mcdonald M, Sachdev M, Jiang YH, Mikati MA. Does age affect response to quinidine in patients with KCNT1 mutations? Report of three new cases and review of the literature. Seizure. 2018;55:1–3. | ||
Nobili L, Sartori I, Terzaghi M, et al. Relationship of epileptic discharges to arousal instability and periodic leg movements in a case of nocturnal frontal lobe epilepsy: a stereo-EEG study. Sleep. 2006;29(5):701–704. | ||
Nobili L, Cardinale F, Magliola U, et al. Taylor’s focal cortical dysplasia increases the risk of sleep-related epilepsy. Epilepsia. 2009;50(12):2599–2604. | ||
Manni R, Terzaghi M. Comorbidity between epilepsy and sleep disorders. Epilepsy Res. 2010;90(3):171–177. | ||
Malow BA, Foldvary-Schaefer N, Vaughn BV, et al. Treating obstructive sleep apnea in adults with epilepsy: a randomized pilot trial. Neurology. 2008;71(8):572–577. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.