Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 11
Sjögren's syndrome and systemic lupus erythematosus: links and risks
Authors Pasoto SG, Adriano de Oliveira Martins V, Bonfa E ![]()
Received 31 October 2018
Accepted for publication 12 December 2018
Published 29 January 2019 Volume 2019:11 Pages 33—45
DOI https://doi.org/10.2147/OARRR.S167783
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Sandra Gofinet Pasoto,1,2 Victor Adriano de Oliveira Martins,1 Eloisa Bonfa1
1Rheumatology Division, Hospital das Clinicas, Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, Sao Paulo, Brazil; 2Laboratory Division, Hospital das Clinicas, Faculdade de Medicina da Universidade de Sao Paulo (HCFMUSP), Sao Paulo, Sao Paulo, Brazil
Abstract: Systemic lupus erythematosus (SLE) and Sjögren’s syndrome (SS) may coexist, and they are chronic complex disorders, with an autoimmune background, multifactorial etiology, multiple circulating autoantibodies, and variable prognosis. The prominent feature of SS is the impairment of the lacrimal and salivary glands leading to sicca symptoms. This disease may be classified as primary Sjögren’s syndrome (pSS), or secondary Sjögren’s syndrome (sSS) since it is often associated to other autoimmune disorders, principally SLE, rheumatoid arthritis, and systemic sclerosis. Systematic reviews and meta-analyses show an sSS prevalence in SLE patients of about 14%–17.8%. Herein, we updated important aspects of the clinical association between SLE and sSS through a narrative review of the PubMed database in the last 5 years (from July 2013 to October 2018) with the terms “Sjogren syndrome and systemic lupus erythematosus”. The following aspects are addressed: the classification criteria for sSS; differences and similarities between SLE and pSS regarding demographic, clinical, and serological characteristics (including new autoantibodies), as well as comorbidities; the etiopathogenic links between SLE and pSS (including genetic and environmental factors, B-cell activation, and autoantibodies); the predictive factors for sSS onset in SLE patients; the ocular and oral involvements due to sSS in SLE; and the main distinctive demographic, clinical, and serological features of SLE with and without associated SS.
Keywords: systemic lupus erythematosus, primary Sjögren’s syndrome, secondary Sjögren’s syndrome, polyautoimmunity, anti-Ro, anti-SSA, autoantibodies, pathogenesis
Introduction
Systemic lupus erythematosus (SLE) is a complex disorder of autoimmune background, multifactorial etiology, impairment of several organic systems resulting in a widespread spectrum of clinical manifestations, as well as variable prognosis and evolving clinical course characterized by the occurrence of episodes of active disease and remission.1 The prominent SLE feature is the production of multiple circulating autoantibodies. Some of these reactivities, such as anti-dsDNA and anti-Sm, are specific serologic markers of SLE.2,3 In turn, the anti-dsDNA antibody is associated with lupus glomerulonephritis.2–4
It is noteworthy that the prevalence of SLE is increasing in different regions of the world, which have different rates ranging from <40 per 100,000 inhabitants (in the European countries, such as France) to approximately >160 per 100,000 inhabitants (in some regions of the USA and Puerto Rico).5 This enhanced prevalence of SLE may be due to an earlier diagnosis and improved survival rate.5
Interestingly, SLE may be associated with Sjögren’s syndrome (SS), an autoimmune chronic inflammatory clinical condition, which involves principally the lacrimal and salivary glands resulting in decrease of the salivary and lacrimal flows and, consequently, to dry mouth and dry eye symptoms.6 SS may be classified as primary Sjögren’s syndrome (pSS), or secondary Sjögren’s syndrome (sSS) (also called polyautoimmunity, due to its association with other autoimmune disorders, especially SLE, rheumatoid arthritis [RA], and systemic sclerosis [SSc]).6,7 The term secondary SS is most commonly used in the literature, though some authors have a preference for the polyautoimmunity designation.7
SLE is one of the autoimmune diseases most often associated with SS,7 as reported in 9%–33% of SLE patients.6–9 Systematic reviews and meta-analyses published more recently show a prevalence of sSS in SLE patients of about 14%–17.8%.10,11 Of note, sSS constitutes a major cause of ocular and oral involvement in SLE.8,12,13
In this narrative review, we updated important aspects of the clinical association between SLE and SS through a survey in the PubMed database in the last 5 years (from July 2013 to October 2018) with the terms “Sjogren syndrome and systemic lupus erythematosus”. The following aspects were addressed: the classification criteria for sSS; the differences and similarities between SLE and pSS regarding demographic, clinical, and serological characteristics (including new autoantibodies), as well as comorbidities; the etiopathogenic links between SLE and pSS (including genetic and environmental factors, B-cell activation, and autoantibodies); the predictive factors for sSS onset in SLE patients; the ocular and oral involvements due to sSS in SLE; and the main distinctive demographic, clinical, and serological features between SLE with associated SS (sSS-SLE) and without associated SS.
Classification criteria for sSS
The classification criteria of the American-European Consensus Group (2002) for pSS and sSS are based on a combination of the following items: 1) dry eye symptoms; 2) dry mouth symptoms; 3) abnormalities in objective ocular tests; 4) alterations in objective oral tests; 5) positive circulating anti-Ro (SSA) and/or anti-La (SSB) antibodies; and 6) histological analysis of the minor salivary glands revealing a focal lymphocytic sialadenitis (focus score ≥1/4 mm2 of glandular tissue).14
Dry eye symptoms include dry eye complaint for >3 months, and/or feeling the presence of foreign bodies in the eyes, and/or use of artificial tears more than three times a day.14 In turn, symptoms of dry mouth were defined as oral dryness for >3 months, and/or recurrent or persistent parotid gland enlargement, and/or difficulty swallowing solid foods, requiring fluid intake for relief of this symptom.14
Abnormalities in ocular objective tests consist of Schirmer’s I test result ≤5 mm/5 min (Figure 1A), and/or a positive ocular color score, eg, lissamine green (van Bijsterveld’s score ≥4) (Figure 1B).14 Alterations in the objective oral tests comprise the following: an unstimulated salivary flow ≤0.1 mL/min (Figure 1C); and/or salivary gland scintigraphy demonstrating decrease/delay in the uptake and/or excretion of the radioactive tracer; and/or sialography-imaging test of the parotid glands revealing the presence of characteristic diffuse sialectasias.14
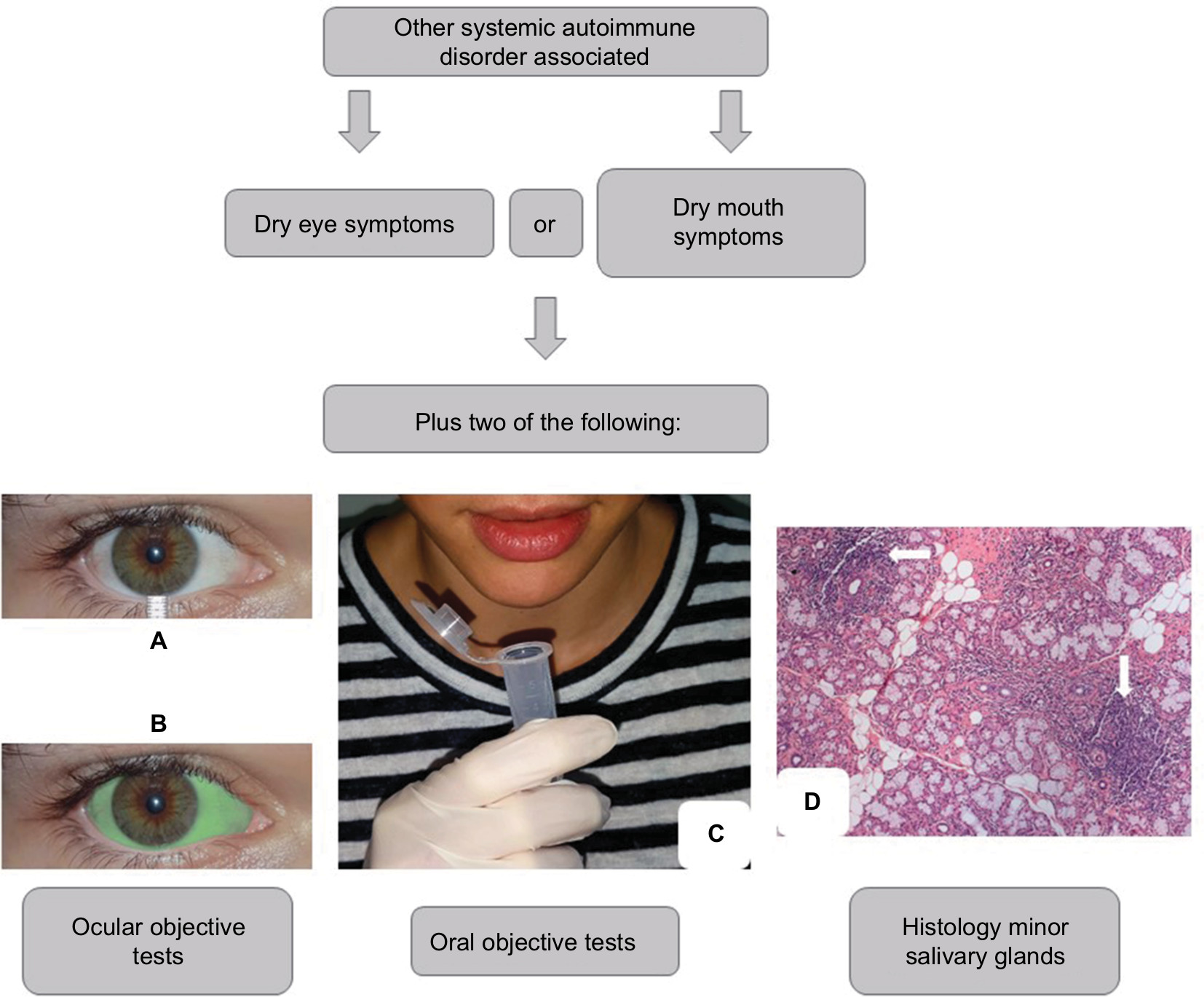 | Figure 1 Scheme illustrating the classification criteria (according to The American-European Consensus Group14) for secondary Sjögren’s syndrome. Notes: Ocular objective tests: Schirmer’s I test (A) and ocular color score, eg, lissamine green (B). Oral objective tests: unstimulated salivary flow (C), salivary gland scintigraphy, and sialography-imaging test of the parotid glands. Histological image of the minor salivary glands (H&E; image courtesy: Professor Daniela Assis do Vale, Oral and Maxillofacial Pathology Department, School of Dentistry, University of Sao Paulo, Sao Paulo, Brazil) (D) showing foci of lymphocytes (arrows). |
Histological analysis of the minor salivary glands (lip biopsy) showing a focal lymphocytic sialadenitis with the presence of ≥1 foci of infiltrating lymphocytes per 4 mm2 of glandular tissue (focus score ≥1/4 mm2 of glandular tissue) favors an SS diagnosis (Figure 1D).14 One focus is set to an aggregate of ≥50 lymphocytes.14
According to these criteria, four of the above-mentioned six items are required for pSS classification, necessarily with positive autoantibodies or abnormal histological analysis, and the absence of any other associated autoimmune disease.14 On the other hand, the requirements for sSS classification are shown in Figure 1. The exclusion criteria are prior head/neck radiotherapy, hepatitis C, AIDS, prior lymphoma, sarcoidosis, graft vs host disease, and the utilization of anticholinergic drugs.14
Subsequently, the American College of Rheumatology (ACR) published the classification criteria for SS (2012).15 To fulfill these criteria, two of the following items are necessary: 1) anti-Ro (SSA) or anti-La (SSB), or rheumatoid factor (RF) plus antinuclear antibody (ANA) test with titer >1:320; 2) ocular staining score (OSS) ≥3; and 3) minor salivary gland biopsy showing a focal lymphocytic sialadenitis (focus score ≥1 focus/4 mm2 of glandular tissue).15 The exclusion criteria are similar to those of the American-European Consensus Group (2002),14 but IgG4-related disease was added and lymphoma was no longer a cause for exclusion.15 However, these criteria do not differentiate the primary and secondary forms of SS, and they were validated in a cohort of pSS patients (Sjögren’s International Collaborative Clinical Alliance cohort).15
Currently, ACR/European League Against Rheumatism (EULAR) have proposed new classification criteria for pSS (2016).16 In these criteria, the symptoms of sicca syndrome were not included.16 Furthermore, the most important components, with higher classification weight, were positive anti-Ro (SSA) antibody (weight 3) and histological analysis of the minor salivary glands evidencing a focal sialadenitis with focus score ≥1 focus/4 mm2 of glandular tissue (weight 3).16 Anti-La (SSB) antibody was excluded from these novel classification criteria, as when present alone (without anti-Ro [SSA]) it was not associated with the SS phenotype.17 The other items of these criteria include OSS ≥5 (or van Bijsterveld’s score ≥4) (weight 1), Schirmer’s test ≤5 mm/5 min (weight 1), and an unstimulated salivary flow ≤0.1 mL/min (weight 1).16 For classification as pSS, the patient must achieve a total score (sum of the weights obtained) ≥4.16 Moreover, these criteria should be applied to patients with symptoms of sicca syndrome (as defined in the classification criteria of the American-European Consensus Group, 2002) and/or systemic manifestations suggestive of SS (defined according to the EULAR Sjögren’s Syndrome Disease Activity Index [ESSDAI]).18 The exclusion criteria are similar to those proposed by the ACR (2012),15 but hepatitis C is only considered a cause of exclusion if the PCR test is positive.16 Patients using anticholinergic drugs should undergo objective ocular and oral tests after temporary withdrawal of these drugs for a sufficient time.16
One of the major advantages of these novel criteria was probably to allow an earlier diagnosis of the disease.16 In addition, these criteria were a paradigm break since they do not necessarily require the presence of symptoms of ocular and/or oral dryness.16 On the contrary, systemic manifestations of pSS are equally appreciated.16
Importantly, the ACR/EULAR classification criteria for pSS were validated specifically for pSS.16 Therefore, sSS is still classified using the criteria of the American-European Consensus Group (2002)14 (Figure 1).
Demographic, clinical, and serological features of pSS and SLE
Demographical features
SLE and pSS have several similar and distinct epidemiologic and clinical aspects. Although both disorders affect mostly women,19,20 the female:male ratio is up to 20:1 in pSS,19 and lower in SLE (up to 15:1).20 Likewise, a 40–60 years old peak incidence is observed in pSS,19 whereas in SLE it occurs in the fertile phase of women.20 Concerning ethnicity, SLE incidence is higher in Blacks than in White individuals,20 while pSS predominates in Caucasians.19
Clinical features
SLE and pSS have various comparable clinical manifestations and organic involvements, such as fever, fatigue, adenomegalies, photosensitivity, Raynaud’s phenomenon, palpable purpura, arthralgia, nonerosive symmetric polyarthritis, myositis, hemolytic anemia, leukopenia, lymphopenia, thrombocytopenia, interstitial pneumonitis, renal tubular acidosis, and peripheral neuropathy.18,21–24 However, the frequencies of clinical manifestations are distinct in both diseases, particularly for serositis, glomerulonephritis, seizures, and psychosis, which are more frequently reported in SLE than in pSS.18,21–25
Serological features
Serum antinuclear antibodies (ANAs) detected by the indirect immunofluorescence test on HEp-2 cells is positive in ~99% of SLE patients, and they were included in the classification criteria of this disease.21,23 On the other hand, a positive ANA test is observed in >70% of pSS patients, often showing a nuclear fine speckled pattern (Figure 2A), which is suggestive of anti-Ro (SSA) and/or anti-La (SSB) autoantibodies26 (Figure 2B).
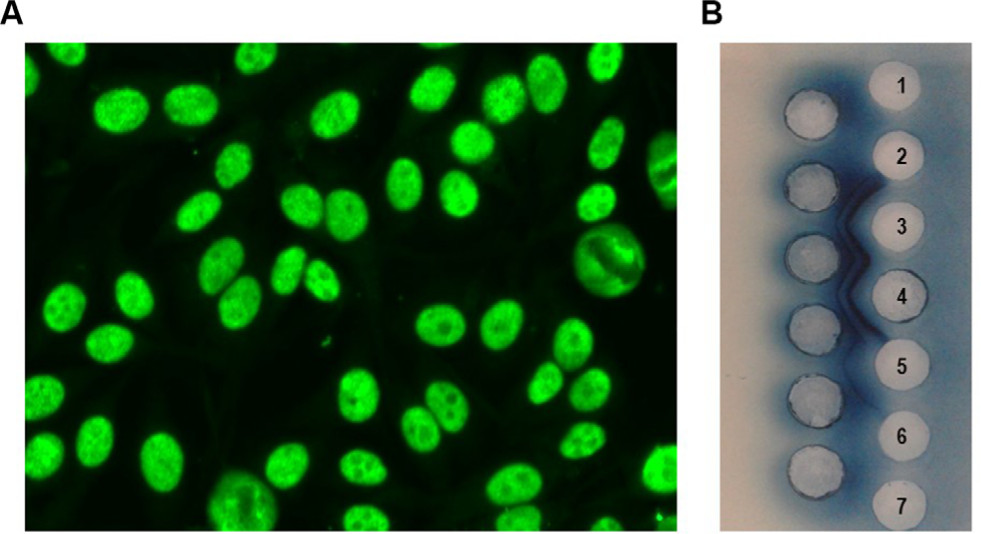 | Figure 2 Antinuclear antibodies test (indirect immunofluorescence on HEp-2 cells) showing the nuclear fine speckled pattern (A). Illustrative image of anti-Ro (SSA) and anti-La (SSB) antibodies detected by counterimmunoelectrophoresis (B). 1, 2, 6, 7: negative serum samples; 3, 4: serum samples positive for anti-Ro (SSA) and anti-La (SSB); 5: serum sample positive for other reactivity (anti-U1RNP). |
The frequencies of these two autoantibodies in pSS patients are 50%–90% and 25%–60%, respectively, depending on the applied laboratorial technique and particular features of the assessed cohort.26 Their presences are correlated with higher rates of parotiditis and extraglandular involvements in pSS, compared to those patients negative for these reactivities.26,27 Ro 52 kDa, Ro 60 kDa, and La proteins are major target antigens for these antibodies in pSS, but they are not specific serological markers for this disorder.2,4 These proteins are associated with RNA particles (small RNAs), constituting a highly conserved antigenic complex localized in the nucleus and cytoplasm of several cell types, and they seem to play a role in the cell division process.2,26
Anti-Ro (SSA) and/or anti-La (SSB) antibodies are also detected in sera from individuals with different autoimmune disorders, such as SLE, subacute cutaneous lupus, neonatal lupus, inflammatory myopathies (polymyositis [PM] and dermatomyositis [DM]), SSc, mixed connective tissue disease, RA, and primary biliary cholangitis (CBP).2,4,28–30 In SLE, these antibodies are found in ~30%–40% and 10%–15% of cases, respectively, and they are not correlated with lupus disease activity.2–4,28 In SLE patients, anti-Ro (SSA) antibody associates to hematological manifestations (anemia, leucopenia, lymphopenia, and thrombocytopenia), palpable purpura, and sSS.3,4,28
A large cohort study assessed serum anti-Ro (SSA) and anti-La (SSB) antibodies in 2,082 individuals with several autoimmune disorders, and in 524 sera from geographically matched healthy control subjects.31 Anti-Ro (SSA) antibody was significantly more frequent and demonstrated high titers in five of these disorders: pSS, SLE, antiphospholipid syndrome (APS) (both primary and secondary to SLE), SSc, and CBP.31 Anti-La (SSB) was also more common and showed high titers in pSS, SLE, APS secondary to SLE, and CBP.31 The frequencies (and not the titers) of anti-Ro (SSA) and anti-La (SSB) antibodies were also higher in PM.31
Recent studies revealed that the anti-Ro (SSA) antibody is usually reactive with Ro52 (also termed Ro52/tripartite motif-containing 21) and not with Ro60 protein in the idiopathic inflammatory myopathies (PM, DM).32 In pSS patients, 63.2% of anti-Ro52 positive sera were also positive for anti-Ro60.33 Interestingly, the epitopes of Ro52 protein are mainly linear, whereas the epitopes of Ro60 have a conformational structure.32,34 Therefore, the anti-Ro52 antibody is neither detected by usual precipitation immunoassays such as counterimmunoelectrophoresis (Figure 2B) nor by the conventional ELISA tests using native Ro/SSA protein as an antigenic substrate.32,34 Thus, it is recommended that the anti-Ro52 and anti-Ro60 antibodies must be assayed through separate assays.32,34 Furthermore, anti-Ro52 alone (without anti-Ro60) may produce a cytoplasmic indirect immunofluorescence pattern in the ANA test on HEp-2 cells.34
RF is detected in 36%–74% of the sera from pSS patients, and it was associated with the severity of the salivary gland injury and extraglandular manifestations.26 In SLE, RF is reported in 15%–30% of cases.2
In a large international multicenter study, cryoglobulins were detected in 7.2% of pSS patients at the time of diagnosis.27 Importantly, cryoglobulins may be observed in up to 20% of pSS patients, and they are closely associated with the occurrence of vasculitis and lymphoma in these patients.24,26 Moreover, reduced C3 and C4 complement fractions at the pSS diagnosis were observed in 13.4% and 14.4% of the cases, respectively.27 These findings and the prevalence of cryoglobulins were associated with high ESSDAI score values.27
Hypocomplementemia is known to be associated with flare episodes in SLE.22 The decrease in C3 and C4 complement fractions was detected in 41.3% and 48.8% of SLE patients, respectively.35 Cryoglobulins were also found in 48.8% of SLE patients, and they were correlated with the total hemolytic complement levels and anti-dsDNA titres.35 Nevertheless, there was no significant association of cryoglobulins with disease flare measured through the Systemic Lupus Erythematosus Disease Activity Index.35
Antibodies to α-fodrin, a structural cytoskeleton protein that participates in cellular exocrine secretion, may be detected in 38%–42% of sera from pSS patients.36 These antibodies, however, are not specific serological markers for pSS.36 Antibodies directed to α-fodrin may also be detected in the sera from SLE and RA patients with or without sSS.36 The associations of anti-α-fodrin + RF, or anti-α-fodrin + ANA >1:320, or at least two of three may favor pSS diagnosis in patients negative for anti-Ro (SSA) and anti-La (SSB) antibodies.37
In contrast, antibodies directed to cyclic type 3 muscarinic acetylcholine receptor peptides were detected in 62.2% of SLE patients and only in 7.1% of pSS patients.38
Antiphospholipid antibodies, such as anticardiolipin (aCL), lupus anticoagulant, and/or anti-β2-glycoprotein-1, may be found in 16% of pSS patients,39 and in ~40% of SLE patients.4
In summary, the numerous clinical and serological similarities between pSS and SLE often hamper the distinction of both disorders. This aspect was recently assessed in a large cohort study including 1,175 patients with sicca syndrome symptoms.40 Five hundred and twenty four of these patients (44.6%) referred a prior diagnosis or possible diagnosis of RA, SLE, or SSc.40 These diagnoses were endorsed in ~1/4 of these patients (130/524 [24.8%]), and they were ruled out in 394/524 (75.2%) of them (using the respective classification criteria for these diseases).40 Of note, >1/3 (183/524 [34.9%]) of the individuals with prior diagnosis/doubt of RA, SLE, or SSc fulfilled the classification criteria for pSS (American-European Consensus Group, 2002).40 A positive ANA test was correlated to a prior incorrect diagnosis of SLE, while a positive RF test was correlated to a misdiagnosis of RA.40 These findings reinforce the importance of careful investigation of pSS in patients with suspicion of diffuse connective tissue diseases, especially in cases with symptoms of sicca syndrome. Table 1 summarizes the main serological features observed in pSS and SLE.
 | Table 1 Main serological features observed in pSS and SLE Abbreviations: SLE, systemic lupus erythematosus; pSS, primary Sjögren’s syndrome; anti-c2M3RP, anti-cyclic type 3 muscarinic acetylcholine receptor peptides. |
New autoantibodies
New autoantibodies directed to salivary gland protein 1, carbonic anhydrase 6, and parotid secretory protein (PSP) were described in an experimental model of SS, as well as in sera from pSS patients.41,42 Importantly, these three autoantibodies may also be detected in patients with sSS, including SLE patients.43 Of note, the prevalence of anti-PSP appears to be higher in sSS-SLE patients than in SLE patients without sSS.43 However, this finding needs to be confirmed in future studies.
Our group recently described antibodies to DNase I, an enzyme that plays a role in the clearance of extracellular DNA derived from apoptosis, in 43.5% of the sera from pSS patients.44 This reactivity was formerly reported in 62% of SLE patients.45 Of note, it was recently demonstrated that sera from pSS patients have diminished DNase I activity,46 and it is possible that anti-DNase I antibodies underlie the impaired function of this enzyme.45
Comorbidities
A recent large multicenter study, which included 437 pSS patients and 2,926 SLE patients, evaluated the frequency of cardiovascular events, severe infections, and fibromyalgia (FM) in pSS patients in comparison to SLE patients without sSS.47 This comparative analysis was adjusted for demographical features, disease duration, and previous therapies with glucocorticoids, and antimalarial and immunosuppressive drugs.47 Traditional cardiovascular risk factors, eg, arterial hypertension (OR 0.51; 95% CI 0.39–0.66), dyslipidemia (OR 0.77; 95% CI 0.61–0.98), and smoking (OR 0.39; 95% CI 0.29–0.51), were less frequent in the pSS group than in SLE patients without sSS.47 A similar finding was observed for stroke/myocardial infarction (OR 0.61; 95% CI 0.38–0.93).47 The adjusted analysis for demographical characteristics, and disease duration revealed that serious infections were also less commonly observed in the pSS group (OR 0.62; 95% CI 0.44–0.86) than in SLE patients without sSS.47 However, after adjustment for the use of glucocorticoids, hydroxychloroquine, and immunosuppressants, the risk of severe infections was comparable between the groups with pSS and SLE without pSS.47 In contrast, lymphoma (adjusted OR 4.41; 95% CI 1.35–14.43) and FM (adjusted OR 2.43; 95% CI 1.70–3.49) occurred more often in pSS than in SLE patients without sSS.47
Etiopathogenic links between SLE and pSS
The etiopathogenesis of pSS and SLE are not yet completely clarified, but they seem to involve genetic susceptibility associated with epigenetic, environmental, and hormonal factors.6,48 In this regard, new evidences suggest that both disorders share many etiopathogenic links, which are presented below.
Genetic factors
Interestingly, a family history of SLE increases the risk of evolving with this disease, as well as the risk for other autoimmune diseases, including pSS.49,50
Additionally, the frequency of karyotype 47, XXX (Klinefelter syndrome) is significantly greater in SLE patients (OR 8.78; 95% CI 1.67–86.79) and pSS patients (OR 10.29; 95% CI 1.18–123.47) than in the control population without rheumatic diseases.51 Interestingly, sex steroids are in the normal range in 47, XXX patients.51 Therefore, such findings indicate that SLE and pSS susceptibility is augmented by the X chromosome number.51
However, X chromosome trisomy in women is rarely observed in the general population.51 On the other hand, there are evidences suggesting that hormonal factors, such as the imbalance of sex hormones, altered serum prolactin levels, and the decrease of serum concentrations of vitamin D, play a role in the pathogenesis of SLE and pSS.1,6,48
Various susceptibility loci were also associated with the development of SLE and pSS, suggesting a complex polygenic pattern.6,48 Moreover, studies of genome-wide associations identified alleles shared between SLE and pSS patients, such as STAT4, interferon (IFN) regulatory factor 5 (IRF5), B lymphoid tyrosine kinase, tumor necrosis factor (TNF) α-induced protein 3-interacting protein 1, human leukocyte antigen (HLA) complex group 22, chromosome 7 open reading frame 72 (C7orf72), C6orf15, C6orf10, neurogenic locus notch homolog protein 4, butyrophilin-like protein 2, PR domain zinc finger protein 10, autophagy-related 5 protein, Ikaros family zinc finger protein 1, as well as HLA molecules including HLA-DRA, HLA-DQA1, HLA-DQB1, HLA-DQA2, HLA-DPA1, and HLA-DPB1.52
Interestingly, many of these genes are very important for normal activity of the immune system.6,48 For example, the upregulation of transcription factors related to type I IFN regulated genes, the so-called IFN signature, including STAT4, IRF5, IRF7, and IRF8, may lead to abnormal activation of B lymphocytes.53,54 B-cell signaling proteins may be also overexpressed, such as LYN (proto-oncogene Src family tyrosine kinase) and BANK1 (B-cell scaffold protein with ankyrin repeats 1), as well as several cytokines and receptors including IL-10, CD44, and TNF superfamily member 4.53,54 Moreover, non-deleted (functional) leukocyte immunoglobulin-like receptor A3 conferred high susceptibility for SLE and pSS.55
LncRNAs, that are RNA molecules with >200 nucleotides with little or without protein synthesis capability, are important for regulation of gene expression.56 Recently, abnormal expression of several LncRNAs was demonstrated in SLE and pSS patients.56 Particularly, LncRNA Theiler’s murine encephalomyelitis virus persistence candidate gene 1 participates in IFN-γ overexpression in SLE and pSS patients.56
Epigenetic alterations
Long interspersed nuclear elements (LINEs) are endogenous DNA sequences transcribed into mRNA and translated into proteins that act as reverse transcriptases.57 The reverse transcriptase makes a DNA copy of the LINE RNA that may be integrated into the genome at a novel site.57 Recently, it was demonstrated that abnormal DNA methylation leads to altered expression of LINE1 (L1) in samples of minor salivary gland tissue from pSS patients and renal biopsy specimens from SLE patients.58 This mechanism might increase the activation of potentially pathogenic LINEs.58
Activation of B-cells
T helper (Th) lymphocytes CD4(+), Th1, Th2, Th17, and follicular helper T are important in the pathogenesis of SLE and pSS.1,6,48 In addition, B-cell activation is a remarkable finding in both diseases.1,6,48
Several mechanisms are important for regulating B lymphocyte activity in SLE and pSS.6,48 Recent evidences suggest that activation of these cells and of long-lived plasma cells via signaling of toll-like receptors (TLRs) promotes the arrangement of ectopic lymphoid aggregates (germinal centers) into the kidneys (SLE) and salivary glands (pSS) of these patients.59 High concentrations of type I IFN are detected in sera and tissue samples from SLE and pSS patients, indicating the upregulation of IFN regulatory factors (eg, IRF8 and IRF9) and the activation of innate immune response cells.6,48,59 In fact, numerous genetic polymorphisms associated with the activation of type I IFNs confer augmented susceptibility to SLE and pSS.52–54 Of note, immune complexes carrying nucleic acids may induce IFN-α release by plasmacytoid dendritic cells (pDCs) through the signaling of TLRs.59 Likewise, RNA from endogenous viral retro elements of the human genome, which may be found in tissues of SLE and pSS patients, might also trigger type I IFN production.57,58
B-cell stimulatory factors (eg, B-cell activating factor and IL-6) and chemotactic cytokines for B-cells and plasma cells (eg, CXCL13 and CXCL12) are increased in the kidney of lupus-prone mice concomitantly with the proliferation of anti-dsDNA secreting cells.60 Moreover, TLR7 and TLR9 seem important for autoantibody production and disease progression in murine models of SLE.61 In pSS, the development of germinal centers seems to be a consequence of the signaling through lymphotoxins CXCL13, CXCL12, chemokine C-C motif ligand (CCL)19, and CCL21 produced in the target tissues by immune and epithelial cells.61
Environmental factors and the possible role of the anti-Ro antibody in the pathogenesis of SLE and pSS
Infectious agents may potentially trigger the development of SLE and pSS in genetically predisposed subjects.62 Different mechanisms may lead to loss of the immunological tolerance to self-antigens, and to the increased production of circulating autoantibodies, including molecular mimicry, polyclonal lymphocyte activation, and epitope spreading.62 In this respect, several evidences suggest a possible role of the Epstein–Barr virus (EBV) in the pathogenesis of SLE and pSS,62 including the presence of EBV early antigen diffuse (anti-EA-D), an indicator of EBV replication, in the sera from SLE and pSS patients.63,64
There are also evidences suggesting that normal human microbial imbalance may be an important etiopathogenic factor for SLE and pSS.65 This imbalance in the intestinal microbiota, also called intestinal dysbiosis, may result in increased epithelial permeability, allowing fragments and microbial products to enter the subepithelial space and the lamina propria. Then, these molecules bind to specific receptors on the surface of antigen-presenting cells and proinflammatory T cells (including Th1 and Th17), resulting in B-cell differentiation and autoantibody production.65 In this regard, it was demonstrated that peptides from natural oral and gut microbiota may activate Ro60-reactive T cells.66
In addition, the anti-Ro (SSA) antibody may play a role in tissue damage in autoimmune diseases. Reinforcing this hypothesis, antibodies directed to Ro60 273-289 (Ro274) may induce autoimmune sialadenitis in an animal model.67
The proposed mechanism is briefly described below. Ro60 is exposed to the respective autoantibody through the apoptosis process.68 Then, opsonized apoptotic cell debris and immune complexes derived from these cells are phagocytosed by macrophages and pDCs.68 Later, the complex (consisting of anti-Ro60, Ro60, and Ro60-associated RNA) is transported to endosomes.68 In these organelles, the binding of Ro60-RNA to TLR7/TLR8 occurs, triggering the inflammatory process.68
Furthermore, immunotoxins that specifically bind to epitopes of the Ro60 molecule (aa482-493, aa310-323, or aa230-241) may cause lymphocytopenia in SLE and pSS patients.69
Figure 3 shows the main etiopathogenic links between pSS and SLE.
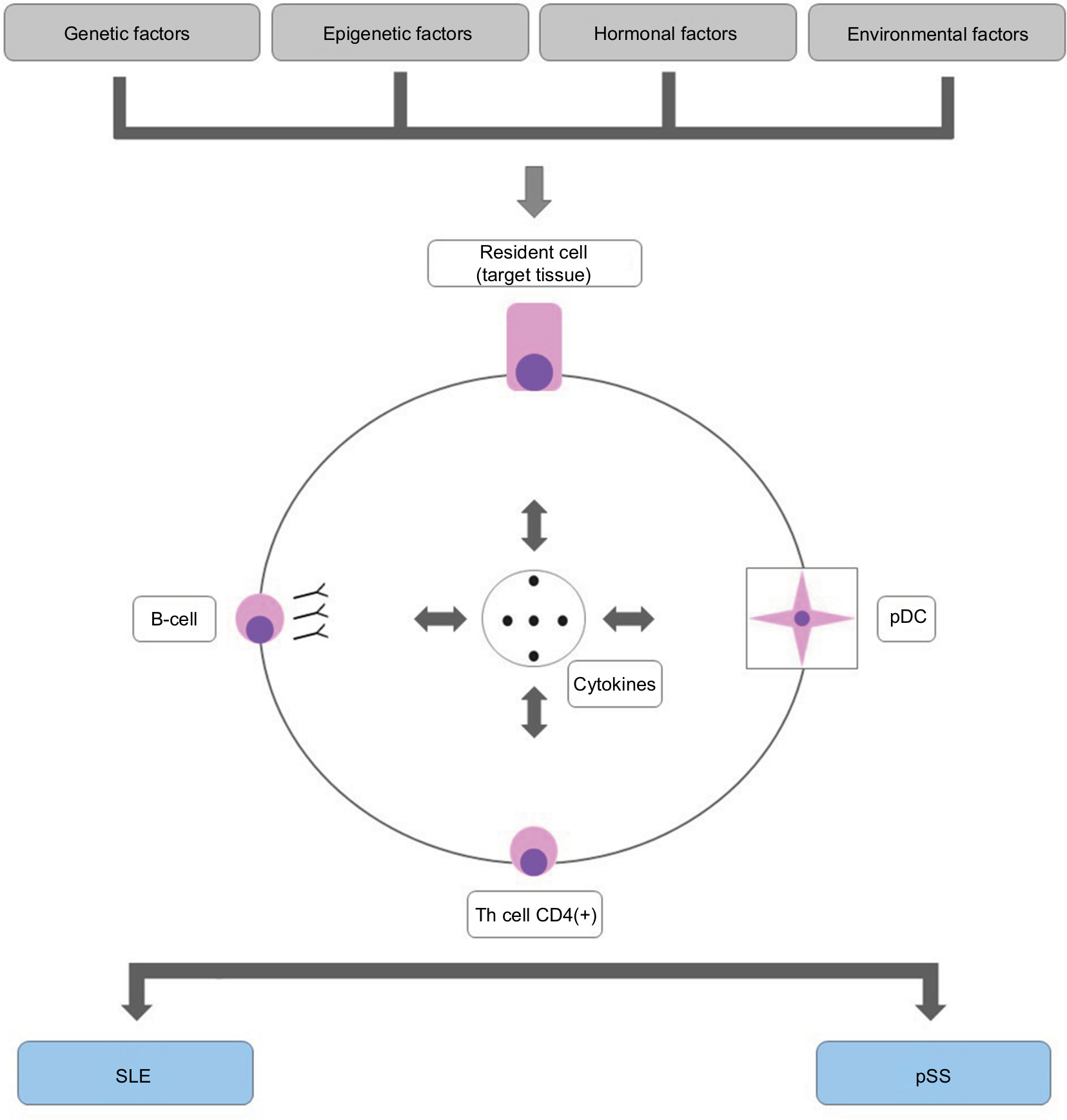 | Figure 3 Main etiopathogenic links between SLE and pSS. Abbreviations: pDC, plasmacytoid dendritic cell; Th, T helper lymphocyte; SLE, systemic lupus erythematosus; pSS, primary Sjögren’s syndrome. |
Predictive factors for sSS onset in SLE patients
In a cohort study including 103 SLE patients with recent onset (90.3% females; mean age at time of diagnosis =25.9±8.9 years), 18.5% of them developed sSS.70 These patients were significantly older at the SLE diagnosis than those without sSS (30.8±9.3 vs 24±8.8 years, respectively; P=0.004), and they had higher frequency of anti-Ro (SSA) antibody.70 The main risk factors for sSS development were disease onset at ≥25 years in addition to positive anti-Ro (SSA) antibody at diagnosis.70
Ocular involvement due to sSS in SLE
Approximately 30% of SLE patients may present with ocular manifestations caused by inflammation, ischemia due to vasculopathy/vasculitis, or toxicity by the use of glucocorticoids and hydroxychloroquine.12,71 Furthermore, different regions of the eye may be affected including cornea, conjunctiva, lens, retina, as well as the intrinsic and extrinsic ocular musculature.12,71 It is very important to investigate symptoms suggestive of ocular impairment at disease onset and during the follow-up of these patients, in addition to adequately monitoring drug toxicity, aiming at the early diagnosis and treatments to prevent complications such as vision loss. In this regard, the most frequent ocular involvement in SLE was keratoconjunctivitis sicca,12,72 and ~1/3 of SLE patients had dry eye symptoms.73
Importantly, dry eye symptoms are often more severe and refractory to treatment in individuals with associated systemic autoimmune rheumatic diseases (SARDs), such as pSS and sSS to SLE or RA, than those with dry eyes only.74 Accordingly, decrease in Schirmer’s I test result is more significant in patients with dry eye associated with SARDs comparatively to those without SARDs, suggesting a more significant dysfunction/destruction of the lacrimal glandular tissue in the first group.74 A similar finding was observed with corneal fluorescein staining, indicating increased epithelial damage of the cornea in patients with dry eye due to SARDs.74
The frequency of xerophthalmia is significantly higher in sSS-SLE patients than in SLE patients without sSS.8 Nevertheless, dry eyes in SLE do not always arise from sSS. Dry eye without sSS was reported in SLE patients, and the severity of this clinical manifestation was associated with serum anti-dsDNA titers.73
Oral involvement due to sSS in SLE
Oral manifestations in SLE are common and often are useful for the diagnosis of this disease. Furthermore, ulcerations, and erythematous and discoid lesions in the oral cavity are correlated with systemic disease activity.13 Dry mouth, candidiasis, periodontal disease, and squamous cell carcinoma may also occur.13
It is also interesting that salivary flow and saliva composition may reflect the changes resulting from the systemic activity of SLE. Salivary flow and the pH of saliva are lower in disease flare compared to remission periods of the disease.75 Levels of α-amylase and anti-chromatin antibodies in saliva from SLE patients are also associated with systemic disease activity.76 Additionally, the glutathione levels are diminished in saliva from SLE patients, indicating an impaired oxidative capacity.77
The frequency of xerostomia is significantly higher in sSS-SLE patients than in those without sSS.8
Comparison of demographic, clinical, and serological features of sSS-SLE and SLE without associated SS
The prevalence of sSS appears increased in female SLE patients,8,78 and in patients with late-onset (age ≥50 years) SLE.79 The higher prevalence of females in sSS-SLE patients compared to those without sSS was confirmed in a recently published systematic review and meta-analysis.11 This study also showed higher thyroiditis frequency in sSS-SLE patients than in those without sSS.11
A meta-analysis that included 444 sSS-SLE patients and 2,489 SLE patients without sSS demonstrated that the first group was significantly older, and it also had higher frequencies of oral ulcers and arthritis, as well as lower rates of proteinuria.10 The anti-Ro (SSA) and anti-La (SSB) antibodies were also more common in sSS-SLE patients than in those without sSS.10 The frequencies of anti-dsDNA were comparable in both groups.10 On the other hand, the anti-Sm and aCL antibodies were more frequent in the second group.10
Other significant clinical associations reported in case series were longer disease duration, and higher frequencies of interstitial pneumonitis, renal tubular acidosis, elevated serum IgG, and positive RF test in sSS-SLE patients than in those without associated sSS.8 In contrast, a significant lower frequency of involvement of the central nervous system was observed in the sSS-SLE group compared to the SLE patients without associated sSS.8
sSS was more prevalent in patients with subacute cutaneous lupus and SLE than in those with discoid lupus erythematosus.80
To date, there is no evidence of a higher prevalence of lymphoma in sSS-SLE patients compared to those without sSS.11 The mortality rate was lower in sSS-SLE patients than in SLE patients without sSS.8,81
Table 2 illustrates the main demographic, clinical, and serological features of SLE patients with and without sSS.
 | Table 2 Main demographic, clinical, and serological features of SLE patients with and without sSS Abbreviations: SLE, systemic lupus erythematosus; sSS, secondary Sjögren’s syndrome; sSS-SLE, SLE patients with sSS. |
Comparison of demographic, clinical, and serological features of pSS and sSS
The comparative analysis between pSS and sSS is relevant to clinical practice, since an epidemiological study showed a pSS:sSS ratio of about 60% to 40%.82
Interestingly, relatives of SLE patients may develop pSS and sSS, showing a similar genetic susceptibility for both syndromes.50 On the other hand, HLA-DRB1*14:06 was significantly more frequent in pSS patients than in those with sSS.83
Subpopulations of infiltrating lymphocytes in salivary gland tissues from patients with pSS and sSS are also comparable, including CD4(+)/IFN-γ(+), CD4(+)/IL-4(+), and IL-22(+) cells.84 Regarding histological characteristics of the minor salivary glands, SLE patients with xerostomia may have, additional to the typical lymphocytic foci of SS, other alterations such as mild/moderate sialadenitis, and thickening and hyalinization of the ductal basal membrane.85
Although these findings may explain the presence of a similar profile of clinical and laboratory characteristics in both syndromes, some subtle differences were noted. In this aspect, pSS patients had high frequencies of dry mouth (92% vs 84%; P=0.02), parotid enlargement (56% vs 9.2%; P<0.001), and positive anti-Ro (SSA)/La (SSB) antibodies (82% vs 41%; P<0.001) than those with sSS.86 In contrast, Raynaud’s phenomenon was more frequent in sSS patients (16% vs 41%; P<0.001).86 Table 3 presents the main similarities and differences between pSS and sSS-SLE.
 | Table 3 Main demographic, clinical, and serological features of pSS and sSS-SLE Abbreviations: SLE, systemic lupus erythematosus; pSS, primary Sjögren’s syndrome; sSS, secondary Sjögren’s syndrome. |
In addition to the usual autoantibodies, a novel reactivity appears to be promising for the differential diagnosis between pSS and sSS. The anti-self-vimentin specific antibody (anti-3S-P) was detected in 68.2% sera from sSS patients (66.2% with sSS-RA and 76.5% with sSS-SLE), and only in 1.8% of pSS, 1.3% of RA, and 4.2% of SLE patients.87
Conclusion
- SLE and pSS are closely related chronic inflammatory clinical conditions of autoimmune nature. They share several possible underlying etiopathogenic aspects, including numerous genetic factors, epigenetic, environmental, and hormonal factors, as well as immunological characteristics, which may account for the comparable clinical and autoantibody spectrum. Thus, careful investigation of pSS in patients with SLE diagnosis is recommended, particularly in cases with sicca syndrome symptoms.
- The occurrence of sSS in patients with SLE is around 14.0%–17.8% of cases.
- The possible development of sSS in SLE patients should be considered, especially in those aged 25 years at the onset of disease and with positive anti-Ro (SSA) antibody.
- Compared to SLE without sSS, sSS-SLE patients have a peculiar profile of clinical and serological manifestations, such as older age of disease onset, longer disease duration, higher prevalence of females and patients with SLE late-onset, greater frequencies of thyroiditis, oral ulcers, arthritis, interstitial pneumonitis, renal tubular acidosis, elevated serum IgG, RF, anti-Ro (SSA) and anti-La (SSB) antibodies, as well as lower frequencies of proteinuria, involvement of central nervous system, mortality rate, anti-Sm, and aCL antibodies.
- pSS and sSS present similar clinical manifestations. Careful analyses of the clinical features and autoantibody profile are important for the differential diagnosis between both syndromes.
Acknowledgments
We thank Professor Daniela Assis do Vale of the Oral and Maxillofacial Pathology Department, School of Dentistry, University of Sao Paulo, Sao Paulo, Brazil, for her courtesy with the histological image of the minor salivary glands (Figure 1D). We also thank Nicole Andressa Gomes Fontoura, Margarete Borges Galhardo Vendramini, and Cleonice Bueno for their support in obtaining the images for the figures.
This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (#2015/03756-4 and 2018/09937-9) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (303422/2015-7).
Disclosure
The authors report no conflicts of interest in this work.
References
Di Battista M, Marcucci E, Elefante E, et al. One year in review 2018: systemic lupus erythematosus. Clin Exp Rheumatol. 2018;36(5):763–777. | ||
Aggarwal A. Role of autoantibody testing. Best Pract Res Clin Rheumatol. 2014;28(6):907–920. | ||
Yaniv G, Twig G, Shor DB, et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev. 2015;14(1):75–79. | ||
Cozzani E, Drosera M, Gasparini G, Parodi A. Serology of lupus erythematosus: correlation between immunopathological features and clinical aspects. Autoimmune Dis. 2014;2014:321359:1–13. | ||
Lewis MJ, Jawad AS. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology (Oxford). 2017;56(suppl_1):i67–i77. | ||
Psianou K, Panagoulias I, Papanastasiou AD, et al. Clinical and immunological parameters of Sjögren’s syndrome. Autoimmun Rev. 2018;17(10):1053–1064. | ||
Anaya JM, Rojas-Villarraga A, Mantilla RD, Arcos-Burgos M, Sarmiento-Monroy JC. Polyautoimmunity in Sjögren Syndrome. Rheum Dis Clin North Am. 2016;42(3):457–472. | ||
Xu D, Tian X, Zhang W, Zhang X, Liu B, Zhang F. Sjogren’s syndrome-onset lupus patients have distinctive clinical manifestations and benign prognosis: a case-control study. Lupus. 2010;19(2):197–200. | ||
Baer AN, Maynard JW, Shaikh F, Magder LS, Petri M. Secondary Sjogren’s syndrome in systemic lupus erythematosus defines a distinct disease subset. J Rheumatol. 2010;37(6):1143–1149. | ||
Yao Q, Altman RD, Wang X. Systemic lupus erythematosus with Sjögren syndrome compared to systemic lupus erythematosus alone: a meta-analysis. J Clin Rheumatol. 2012;18(1):28–32. | ||
Alani H, Henty JR, Thompson NL, Jury E, Ciurtin C. Systematic review and meta-analysis of the epidemiology of polyautoimmunity in Sjögren’s syndrome (secondary Sjögren’s syndrome) focusing on autoimmune rheumatic diseases. Scand J Rheumatol. 2018;47(2):141–154. | ||
Silpa-Archa S, Lee JJ, Foster CS. Ocular manifestations in systemic lupus erythematosus. Br J Ophthalmol. 2016;100(1):135–141. | ||
Menzies S, O’Shea F, Galvin S, Wynne B. Oral manifestations of lupus. Ir J Med Sci. 2018;187(1):91–93. | ||
Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002;61(6):554–558. | ||
Shiboski SC, Shiboski CH, Criswell LA, et al. American College of Rheumatology classification criteria for Sjögren’s syndrome: a data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance Cohort. Arthritis Care Res. 2012;64(4):475–487. | ||
Shiboski CH, Shiboski SC, Seror R, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis. 2017;76(1):9–16. | ||
Baer AN, Mcadams Demarco M, Shiboski SC, et al. The SSB-positive/SSA-negative antibody profile is not associated with key phenotypic features of Sjögren’s syndrome. Ann Rheum Dis. 2015;74(8):1557–1561. | ||
Seror R, Ravaud P, Bowman SJ, et al. EULAR Sjogren’s syndrome disease activity index: development of a consensus systemic disease activity index for primary Sjogren’s syndrome. Ann Rheum Dis. 2010;69(6):1103–1109. | ||
Patel R, Shahane A. The epidemiology of Sjögren’s syndrome. Clin Epidemiol. 2014;6:247–255. | ||
Rees F, Doherty M, Grainge MJ, Lanyon P, Zhang W. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatology (Oxford). 2017;56(11):1945–1961. | ||
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. | ||
Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29(2):288–291. | ||
Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–2686. | ||
Mariette X, Criswell LA. Primary Sjögren’s syndrome. N Engl J Med. 2018;378(10):931–939. | ||
Bougea A, Anagnostou E, Konstantinos G, George P, Triantafyllou N, Kararizou E. A systematic review of peripheral and central nervous system involvement of rheumatoid arthritis, systemic lupus erythematosus, primary Sjögren’s syndrome, and associated immunological profiles. Int J Chronic Dis. 2015;2015:910352. | ||
Fayyaz A, Kurien BT, Scofield RH. Autoantibodies in Sjögren’s syndrome. Rheum Dis Clin North Am. 2016;42(3):419–434. | ||
Brito-Zerón P, Acar-Denizli N, Ng WF, et al. How immunological profile drives clinical phenotype of primary Sjögren’s syndrome at diagnosis: analysis of 10,500 patients (Sjögren Big Data Project). Clin Exp Rheumatol. 2018;36 Suppl 112(3):102–112. | ||
Peng SL, Craft JE, Budd R, Gabriel SE, McInnes IB, O’Dell JR. Anti-nuclear antibodies. In: Firestein GS, Budd R, Gabriel SE, McInnes IB, O’Dell JR, editors. Kelley and Firestein’s Textbook of Rheumatology. 10th ed. Volume 1, Chapter 55. Philadelphia, PA: Elsevier Health Sciences; 2016. | ||
Zuppa AA, Riccardi R, Frezza S, et al. Neonatal lupus: follow-up in infants with anti-SSA/Ro antibodies and review of the literature. Autoimmun Rev. 2017;16(4):427–432. | ||
Vanoni F, Lava SAG, Fossali EF, et al. Neonatal systemic lupus erythematosus syndrome: a comprehensive review. Clin Rev Allergy Immunol. 2017;53(3):469–476. | ||
Agmon-Levin N, Dagan A, Peri Y, et al. The interaction between anti-Ro/SSA and anti-La/SSB autoantibodies and anti-infectious antibodies in a wide spectrum of auto-immune diseases: another angle of the autoimmune mosaic. Clin Exp Rheumatol. 2017;35(6):929–935. | ||
Lee AYS. A review of the role and clinical utility of anti-Ro52/TRIM21 in systemic autoimmunity. Rheumatol Int. 2017;37(8):1323–1333. | ||
Bloch DB. The anti-Ro/SSA and anti-La/SSB antigen-antibody systems. 2017. Available from: https://www.uptodate.com/contents/the-anti-ro-ssa-and-anti-la-ssb-antigen-antibody-systems. Accessed October 31, 2018. | ||
Menor Almagro R, Jurado Roger A, Rodríguez Gutiérrez FJ, Solís Díaz R, Cardiel MH, Salaberri Maestrojuan JJ. Association of anti-Ro52, anti-Ro60 and anti-La antibodies with diagnostic, clinical and laboratory features in a referral hospital in Jerez, Spain. Reumatol Clin. 2016;12(5):256–262. | ||
Karimifar M, Pourajam S, Tahmasebi A, Mottaghi P. Serum cryoglobulins and disease activity in systematic lupus erythematosus. J Res Med Sci. 2013;18(3):234–238. | ||
Shen L, Suresh L. Autoantibodies, detection methods and panels for diagnosis of Sjögren’s syndrome. Clin Immunol. 2017; 182:24–29. | ||
Hernández-Molina G, Nuñez-Alvarez C, Avila-Casado C, et al. Usefulness of IgA anti-α-fodrin antibodies in combination with rheumatoid factor and/or antinuclear antibodies as substitute immunological criterion in Sjögren syndrome with negative Anti-SSA/SSB antibodies. J Rheumatol. 2016;43(10):1852–1857. | ||
He J, Guo JP, Ding Y, et al. Diagnostic significance of measuring antibodies to cyclic type 3 muscarinic acetylcholine receptor peptides in primary Sjogren’s syndrome. Rheumatology (Oxford). 2011;50(5):879–884. | ||
Pasoto SG, Chakkour HP, Natalino RR, et al. Lupus anticoagulant: a marker for stroke and venous thrombosis in primary Sjögren’s syndrome. Clin Rheumatol. 2012;31(9):1331–1338. | ||
Rasmussen A, Radfar L, Lewis D, et al. Previous diagnosis of Sjögren’s syndrome as rheumatoid arthritis or systemic lupus erythematosus. Rheumatology (Oxford). 2016;55(7):1195–1201. | ||
Shen L, Kapsogeorgou EK, Yu M, et al. Evaluation of salivary gland protein 1 antibodies in patients with primary and secondary Sjogren’s syndrome. Clin Immunol. 2014;155(1):42–46. | ||
Suresh L, Malyavantham K, Shen L, Ambrus JL. Investigation of novel autoantibodies in Sjogren’s syndrome utilizing sera from the Sjogren’s international collaborative clinical alliance cohort. BMC Ophthalmol. 2015;15:38. | ||
de Langhe E, Bossuyt X, Shen L, Malyavantham K, Ambrus JL, Suresh L. Evaluation of autoantibodies in patients with primary and secondary Sjogren’s syndrome. Open Rheumatol J. 2017;11:10–15. | ||
Griffo P, Viana VST, Pasoto SG, Leon EP, Bonfa E. Anti-DNase I antibody: a new serological reactivity in primary Sjögren syndrome. J Clin Rheumatol. Epub 2018 Sep 27. | ||
Yeh TM, Chang HC, Liang CC, Wu JJ, Liu MF. Deoxyribonuclease-inhibitory antibodies in systemic lupus erythematosus. J Biomed Sci. 2003;10(5):544–551. | ||
Fragoulis GE, Vakrakou AG, Papadopoulou A, et al. Impaired degradation and aberrant phagocytosis of necrotic cell debris in the peripheral blood of patients with primary Sjögren’s syndrome. J Autoimmun. 2015;56:12–22. | ||
Rúa-Figueroa I, Fernández Castro M, Andreu JL, et al. Comorbidities in patients with primary Sjögren’s syndrome and systemic lupus erythematosus: a comparative registries-based study. Arthritis Care Res. 2017;69(1):38–45. | ||
Kwok SK, Tsokos GC. New insights into the role of renal resident cells in the pathogenesis of lupus nephritis. Korean J Intern Med. 2018;33(2):284–289. | ||
Kuo CF, Grainge MJ, Valdes AM, et al. Familial aggregation of systemic lupus erythematosus and coaggregation of autoimmune diseases in affected families. JAMA Intern Med. 2015;175(9):1518–1526. | ||
Aggarwal R, Anaya JM, Koelsch KA, Kurien BT, Scofield RH. Association between secondary and primary Sjögren’s syndrome in a large collection of lupus families. Autoimmune Dis. 2015;2015:298506. | ||
Liu K, Kurien BT, Zimmerman SL, et al. X chromosome dose and sex bias in autoimmune diseases: increased prevalence of 47,XXX in systemic lupus erythematosus and Sjögren’s syndrome. Arthritis Rheumatol. 2016;68(5):1290–1300. | ||
Li Y, Zhang K, Chen H, et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjögren’s syndrome at 7q11.23. Nat Genet. 2013;45(11):1361–1365. | ||
Burbelo PD, Ambatipudi K, Alevizos I. Genome-wide association studies in Sjögren’s syndrome: what do the genes tell us about disease pathogenesis? Autoimmun Rev. 2014;13(7):756–761. | ||
Toro-Domínguez D, Carmona-Sáez P, Alarcón-Riquelme ME. Shared signatures between rheumatoid arthritis, systemic lupus erythematosus and Sjögren’s syndrome uncovered through gene expression meta-analysis. Arthritis Res Ther. 2014;16(6):489. | ||
du Y, Su Y, He J, et al. Impact of the leucocyte immunoglobulin-like receptor A3 (LILRA3) on susceptibility and subphenotypes of systemic lupus erythematosus and Sjögren’s syndrome. Ann Rheum Dis. 2015;74(11):2070–2075. | ||
Gao Y, Li S, Zhang Z, Yu X, Zheng J. The role of long non-coding RNAs in the pathogenesis of RA, SLE, and SS. Front Med. 2018;5:193. | ||
Belancio VP, Roy-Engel AM, Pochampally RR, Deininger P. Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res. 2010;38(12):3909–3922. | ||
Mavragani CP, Nezos A, Sagalovskiy I, Seshan S, Kirou KA, Crow MK. Defective regulation of L1 endogenous retroelements in primary Sjogren’s syndrome and systemic lupus erythematosus: role of methylating enzymes. J Autoimmun. 2018;88:75–82. | ||
Thorlacius GE, Wahren-Herlenius M, Rönnblom L. An update on the role of type I interferons in systemic lupus erythematosus and Sjögren’s syndrome. Curr Opin Rheumatol. 2018;30(5):471–481. | ||
Wang W, Rangel-Moreno J, Owen T, et al. Long-term B cell depletion in murine lupus eliminates autoantibody-secreting cells and is associated with alterations in the kidney plasma cell niche. J Immunol. 2014;192(7):3011–3020. | ||
Bird AK, Meednu N, Anolik JH. New insights into B cell biology in systemic lupus erythematosus and Sjögren’s syndrome. Curr Opin Rheumatol. 2015;27(5):461–467. | ||
Pasoto SG, Ribeiro AC, Bonfa E. Update on infections and vaccinations in systemic lupus erythematosus and Sjögren’s syndrome. Curr Opin Rheumatol. 2014;26(5):528–537. | ||
Hanlon P, Avenell A, Aucott L, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between Epstein-Barr virus and systemic lupus erythematosus. Arthritis Res Ther. 2014;16(1):R3. | ||
Pasoto SG, Natalino RR, Chakkour HP, et al. EBV reactivation serological profile in primary Sjögren’s syndrome: an underlying trigger of active articular involvement? Rheumatol Int. 2013;33(5):1149–1157. | ||
Zhong D, Wu C, Zeng X, Wang Q. The role of gut microbiota in the pathogenesis of rheumatic diseases. Clin Rheumatol. 2018;37(1):25–34. | ||
Szymula A, Rosenthal J, Szczerba BM, Bagavant H, Fu SM, Deshmukh US. T cell epitope mimicry between Sjögren’s syndrome antigen A (SSA)/Ro60 and oral, gut, skin and vaginal bacteria. Clin Immunol. 2014;152(1–2):1–9. | ||
Maier-Moore JS, Kurien BT, D’Souza A, et al. Passive transfer of antibodies to the linear epitope 60 kD Ro 273-289 induces features of Sjögren’s syndrome in naive mice. Clin Exp Immunol. 2015;180(1):19–27. | ||
Reed JH, Gordon TP. Autoimmunity: Ro60-associated RNA takes its toll on disease pathogenesis. Nat Rev Rheumatol. 2016;12(3):136–138. | ||
Shuai ZW, Huang Y, Zhang L, Cai J, Li M. Role of autoantibodies to various Ro60 epitopes in the decrease of lymphocytes seen in systemic lupus erythematosus and primary Sjögren’s syndrome. Genet Mol Res. 2015;14(3):10096–10102. | ||
Hernández-Molina G, Zamora-Legoff T, Romero-Díaz J, et al. Predicting Sjögren’s syndrome in patients with recent-onset SLE. Rheumatology (Oxford). 2013;52(8):1438–1442. | ||
Conigliaro P, Triggianese P, Draghessi G, et al. Evidence for the detection of subclinical retinal involvement in systemic lupus erythematosus and Sjögren syndrome: a potential association with therapies. Int Arch Allergy Immunol. 2018;177(1):45–56. | ||
Dammacco R. Systemic lupus erythematosus and ocular involvement: an overview. Clin Exp Med. 2018;18(2):135–149. | ||
Chen A, Chen HT, Hwang YH, Chen YT, Hsiao CH, Chen HC. Severity of dry eye syndrome is related to anti-dsDNA autoantibody in systemic lupus erythematosus patients without secondary Sjogren syndrome: a cross-sectional analysis. Medicine (Baltimore). 2016;95(28):e4218. | ||
Wang H, Wang PB, Chen T, et al. Analysis of clinical characteristics of immune-related dry eye. J Ophthalmol. 2017;2017:8532397. | ||
Qin R, Steel A, Fazel N. Oral mucosa biology and salivary biomarkers. Clin Dermatol. 2017;35(5):477–483. | ||
Jung JY, Nam JY, Kim HA, Suh CH. Elevated salivary alpha-amylase level, association between depression and disease activity, and stress as a predictor of disease flare in systemic lupus erythematosus: a prospective case-control study. Medicine (Baltimore). 2015;94(30):e1184. | ||
Moori M, Ghafoori H, Sariri R. Nonenzymatic antioxidants in saliva of patients with systemic lupus erythematosus. Lupus. 2016;25(3):265–271. | ||
Alonso MD, Martínez-Vázquez F, Riancho-Zarrabeitia L, et al. Sex differences in patients with systemic lupus erythematosus from Northwest Spain. Rheumatol Int. 2014;34(1):11–24. | ||
Alonso MD, Martinez-Vazquez F, de Teran TD, et al. Late-onset systemic lupus erythematosus in Northwestern Spain: differences with early-onset systemic lupus erythematosus and literature review. Lupus. 2012;21(10):1135–1148. | ||
Koskenmies S, Järvinen TM, Onkamo P, et al. Clinical and laboratory characteristics of Finnish lupus erythematosus patients with cutaneous manifestations. Lupus. 2008;17(4):337–347. | ||
Nossent JC, Swaak AJ. Systemic lupus erythematosus VII: frequency and impact of secondary Sjøgren’s syndrome. Lupus. 1998;7(4):231–234. | ||
Tsuboi H, Asashima H, Takai C, et al. Primary and secondary surveys on epidemiology of Sjögren’s syndrome in Japan. Mod Rheumatol. 2014;24(3):464–470. | ||
Hernández-Molina G, Vargas-Alarcón G, Rodríguez-Pérez JM, Martínez-Rodríguez N, Lima G, Sánchez-Guerrero J. High-resolution HLA analysis of primary and secondary Sjögren’s syndrome: a common immunogenetic background in Mexican patients. Rheumatol Int. 2015;35(4):643–649. | ||
Furuzawa-Carballeda J, Sánchez-Guerrero J, Betanzos JL, et al. Differential cytokine expression and regulatory cells in patients with primary and secondary Sjögren’s syndrome. Scand J Immunol. 2014;80(6):432–440. | ||
Fernandes JD, Nico MM, Aoki V, et al. Xerostomia in Sjögren’s syndrome and lupus erythematosus: a comparative histological and immunofluorescence study of minor salivary glands alterations. J Cutan Pathol. 2010;37(4):432–438. | ||
Hernández-Molina G, Avila-Casado C, Cárdenas-Velázquez F, et al. Similarities and differences between primary and secondary Sjögren’s syndrome. J Rheumatol. 2010;37(4):800–808. | ||
Li YH, Gao YP, Dong J, et al. Identification of a novel autoantibody against self-vimentin specific in secondary Sjögren’s syndrome. Arthritis Res Ther. 2018;20(1):30. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.