Back to Journals » Journal of Inflammation Research » Volume 14
SIRT1 is Required for Exercise-Induced Beneficial Effects on Myocardial Ischemia/Reperfusion Injury
Authors Wang D, Cao H, Wang X ![]() , Wang J, Wang M, Zhang J, Wang L
, Wang J, Wang M, Zhang J, Wang L
Received 8 January 2021
Accepted for publication 24 March 2021
Published 7 April 2021 Volume 2021:14 Pages 1283—1296
DOI https://doi.org/10.2147/JIR.S300997
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
This paper has been retracted.
Dawei Wang,1,* Hongyan Cao,2,* Xu Wang,3 Jinchun Wang,4 Manli Wang,5 Jian Zhang,6 Lin Wang6
1Department of Emergency, The First Hospital of Jilin University, Changchun, 130021, People’s Republic of China; 2Department of Cardiology, The First Hospital of Jilin University, Changchun, 130021, People’s Republic of China; 3Department of Colorectal and Anal Surgery, The First Hospital of Jilin University, Changchun, 130021, People’s Republic of China; 4Department of Emergency, Jilin Provincial FAW General Hospital, Changchun, 130011, People’s Republic of China; 5Department of Geriatrics, Changchun Central Hospital, Changchun, 130000, People’s Republic of China; 6Department of Cardiovascular Center, The First Hospital of Jilin University, Changchun, 130021, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Zhang; Lin Wang
Department of Cardiovascular Center, The First Hospital of Jilin University, No. 71, Xinmin Street, Changchun, 130021, Jilin Province, People’s Republic of China
Tel +86-0431-88783417
Email [email protected]; [email protected]
Background: Exercise training has been regarded as an effective mean of prevention and treatment of cardiovascular diseases (CVD), and exercise can improve the antioxidant capacity of the myocardial. While SIRT1 has been proved to protects the heart from myocardial ischemia/reperfusion (MI/R) injury and apoptosis, less is known about the association between exercise-induced cardioprotection and SIRT1.
Methods and Results: MI/R injury model was constructed after swimming training in mice. Significantly reduced myocardial infarct size, decreased apoptosis ratio and upregulated SIRT1 protein expression in heart were found in swam mice by 2,3,5-triphenyltetrazolium chloride (TTC) staining of heart sections, TUNEL staining of frozen sections and Western blotting. The results of TUNEL staining and Western blotting suggested activation of SIRT1 using resveratrol (RSV) or inhibition of SIRT1 using EX527 in vitro blocked or accelerated cardiomyocytes apoptosis which induced by hypoxia/reoxygenation (H/R) respectively and regulated the expression of antioxidants in vitro. PGC-1α has been identified as one of the downstream genes of SIRT1 modulating oxidative stress and apoptosis. Importantly, the data of TTC staining, TUNEL staining, Western blotting, echocardiography and histopathological staining revealed that mice with inducible cardiac SIRT1-knockout blocked the protective effects of exercise preconditioning on myocardial infarct size, myocardial apoptosis, adverse ventricular remodeling, cardiac fibrosis and cardiac dysfunction after MI/R injury, simultaneously exercise-induced expression of myocardial antioxidant stress factors was hindered which was detected by immunohistochemical analysis.
Conclusion: SIRT1 protects against oxidative stress after MI/R injury by activating downstream PGC-1α and promoting the production of antioxidant enzymes. SIRT1 is required for exercise to protect against myocardial apoptosis and maladaptive ventricular remodelling induced by myocardial ischemia/reperfusion injury.
Keywords: exercise, myocardial ischemia/reperfusion injury, oxidative stress, SIRT1
Introduction
Cardiovascular diseases (CVD) are the main diseases that seriously endanger the health of human beings worldwide.1 The burden of CVD will increase with an aging population.2 As one of the most common CVD, myocardial infarction (MI) leads to acute and persistent myocardial ischemia resulting in massive cardiomyocyte death due to necrosis and apoptosis which appear to be crucial for cardiac dysfunction.3–5 Early reperfusion therapy will inevitably lead to reperfusion injury to a certain extent when it resists myocardial ischemia,6 it has become a critical factor affecting the therapeutic effect of MI. Thus, exploring the pathways and mechanisms that alleviate myocardial ischemia reperfusion injury could be of great significance to fundamental research and clinical treatment implications.
Previous studies have shown that myocardial ischemia reperfusion (MI/R) injury is mainly associated with increased oxidative stress levels.7–9 The accelerated production of excessive oxygen free radicals and reactive oxygen species (ROS) induced by reperfusion is widely accepted to be one of the key pathophysiological mechanisms in the mitochondrial oxidative stress injury and myocardial apoptosis.10,11 In addition, dysregulated expression of endogenous antioxidant key regulators also accelerate the apoptosis of cardiomyocytes.12–14 These studies suggested that it is essential to finding effective means or agents that alleviate oxidative stress and suppress excess ROS may represent as a practicable therapy for ischemia reperfusion injury.
Exercise training has been regarded as an effective mean of prevention and treatment of CVD.15,16 The findings of the previous study proved that exercise protects against MI/R injury-induced apoptosis and ventricular remodeling by regulating physiological cardiac growth.17 Meanwhile, exercise training has been proved to improve the antioxidant capacity of the heart.18 For instance, recent research has confirmed that exercise might exert a positive effect on myocardial antioxidant capacity by phosphorylation and activation of eNOS.19 And there is an inevitable relationship between the up-regulated Nrf2 as an adaptive response to exercise and the high expression of cardiac antioxidants.20,21 In addition, exercise can also exert heart protective benefits by inducing minimal cardiomyocyte proliferation,22 suppressing inflammation23,24 and promoting mitochondria energy metabolism.25,26
Silent information regulator 2 homolog 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD+) dependent histone deacetylase.27 The results of Alcendor et al showed that SIRT1 can delay the pace of cardiac aging and protect against cardiac oxidative stress.28 What is even more refreshing is the activation of SIRT1 can protect the heart from ischemia/reperfusion injury and cardiomyocyte apoptosis.29 In keeping with these observations, prior work from our group revealed that cardiomyocyte-specific deletion of SIRT1 gene sensitizes myocardium to ischaemia and reperfusion injury in mice.30 However, little is known about whether there is an association between exercise-induced cardioprotection and SIRT1.
In the present study, we sought to investigate the role of SIRT1 in exercise-induced beneficial effects against myocardial ischemia-reperfusion injury. Reduced expression of SIRT1 protein was observed in a mice model of MI/R injury, however, swimming exercise was found to reverse the downregulation of SIRT1. All these results suggest that SIRT1 may not only be involved in MI/R injury but also its protein expression level is closely related to exercise. Since the current research status shows that SIRT1 can participate in the regulation of cardiac oxidative stress, we attempted to explore whether the exercise can regulate oxidative stress injury induced by MI/R injury through SIRT1. Our present experimental results suggest that activation of SIRT1 blocked cardiomyocytes apoptosis induced by H/R and regulated the expression of antioxidants in vitro by activating downstream PGC-1α. Moreover, mice with icSIRT1 KO blocked the resistance of exercise to MI/R injury, simultaneously hindered exercise-induced expression of myocardial antioxidant stress factors. These data indicate a potential correlation between SIRT1 and exercise-induced beneficial effects on myocardial ischemia/reperfusion injury.
Materials and Methods
Mice Exercise Models
All animal experiments were performed according to the approved protocols of the Animal Care and Use Committee at Jilin University. And this experiment study was approved by the Ethics Committee of Jilin University (ethical approval number: 20200063). SIRT1flox/flox mice and CreERT2 mice were purchased from Jackson. 8-week-old male C57BL/6 mice were purchased from Viton Lihua. According to the methods given in the references, 8-week-old male C57BL/6 mice and transgenic mice swam in water pool (50 cm in diameter), starting with 10 minutes and increase by 10 minutes a day to 90 minutes, twice a day, approximately 6 h apart.3,31 MI/R surgery was performed on mice at the end of 21 days swimming training. At the endpoint of each experiment, mice ventricular tissues were harvested and frozen or for frozen section preparation for subsequent analysis.
Heart MI/R Injury Models and Analyses
Mice were anesthetized with isoflurane (2%) via inhalation and kept ventilated during surgeries. Mice heart IRI models were induced by ligation of the left anterior descending artery (LAD) with an 8–0 nylon suture for 45 minutes, followed by cardiac reperfusion for 24 h or 3 weeks, the sham operation does not involve ligation. The detailed process were described in our previous report.30 After 24 h of reperfusion, mice were sacrificed by cervical dislocation under 2% isoflurane anesthesia. Then, myocardial infarct size was assessed. Briefly speaking, the heart sections were stained with 2,3,5-triphenyltetrazolium (TTC) and Evans blue to analyse the area at risk (AAR/LV) and infarct size (INF/AAR).32 In addition, hearts from another group of mice sacrificed after 24 h of reperfusion were frozen in OCT and cut in 10 mm sections. On the other hand, to detect long-term ventricular remodelling after ischemia/reperfusion injury, echocardiography was performed on conscious mice by Vevo2100 (VisualSonics, Ontario, Canada) to measure heart function in mice after 3 weeks of reperfusion. Briefly, parameters were measured from M-mode images of parasternal short-axis view at papillary muscle level: ejection fraction (EF). The measurements performed in triplicate and took the average of three measurements as the final data for each mouse.3
Primary Cardiomyocytes Isolation, Culture, and Treatment
Primary neonatal rat cardiomyocytes (NRCMs) were isolated from neonatal rat (1–3 day old Sprague-Dawley) ventricular and cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose supplemented with 5% fetal bovine serum. The hypoxia/reoxygenation (H/R) model was established according to Song et al description.33 Briefly, cardiomyocytes were treated with 4 h of hypoxia (1% O2, 5% CO2 and 94% N2) and then followed by 3 h of reoxygenation (21% O2, 5% CO2 and 74% N2) in an air-tight chamber at 37 °C. To explore the effect of SIRT1 in regulating cardiomyocytes apoptosis, NRCMs were treated with 10 μM SIRT1 inhibitor (EX527, Sigma-Aldrich, USA), or 15 μM SIRT1 activator (Resveratrol, RSV, Sigma-Aldrich, USA) for 24 h, respectively. Next, the treated NRCMs were used to construct the H/R model. To investigate the effect of inhibition of PGC1-α on the anti-apoptotic effect of RSV, NRCMs were first incubated with RSV for 24 h, then siRNAs for PGC1-α (50 nM, 48h) (GenePharma, China)34 were transfected to cardiomyocytes for 48 h using lipofectamine 2000 as recommended by the manufacturer to silence PGC1-α expression. Next, the treated NRCMs were used to construct the H/R model. In all in vitro experiments, the H/R-induced apoptosis (4 h hypoxia/3 h reoxygenation) was conducted in the last 7 h of cell treatment.
Immunofluorescent and TUNEL Staining
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labelling (TUNEL) staining was conducted to detect apoptotic nuclei by fluorescence microscopy in α-actinin-labeled NRCMs. Briefly, NRCMs or heart frozen sections were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature, and then washed by PBS three times. Then, NRCMs or heart frozen sections were incubated with 0.5% Triton X-100 to It makes the cell membrane permeable at room temperature. Next, NRCMs or heart frozen sections were blocked with 5% bovine serum album (BSA) for 1 h at 37°C. After washed by PBS, NRCMs or heart frozen sections were incubated with anti-α-actinin (1:200, Sigma-Aldrich, USA) in a humid chamber overnight at 4°C. On the next day, NRCMs or heart frozen sections were incubated with Cy3-AffiniPure goat anti-mouse IgG (H+L) second antibodies (1:400, Jackson, USA) for 2 h at 37°C. Then, NRCMs or heart frozen sections were stained with TUNEL FITC Apoptosis Detection Kit (Vazyme, China) according to the manufacturer’s instructions. Finally, Nuclei were labeled with 4,6-diamidino-2-phenylindole (DAPI) staining. Nuclei were counterstained with DAPI. The TUNEL and α-actinin co-labeled positive cells were calculated using fluorescence microscopy to determine apoptosis induced by H/R or MI/R injury.
Western Blotting
Western blotting was performed as previously described.30 The equivalent amount of proteins were separated by electrophoretic techniques. Cleaved-Caspase 3 (1:1000, CST, USA), Bax (1:1000, CST, USA), Bcl-2 (1:1000, CST, USA), SIRT1 (1:1000, CST, USA), Nrf2 (1:1000, Santa Cruz Biotechnology, USA), HO-1 (1:1000, CST, USA), SOD1 (1:1000, CST, USA), PGC1-α (1:1000, CST, USA), GAPDH (1:2000, Bioworld, USA) were used as primary antibodies. Anti-rabbit or mouse IgG antibodies coupled to horseradish peroxidase were used as the secondary antibodies. GAPDH was used as loading control.
Immunohistochemical Staining and Histological Analysis
Antioxidant molecules in myocardial tissues were determined by immunohistochemical with antibodies reacting to HO-1, Nrf2, SOD1. Briefly, following euthanasia, mice hearts were immediately harvested and rinsed in PBS, fixed in buffered paraformaldehyde solution (4%) and embedded in paraffin. After cutting the heart tissue paraffin samples into 3 μm thick slices, the paraffin sections were dried in an oven at 60 °C for 1 h and then dewaxed in xylene twice (5 min for each xylene solution), rinsed in alcohol, rehydrated in descending gradient alcohol solutions (100, 95, 85, 80 and 75%, 10 min for each alcohol solutions). After rehydrated, paraffin sections were subjected to heat-induced antigen retrieval in a 10 mM citrate buffer (pH 6.0) for 20 min with a 20 min cool down. All paraffin sections were incubated with 2% H2O2 for 15 min to quench endogenous peroxidases. Following washed 3 times for 3 min and incubated in 0.3% Triton X-100 in PBS for 2 h. And then the paraffin sections were blocked by incubation with 5% BSA for 60 min. To detect the expression of target proteins, the three batches of paraffin sections were incubated with the anti-HO-1, Nrf2, SOD1 antibody at 4°C for 12 h, respectively, and then with the secondary antibody for 2 h at normal temperature followed by three additional washes with PBS for 5 min. The secondary antigen was visualized with 3.3-diaminobenzidine tetrahydrochloride for 4 min (YEASEN, China). Finally, the regions of positive staining appear brown with hematoxylin counterstaining. For morphological and fibrosis measurement, hematoxylin and eosin and Masson’s Trichrome staining was performed. Briefly, after rehydrated, paraffin sections were immersed in Haematoxylin solution for 5 min, washed in running tap water. After that, paraffin sections were stained for 6 min with Acid Fuchsin, rinsed in distilled water. For Masson’s Trichrome staining, the paraffin sections were incubated for 3 min with a phosphomolybdic acid solution and then immediately stained for 4 min with methyl blue solution, rinsed in distilled water. Then, paraffin sections were treated with 1% acetic acid solution for 3 min. The collagen fibers appeared green, the myocardial fibers were red. Finally, sealed with neutral resin and the tissue sections were visualized by fluorescence microscopy at 20x (hematoxylin and eosin and Masson’s Trichrome staining) or 40x (immunohistochemical staining) magnification. For evaluation of target proteins expression and fibrosis, the densitometry analysis of immunohistochemical staining and fibrosis area were performed using the ImageJ software.
Statistical Analysis
All experimental data were analyzed using SPSS (version 20.0) and presented as mean ± SD using GraphPad Prism 8.0 unless otherwise stated. Comparisons were performed using either a two-tailed, unpaired Student’s t-test between two groups. One-way ANOVA followed by Bonferroni’s post hoc test was used for comparison among more than three groups. P values <0.05 was considered statistically significant.
Results
Exercise Protects Against Myocardial Ischemia/Reperfusion Injury in Mice
At the beginning of the experiment, we constructed a mice swimming training model. After 3-weeks exercise, myocardial ischemia/reperfusion (MI/R) injury surgery was performed on the mice. After 24 h of reperfusion, the area of myocardial infarction was detected by Evans blue-2,3,5-triphenyltetrazolium chloride (TTC) double staining. Significantly decreased infarct size was observed in mice model after 3-week exercise compared with control (CON) mice (Figure 1A), and there were no differences in area at risk (AAR/LV) between the groups represented qualified surgical outcome and uniformity. Meanwhile, as shown in Figure 1B, MI/R injury induced significant apoptosis of cardiomyocytes as determined by TUNEL immunofluorescence staining. However, reduced TUNEL positive cardiomyocytes were observed in swam mice (Figure 1B). Moreover, as measured by Western blotting, exercise preconditioning could markedly reduce cellular apoptosis with reduced apoptosis-related protein indicators including Bax/Bcl2 and cleaved-caspase-3/caspase-3 ratios (Figure 1C). Taken together, these data indicate that swimming preconditioning can protect the cardiomyocytes apoptosis induced by myocardial ischemia/reperfusion injury in mice.
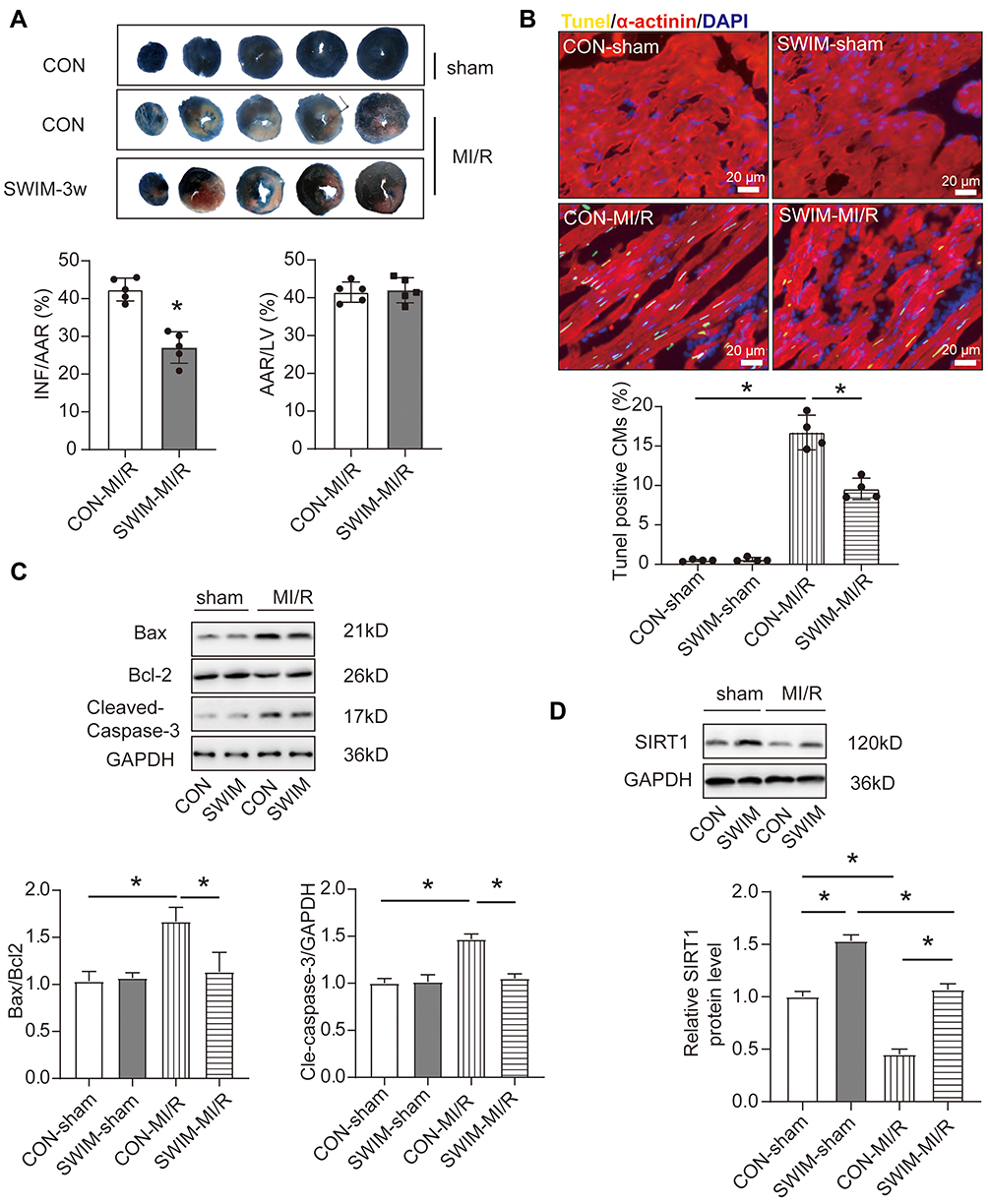 |
Figure 1 Exercise training reduces MI/R-induced myocardial apoptosis. (A) Representative photographs of heart sections 2,3,5-triphenyltetrazolium chloride (TTC) staining and quantified data demonstrate that swimming training decreased infarct size in mice subjected to MI/R (45 min/24 h) (n=5) AAR: area at risk, INF: infarct size, LV: left ventricle. (B) Representative photographs of heart sections TUNEL-immunofluorescent staining and quantified data. TUNEL-positive nuclei in α-actinin-labeled cells were calculated to determine myocardial apoptosis (n=4) Scale bar=20 μm, CM: cardiomyocytes (C) Western blotting for Bax, Bcl-2 and Cleaved-Caspase-3 in MI/R injury heart samples (n=3). (D) Representative blots and quantified data showing SIRT1 expression in heart of mice treated with swum or MI/R surgery (n=3). Data were expressed as mean ± SD. *P<0.05. |
Recent studies have suggested that the expression and activation of SIRT1 are correlated with myocardial ischemia-reperfusion injury.35,36 In addition, our previous research also revealed that cardiomyocyte-specific deletion of SIRT1 gene sensitizes the tolerance of the heart to ischemia stress.30 Therefore, we wondered whether the cardioprotective effects of exercise could be related to the expression of SIRT1. Western blotting experiments were subsequently carried out to measure the expression of SIRT1. As shown in Figure 1D, compared with the con-sham group, swum significantly increased the protein expression of SIRT1 in the heart, while SIRT1 was significantly down-regulated in the heart of the mice undergoing MI/R surgery. What is particularly noteworthy is that the long-term exercise training has resisted the down-regulation of SIRT1 during MI/R injury (Figure 1D). These results suggested that SIRT1 expression was negatively correlated with the myocardial infarction size and extent of myocardial apoptosis.
SIRT1 Regulates Apoptosis of Cardiomyocytes Induced by H/R in vitro
Accordingly, we investigated whether SIRT1 has a regulatory effect on apoptosis of cardiomyocytes. Gain- and loss-of-function were used to investigate SIRT1’s roles in neonatal cardiomyocytes. EX527 (SIRT1 inhibitor)37 and resveratrol (RSV, SIRT1 activator)38 were introduced into NRCMs to regulate SIRT1 activity. We measured the degree of apoptosis in neonatal rat cardiomyocytes (NRCMs) treated with hypoxia/reoxygenation (H/R), a cell model to simulate cardiomyocyte apoptosis and MI/R injury in vitro. As we expected, after RSV treatment, H/R-induced apoptosis of NRCMs was decreased, while EX527 further increased the apoptosis ratio significantly (Figure 2A and C). Activation of the SIRT1/Nrf2 signaling pathway has been reported to promote the production of antioxidant.39,40 Furthermore, regular exercise can stimulate the activation of Nrf2 to enhance the endogenous antioxidant ability to resist the destructive effect of reactive oxygen species.20 Therefore, we explored the expression of Nrf2 and its downstream cytoprotective proteins through Western blotting. As shown in Figure 2B, activation of SIRT1 rescued HO-1 and SOD1 reduction induced by H/R insult. However, the opposite results were found in inhibition of SIRT1 activity (Figure 2D). Collectively, these data revealed that activation of SITR1 protects against H/R-induced cardiomyocytes apoptosis by regulating Nrf2-mediated antioxidant regulation.
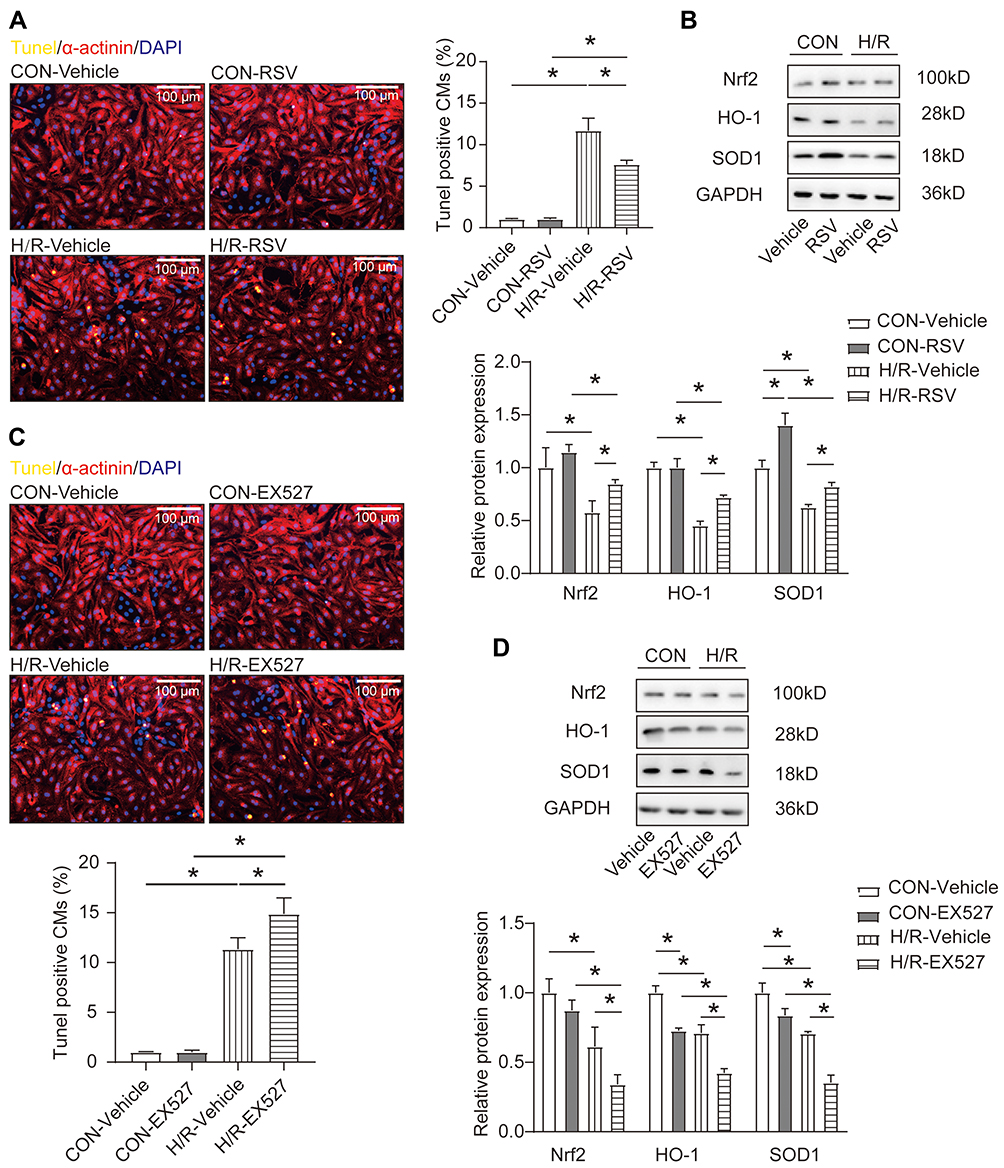 |
Figure 2 SIRT1 regulates apoptosis of cardiomyocytes through antioxidant stress factors. (A) The ratio of apoptosis after RSV stimulation in H/R-treated neonatal rat cardiomyocytes (NRCMs) as determined by TUNEL staining (n=4) Scale bar = 100 μm. (B) Western blotting analysis for Nrf2, HO-1 and SOD1 after treatment of NRCMs with RSV in the presence or absence of H/R treatment (n=3). (C) The ratio of apoptosis after EX527 stimulation in H/R-treated neonatal rat cardiomyocytes (NRCMs) as determined by TUNEL staining (n=4) Scale bar=100 μm. (D) Western blotting analysis for Nrf2, HO-1 and SOD1 after treatment of NRCMs with EX527 in the presence or absence of H/R treatment (n=3). Data were expressed as mean ± SD. *P < 0.05. |
SIRT1 Suppresses Oxidative Stress Injury of Cardiomyocytes Through PGC-1α
The molecular biology of Peroxisome proliferator-activated receptors gamma coactivator-1 alpha (PGC-1α) has been extensively explored in cardiovascular disease.41 Once PGC-1α is activated, it is recruited to the chromatin through interaction with transcription factors to activate certain genes expression, and Nrf2 is one of these transcription factors.42 Further more, SIRT1 stimulates the deacetylation of PGC-1α, leading to the promotion of PGC-1α activities.8 Based on the above experimental results, we hypothesised that PGC-1α mediated the regulation of SIRT1 on Nrf2 and antioxidant enzymes as well as apoptosis. Next, Western blotting experiments were performed under different treatment conditions. Consistent with SIRT1, we found that H/R treatment could down-regulate the expression of PGC-1α in NRCMs (Figure 3A). Moreover, with the activation or inhibition of SIRT1, the protein expression of PGC-1α also showed a consistent expression trend (Figure 3A). In order to verify that the activation of SIRT1 exerts an anti-apoptotic effect by stimulating the activity of PGC-1α, we designed a rescue experiment in which PGC-1α siRNA was transfected while the H/R NRCMs were treated with RSV. Immunofluorescent TUNEL staining showed that transfection of PGC-1α siRNA reversed the anti-apoptotic effect of RSV (Figure 3B). These experimental results suggest that SIRT1 plays an important role in the regulation of oxidative stress injury through directly activating PGC-1α.
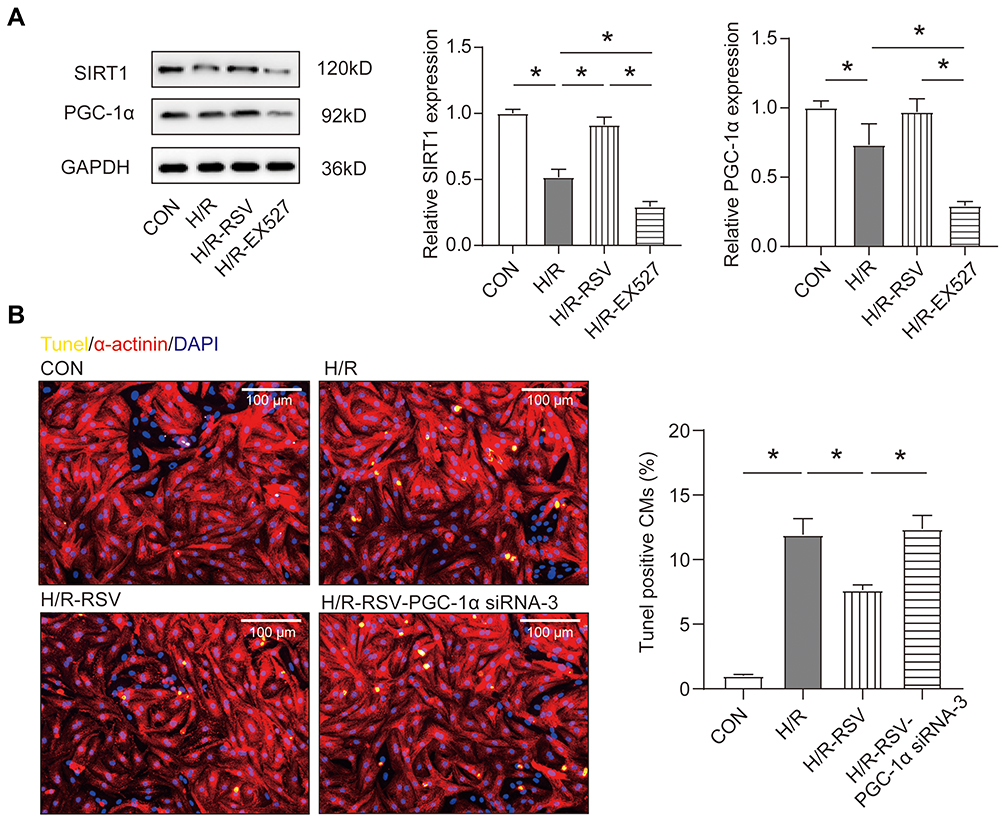 |
Figure 3 Activation of SIRT1 reduces apoptosis through the SIRT1-PGC-1α pathway. (A) The protein levels of SIRT1 and PGC-1α were analyzed by Western blotting in NRCMs treated with H/R after stimulation with the RSV or EX527 (n=3). (B) The cardiomyocyte apoptosis was measured by TUNEL immunofluorescent staining after transfection with siRNAs targeting PGC-1α in NRCMs treated with H/R and RSV (n=4) Scale bar=100 μm. Data were expressed as mean ± SD. *P < 0.05. |
Deletion of the SIRT1 Gene Blocks Exercise-Induced Cardioprotection in vivo
In the above experimental results (Figure 1D and 2A), we found that SIRT1 protein expression was elevated in the heart of mice after swimming training, and the activation of SIRT1 could resist H/R-induced NRCMs apoptosis in vitro. To investigate if SIRT1 contributes to exercise-induced cardioprotection, we constructed a mice swimming model upon inducible cardiac SIRT1 knock-out mice by tamoxifen (icSIRT1 KO) and SIRT1flox/flox mice (Figure 4A). Then, MI/R injury surgery was performed on both sedentary and swim preconditioning mice. Interestingly, SIRT1 knocked out at least partly abolished exercise-induced cardioprotection as evidenced by significantly increased infarct size (Figure 4B). In addition, immunofluorescent TUNEL staining suggested that SIRT1 knockout also blocked the decreased apoptosis ratio (Figure 4C). Meanwhile, the apoptotic levels were also confirmed by increased Bax/Bcl2 and cleaved-caspase-3/caspase-3 ratios (Figure 4D).
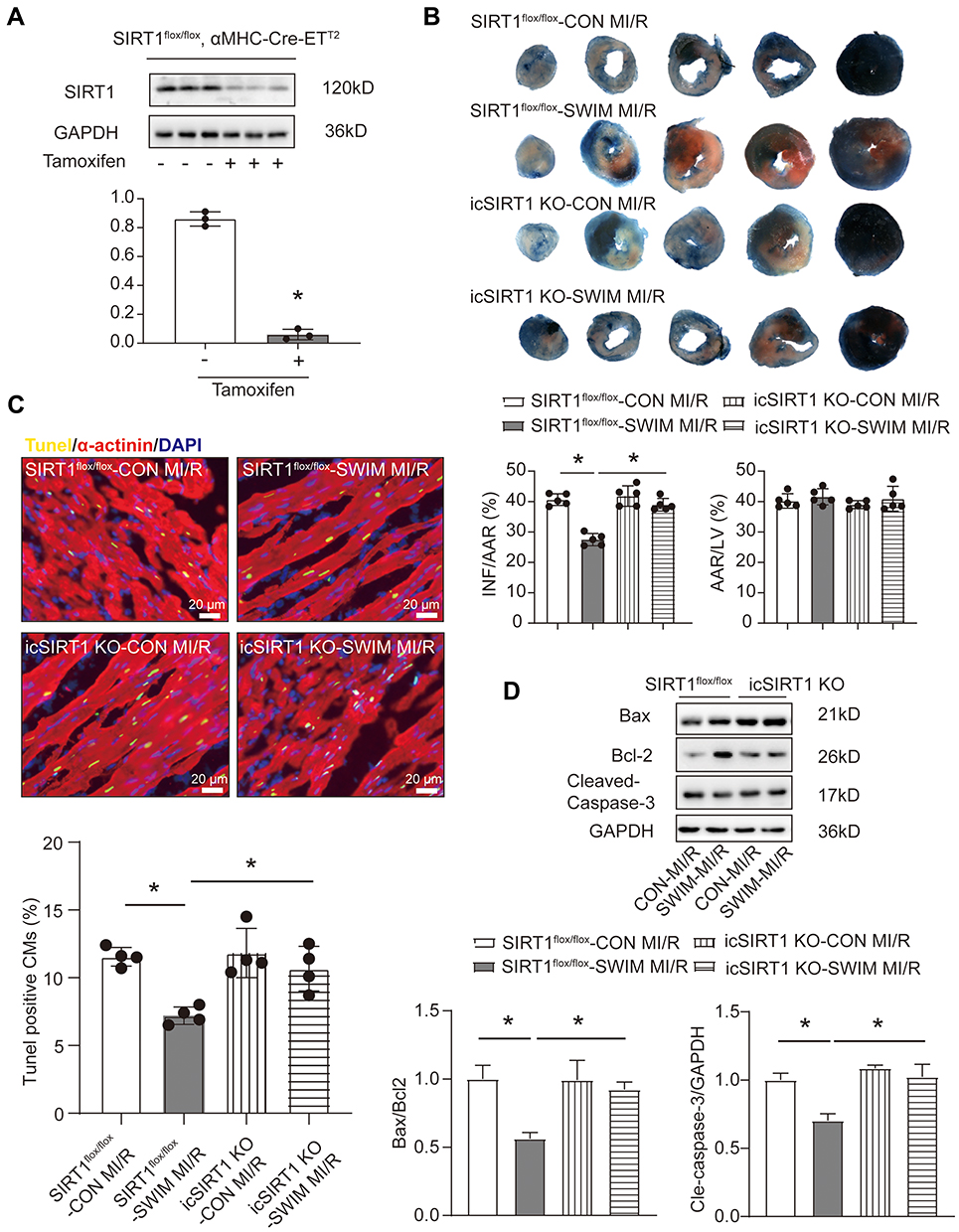 |
Figure 4 SIRT1 is required for exercise-induced cardioprotection in vivo. (A) Western blotting shows the down-regulation of heart SIRT1 after 10 days of Tamoxifen injection (0.08 mg/g, i.p. 5 days, n=3). (B) The infarct size was measured upon MI/R injury (45 min/24 h) of icSIRT1 KO and SIRT1flox/flox mice in the presence or absence of swimming training as determined by TTC staining (n=5) AAR: area at risk, INF: infarct size, LV: left ventricle. (C) The myocardial apoptosis was measured upon MI/R injury (45 min/24 h) of icSIRT1 KO and SIRT1flox/flox mice in the presence or absence of swimming training as determined by TUNEL staining (n=4), immunofluorescent staining for α-actinin was used to label cardiomyocytes. Scale bar=20 μm. (D) Western blotting for Bax, Bcl-2 and Cleaved-Caspase-3 in MI/R injury heart samples (n=3). Data were expressed as mean ± SD. *P < 0.05. |
It has been reported that exercise training can prevent ventricular remodeling and maintain cardiac function after myocardial infarction.3 The cardiac function was detected by echocardiography after 3 weeks myocardial reperfusion in mice. Consistent with previous reports, swum can improve the cardiac ejection fraction (Figure 5A) after MI/R injury and against both ventricular remodeling and cardiac fibrosis in mice (Figure 5B). However, in icSIRT1 KO mice, increased ejection fraction and reduced fibrotic area were abolished (Figure 5A and B). Collectively, in swum mice, the icSIRT1 KO mice showed the larger myocardial infarct size, higher apoptosis ratio, higher level of apoptotic proteins expression, lower ejection fraction and greater degree of fibrosis compared with SIRT1flox/flox mice. These data indicate that SIRT1 is necessary for exercise-induced beneficial effects on MI/R injury in vivo.
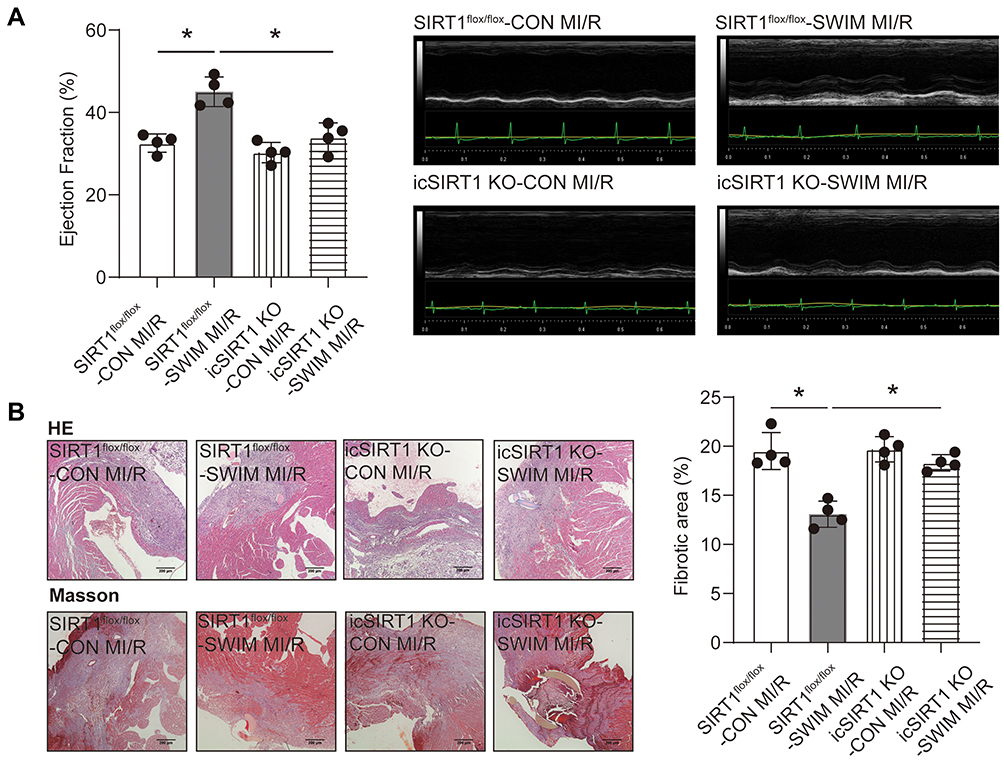 |
Figure 5 Deletion of the SIRT1 abolishes resistance of exercise to cardiac dysfunction after MI/R. (A) Representative echocardiographic images 21 days after reperfusion and quantified data of ejection fraction upon MI/R injury of icSIRT1 KO and SIRT1flox/flox mice in the presence or absence of swimming training (n=4). (B) Representative histopathological analysis of cardiac sections of the left ventricle stained with hematoxylin and eosin (upper, n=4, Scale bar=200 μm.); Representative images of Masson’s trichrome staining of the cardiac left ventricle areas (under, n=4, Scale bar=200 μm) and quantitative data of fibrotic area. Data were expressed as mean ± SD. *P < 0.05. |
SIRT1 Deficiency Hinders Exercise-Induced Expression of Myocardial Antioxidant Stress Factors
Subsequent in vivo experiments were carried out to verify the results obtained in vitro. The above results in Figure 2B showed that activation of SIRT1 by RSV in H/R treated NRCMs significantly up-regulated the proteins expression of Nrf2 and the antioxidants including SOD1 and HO-1 in vitro. Consistent with these results, immunohistochemical staining of Nrf2, HO-1 and SOD1 in myocardial sections of hearts from MI/R injury mice suggested that swum could up-regulate the expression of Nrf2, HO-1 and SOD1 proteins in myocardium, but after myocardial specific SIRT1 knocked out, the expression of these antioxidants was reduced to the basic level (Figure 6A). On the other hand, swimming preconditioning up-regulated the expression of SIRT1 and PCG-1α in myocardial ischemia zone (Figure 6B), which is consistent with the results of our in vitro experiments. Moreover, swum did not change the expression of PCG-1α in the hearts of icSITR1 KO mice (Figure 6B). Collectively, these in vivo results further confirm that SIRT1 exerts its anti-oxidative stress effect on the myocardium after MI/R injury through its downstream target gene PCG-1α, to a certain extent.
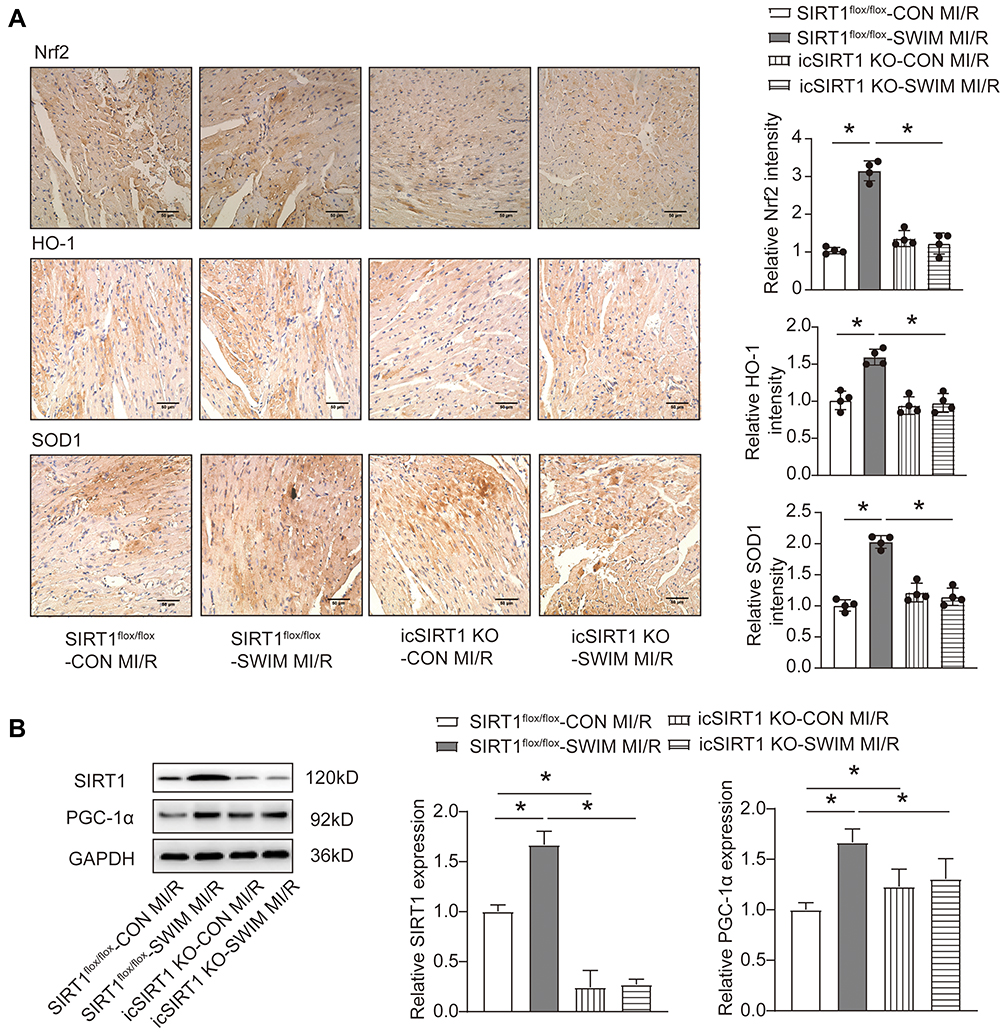 |
Figure 6 SIRT1 deficiency hinders exercise induced expression of myocardial antioxidant stress enzymes. (A) Representative images of immunohistochemical analysis for Nrf2, HO-1 and SOD1 expression in myocardial tissues sections and quantitative evaluation of the images. Scale bar=50 μm. (B) Western blotting analysis for SIRT1and PGC1-α upon MI/R injury (45 min/24 h) of icSIRT1 KO and SIRT1flox/flox mice in the presence or absence of swimming training (n=4). Data were expressed as mean ± SD. *P < 0.05. |
Discussion
MI/R injury results in the cardiomyocytes necrosis, apoptosis, ventricular systolic dysfunction, and cardiac fibrosis.5 Excessive production of reactive oxygen species, mitochondrial dysfunction, oxidative stress and increased expression of inflammatory factors are the main responses during ischemia-reperfusion.12 Currently, there are no standard therapy for MI/R injury. It is known that regular exercise improves both physical and mental health. Despite a wealth of evidence supporting the cardiovascular benefits of exercise, little is known about the mechanisms by which exercise prevents myocardial damage and reduces ventricular dysfunction. SIRT1 was first discovered in the nucleus and the activation of transcriptional factors via the deacetylation is thought to be the main role of SIRT1. As a transcriptional co-activator, PCG-1α has been shown to activate the transcription and translation of antioxidant genes by binding to Nrf2 transcription factor, a major regulator of antioxidant defenses and cellular stress resistance.20,43 Interestingly, SIRT1 was determined to act as a deacetylating enzyme via direct deacetylation of PGC-1α, leading to the promotion of PGC-1α activities in cardiomyocytes.8 On the other hand, SIRT1 interacts with its substrate PGC-1α to regulate energy metabolism in various aspects of the body through mitochondria.44 Jia et al found that exercise training after myocardial infarction improved cardiac function and against ventricular remodeling in mice through mitochondrial biogenesis and the SIRT1/PGC-1α signaling pathway.45 Our previous research has provided evidence that SIRT1 has become a critical regulator of cardiac MI/R injury pathologic processes.30 Coincidentally, Hsu et al proved that SIRT1 can resist myocardial ischemia injury by inducing Mn-SOD up-regulation and inhibiting oxidative stress in cardiomyocytes.46 These reported findings prompt us to explore the link between exercise-induced cardiac benefits and SIRT1.
Exercise training has been recommended as an effective mean of prevention and treatment of CVD. Previous reports have mentioned that long-term aerobic exercise can play a cardiac protective role by promoting proliferation of cardiomyocytes, inhibiting myocardial inflammation, reducing oxidative stress injury and protecting against ventricular remodeling.47 Our present data of Figure 1 suggest that exercise preconditioning significantly reduced myocardial infarct size and apoptosis in mice which are consistent with previous reports.48 Moreover, we also found that exercise preconditioning can improve the cardiac ejection fraction after MI/R injury and against both ventricular remodeling and cardiac fibrosis in mice. Interestingly, exercise training after myocardial ischemia also improved cardiac function and protected against poor ventricular remodeling.45 Oxidative stress resulted from the MI/R-induced massive ROS generation. Recently, suppression of oxidative stress and inflammatory pathways has been suggested as potential method to reduce myocardial fibrosis and cardiomyocyte death.49,50 In this study, we have evaluated the effects of resveratrol (RSV, SIRT1 activator) on the cell model of NRCMs in the presence of H/R. The Western blotting and immunofluorescence staining results showed that RSV treatment can significantly up-regulate the proteins expression of antioxidant enzymes including HO-1 and SOD1 and inhibit apoptosis of cardiomyocytes. While EX527 mediated inhibition of SIRT1 reduced HO-1 and SOD1 production.
One of the important points of our experiment was that we found that swimming preconditioning reversed down-regulation of SIRT1 in the heart of MI/R injury mice (Figure 1D). Similarly, Maria Donniacuo et al also found that running training increased the expression of SIRT1 in the heart border zone of rats.51 This finding is the basis for us to explore the relationship between exercise-induced heart benefits and SIRT1 in mice. To investigate if SIRT1 contributes to exercise-induced cardioprotection, we constructed an inducible cardiac-specific SIRT1 knockout mice. Interestingly, SIRT1 knocked out abolished the effects of swimming on decreased apoptotic cell ratio and heart infarct size (Figure 4). These results suggest that the antiapoptotic benefit of swimming cannot be achieved without the presence of SIRT1. We isolated neonatal rat cardiomyocytes for in vitro experiments and found that activation or inhibition of SIRT1 can alter the expression of antioxidant enzymes in cardiomyocytes during hypoxia/reoxygenation treatment (Figure 2). In addition, we treated cardiomyocytes with PGC-1α siRNA and found that silenced PGC-1α reversed the antiapoptotic effect of SIRT1 agonist RSV (Figure 3). Based on the above results in vitro, we speculated whether activation of SIRT1 could also resist myocardial injury by regulating the expression of antioxidant enzymes in vivo. To test our hypothesis, we detected the expression of antioxidant enzymes in myocardial tissue by immunohistochemical staining at the end of the experiment. These results confirm our conjecture, after myocardial-specific SIRT knocked out, swimming training had no effect on the proteins expression of Nrf2, HO-1 and SOD1 in myocardial tissue anymore (Figure 6). The above results indicated that SIRT1 protects against oxidative stress after MI/R injury by activating downstream PGC-1α and promoting the production of antioxidant enzymes. To further verify whether SIRT1 is necessary for exercise training to protect against pathological myocardial remodeling, cardiac function and histopathological analysis were evaluated by echocardiography and tissue sections staining in mice following 3 weeks of ischemia/reperfusion. As we expected, SIRT1 is also required for exercise resistance to myocardial remodeling (Figure 5). Based on the above experimental results, we strongly believe that SIRT1 is required for exercise-induced beneficial effects on myocardial ischemia/reperfusion injury.
In the experiment, only one substrate of SIRT1, PGC-1α was included in our study, which was not comprehensive. Subsequent work will focus on other target molecules downstream of SIRT1 to explore the relationship between SIRT1 and MI/R injury. Moreover, more indicators of oxidation and inflammation should be more widely tested.
Conclusion
We demonstrated that SIRT1 activated during swimming is protective against myocardial apoptosis upon MI/R injury. However, once SIRT1 was knocked out in cardiomyocytes, the protective effects of exercise preconditioning is disappeared. Therefore, our experimental results reveal that SIRT1 is required for exercise-induced beneficial effects on myocardial ischemia/reperfusion injury. Moreover, we found that PGC-1α, as a downstream gene of SIRT1, mediated the antioxidant stress effects in the myocardium of mice subjected to exercise training. In conclusion, SIRT1 is necessary for exercise-induced cardioprotection and its agonists may have therapeutic potential for treatment of ischemia heart disease and promoting functional recovery after cardiac ischemia/reperfusion.
Acknowledgment
We give our sincere gratitude to the reviewers for their valuable suggestions.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no competing interests exist.
References
1. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):e240–327. doi:10.1161/CIR.0b013e31829e8776
2. Pagidipati NJ, Gaziano TA. Estimating deaths from cardiovascular disease: a review of global methodologies of mortality measurement. Circulation. 2013;127(6):749–756. doi:10.1161/CIRCULATIONAHA.112.128413
3. Liu X, Xiao J, Zhu H, et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21(4):584–595. doi:10.1016/j.cmet.2015.02.014
4. Yan W, Lin C, Guo Y, et al. N-cadherin overexpression mobilizes the protective effects of mesenchymal stromal cells against ischemic heart injury through a beta-catenin-dependent manner. Circ Res. 2020;126(7):857–874. doi:10.1161/CIRCRESAHA.119.315806
5. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123(1):92–100. doi:10.1172/JCI62874
6. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi:10.1038/nm.2507
7. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301(6):H2181–90. doi:10.1152/ajpheart.00554.2011
8. Li D, Wang X, Huang Q, et al. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-kappaB pathway in vivo and in vitro. Redox Biol. 2018;15:62–73. doi:10.1016/j.redox.2017.11.023
9. Simon JN, Vrellaku B, Monterisi S, et al. Oxidation of Protein Kinase A regulatory subunit PKARIalpha protects against myocardial ischemia-reperfusion injury by inhibiting lysosomal-triggered calcium release. Circulation. 2021;143(5):449–465. doi:10.1161/CIRCULATIONAHA.120.046761
10. Hou L, Guo J, Xu F, et al. Cardiomyocyte dimethylarginine dimethylaminohydrolase1 attenuates left-ventricular remodeling after acute myocardial infarction: involvement in oxidative stress and apoptosis. Basic Res Cardiol. 2018;113(4):28. doi:10.1007/s00395-018-0685-y
11. Pisarenko O, Shulzhenko V, Studneva I, et al. Structural apelin analogues: mitochondrial ROS inhibition and cardiometabolic protection in myocardial ischaemia reperfusion injury. Br J Pharmacol. 2015;172(12):2933–2945. doi:10.1111/bph.13038
12. Dhalla NS, Elmoselhi AB, Hata T, et al. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res. 2000;47(3):446–456. doi:10.1016/S0008-6363(00)00078-X
13. Zhao W, Fan G-C, Zhang Z-G, et al. Protection of peroxiredoxin II on oxidative stress-induced cardiomyocyte death and apoptosis. Basic Res Cardiol. 2009;104(4):377–389. doi:10.1007/s00395-008-0764-6
14. Zhao XR, Zhang Z, Gao M, et al. MicroRNA-27a-3p aggravates renal ischemia/reperfusion injury by promoting oxidative stress via targeting growth factor receptor-bound protein 2. Pharmacol Res. 2020;155:104718. doi:10.1016/j.phrs.2020.104718
15. Fleg JL, Cooper LS, Borlaug BA, et al. Exercise training as therapy for heart failure: current status and future directions. Circ Heart Fail. 2015;8(1):209–220. doi:10.1161/CIRCHEARTFAILURE.113.001420
16. Akyuz A. Exercise and coronary heart disease. Adv Exp Med Biol. 2020;1228:169–179.
17. Bostrom P, Mann N, Wu J, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143(7):1072–1083. doi:10.1016/j.cell.2010.11.036
18. Powers SK, Sollanek KJ, Wiggs MP, et al. Exercise-induced improvements in myocardial antioxidant capacity: the antioxidant players and cardioprotection. Free Radic Res. 2014;48(1):43–51. doi:10.3109/10715762.2013.825371
19. Farah C, Kleindienst A, Bolea G, et al. Exercise-induced cardioprotection: a role for eNOS uncoupling and NO metabolites. Basic Res Cardiol. 2013;108(6):389. doi:10.1007/s00395-013-0389-2
20. Done AJ, Traustadottir T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016;10:191–199. doi:10.1016/j.redox.2016.10.003
21. Muthusamy VR, Kannan S, Sadhaasivam K, et al. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol Med. 2012;52(2):366–376. doi:10.1016/j.freeradbiomed.2011.10.440
22. Bernardo BC, Ooi JYY, Weeks KL, et al. Understanding key mechanisms of exercise-induced cardiac protection to mitigate disease: current knowledge and emerging concepts. Physiol Rev. 2018;98(1):419–475. doi:10.1152/physrev.00043.2016
23. Pedersen LR, Olsen RH, Anholm C, et al. Effects of 1 year of exercise training versus combined exercise training and weight loss on body composition, low-grade inflammation and lipids in overweight patients with coronary artery disease: a randomized trial. Cardiovasc Diabetol. 2019;18(1):127. doi:10.1186/s12933-019-0934-x
24. Pedersen BK. The physiology of optimizing health with a focus on exercise as medicine. Annu Rev Physiol. 2019;81(1):607–627. doi:10.1146/annurev-physiol-020518-114339
25. Morra EA, Rodrigues PL, Jesus ICGD, et al. Endurance training restores spatially distinct cardiac mitochondrial function and myocardial contractility in ovariectomized rats. Free Radic Biol Med. 2019;130:174–188. doi:10.1016/j.freeradbiomed.2018.10.406
26. Boulghobra D, Coste F, Geny B, et al. Exercise training protects the heart against ischemia-reperfusion injury: a central role for mitochondria? Free Radic Biol Med. 2020;152:395–410.
27. Pillarisetti S. A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat Cardiovasc Drug Discov. 2008;3(3):156–164. doi:10.2174/157489008786263989
28. Alcendor RR, Gao S, Zhai P, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100(10):1512–1521. doi:10.1161/01.RES.0000267723.65696.4a
29. Tong C, Morrison A, Mattison S, et al. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress. FASEB J. 2013;27(11):4332–4342. doi:10.1096/fj.12-216473
30. Wang L, Quan N, Sun W, et al. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc Res. 2018;114(6):805–821. doi:10.1093/cvr/cvy033
31. Shi J, Bei Y, Kong X, et al. miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. Theranostics. 2017;7(3):664–676. doi:10.7150/thno.15162
32. Morrison A, Yan X, Tong C, et al. Acute rosiglitazone treatment is cardioprotective against ischemia-reperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice. Am J Physiol Heart Circ Physiol. 2011;301(3):H895–902. doi:10.1152/ajpheart.00137.2011
33. Song H, Feng X, Zhang H, et al. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. 2019;15(8):1419–1437. doi:10.1080/15548627.2019.1586246
34. Shoag J, Haq R, Zhang M, et al. PGC-1 coactivators regulate MITF and the tanning response. Mol Cell. 2013;49(1):145–157. doi:10.1016/j.molcel.2012.10.027
35. Yu L, Liang H, Dong X, et al. Reduced silent information regulator 1 signaling exacerbates myocardial ischemia-reperfusion injury in type 2 diabetic rats and the protective effect of melatonin. J Pineal Res. 2015;59(3):376–390. doi:10.1111/jpi.12269
36. Wang YH, Li S-A, Huang C-H, et al. Sirt1 activation by post-ischemic treatment with lumbrokinase protects against myocardial ischemia-reperfusion Injury. Front Pharmacol. 2018;9:636. doi:10.3389/fphar.2018.00636
37. Yu L, Sun Y, Cheng L, et al. Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: role of SIRT1. J Pineal Res. 2014;57(2):228–238. doi:10.1111/jpi.12161
38. Planavila A, Iglesias R, Giralt M, et al. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. 2011;90(2):276–284. doi:10.1093/cvr/cvq376
39. Zhang B, Zhai M, Li B, et al. Honokiol ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by reducing oxidative stress and apoptosis through activating the SIRT1-Nrf2 signaling pathway. Oxid Med Cell Longev. 2018;2018:3159801. doi:10.1155/2018/3159801
40. Ma W, Guo W, Shang F, et al. Bakuchiol alleviates hyperglycemia-induced diabetic cardiomyopathy by reducing myocardial oxidative stress via activating the SIRT1/Nrf2 signaling pathway. Oxid Med Cell Longev. 2020;2020:3732718.
41. Riehle C, Abel ED. PGC-1 proteins and heart failure. Trends Cardiovasc Med. 2012;22(4):98–105. doi:10.1016/j.tcm.2012.07.003
42. Puigserver P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-alpha. Int J Obes (Lond). 2005;29(Suppl 1):S5–9. doi:10.1038/sj.ijo.0802905
43. Waldman M, Cohen K, Yadin D, et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving ‘SIRT1 and PGC-1alpha’. Cardiovasc Diabetol. 2018;17(1):111. doi:10.1186/s12933-018-0754-4
44. Tang BL. Sirt1 and the mitochondria. Mol Cells. 2016;39(2):87–95.
45. Jia D, Hou L, Lv Y, et al. Postinfarction exercise training alleviates cardiac dysfunction and adverse remodeling via mitochondrial biogenesis and SIRT1/PGC-1alpha/PI3K/Akt signaling. J Cell Physiol. 2019;234(12):23705–23718. doi:10.1002/jcp.28939
46. Hsu CP, Zhai P, Yamamoto T, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122(21):2170–2182. doi:10.1161/CIRCULATIONAHA.110.958033
47. Piepoli MF, Conraads V, Corrà U, et al. Exercise training in heart failure: from theory to practice. A consensus document of the heart failure association and the European Association for cardiovascular prevention and rehabilitation. Eur J Heart Fail. 2011;13(4):347–357. doi:10.1093/eurjhf/hfr017
48. Hou Z, Qin X, Hu Y, et al. Longterm exercise-derived exosomal miR-342-5p: a novel exerkine for cardioprotection. Circ Res. 2019;124(9):1386–1400. doi:10.1161/CIRCRESAHA.118.314635
49. Cavalera M, Wang J, Frangogiannis NG. Obesity, metabolic dysfunction, and cardiac fibrosis: pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl Res. 2014;164(4):323–335. doi:10.1016/j.trsl.2014.05.001
50. Kang P, Wang J, Fang D, et al. Activation of ALDH2 attenuates high glucose induced rat cardiomyocyte fibrosis and necroptosis. Free Radic Biol Med. 2020;146:198–210. doi:10.1016/j.freeradbiomed.2019.10.416
51. Donniacuo M, Urbanek K, Nebbioso A, et al. Cardioprotective effect of a moderate and prolonged exercise training involves sirtuin pathway. Life Sci. 2019;222:140–147. doi:10.1016/j.lfs.2019.03.001
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.