Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Sirt6-Mediated Endothelial-to-Mesenchymal Transition Contributes Toward Diabetic Cardiomyopathy via the Notch1 Signaling Pathway
Authors Zhang Y ![]() , Dong Y, Xiong Z, Zhu Z, Gao F, Wang T, Man W, Sun D, Lin J, Li T, Li C, Zhao Z, Shen M, Sun D, Fan Y
, Dong Y, Xiong Z, Zhu Z, Gao F, Wang T, Man W, Sun D, Lin J, Li T, Li C, Zhao Z, Shen M, Sun D, Fan Y
Received 17 October 2020
Accepted for publication 21 November 2020
Published 7 December 2020 Volume 2020:13 Pages 4801—4808
DOI https://doi.org/10.2147/DMSO.S287287
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Antonio Brunetti
Yan Zhang,1,* Yuan Dong,1,* Zhenyu Xiong,1,* Zhengru Zhu,2 Fanya Gao,3 Tingting Wang,1 Wanrong Man,1 Dong Sun,1 Jie Lin,1 Tongbin Li,1 Congye Li,1 Zhijing Zhao,1 Min Shen,1 Dongdong Sun,1 Yanhong Fan1
1Department of Cardiology, Xijing Hospital, Fourth Military Medical University, Xi’an, People’s Republic of China; 2Department of Otolaryngology Head and Neck Surgery, First Hospital of Lanzhou University, Lanzhou, People’s Republic of China; 3Department of Cardiology, Shaanxi Provincial People’s Hospital, Xi’an Medical University, Xi’an, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dongdong Sun; Yanhong Fan
Department of Cardiology, Xijing Hospital, Fourth Military Medical University, 127 West Changle Road, Xi’an, Shaanxi 710032, People’s Republic of China
Tel +86 18691569930; +86 18829395402
Email [email protected]; [email protected]
Background: Endothelial-to-mesenchymal transition (EndMT) is an important source of myofibroblasts that directly affects cardiac function in diabetic cardiomyopathy (DCM) via an unknown underlying mechanism. Sirt6 is a member of the Sirtuin family of NAD(+)-dependent enzymes that plays an important role in glucose and fatty acid metabolism. In this study, we investigated whether Sirt6 participates in EndMT during the development of T2DM and the possible underlying regulatory mechanisms.
Methods: Endothelium-specific Sirt6 knockout (Sirt6-KOEC) mice (C57BL/6 genetic background) were generated using the classic Cre/loxp gene recombination system. T2DM was induced in eight-week-old male mice by feeding with a high-fat diet for three weeks followed by i.p. injection with 30 mg/kg of streptozotocin. The weight, lipids profiles, insulin, food intake and water intake of experimental animals were measured on a weekly basis. Cardiac microvascular endothelial cells (CMECs) were obtained from adult male mice; the isolated cells were cultured with high glucose (HG; 33 mmol/L) and palmitic acid (PA; 500 μmol/L) in DMEM for 24 h, or with normal glucose (NG; 5 mmol/L) as the control.
Results: Sirt6 expression is significantly downregulated in CMECs treated with HG+PA. Additionally, Sirt6-KOEC was found to worsen DCM, as indicated by aggravated perivascular fibrosis, cardiomyocyte hypertrophy, and decreased cardiac function. In vitro, Sirt6 knockdown exacerbated the proliferation, and migration of CMECs exposed to HG+PA. Mechanistically, Sirt6 knockdown significantly enhanced Notch1 activation in CMECs treated with HG+PA, whereas Notch1 adenoviral interference significantly blunted the effects of Sirt6 knockdown on CMECs.
Conclusion: This study is the first to demonstrate that Sirt6 participates in EndMT via the Notch1 signaling pathway in CMECs stimulated with HG+PA. Therefore, the findings of this study suggest that Sirt6 could provide a potential treatment strategy for DCM.
Keywords: diabetic cardiomyopathy, endothelial-to-mesenchymal transition, cardiac function, fibrosis, Sirt6
Introduction
Type 2 diabetes mellitus (T2DM) is a highly prevalent disease that can cause cardiovascular diseases, which are the leading cause of T2DM-associated death.1 Diabetic cardiomyopathy (DCM) is a complex metabolic disease characterized by microvascular injury, cardiac fibrosis, and associated dysfunction.2,3 Myofibroblast-mediated extracellular matrix remodeling is the key pathological basis for the occurrence and development of DCM, since increased extracellular matrix deposition leads to cardiac stiffness and diastolic dysfunction, which can ultimately result in cardiomyocyte hypertrophy and left ventricular systolic dysfunction.4,5 In addition, perivascular fibrosis can affect the oxygen supply to cardiomyocytes and thus exacerbate myocardial ischemia.
Recent studies have shown that endothelial cells are an important source of myofibroblasts and that the endothelial-to-mesenchymal transition (EndMT) is a key pathway in this process.6,7 During EndMT, endothelial cells undergo enhanced proliferation and migration, acquire mesenchymal characteristics (α-SMA) and lose endothelial cell markers (CD31).8 This process can be promoted by various changes in diabetes, such as hyperglycemia, fatty acid oxidation, and inflammation activation; therefore, it has been suggested that this process could serve as a potential therapeutic target for cardiac fibrosis in DCM.
Cardiac microvascular endothelial cells (CMECs) are a single layer of cells arranged on the inner surface of cardiac microvasculature, forming an important barrier between circulation and cardiomyocytes. In addition, CMECs act as an important endocrine organ within the heart and are an initial target for hyperglycemia.9,10 Considerable evidence has indicated that cardiac microvasculature is closely related to cardiac function,11,12 and our previous study suggested that endothelial cell-specific gene editing can affect cardiac function in diabetic mice.13 Consequently, protecting cardiac microvessels could be a potential treatment strategy for DCM.
Sirt6 is a member of the Sirtuin family of NAD(+)-dependent enzymes. It has been shown to play an important role in physiological and pathological processes, particularly glucose and fatty acid metabolism, and a protective role in maintaining cardiac homeostasis.14,15 However, it remains unclear whether Sirt6 is involved in T2DM-induced EndMT in CMECs. In this study, we investigated whether Sirt6 participates in EndMT during the development of T2DM and the possible underlying regulatory mechanisms.
Materials and Methods
All experiments were performed according to the National Institutes of Health Guidelines on the Use of Laboratory Animals and were approved by the Fourth Military Medical University Ethics Committee on Animal Care (Approval ID: 2,018,103).
Animals
Endothelium-specific Sirt6 knockout (Sirt6-KOEC) mice (C57BL/6 genetic background) were generated using the classic Cre/loxp gene recombination system, as described previously.16 Briefly, Tie2-Cre transgenic mice that have the mouse endothelium-specific receptor were used to deleting floxed sequences from endothelial cells of adult mice. We crossed Sirt6fl/fl mice with Tie2-cre mice to establish (Sirt6fl/fl; Tie2-cre+) mice. Sirt6-KOEC mice were induced by intraperitoneal (i.p.) injection with tamoxifen (40 mg/KG/day) for seven days. The experimental mice were genotyped using PCR. T2DM was induced in eight-week-old male mice by feeding with a high-fat diet for three weeks followed by i.p. injection with 30 mg/kg of streptozotocin (STZ; Sigma–Aldrich, St. Louis, MO) dissolved in citrate buffer (0.1 M citrate buffer, pH 4.5) for five days. The mice were fed a high-fat diet for 12 weeks after the course of injections was completed. The control group mice were injected with the same amount of citrate buffer and were fed a normal diet. The normal diet (10% of kcal from fat, D12450) and high-fat diet (60% of kcal from fat, D12492) were purchased from Research Diets, Inc. (New Brunswick, USA). Blood glucose levels were monitored every week after the last STZ injection.
Echocardiography
Echocardiography was conducted using an echocardiography system with a 15-MHz linear transducer (VisualSonics, Toronto, ON, Canada). The left ventricular ejection fraction (LVEF), left ventricular fraction shortening (LVFS), left ventricular end-diastolic diameter (LVEDD), and left ventricular end-systolic diameter (LVESD) were calculated using Vevo 2100 software algorithms (VisualSonics, Toronto, ON, Canada).
CMEC Isolation, Cultivation, and Treatment
CMECs were obtained from adult male mice, as described previously.13 Briefly, adult mouse hearts were removed under aseptic conditions, digested with 0.2% type II collagenase (Sigma–Aldrich) for 30 min after the atria had been removed, and then digested with 0.25% trypsin (Sigma–Aldrich) for further 10 min. CMECs were collected by centrifugation (1000 × g for 10 min) and cultured with Dulbecco’s Modified Eagle’s medium (DMEM) containing 20% fetal bovine serum (HyClone, UT, USA) at 37 °C in the presence of 75% N2, 20% O2, and 5% CO2. The isolated cells were then cultured with high glucose (HG; 33 mmol/L) and palmitic acid (PA; 500 μmol/L) in DMEM for 24 h, or with normal glucose (NG; 5 mmol/L) as the control, as described previously.17
Adenovirus Management
As described previously,16 adenoviruses expressing short hairpin (sh) RNAs directed against Sirt6 (Ad-shSirt6), Notch1 (Ad-shNotch1), and a control vector (Ad-scramble) were purchased from Hanbio Technology Ltd (Shanghai, China).
Histological Analysis and Immunofluorescence (IF) Staining
Mouse hearts were fixed in 4% paraformaldehyde overnight at 4 °C, embedded in paraffin, and cut into 4 μm thick slices. Fibrosis and collagen content were assessed using Masson trichrome staining. Myocyte size was evaluated using wheat germ agglutinin staining. Histological analysis and IF staining were performed as described previously.18,19
Scratch-Migration Assay
CMECs were cultured in six-well plates containing serum-deprived medium for 24 h and scratched, and the medium was replaced with a fresh serum-deprived medium. Images were captured at 6 h intervals, and the migration area was quantified using ImageJ software (version 1.46; National Institutes of Health, MA, USA,).
The 5-Ethynyl-2ʹ-Deoxyuridine (EdU) Cell Proliferation Assay
Cell proliferation was detected using an EdU kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Briefly, EdU (10 μmol/L) was added to the culture medium and the CMECs were incubated for 2 h before being fixed for detection. The proliferating cells were indicated by bright red fluorescence under a laser confocal microscope (FV-10i, Olympus, Japan).
Western Blotting (WB)
Proteins were isolated from CMECs in heart tissue or cultured in DMEM as described above. Next, 40 μg of each protein sample was separated via 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; Cwbiotech, Beijing, China), transferred onto 0.22 μm nitrocellulose blotting membranes (Millipore, MA, USA), and blocked with 5% milk for 1 h at 37 °C. The membranes were then incubated with the following primary antibodies at 4 °C overnight: anti-Notch-1 (Santa Cruz Biotechnology, USA), anti-NICD and anti-HES-1 (Cell Signaling Technology Inc., Danvers, MA), and anti-CD31, anti-α-SMA, anti-Sirt6, and anti-GAPDH (Abcam Co, Cambridge, UK). Antigen-antibody complexes were detected and imaged by a chemiluminescence system (Amersham Bioscience, Buckinghamshire, UK) after incubation with secondary antibodies for 1 h at 37 °C.
Statistical Analysis
Data are presented as the mean ± SD. Differences were analyzed using Student’s t-tests and one-way Analysis of Variance (ANOVA) followed by the Bonferroni comparison test. P values of <0.05 were considered statistically significant.
Results
Sirt6 is Downregulated in CMECs in Response to EndMT Induced by HG+PA
To evaluate the role of Sirt6 and EndMT in CMECs, we constructed a mouse model of T2DM through a high-fat diet and low-dose STZ. We measured the weight, lipids profiles, insulin, food intake and water intake of experimental animals on a weekly basis (s-Figure 1A–G), the diabetic mice displayed glucose intolerance characterized by intra-peritoneal glucose tolerance test (IPGTT) at 15 weeks (Figure 1A). To evaluate whether CMECs underwent EndMT in T2DM, we detected the expression of mesenchymal and endothelial cell markers. IF staining revealed downregulated microvascular CD31 and upregulated α-SMA (Figure 1B), consistent with the characteristics of EndMT. We then isolated CMECs from six-week-old male mice and cultured them in HG+PA or NG to further verify whether the EndMT process was caused by HG+PA. WB and IF staining indicated decreased CD31 expression, upregulated α-SMA expression, and decreased Sirt6 expression in the HG+PA group (Figure 1E) compared to the control group (Figure 1C and D). Taken together, these results confirm that HG+PA induced EndMT in CMECs and significantly downregulated Sirt6.
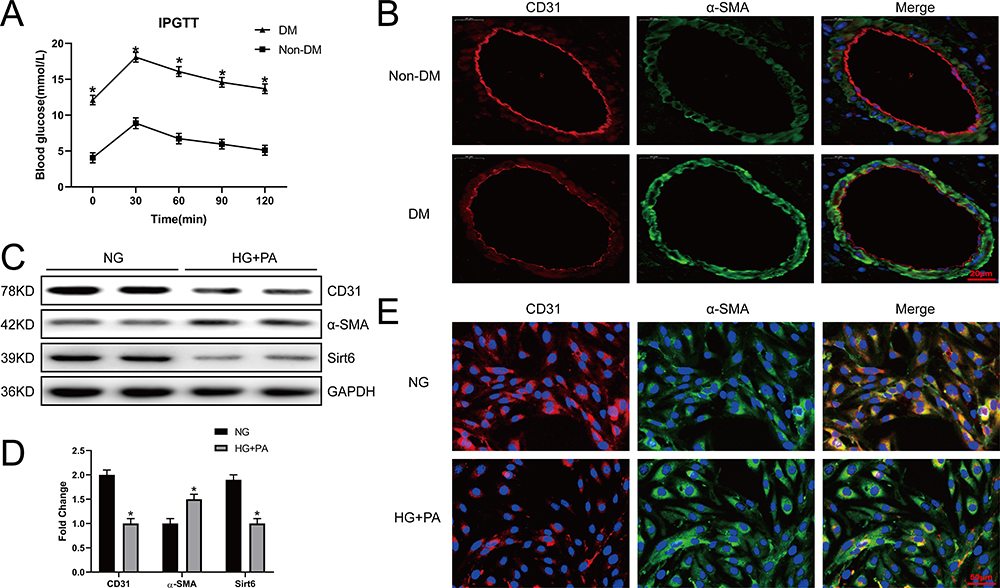 |
Figure 1 Sirt6 is downregulated in CMECs in response to EndMT induced by HG+PA. (A) A mouse mode of T2DM was constructed, and 2 h post-IPGTT hyperglycemia was performed (n = 20). (B) Representative images of IF staining in different vessels. Red fluorescence represents CD31, green fluorescence represents α-SMA, and blue fluorescence represents the nucleus (n = 5). (C and D) Western blotting and quantitative analysis of CD31, α-SMA, and Sirt6 protein levels in CMECs treated with HG+PA. (E) Representative images of IF staining in CMECs from the NG and HG+PA groups. Red fluorescence represents CD31, green fluorescence represents α-SMA, and blue fluorescence represents the nucleus (n = 5). *P < 0.05 vs NG. |
Endothelium-Specific Sirt6 Knockout Worsens Perivascular Fibrosis and DCM
Having confirmed that Sirt6 was significantly downregulated in CMECs during EndMT, we investigated whether Sirt6 downregulation contributes toward or protects against EndMT. Sirt6-KOEC mice and wild-type (WT) mice were subjected to low-dose STZ and a high-fat diet for 16 weeks to induce T2DM. First, we evaluated perivascular fibrosis, with Masson staining revealing that T2DM significantly increased extracellular matrix deposition in Sirt6-KOEC mice compared to WT mice (Figure 2A and D). Moreover, mice with T2DM exhibited a lower LVEF and LVFS than the WT mice accompanied by a higher LVEDD and LVESD, as determined by echocardiography (Figure 2C), while endothelium-specific Sirt6 knockout reduced cardiac function (Figure 2F–I). In addition, wheat germ agglutinin staining revealed that Sirt6-KOEC mice had larger myocytes than WT mice with T2DM (Figure 2B and E). Thus, endothelium-specific Sirt6 knockout appears to worsen perivascular fibrosis and DCM in T2DM.
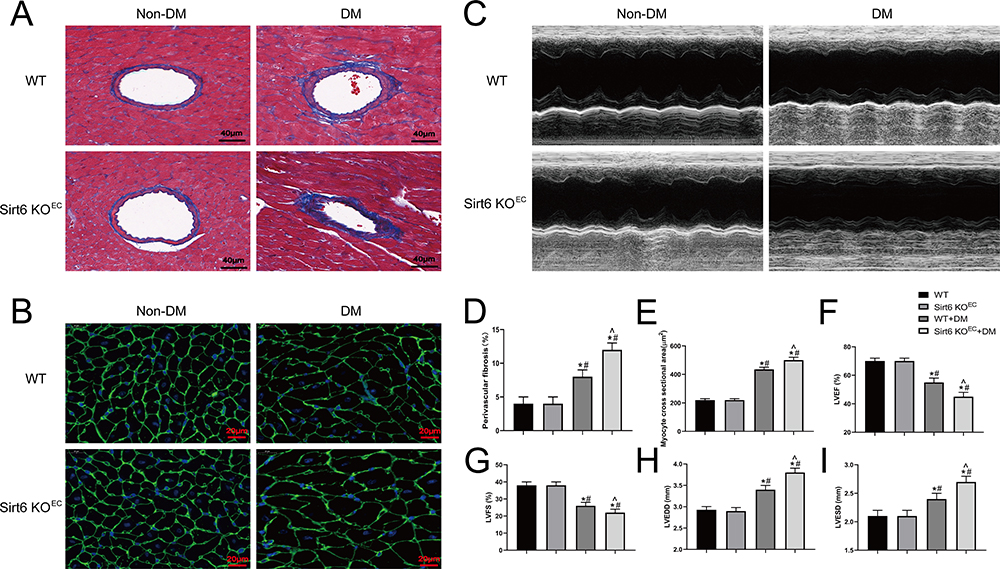 |
Figure 2 Endothelium-specific Sirt6 knockout worsens perivascular fibrosis and diabetic cardiomyopathy in T2DM. (A) Representative images of Masson trichrome staining in mouse hearts from different groups (n = 6). (B) Representative images of wheat germ agglutinin staining in different groups (n = 6). (C) Cardiac function evaluated using M-mode echocardiograms (n = 8). (D) Quantitative analysis of perivascular Masson trichrome staining (n = 6). (E) Quantitative analysis of wheat germ agglutinin staining in different groups (n = 6). (F–I) Calculated cardiac function indices: LVEDD, LVESD, LVEF, and LVFS (n = 8). *P < 0.05 vs WT+Non-DM; #P < 0.05 vs Sirt6 KOEC+Non-DM; ^P < 0.05 vs WT+DM. |
Sirt6 Knockout Affects the Proliferation, Migration, and Mesenchymal Features of CMECs Exposed to HG+PA
EndMT enhances the migration and proliferation capabilities of endothelial cells and induces the expression of mesenchymal markers.20 To investigate the effect of Sit6 on EndMT, we cultured CMECs in vitro with Ad-sh-Sirt6 or Ad- scramble in HG+PA or NG for 48 h. The EdU proliferation assay revealed an 8% positive rate in the NG group that did not change significantly with Ad-sh-Sirt6 transfection. Conversely, the HG+PA group displayed a positive rate of 13% that was increased to 17% by Ad-sh-Sirt6 transfection (Figure 3A and D). These results indicate that HG+PA effectively promotes CMEC proliferation, which is significantly increased by Sirt6 knockout.
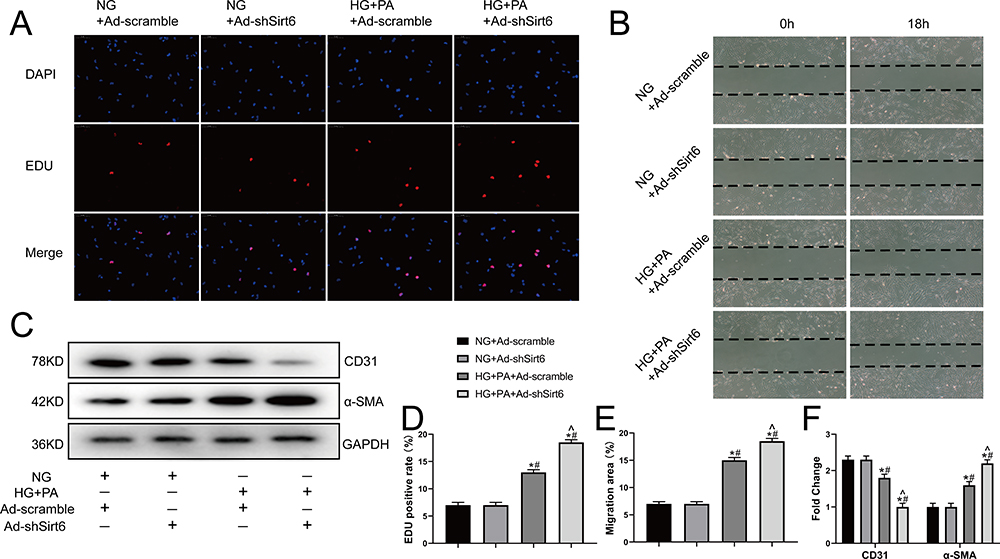 |
Figure 3 Sirt6 knockout contributes toward proliferation, migration, and mesenchymal features expressing in CMECs exposed to HG+PA. (A) Representative images from the EdU proliferation assay in CMECs treated as indicated. Red fluorescence represents proliferating cells (n = 5). (B) Representative images from the scratch-migration assay in CMECs (n = 5). (C) Western blotting and quantitative analysis of α-SMA, and CD31 protein levels in CMECs treated as indicated (n = 5). (D) Quantitative analysis of EdU proliferation assay results (n = 5). (E) Quantitative analysis of scratch-migration assay results (n = 5). (F) Quantitative analysis of α-SMA, and CD31 protein levels in CMECs treated as indicated (n = 5). *P < 0.05 vs NG+Ad-scramble; #P < 0.05 vs NG+Ad-shSirt6; ^P < 0.05 vs HG+PA+Ad-scramble. |
Consistently, the scratch-migration assay revealed no significant difference between the recovered area in the NG group 18 h after scratching with or without Ad-sh-Sirt6 transfection. However, the recovered area in the HG+PA group was 14% and increased to 18% with Ad-sh-Sirt6 transfection (Figure 3B and E), indicating that Sirt6 knockout affects the migration of CMECs treated with HG+PA. In addition, Sirt6 knockout promoted α-SMA expression and inhibited CD31 expression under HG+PA exposure, as confirmed by WB (Figure 3C and F). Together, these results indicate that Sirt6 knockout contributes toward EndMT in CMECs.
Sirt6 Mediates EndMT via the Notch1 Signaling Pathway in CMECs
Finally, we explored how Sirt6 mediates EndMT in CMECs. As the Notch signaling pathway plays a key role in cardiac development by regulating vascular endothelial cell differentiation and proliferation,21,22 we detected all Notch signaling receptors (Notch1–4) and found that HG+PA exposure significantly activated Notch1. In addition, Sirt6 knockdown enhanced Notch1, NICD, and HES-1 activation (Figure 4A and B), indicating that Notch1 signaling may participate in EndMT. To further elucidate the role of Notch1 in HG+PA-induced EndMT regulation, we cultured CMECs in four groups under different conditions: (1) HG+PA, (2) HG+PA+Ad-shNotch1, (3) HG+PA+Ad-shSirt6, and (4) HG+PA+Ad-shSirt6+Ad-shNotch1. Interestingly, Ad-shNotch1 reduced mesenchymal marker expression and enhanced the expression of endothelial cell markers in the HG+PA group treated with Ad-shSirt6 (Figure 4C and D). Moreover, Notch1 knockdown reduced the migration area from 18% to 15% in the HG+PA group treated with Ad-shSirt6 but had no significant effect in the HG+PA group treated without Ad-shSirt6 (Figure 4G and F). Furthermore, Ad-shNotch1 reduced the EdU positive rate from 34% to 26% in the HG+PA group treated with Ad-shSirt6 but had no significant effect in the HG+PA group treated without Ad-shSirt6 (Figure 4H and E). Taken together, these results indicate that Sirt6 mediates EndMT via the Notch1 signaling pathway in CMECs.
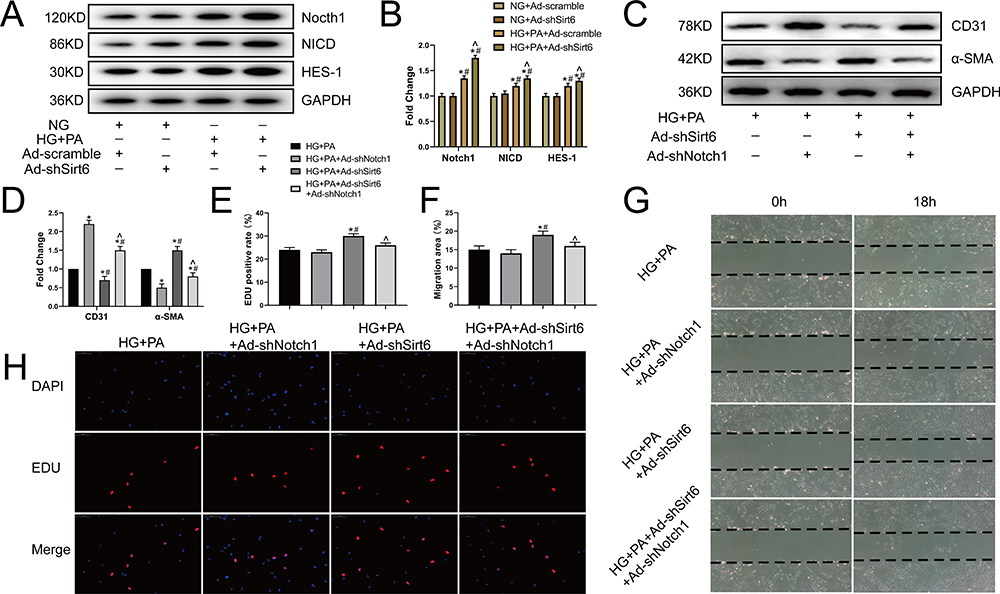 |
Figure 4 Sirt6 mediates EndMT via Notch1 signaling in CMECs. (A and B) Western blotting and quantitative analysis of Notch1, NICD, and HES-1 protein levels in CMECs treated as indicated (n = 5). (C and D) Western blotting and quantitative analysis of α-SMA and CD31 protein levels in CMECs treated as indicated (n = 5). (G and F) Scratch-migration assay and quantitative analysis in CMECs (n = 5). (H and E) EdU proliferation assay and quantitative analysis in CMECs (n = 5). *P < 0.05 vs HG+PA; #P < 0.05 vs HG+PA+ Ad-shNotch1; ^P < 0.05 vs HG+PA+Ad-shSirt6. |
Discussion
This study investigated the EndMT of CMECs in T2DM. As reviewed in the introduction, EndMT is an important cellular phenomenon that induces microvascular dysfunction and acts as an important source of myofibroblasts that directly affect cardiac function in DCM.23,24 EndMT has also been implicated in the pathogenesis of diabetic fibrosis; for instance, Wang et al reported that HG levels can induce EndMT in human umbilical vein endothelial cells, including changes in corresponding protein expression and morphology.25 These evidences indicate the sensitivity of endothelial cells to hyperglycemia, but there was no direct evidence to support the EndMT in CMECs caused by T2DM. Therefore, we cultivated CMECs with HG+PA to simulate T2DM in vitro and demonstrated the promotion of EndMT by HG+PA in CMECs.
Sirt6 is known to play an important role in glucose and fatty acid metabolism, and previous studies have demonstrated that Sirt6 levels decrease in aging-induced EndMT,26 confirming a link between SIRT6 and EndMT. Therefore, we hypothesized that Sirt6 plays a key role in mediating HG+PA-induced EndMT. Consistently, the findings of this study demonstrated that Sirt6 was downregulated in CMECs in response to EndMT induced by HG+PA. To further determine whether Sirt6 downregulation contributes toward or protects against EndMT, we constructed Sirt6-KOEC mice with or without T2DM. We found that Sirt6-KOEC worsens perivascular fibrosis in T2DM, and Sirt6-KOEC mice displayed poorer cardiac function, demonstrating that endothelial cells play an important role in DCM through endotheliocyte- cardiomyocyte or endotheliocyte-fibroblast crosstalk fashion, and the protection of endothelial cells may be an important measure in the prevention of diabetic injury. Consistently, the CMECs cultured in vitro in HG+PA with Ad-shSirt6 displayed stronger migration, proliferation, and mesenchymal features; therefore, we investigated the underlying mechanisms.
Notch signaling has been reported to induce EndMT in many normal or malignant cells, whereas recent studies have reported that Notch signaling is activated in human CMECs undergoing hypoxia-induced EndMT.27 Notch signaling activation has also been shown to induce EndMT progression in human coronary artery endothelial cells and promote the development of atherosclerotic lesions.28 In addition, the Sirt6 and Notch signaling pathways have been reported to regulate podocyte injury and proteinuria,29 whereas Sirt6 deficiency was found to impact corneal epithelial wound healing via Notch signaling.30 Consistently, our study demonstrated that Sirt6 deficiency contributes toward EndMT induced by HG+PA via Notch1 signaling.
Despite these important findings, our study has the following limitations. Although our results suggest that Sirt6 plays an important regulatory role in EndMT, the protective effects of Sirt6 on CMECs need to be confirmed in endothelium-specific Sirt6 transgenic mice. In addition, the relationship between Sirt6 and the Notch1 pathway did not display one-to-one stoichiometry; therefore, we can only confirm that Notch1 plays a role in this process and cannot rule out the possibility that Sirt6 promotes EndMT by simultaneously targeting other genes involved in diabetes. Previous results suggested that Sirt6 mediated transcriptional suppression of MALAT1 is a key mechanism for EndMT in a model of ECs aging,26 future research should focus on the role of Sirt6 and target genes besides Notch1 in EndMT.
In conclusion, our results highlight the importance of CMECs in the development of DCM, provide a molecular basis for HG+PA-induced EndMT in CMECs, and suggest that the Sirt6/Notch1 pathway might serve as a new therapeutic target for treating DCM.
Abbreviations
EndMT, endothelial-to-mesenchymal transition; DCM, diabetic cardiomyopathy; CMEC, cardiac microvascular endothelial cell; STZ, streptozotocin; NG, normal glucose; HG, high glucose; PA, palmitic acid; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; LVEF, left ventricular ejection fraction; LVFS, left ventricular fraction shortening.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81922008, No. 81770224, No. 81670204), the National Key Research and Development Plan (2018YFA0107400), and the Top Young Talents Special Support Program in Shaanxi Province (2020).
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
All authors declare that there is no conflict of interest related to this manuscript.
References
1. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88–98. doi:10.1038/nrendo.2017.151
2. Dillmann WH. Diabetic cardiomyopathy. Circ Res. 2019;124(8):1160–1162. doi:10.1161/circresaha.118.314665
3. Riehle C, Bauersachs J. Of mice and men: models and mechanisms of diabetic cardiomyopathy. Basic Res Cardiol. 2018;114(1):2. doi:10.1007/s00395-018-0711-0
4. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122(4):624–638. doi:10.1161/circresaha.117.311586
5. Tong M, Saito T, Zhai P, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 2019;124(9):1360–1371. doi:10.1161/circresaha.118.314607
6. Piera-Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol Rev. 2019;99(2):1281–1324. doi:10.1152/physrev.00021.2018
7. Pérez L, Muñoz-Durango N, Riedel CA, et al. Endothelial-to-mesenchymal transition: cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017;33:41–54. doi:10.1016/j.cytogfr.2016.09.002
8. Li Y, Lui KO, Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. 2018;15(8):445–456. doi:10.1038/s41569-018-0023-y
9. Segers VFM, Brutsaert DL, De Keulenaer GW. Cardiac remodeling: endothelial cells have more to say than just NO. Front Physiol. 2018;9:382. doi:10.3389/fphys.2018.00382
10. Talman V, Kivelä R. Cardiomyocyte-endothelial cell interactions in cardiac remodeling and regeneration. Front Cardiovasc Med. 2018;5:101. doi:10.3389/fcvm.2018.00101
11. Potente M, Mäkinen T. Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol. 2017;18(8):477–494. doi:10.1038/nrm.2017.36
12. Brutsaert DL. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. 2003;83(1):59–115. doi:10.1152/physrev.00017.2002
13. Hu J, Wang S, Xiong Z, et al. Exosomal Mst1 transfer from cardiac microvascular endothelial cells to cardiomyocytes deteriorates diabetic cardiomyopathy. Biochim Biophys Acta Mol Basis Dis. 2018;1864(11):3639–3649. doi:10.1016/j.bbadis.2018.08.026
14. Chang AR, Ferrer CM, Mostoslavsky R. SIRT6, a mammalian deacylase with multitasking abilities. Physiol Rev. 2020;100(1):145–169. doi:10.1152/physrev.00030.2018
15. Kuang J, Chen L, Tang Q, Zhang J, Li Y, He J. The role of Sirt6 in obesity and diabetes. Front Physiol. 2018;9:135. doi:10.3389/fphys.2018.00135
16. Wang T, Sun C, Hu L, et al. Sirt6 stabilizes atherosclerosis plaques by promoting macrophage autophagy and reducing contact with endothelial cells. Biochem Cell Biol. 2020;98(2):120–129. doi:10.1139/bcb-2019-0057
17. Kuwabara Y, Horie T, Baba O, et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res. 2015;116(2):279–288. doi:10.1161/circresaha.116.304707
18. Cheng Z, Zhang M, Hu J, et al. Mst1 knockout enhances cardiomyocyte autophagic flux to alleviate angiotensin II-induced cardiac injury independent of angiotensin II receptors. J Mol Cell Cardiol. 2018;125:117–128. doi:10.1016/j.yjmcc.2018.08.028
19. Xiong Z, Li Y, Zhao Z, et al. Mst1 knockdown alleviates cardiac lipotoxicity and inhibits the development of diabetic cardiomyopathy in db/ db mice. Biochim Biophys Acta Mol Basis Dis. 2020;1866(8):165806. doi:10.1016/j.bbadis.2020.165806
20. Hulshoff MS, Xu X, Krenning G, Zeisberg EM. Epigenetic regulation of endothelial-to-mesenchymal transition in chronic heart disease. Arterioscler Thromb Vasc Biol. 2018;38(9):1986–1996. doi:10.1161/atvbaha.118.311276
21. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116(7):1231–1244. doi:10.1161/circresaha.116.302855
22. Tian DY, Jin XR, Zeng X, Wang Y. Notch signaling in endothelial cells: is it the therapeutic target for vascular neointimal hyperplasia? Int J Mol Sci. 2017;18(8):1615. doi:10.3390/ijms18081615
23. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263–271. doi:10.1016/j.jacc.2013.02.092
24. Jiang F, Mohr F, Ullrich ND, Hecker M, Wagner AH. Endothelial cell modulation of cardiomyocyte gene expression. Exp Cell Res. 2019;383(2):111565. doi:10.1016/j.yexcr.2019.111565
25. Wang B, Wu Y, Ge Z, Zhang X, Yan Y, Xie Y. NLRC5 deficiency ameliorates cardiac fibrosis in diabetic cardiomyopathy by regulating EndMT through Smad2/3 signaling pathway. Biochem Biophys Res Commun. 2020;528(3):545–553. doi:10.1016/j.bbrc.2020.05.151
26. Qin W, Zhang L, Li Z, et al. SIRT6-mediated transcriptional suppression of MALAT1 is a key mechanism for endothelial to mesenchymal transition. Int J Cardiol. 2019;295:7–13. doi:10.1016/j.ijcard.2019.07.082
27. Liu Y, Zou J, Li B, et al. RUNX3 modulates hypoxia-induced endothelial-to-mesenchymal transition of human cardiac microvascular endothelial cells. Int J Mol Med. 2017;40(1):65–74. doi:10.3892/ijmm.2017.2998
28. Lin QQ, Zhao J, Zheng CG, Chun J. Roles of notch signaling pathway and endothelial-mesenchymal transition in vascular endothelial dysfunction and atherosclerosis. Eur Rev Med Pharmacol Sci. 2018;22(19):6485–6491. doi:10.26355/eurrev_201810_16062
29. Liu M, Liang K, Zhen J, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat Commun. 2017;8(1):413. doi:10.1038/s41467-017-00498-4
30. Hu X, Zhu S, Liu R, et al. Sirt6 deficiency impairs corneal epithelial wound healing. Aging. 2018;10(8):1932–1946. doi:10.18632/aging.101513
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.