Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 18
Single-Cell Sequencing Uncovers a TMSB10-Expressing Fibroblast Subpopulation Driving Renal Fibrosis in Diabetic Nephropathy
Authors Qin Z ![]() , Huang X
, Huang X ![]() , Du K
, Du K ![]() , Zhang L
, Zhang L ![]() , Xu X
, Xu X ![]() , Fang Y
, Fang Y
Received 5 September 2025
Accepted for publication 13 December 2025
Published 31 December 2025 Volume 2025:18 Pages 4913—4929
DOI https://doi.org/10.2147/DMSO.S559695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rebecca Baqiyyah Conway
Zihan Qin,1 Xiaoli Huang,2 Ke Du,3 Liexiang Zhang,4 Xiaohong Xu,5 Yuepeng Fang6
1Department of Endocrinology & Geriatrics, Shandong Provincial Hospital, Shandong University, Jinan, People’s Republic of China; 2Department of Endocrinology, Affiliated Hospital 2 of Nantong University and First People’s Hospital of Nantong City, Nantong, People’s Republic of China; 3Department of Endocrinology and Metabolism, Jurong People’s Hospital, Jiangsu University, Zhenjiang, People’s Republic of China; 4Department of Neurosurgery, The Affiliated Suqian First People’s Hospital of Nanjing Medical University, Suqian, People’s Republic of China; 5Department of Nephrology, The Affiliated Suqian First People’s Hospital of Nanjing Medical University, Suqian, People’s Republic of China; 6Jinan Central Hospital, Shandong University, Jinan, People’s Republic of China
Correspondence: Xiaohong Xu, Email [email protected] Yuepeng Fang, Email [email protected]
Introduction: Diabetic nephropathy (DN) is a leading cause of end-stage kidney disease (ESKD), with renal fibrosis as a key pathological hallmark. However, the cellular and molecular drivers of fibrosis remain incompletely defined. Here, we employed single-cell RNA sequencing (scRNA-seq) to delineate pro-fibrotic cell subsets and their key regulatory factors in human DN kidneys, providing a higher-resolution view compared to previous fibrosis-related scRNA-seq studies.
Methods: Publicly available scRNA-seq datasets from human DN and control kidneys were analyzed to identify fibrosis-associated fibroblast subsets. A Tmsb10-high fibroblast population was prioritized. Functional validation was performed through Tmsb10 knockdown in NIH-3T3 fibroblasts and in a diabetic mouse model, followed by assessment of fibrosis markers, extracellular matrix (ECM) deposition, and TGF-β/SMAD signaling.
Results: scRNA-seq revealed a significant expansion of Tmsb10-high fibroblasts in DN kidneys, exhibiting strong enrichment of ECM-related and TGF-β/SMAD-responsive genes. Tmsb10 knockdown reduced Fn1, Col1a1, and α-Sma expression by approximately 50– 70% and markedly attenuated ECM accumulation in vivo. Mechanistically, TMSB10 deficiency suppressed phosphorylation of SMAD2/3, mitigating fibroblast activation and matrix deposition.
Discussion: This study identifies TMSB10 as a novel fibroblast-specific regulator of renal fibrosis in DN, acting through the TGF-β/SMAD pathway. These findings expand current understanding of fibroblast heterogeneity and highlight TMSB10 as a potential therapeutic target for DN and other fibrotic diseases. Limitations include validation in a limited sample size and the use of murine fibroblast models, warranting further confirmation in human primary cells.
Keywords: diabetic nephropathy (DN), thymosin beta-10 (TMSB10), single-cell RNA sequencing (scRNA-seq), renal fibrosis, TGF/SMAD signaling pathway
Graphical Abstract:

Introduction
Diabetic nephropathy (DN) stands as one of the most prevalent and severe microvascular complications of diabetes mellitus, accounting for a substantial proportion of end-stage kidney disease (ESKD) cases worldwide.1,2 The progression of DN is characterized by a series of complex pathological changes, among which renal fibrosis is a pivotal and irreversible process that ultimately leads to the loss of renal function.3–6 A better understanding of the molecular and cellular mechanisms underlying fibrosis is therefore essential for developing effective therapeutic strategies to halt or reverse DN.
In the context of renal fibrosis, fibroblasts within the kidney play a pivotal role.7–9 Under normal physiological conditions, they maintain tissue homeostasis by synthesizing and remodeling ECM components. In DN, however, pathological stimuli such as hyperglycemia, oxidative stress, and inflammatory cytokines trigger fibroblast activation.10–13 Activated fibroblasts acquire a myofibroblast-like phenotype, characterized by α-smooth muscle actin (α-Sma) expression and excessive secretion of collagen and fibronectin.13–17 The overproduction of ECM distorts renal architecture, ultimately leading to glomerulosclerosis and renal failure.6,18,19
Among the numerous fibrogenic pathways, the transforming growth factor-β (TGF-β)/SMAD signaling axis serves as a master regulator of fibroblast activation and ECM production.13,20,21 Yet, despite the central role of fibroblasts and TGF-β signaling in DN, the molecular regulators controlling fibroblast heterogeneity and activation remain incompletely defined.
Recent advances in single-cell RNA sequencing (scRNA-seq) have enabled high-resolution dissection of renal cellular landscapes and identification of distinct fibroblast subsets involved in fibrosis.22–24 However, most fibrosis-related scRNA-seq studies to date have primarily characterized fibroblast responses,25 without resolving the specific upstream molecular cues responsible for their pathogenic reprogramming in diabetic nephropathy.
Thymosin beta-10 (TMSB10) is a small acidic peptide implicated in cell migration, proliferation, and cytoskeletal regulation.26–29 While TMSB10 has been linked to tumor progression and tissue remodeling, its role in DN-related renal fibrosis has not been explored. Our preliminary scRNA-seq analysis of DN and control kidney samples revealed a fibroblast subpopulation with high TMSB10 expression, suggesting that TMSB10 may serve as a previously unrecognized regulator of fibroblast activation in DN.
To test this hypothesis, we integrated single-cell transcriptomic analysis with functional validation in vitro and in vivo. Specifically, Tmsb10 knockdown was performed in NIH-3T3 fibroblasts and in a diabetic mouse model to assess its effects on fibroblast activation, ECM deposition, and TGF-β/SMAD signaling activity. Together, this study establishes TMSB10 as a potential pro-fibrotic driver and provides mechanistic insights into fibroblast-mediated renal fibrosis in DN.
Materials and Methods
Single-Cell RNA Sequencing Data Processing
To characterize the cellular landscape and molecular alterations associated with DN, scRNA-seq data were obtained from the publicly available Gene Expression Omnibus (GEO) database (accession number: GSE209781), which contains renal cortical samples from three DN patients and three non-diabetic controls (ND). Raw sequencing data were uniformly preprocessed and subjected to rigorous quality control, normalization, and filtering to ensure data integrity. Specifically, cells with low gene counts, high mitochondrial transcript percentages, or other indicators of poor quality were excluded to eliminate technical artifacts. Following normalization and scaling, high-quality cells were used for dimensionality reduction, clustering, and downstream analyses to delineate distinct renal cell populations and transcriptional alterations associated with DN (Figure 1A).
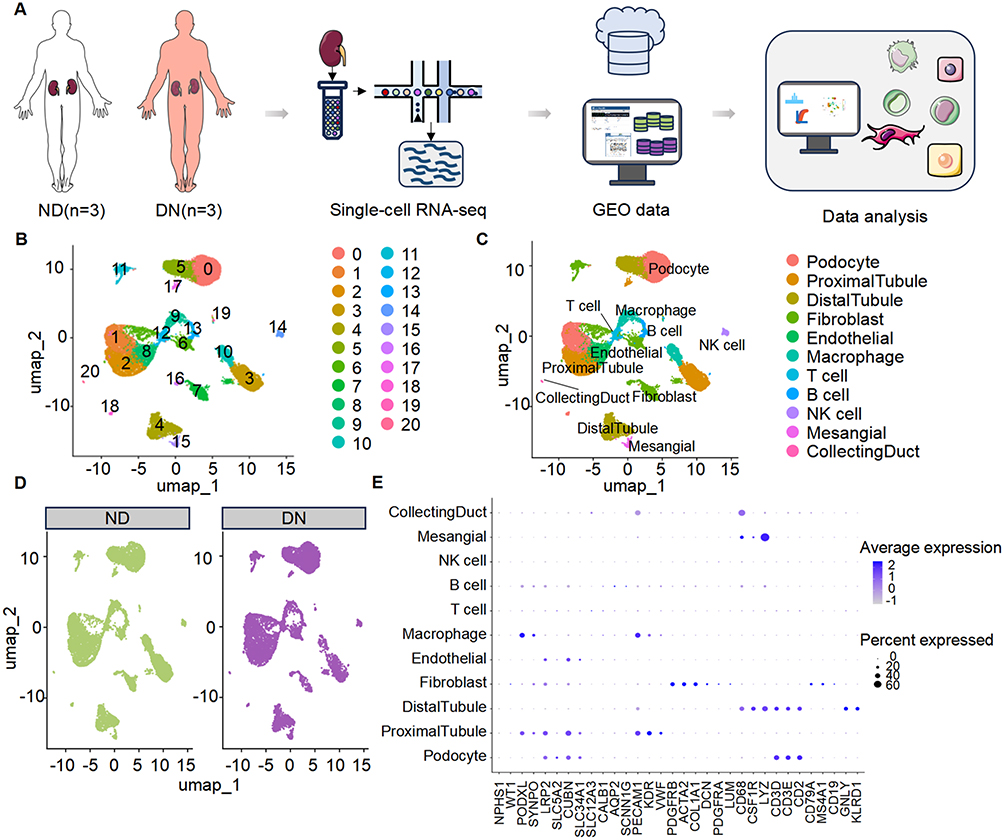 |
Figure 1 Single-cell transcriptomic landscape of kidney tissues from diabetic nephropathy (DN) patients and non-diabetic controls. (A) Schematic overview of the study workflow. Kidney cortical tissues were obtained from non-diabetic control individuals (ND, n = 3) and patients with diabetic nephropathy (DN, n = 3). Single-cell RNA sequencing (scRNA-seq) was performed, followed by integration with publicly available GEO datasets and comprehensive bioinformatics analysis. (B) UMAP dimensionality reduction showing unsupervised clustering of all kidney cells into 21 distinct clusters (labeled 0–20), reflecting the cellular heterogeneity of renal tissues. (C) Annotation of major renal cell populations based on canonical marker genes, including podocytes, proximal and distal tubular cells, fibroblasts, endothelial cells, macrophages, T cells, B cells, NK cells, mesangial cells, and collecting duct cells. (D) UMAP visualization of cell distributions in ND and DN groups, highlighting disease-associated shifts in cellular composition. (E) Dot plot displaying the expression patterns of representative marker genes across annotated cell types. Dot size corresponds to the percentage of cells expressing the gene, and color intensity reflects the average expression level. |
Following normalization and identification of highly variable genes, dimensionality reduction was performed via principal component analysis (PCA). Clustering was conducted at an empirically determined resolution. To minimize batch effects across samples, data integration was performed using a robust alignment algorithm, enabling unified construction of the neighbor graph, clustering, and visualization in a shared embedding space.
Cell type annotation was conducted based on established canonical marker genes reported in the literature, combined with differential expression patterns across clusters. Through manual curation, major renal cell populations were identified, including podocytes, proximal tubular cells, distal tubular cells, fibroblasts, and immune cells.
For cell-cell communication analysis, ligand-receptor interaction networks were independently constructed for the DN and ND groups. Intercellular signaling involving fibroblasts was systematically assessed in terms of interaction number, signaling strength, and pathway-level activity. This analysis identified multiple upregulated signaling pathways associated with ECM remodeling under diabetic conditions.
To investigate the potential dynamic transitions of fibroblasts during disease progression, fibroblast populations were extracted and subjected to pseudotime trajectory analysis. A disease-associated differentiation trajectory was identified, originating from a subpopulation characterized by high Tmsb10 expression. Temporal expression dynamics of fibrosis-associated genes along the pseudotime axis revealed stage-specific transcriptional programs involved in fibroblast activation and fibrogenesis.
Animal Model of Diabetic Nephropathy
Male C57BL/6J mice (8–12 weeks old, specific pathogen-free, genetic background verified by the supplier) were purchased from Shanghai Bikai Keyi Biotechnology Co., Ltd. (Shanghai, China) and housed under SPF conditions at the Animal Center of Shanghai Rat & Mouse Biotech Co., Ltd. Mice were maintained under controlled temperature (22 ± 2°C), humidity (45–65%), and a 12-hour light/dark cycle with ad libitum access to food and water. All experimental procedures adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (Approval No. 20220720(20)). The DN model was established following a previously described protocol.30 Briefly, mice were fed a high-fat/high-sucrose diet for 8 weeks to induce insulin resistance, followed by intraperitoneal streptozotocin (STZ; 50 mg/kg/day for 5 days, Solarbio, China) to induce hyperglycemia and renal injury. Random blood glucose (RBG)≥16.7 mmol/L was considered successful modeling. Mice were randomly assigned to three groups (n=6 per group) using a random number table: (1) Control (normal diet + scrambled shRNA lentivirus, 4×109 PFU); (2) DN (STZ + scrambled shRNA lentivirus, 4×109 PFU); (3) DN + shTMSB10 (STZ + TMSB10-shRNA lentivirus, 4×109 PFU). Allocation was performed by an independent investigator not involved in data analysis.
Sample size was determined based on prior studies showing similar variance in biochemical and histological outcomes in DN models.31 All histological and molecular analyses were performed in a blinded manner; investigators assessing staining and Western blot results were unaware of group allocation. The primary outcome was the degree of renal fibrosis, evaluated by representative images of Masson’s trichrome staining and ECM-related gene/protein expression.Secondary outcomes included renal morphology and TGF-β/SMAD pathway activation. At 28 days post-lentiviral injection, mice were euthanized by intraperitoneal injection of sodium pentobarbital (100 mg/kg) in accordance with the AVMA Guidelines (2020). Kidneys were harvested for histological, molecular, and transcriptomic analyses.
NIH-3T3 Cell Culture and siRNA-Mediated Gene Silencing
The mouse embryonic fibroblast line NIH-3T3 (ATCC) was used for in vitro experiments. This cell line was chosen for its genetic stability, reproducible fibroblast phenotype, and extensive validation in fibrosis-related studies.32,33 Primary renal fibroblasts from human or mouse kidneys were not used because they are limited in availability, heterogeneous, prone to early senescence, and show low transfection efficiency, all of which compromise experimental reproducibility. In contrast, NIH-3T3 cells provide a stable and well-characterized model with consistent responses to high-glucose and TGF-β stimulation, enabling controlled mechanistic analysis of the TGF-β/SMAD pathway.
The mouse fibroblast cell NIH-3T3 was obtained from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 10% calf serum (CS) and 1% penicillin-streptomycin. Cells were maintained at 37°C in a humidified incubator with 5% CO2. For normal glucose (NG) conditions, cells were cultured in medium containing 5.6 mM glucose. To simulate a diabetic-like environment, high glucose (HG) stimulation was performed by culturing cells in medium containing 30 mM glucose for 48 hours.
To investigate the role of TMSB10 in high glucose-induced fibroblast activation, small interfering RNA (siRNA)-mediated knockdown was performed. When NIH-3T3 cells reached 70–80% confluence, transfection was carried out using Lipofectamine™ 3000 (Life Technologies) according to the manufacturer’s protocol. Cells were divided into four experimental groups: (1) NC group, cultured under NG conditions and transfected with siNC; (2) siTMSB10 group, cultured under NG conditions and transfected with siTMSB10; (3) HG group, cultured under HG conditions and transfected with siNC; (4) HG+siTMSB10 group, cultured under HG conditions and transfected with siTMSB10. After 48 hours of transfection, cells were harvested for subsequent analyses, including quantitative real-time PCR, Western blotting, and immunofluorescence staining, to evaluate the regulatory role of TMSB10 in fibroblast phenotypic changes under hyperglycemic conditions.
Quantitative Real-Time PCR (qPCR)
Total RNA was extracted from mouse kidney tissues or NIH-3T3 fibroblasts using Trizol™ reagent (Life Technologies) according to the manufacturer’s instructions. RNA concentration and purity were measured using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized using SuperScript II Reverse Transcriptase (Life Technologies).
Quantitative real-time PCR was performed using a SYBR Green PCR Master Mix (Takara, Otsu, Japan) on an Applied Biosystems AB7300 Real-Time PCR System. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal control. The relative expression levels of target genes were calculated using the 2^-ΔΔCt method. Primer sequences used in this study are listed in Table S1. Each experiment was independently repeated six times (biological replicates), and each measurement (qPCR) was performed in triplicate (technical replicates).
Western Blotting
Total proteins were extracted using ice-cold RIPA lysis buffer (Beyotime, P0013B) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF; Beyotime, ST506). Protein concentrations were quantified using a bicinchoninic acid (BCA) protein assay kit (Beyotime, P0012). After mixing with loading buffer (TransGen, DL101-02) and denaturation at 100 °C, equal amounts of protein were resolved on 10% SDS-PAGE gels and subsequently transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, IPVH00010). For high-molecular-weight protein detection, the GoldBand Plus 3-color High Range Protein Marker (Yeasen, #20347ES) was employed. For low-molecular-weight proteins, the Pierce™ Unstained Protein Molecular Weight Marker, Standard Range (Thermo Fisher Scientific, #26616) was used.
Membranes were blocked in 5% non-fat milk prepared in TBST buffer for 1 hour at room temperature, followed by incubation with specific primary antibodies overnight at 4 °C. After washing, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 hour at room temperature. Protein signals were detected using Immobilon™ Western HRP Substrate Luminol Reagent (Millipore, WBKLS0500) and visualized with the Amersham Imager 600 imaging system (GE Healthcare). Band intensities were quantified using ImageJ software. A detailed list of antibodies used is provided in Table S2. Each experiment was independently repeated six times (biological replicates), and each measurement (Western blot) was performed in triplicate (technical replicates).
Immunofluorescence Staining
Mouse kidney tissues were fixed in 4% paraformaldehyde for 24 hours, followed by graded ethanol dehydration (70–100%), paraffin embedding, and sectioning at a thickness of 5 μm. Antigen retrieval was performed using Tris-EDTA buffer (pH 9.0) in a microwave oven for 20 minutes. Sections were then blocked with 5% bovine serum albumin (BSA) at room temperature for 30 minutes to reduce nonspecific binding.
For cultured cell staining, NIH-3T3 cells were fixed in 4% paraformaldehyde for 20 minutes, rinsed with phosphate-buffered saline (PBS), and permeabilized with 0.5% Triton X-100 for 10 minutes. After blocking with 5% BSA, both tissue sections and cells were incubated with primary antibodies at 4 °C overnight. On the following day, samples were washed with PBS and incubated with fluorescent dye-conjugated secondary antibodies at 37 °C for 30 minutes. Details of the antibodies used are provided in Table S2. Each experiment was independently repeated six times (biological replicates), and each measurement (immunofluorescence) was performed in triplicate (technical replicates).
HE and Masson Staining
Mouse kidney tissues were fixed in 4% paraformaldehyde for 24 hours, followed by dehydration through a graded ethanol series (70–100%), paraffin embedding, and sectioning at a thickness of 5 μm. The tissue sections were subjected to Masson’s trichrome staining and hematoxylin and eosin (H&E) staining. Masson staining was used to evaluate the extent and distribution of fibrotic lesions, while H&E staining was performed to assess glomerulosclerosis. Glomerular images were captured using a digital microscope camera.
Statistical Analysis
Data are presented as the mean ± SEM. All statistical analyses were performed using GraphPad Prism 8 and R. The normality and homogeneity of variances were assessed by the Shapiro–Wilk and Brown-Forsythe tests, respectively. For two-group comparisons, data that passed both tests were analyzed by an unpaired two-tailed Student’s t-test; otherwise, the Mann–Whitney U-test was used. For multi-group comparisons, one-way ANOVA with Tukey’s post hoc test was applied to parametric data, while the Kruskal–Wallis test with Dunn’s post hoc test was used for non-parametric data. A p-value < 0.05 was considered statistically significant. Effect sizes and 95% confidence intervals were computed for primary outcome measures to quantify the magnitude and reliability of group differences. Detailed results are provided in Table S3.
Ethical Approval Statement
This study used only publicly available human single-cell RNA sequencing datasets that were fully de-identified and contained no personally identifiable information. Therefore, according to Article 32, Items 1 and 2 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects (China, February 18, 2023), this study is exempt from Institutional Review Board (IRB) approval.
Results
Single-Cell Transcriptomic Profiling Reveals Abnormal States of Fibroblasts in the Kidneys of Diabetic Nephropathy Patients
Following initial preprocessing of the single-cell data, we applied computational algorithms to perform unsupervised clustering, which identified 21 distinct cell clusters-each likely corresponding to a unique cellular subtype or functional state within the renal tissue microenvironment (Figure 1B). To decipher the identity of these clusters, we utilized a panel of canonical marker genes known to be selectively expressed in specific renal cell types. Through this approach, we accurately annotated the clusters as major renal cell populations, encompassing podocytes, proximal and distal tubular cells, fibroblasts, endothelial cells, and various immune cell subsets (Figure 1C). This meticulous annotation process not only provided a clear roadmap of the cellular composition within the kidneys but also laid the foundation for subsequent comparative analyses.
Our comparative analysis between DN and ND groups unveiled a striking difference in fibroblasts in the renal tissues of DN patients (Figure 1D). This observation was particularly intriguing, given the well-established role of fibroblasts as key effector cells in tissue fibrosis, a hallmark pathological feature of DN. The marker genes employed for cell-type annotation exhibited highly specific expression patterns across different cell types, thereby reinforcing the accuracy and reliability of our annotation process (Figure 1E).
The significant difference in fibroblasts in DN patients, as revealed by our scRNA-seq analysis, strongly suggested their potential involvement in disease progression. This hypothesis was further bolstered by the observation that fibroblasts from DN patients exhibited distinct gene expression profiles compared to their ND counterparts, indicative of an activated or altered phenotypic state. Such alterations in fibroblast behavior could potentially contribute to the excessive deposition of extracellular matrix components, leading to renal fibrosis and subsequent loss of kidney function.
Fibroblasts are Markedly Expanded in DN and Mediate Enhanced Intercellular Communication
To delve deeper into the specific contributions of fibroblasts to the pathogenesis of DN, we undertook a refined reclustering and subgroup analysis of our scRNA-seq data. The results unequivocally confirmed a substantial alteration of fibroblasts within the renal tissues of DN patients, strongly implicating their active participation in the disease’s progression (Figure 2A and B).
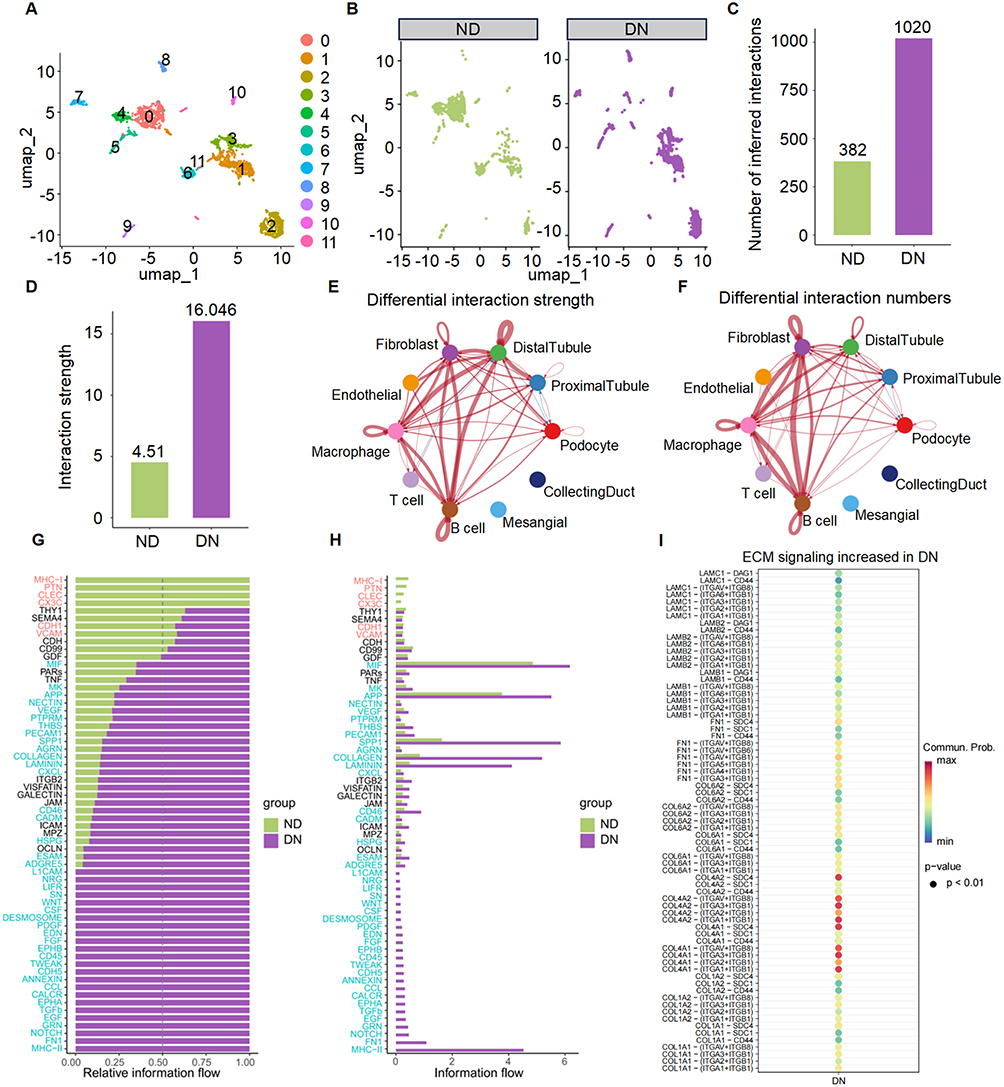 |
Figure 2 Expansion of fibroblasts and enhanced fibroblast-mediated intercellular communication in diabetic nephropathy (DN). (A) UMAP plot of fibroblasts re-clustered from the scRNA-seq dataset, revealing distinct fibroblast subpopulations for downstream intercellular communication analysis. (B) Comparison of fibroblast distribution between ND (left) and DN (right) samples, showing a marked difference in fibroblasts in DN, indicating their potential involvement in disease-associated cell-cell signaling. (C) Total number of inferred ligand-receptor interactions involving fibroblasts in ND and DN groups. (D) Comparison of global interaction strength of fibroblasts between ND and DN conditions, as calculated by CellChat. (E–F) Circle plots showing differential interaction strength (E) and interaction number (F) between fibroblasts and other renal cell populations. Line thickness represents communication magnitude. (G) Relative information flow of individual signaling pathways, indicating a global increase in multiple pathways in DN-derived fibroblasts. (H) Absolute information flow comparison across signaling pathways between ND and DN groups. (I) Dot plot illustrating DN-associated upregulation of extracellular matrix (ECM) signaling pathways. Each dot represents a ligand-receptor pair, with dot size indicating statistical significance (p < 0.01) and color intensity representing communication probability. |
To unravel the complex intercellular communication networks orchestrated by fibroblasts in the context of DN, we employed the sophisticated CellChat algorithm. This analysis enabled us to construct a detailed map of cell-cell interactions within the kidney, revealing a marked increase in both the quantity of ligand-receptor pairings and the overall intensity of communication channels mediated by fibroblasts in the DN cohort (Figure 2C and D).
Differential network analysis further pinpointed that fibroblast interactions with key renal cell types, including proximal tubular cells, endothelial cells, macrophages, and T cells, were significantly augmented in DN kidneys (Figure 2E and F). This heightened communication suggested a coordinated effort among these cells that could drive disease processes.
Pathway analysis focused on signaling cascades related to ECM remodeling-a critical aspect of renal fibrosis in DN-uncovered a pronounced activation of pathways such as ECM, collagen, FN1, and laminin (Figure 2G and H). These pathways are central to the synthesis, deposition, and remodeling of the ECM, processes that are dysregulated in fibrotic conditions. Moreover, a closer examination of the key ligand-receptor pairs within these ECM-related pathways revealed several with high interaction probabilities (Figure 2I). Notably, a more detailed interrogation of the upstream regulators of these dysregulated ECM pathways consistently pointed to the TGF-β signaling pathway as a top candidate master regulator.
TMSB10 Is Highly Expressed in Fibroblasts and Associated with Early Activation States
Differential expression analysis identified TMSB10 as one of the most significantly upregulated genes in DN-associated fibroblasts (Figure 3A). To confirm the transcriptomic findings, we performed quantitative PCR validation in kidney tissues from diabetic mice, which revealed consistent upregulation of Tmsb10 and other top-ranked DEGs compared with normal controls (Figure 3B). These results substantiate the scRNA-seq prediction and demonstrate conservation of the Tmsb10-associated expression pattern across human and murine DN models. Spatial mapping of TMSB10 expression showed that it was concentrated within specific fibroblast subpopulations (Figure 3C and D) and markedly elevated in DN compared to control samples (Figure 3E). Gene set enrichment analysis (GSEA) further indicated that TMSB10-high fibroblasts were enriched in profibrotic functional states, including “cell-matrix adhesion,” “extracellular matrix organization,” and “response to fibroblast growth factor stimulus” (Figure 3F). Collectively, these findings identify TMSB10 as an early activation marker in fibroblasts and link its expression to pathways driving fibrogenic remodeling in DN.
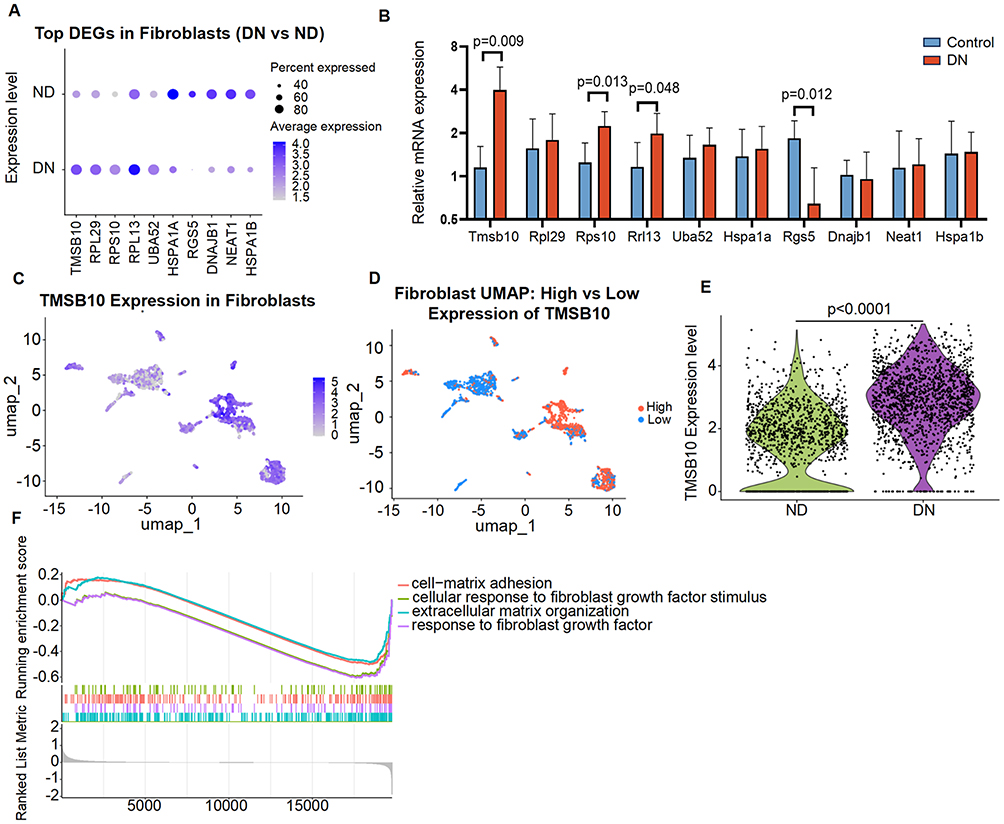 |
Figure 3 TMSB10 is highly expressed in fibroblasts from DN kidneys and associated with profibrotic functional states. (A) Dot plot showing top differentially expressed genes (DEGs) in fibroblasts between DN and ND groups, with TMSB10 ranked among the most upregulated genes in DN. (B) Quantitative validation of the top 10 DEGs identified in (A) in the kidneys of diabetic mice, showing consistent upregulation of Tmsb10 compared with normal controls. mRNA levels were measured by qPCR, normalized to Gapdh, and expressed as mean ± SEM (n = 6 per group). Statistical significance was determined using the unpaired two-tailed Student’s t-test, exact p values are shown within the panel. (C) UMAP plot showing spatial distribution of TMSB10 expression in fibroblasts, with higher expression observed in specific DN-associated clusters. (D) Fibroblast subclusters separated by high (red) vs low (blue) TMSB10 expression, indicating distinct spatial patterns within the fibroblast compartment. (E) Violin plot showing single-cell Tmsb10 expression levels in fibroblasts from ND and DN samples. Each dot represents an individual cell. Data are shown as distributions with median and interquartile range. Statistical significance was assessed using the Wilcoxon rank-sum test; the exact p value is indicated on the plot. (F) Gene set enrichment analysis (GSEA) revealed that fibroblasts with high TMSB10 expression were enriched in pathways related to extracellular matrix organization, fibroblast growth factor response, and cell-matrix adhesion. |
To further elucidate the functional dynamics of TMSB10-expressing fibroblasts, we performed pseudotime trajectory analysis using Monocle3, which delineated distinct differentiation paths and key branch points (Figure 4A and B). Integration of Seurat clusters into the trajectory revealed that clusters 1, 3, and 6 formed the principal developmental continuum. TMSB10 expression was highest at the early pseudotime stage and gradually decreased along the trajectory (Figure 4C and D), suggesting that TMSB10 marks an early, activation-prone fibroblast subset poised to initiate fibrogenic responses. In contrast, pro-fibrotic genes such as FN1 and COL3A1 were progressively upregulated toward the terminal states (Figure 4E and F), reflecting a transcriptional shift from a TMSB10-high “activated precursor” state to a matrix-producing effector phenotype. These findings imply that TMSB10-positive fibroblasts may occupy an upstream niche in the fibrotic hierarchy, acting as early initiators that subsequently give rise to mature myofibroblasts responsible for ECM accumulation. This temporal pattern provides biological insight into how TMSB10 contributes to the initiation and propagation of fibrogenic remodeling in diabetic nephropathy.
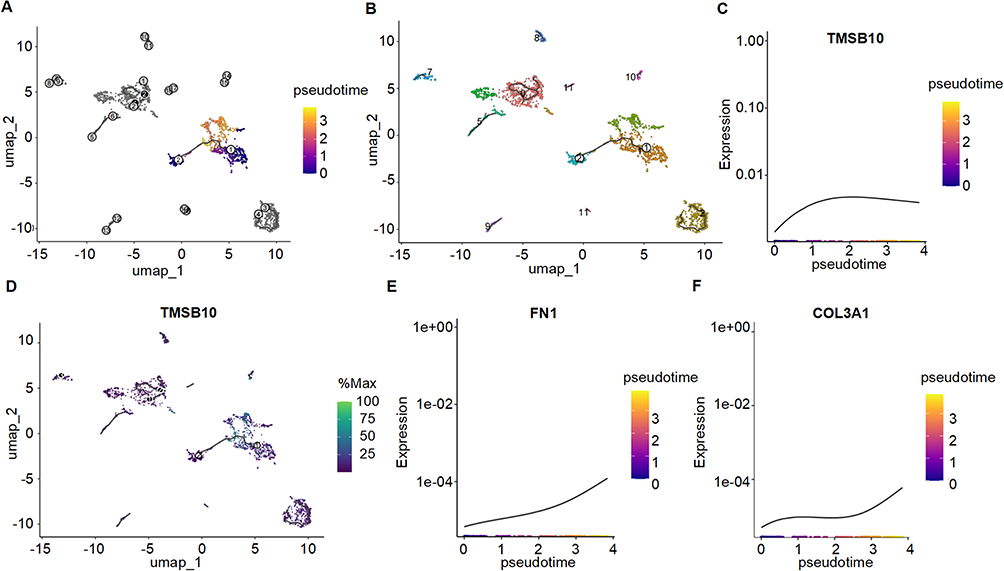 |
Figure 4 Pseudotime trajectory analysis reveals dynamic state transitions of fibroblast subpopulations and temporal regulation of TMSB10 and fibrosis-related genes. (A) Monocle3-inferred pseudotime trajectory of fibroblasts, with color gradient indicating progression from early (purple) to late (Orange) pseudotime states. (B) Same trajectory overlaid with Seurat cluster annotations, highlighting the composition of the main trajectory by clusters 1, 3, and 6. (C) Expression dynamics of TMSB10 along pseudotime, showing a gradual decline from early to late stages. (D) Spatial distribution of TMSB10 expression across the trajectory, with highest expression localized to the trajectory root. (E and F) Pseudotime expression patterns of fibrosis-associated genes FN1 (E) and COL3A1 (F), both showing progressive upregulation along the trajectory, indicating potential roles in fibroblast activation during disease progression. |
In vivo Silencing of Tmsb10 Alleviates Renal Fibrosis in a DN Mouse Model
Our scRNA-seq analysis identified TMSB10 as a fibroblast-enriched gene upregulated in DN. To directly test the pathogenic role of TMSB10 suggested by this discovery, we performed in vivo silencing in a DN mouse model. To substantiate the pathogenic involvement of TMSB10 in vivo, we first demonstrated that the in vivo application of shTMSB10 markedly suppressed Tmsb10 expression (Figure 5A), thereby validating the efficacy of our knockdown intervention in the DN mouse model. PCR analysis further revealed a significant upregulation of Fn1, Col1a1, and α-Sma in DN mice, whereas shTMSB10 treatment substantially attenuated their expression levels (Figure 5B–D).
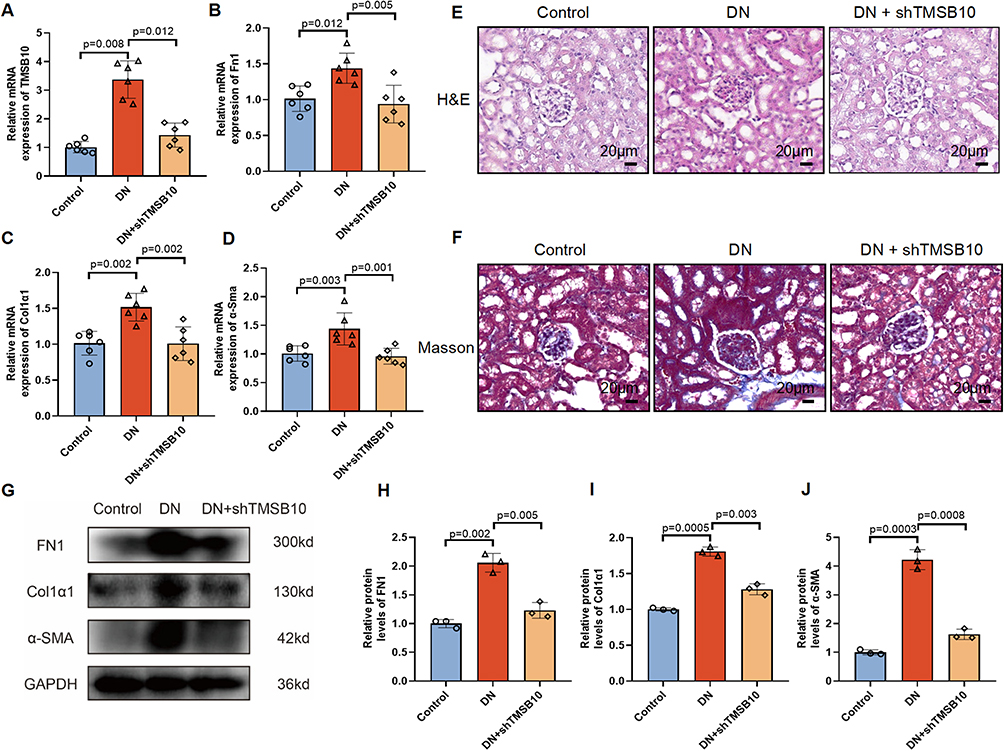 |
Figure 5 In vivo knockdown of Tmsb10 attenuates renal fibrosis in a diabetic nephropathy (DN) mouse model. (A–D) Quantitative RT-PCR analysis of Tmsb10 (A), Fn1 (B) Col1a1 (C) and α-Sma (D) mRNA levels in kidney tissues from control, DN, and DN + shTMSB10 groups. (E–F) Representative images of H&E (E) and Masson’s trichrome (F) staining of renal sections, showing that Tmsb10 knockdown attenuated glomerular and interstitial fibrosis in DN mice. Scale bars=20 µm. (G) Western blot analysis of fibrotic markers FN1, Col1a1, and α-Sma, with GAPDH as the loading control. (H) Quantification of FN1, (I) Quantification of Col1a1, and (J) Quantification of α-Sma protein expression levels from (G). Data are presented as mean ± SEM (n = 6 mice per group). Statistical significance was assessed using one-way ANOVA followed by Tukey’s post hoc test; p values are indicated in the panels. |
Histopathological examination, employing Hematoxylin and Eosin (H&E) as well as Masson’s trichrome staining, demonstrated that knockdown of TMSB10 alleviated both glomerular and interstitial abnormalities, along with collagen deposition characteristic of DN (Figure 5E and F). Western blot analysis corroborated these findings by showing a marked reduction in the protein levels of FN1, Col1a1, and α-Sma within the intervention group (Figure 5G–J). Furthermore, immunofluorescence staining elucidated that TMSB10, Col1a1, and α-Sma were highly expressed in the renal tissues of DN mice, and that shTMSB10 treatment significantly diminished their fluorescence intensity (Figure 6A–C), with statistical significance confirmed by quantitative analysis (Figure 6B, D and E). Collectively, these results underscore that TMSB10 knockdown effectively suppresses pivotal fibrotic mediators and mitigates renal structural impairment in DN.
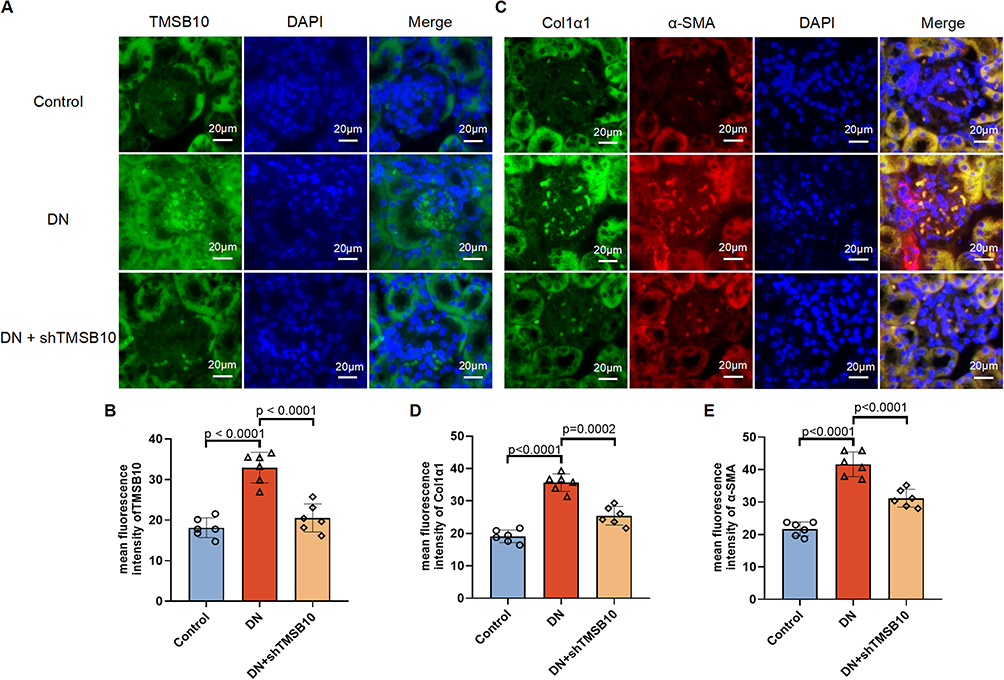 |
Figure 6 Immunofluorescence staining confirms that TMSB10 knockdown reduces renal expression of fibrotic markers in DN mice. (A) Representative immunofluorescence staining of kidney sections showing TMSB10 (green) and nuclei (DAPI, blue) in Control, DN, and DN + shTMSB10 groups. Scale bars=20 µm. (B) Quantification of mean fluorescence intensity of TMSB10 across groups. (C) Representative dual immunofluorescence staining for Col1a1 (green) and α-Sma (red), with DAPI nuclear counterstaining (blue). Scale bars=20 µm. (D–E) Quantification of mean fluorescence intensity of Col1a1 (D) and α-Sma (E). Data are presented as mean ± SEM (n = 6 mice per group). Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test; p values are indicated in the panels. |
TMSB10 Promotes High Glucose-Induced Fibroblast Activation via the TGF-β/SMAD Pathway
Building on our scRNA-seq analysis that implicated TGF-β signaling as a key upstream regulator of ECM dysregulation (Figure 2G–I), we hypothesized that TMSB10 might exert its pro-fibrotic effects through modulation of this central pathway. Furthermore, given the well-recognized role of the TGF-β/SMAD pathway in driving fibrotic processes, we hypothesized that TMSB10 might exert its pro-fibrotic effects through modulation of this signaling axis. In NIH-3T3 fibroblasts, high glucose stimulation, which mimics the hyperglycemic environment in DN, significantly upregulated the expression of Tmsb10 as well as fibrosis-related genes such as Fn1, Col1a1, and α-Sma (Figure 7A–D). Conversely, when we employed siTMSB10 to knock down Tmsb10 expression, the mRNA levels of these fibrotic genes were markedly suppressed (Figure 7A–D), suggesting that TMSB10 is crucial for high glucose-induced fibrotic gene expression in fibroblasts.
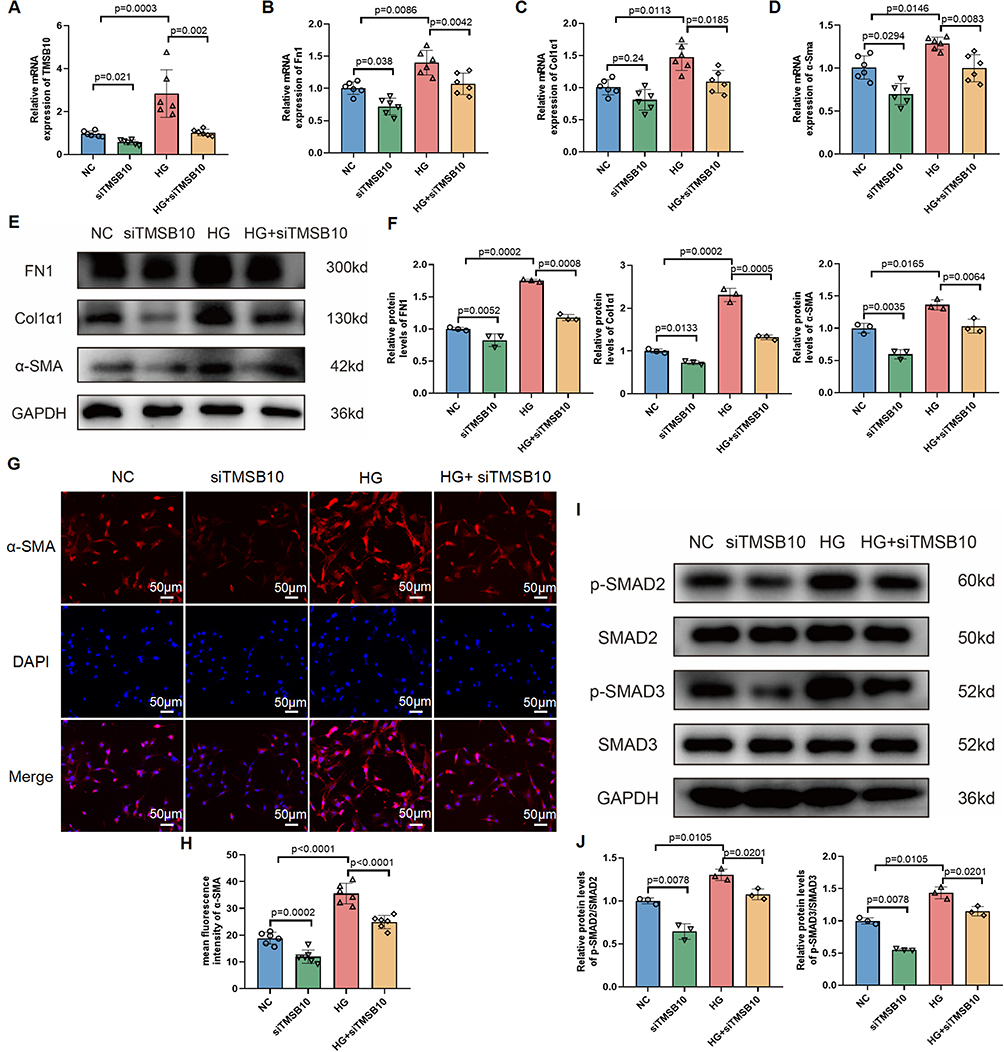 |
Figure 7 TMSB10 knockdown suppresses high glucose-induced fibroblast activation and fibrotic signaling via the TGF-β/SMAD pathway in NIH-3T3 cells. (A–D) Quantitative RT-PCR analysis of Tmsb10 (A), Fn1 (B), Col1a1 (C), and α-Sma (D) mRNA levels in NIH-3T3 fibroblasts under normal control (NC), siTMSB10, high glucose (HG), and HG + siTMSB10 conditions. Data are presented as mean ± SEM (n = 6 per group). Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test; p values are indicated in the panels. (E) Western blot analysis of FN1, Col1a1, and α-Sma protein levels, with GAPDH as loading control. (F) Quantification of protein expression shown in (E). Data are presented as mean ± SEM (n = 3 per group). Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test; p values are indicated in the panels. (G) Immunofluorescence staining of α-Sma (red) and nuclei (DAPI, blue) in each group. (H) Quantification of α-Sma fluorescence intensity. Scale bars = 50 µm. Data are presented as mean ± SEM (n = 6 per group). Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test; p values are indicated in the panels. (I) Western blot analysis of total and phosphorylated SMAD2 and SMAD3, showing that TMSB10 knockdown inhibits HG-induced SMAD activation. (J) Quantification of p-SMAD2 and p-SMAD3 protein levels. Data are presented as mean ± SEM (n = 3 per group). Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test; p values are indicated in the panels. |
To extend these findings to the protein level, we performed protein-level analysis and immunofluorescence staining. The results demonstrated that siTMSB10 effectively reduced the high glucose-induced upregulation of FN1, CoL1a1, and α-Sma proteins, as evidenced by the decreased protein expression and fluorescence signal intensity (Figure 7E–H). This indicates that TMSB10 knockdown can counteract the fibrotic effects of high glucose at both the transcriptional and translational levels.
Mechanistically, we explored the potential signaling pathways involved in TMSB10-mediated fibroblast activation and fibrogenesis. Western blot analysis revealed that high glucose conditions led to increased phosphorylation of SMAD2 and SMAD3, which are key downstream effectors of the TGF-β signaling pathway known to promote fibrosis. Importantly, Tmsb10 knockdown inhibited the activation of SMAD2 and SMAD3 (Figure 7I–J), suggesting that our hypothesis was correct-TMSB10 may promote fibroblast activation and fibrogenesis by modulating the TGF-β/SMAD signaling axis.
Discussion
Fibroblasts as Central Players in Diabetic Nephropathy Fibrosis
DN represents a major cause of ESKD, with renal fibrosis being a critical pathological feature that drives disease progression.5,13,34 Fibroblasts act as key effector cells orchestrating ECM accumulation and tissue scarring in DN. In DN, the kidney undergoes significant structural and functional alterations, with fibroblasts being the primary cells responsible for the excessive synthesis and deposition of ECM components, such as collagen, fibronectin, and laminin.35 This fibroblast-driven ECM accumulation progressively disrupts renal architecture, leading to glomerulosclerosis and tubulointerstitial fibrosis that culminate in renal dysfunction.13,35
Numerous studies in the literature have firmly established the TGF-β/SMAD signaling pathway as a central mediator in the process of tissue fibrosis across various organs and diseases.21,36,37 For instance, previous research has demonstrated that TGF-β is a potent pro-fibrotic cytokine that can stimulate the production and deposition of extracellular matrix components, such as collagen, fibronectin, and α-Sma, which are hallmark features of fibrotic tissues.38,39 Upon receptor binding, TGF-β triggers phosphorylation of SMAD2/3, which translocate into the nucleus to regulate fibrosis-related genes such as Fn1, Col1a1, and α-Sma.40–43 Our study extends this paradigm by demonstrating that fibroblast populations are markedly expanded and transcriptionally activated in DN kidneys, contributing to the fibrotic microenvironment through enhanced ECM secretion.
NIH-3T3 fibroblasts were employed as an in vitro model to explore fibrotic mechanisms in DN due to their well-characterized pro-fibrotic phenotype. However, as a 2D murine cell system, NIH-3T3 cells lack the complexity and species-specific context of human renal fibroblasts. Therefore, these findings should be interpreted cautiously and validated in patient-derived fibroblast cultures or 3D kidney organoids.
Summary of Key Findings
In this study, we employed a multi-faceted approach to investigate the role of TMSB10 in DN-related renal fibrosis. Through scRNA-seq analysis of renal tissues, we identified a significant upregulation of TMSB10 expression in fibroblasts from DN patients. Subsequent in vitro and in vivo studies demonstrated that silencing TMSB10 reduced fibroblast activation, ECM accumulation, and overall renal fibrotic remodeling. Mechanistically, Tmsb10 knockdown attenuated the TGF-β/SMAD pathway, revealing that TMSB10 positively regulates this canonical fibrotic signaling cascade. Collectively, these findings identify TMSB10 as a previously unrecognized pro-fibrotic driver in DN.
Comparison with Existing Literature
Our results are in line with previous studies that have highlighted the importance of fibroblasts and the TGF/SMAD signaling pathway in renal fibrosis. It is well-established that activated fibroblasts are the main source of ECM components in fibrotic tissues, and the TGF/SMAD pathway is a central regulator of fibroblast activation and ECM production.39,44,45 However, our data introduce TMSB10 as a novel upstream regulator modulating fibroblast activation and matrix production, extending current understanding of fibrotic control mechanisms in DN. While some studies have reported the involvement of TMSB10 in other biological processes such as cell migration,26–28,46–48 its role in DN-related fibrosis has not been previously described. This indicates that our research has uncovered a previously unrecognized mechanism contributing to renal fibrosis in DN.
Moreover, the integration of single-cell transcriptomics provided cell-type-specific resolution, enabling us to pinpoint fibroblast-restricted TMSB10 upregulation that bulk RNA analyses could not capture. Our scRNA-seq analysis revealed that TMSB10 upregulation is specifically associated with fibroblasts in DN patients, providing strong evidence for its role in fibroblast-mediated fibrosis.
Implications for Clinical Practice
Our findings offer potential translational implications but must be interpreted with caution. Targeting TMSB10 may provide a new therapeutic avenue to mitigate renal fibrosis in DN, but its feasibility and safety remain to be established. Pharmacologic inhibition of TMSB10, or promotion of its degradation may suppress fibroblast activation and ECM accumulation, offering a conceptual framework for anti-fibrotic therapy. Furthermore, scRNA-seq-guided patient stratification may help identify individuals with fibroblast TMSB10 overexpression who could benefit from targeted therapy, paving the way for personalized treatment approaches.
Limitations and Future Directions
Despite the significance of our findings, several limitations should be acknowledged. To begin with, our in vitro experiments relied on NIH-3T3 fibroblasts-a well-established yet simplified murine model that does not fully recapitulate the molecular and phenotypic complexity of human renal fibroblasts. Future investigations employing patient-derived primary renal fibroblasts or kidney organoids will be indispensable to substantiate and refine these observations. Furthermore, although our in vivo data were derived from a preclinical mouse model of DN, species-specific differences in renal physiology and fibrotic signaling may restrict direct translational extrapolation. Validation in higher-order mammalian models and human biopsy cohorts will be crucial for determining the clinical applicability of TMSB10-targeted interventions. At the mechanistic level, although our data implicate the TGF-β/SMAD signaling cascade as a downstream effector of TMSB10, the molecular interface connecting these pathways remains insufficiently defined. Future work should delineate the interacting partners, post-translational modifications, and upstream regulators that modulate TMSB10 stability, localization, and signaling output. In addition, the single-cohort scRNA-seq dataset employed in this study may not fully capture inter-patient heterogeneity. Integrating multi-omics datasets and expanding validation across larger cohorts would enhance the reproducibility and generalizability of our findings. Of translational significance, targeting TMSB10 represents a promising yet preliminary therapeutic avenue. Small-molecule modulators that induce TMSB10 degradation-such as urolithin A-should be systematically evaluated for selectivity, potency, and safety in diabetic models. Finally, beyond fibrotic regulation, probing the role of TMSB10 in inflammatory signaling and oxidative stress networks may uncover broader contributions to DN pathogenesis and progression.
Conclusions
In summary, our study provides a comprehensive single-cell transcriptomic landscape of DN, revealing pronounced alterations in fibroblasts and underscoring their central role in disease progression. This fibroblast enrichment, accompanied by altered intercellular communication networks, suggests that fibroblasts actively reshape the renal microenvironment, thereby amplifying fibrogenic signaling and contributing to the progressive decline in renal function. Integrating in vitro and in vivo evidence, we further demonstrate that TMSB10 acts as a novel regulator of fibroblast activation and extracellular matrix deposition, likely through modulation of the TGF-β/SMAD signaling pathway. The identification of TMSB10 as a pro-fibrotic driver expands our mechanistic understanding of DN and introduces a potential molecular target for therapeutic intervention. While these findings highlight the translational potential of TMSB10 inhibition, they should be interpreted with caution. Validation using patient-derived renal fibroblasts, larger clinical cohorts, and multi-omics approaches will be essential to confirm its clinical relevance and safety. Collectively, this work establishes a cellular and molecular framework for exploring fibroblast-directed strategies to mitigate renal fibrosis in diabetic nephropathy.
Data Sharing Statement
Data supporting the present study are available from the corresponding author upon reasonable request. All datasets used in this study are publicly accessible through the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/) (accession number: GSE209781) or via the original publications. Analysis scripts for quality control, normalization, and visualization are available upon request from the corresponding author.
Acknowledgments
This work was comprehensively supported by Suqian Sci&Tech Program (grant no. K202217 and K202326).
Author Contributions
Zihan Qin: Investigation; Formal Analysis; Data Curation.
Xiaoli Huang: Resources; Investigation; Data Curation.
Ke Du: Software; Formal Analysis.
Liexiang Zhang: Conceptualization; Supervision; Project Administration; Writing-Review & Editing.
Xiaohong Xu: Conceptualization; Supervision; Project Administration; Funding Acquisition.
Yuepeng Fang: Conceptualization; Methodology; Supervision; Writing-Original Draft; Writing-Review & Editing; Project Administration.
All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflict of interest.
References
1. Zoja C, Xinaris C, Macconi D. Diabetic nephropathy: novel molecular mechanisms and therapeutic targets. Front Pharmacol. 2020;11:586892. doi:10.3389/fphar.2020.586892
2. Pillai A, Fulmali D. A narrative review of new treatment options for diabetic nephropathy. Cureus. 2023;15(1):e33235. doi:10.7759/cureus.33235
3. Remuzzi A, Nangaku M. Another piece in the puzzle of kidney fibrosis. Kidney Int. 2023;103(6):1020–1023. doi:10.1016/j.kint.2023.03.028
4. Cao Y, Lin JH, Hammes HP, Zhang C. Cellular phenotypic transitions in diabetic nephropathy: an update. Front Pharmacol. 2022;13:1038073. doi:10.3389/fphar.2022.1038073
5. Du L, Liu S, Lu Y, et al. GABP promotes mesangial cell proliferation and renal fibrosis through GLI1 in diabetic nephropathy. Adv Sci. 2025;12(15):e2407462. doi:10.1002/advs.202407462
6. Yin R, Wang Y, Zhang P, et al. Promoting ubiquitin-dependent Drp1 degradation contributes to the protective effect of Astragalin against diabetic renal fibrosis. Biochem Pharmacol. 2025;241:117158. doi:10.1016/j.bcp.2025.117158
7. Saraswati S, Martínez P, Serrano R, et al. Renal fibroblasts are involved in fibrogenic changes in kidney fibrosis associated with dysfunctional telomeres. Exp Mol Med. 2024;56(10):2216–2230. doi:10.1038/s12276-024-01318-8
8. Sato Y, Yanagita M. Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflamm regen. 2017;37(1):17. doi:10.1186/s41232-017-0048-3
9. Wang Y, Wu Y, Xiang L, et al. Inhibition of the YAP-E2F2-FGF2 axis in renal tubular cells ameliorates renal fibrosis in chronic kidney disease. Sci China Life Sci. 2025. doi:10.1007/s11427-024-2880-1
10. DeLeon-Pennell KY, Barker TH, Lindsey ML. Fibroblasts: the arbiters of extracellular matrix remodeling. Matrix Biol. 2020;91-92:1–7. doi:10.1016/j.matbio.2020.05.006
11. Penke LR, Peters-Golden M. Molecular determinants of mesenchymal cell activation in fibroproliferative diseases. Cellular and molecular life sciences. CMLS. 2019;76(21):4179–4201. doi:10.1007/s00018-019-03212-3
12. Jain M, Sayyed AA, Gondaliya P, et al. Imidazopyrimidine-based pyruvate kinase M2 activator halts diabetic nephropathy progression via modulating epithelial-to-mesenchymal transition and fibrosis. Chem Biol Interact. 2025;420:111673. doi:10.1016/j.cbi.2025.111673
13. Ansari Z, Chaurasia A, Neha, Sharma N, Bachheti RK, Gupta PC, Gupta PC. Exploring inflammatory and fibrotic mechanisms driving diabetic nephropathy progression. Cytokine Growth Factor Rev. 2025;84:120–134. doi:10.1016/j.cytogfr.2025.05.007
14. Li J, Zhang J, Zhao X, Tian L. MSU crystallization promotes fibroblast proliferation and renal fibrosis in diabetic nephropathy via the ROS/SHP2/TGFβ pathway. Sci Rep. 2024;14(1):20251. doi:10.1038/s41598-024-67324-y
15. Li L, Tao M, Gao X, et al. Uncovering key markers and therapeutic targets for renal fibrosis in diabetic kidney disease through bulk and single-cell RNA sequencing. J Transl Med. 2025;23(1):742. doi:10.1186/s12967-025-06554-8
16. Kuppe C, Ibrahim MM, Kranz J, et al. Decoding myofibroblast origins in human kidney fibrosis. Nature. 2021;589(7841):281–286. doi:10.1038/s41586-020-2941-1
17. Gerrits T, Zandbergen M, Wolterbeek R, Bruijn JA, Baelde HJ, Scharpfenecker M. Endoglin promotes myofibroblast differentiation and extracellular matrix production in diabetic nephropathy. Int J Mol Sci. 2020;21(20):7713. doi:10.3390/ijms21207713
18. Gerrits T, Brouwer IJ, Dijkstra KL, et al. Endoglin is an important mediator in the final common pathway of chronic kidney disease to end-stage renal disease. Int J Mol Sci. 2022;24(1):646. doi:10.3390/ijms24010646
19. Sakuma H, Hagiwara S, Kantharidis P, Gohda T, Suzuki Y. Potential targeting of renal fibrosis in diabetic kidney disease using MicroRNAs. Front Pharmacol. 2020;11:587689. doi:10.3389/fphar.2020.587689
20. Feng L, Chen C, Xiong X, et al. PS-MPs promotes the progression of inflammation and fibrosis in diabetic nephropathy through NLRP3/Caspase-1 and TGF-β1/Smad2/3 signaling pathways. Ecotoxicol Environ Saf. 2024;273:116102. doi:10.1016/j.ecoenv.2024.116102
21. Jung SW, Kim SM, Kim A, Park SH, Moon JY, Lee SH. Midbody plays an active role in fibroblast-myofibroblast transition by mediating TGF-β signaling. FASEB J. 2022;36(5):e22272. doi:10.1096/fj.202101613R
22. Wang Y, Wang JY, Schnieke A, Fischer K. Advances in single-cell sequencing: insights from organ transplantation. Military Med Res. 2021;8(1):45. doi:10.1186/s40779-021-00336-1
23. Jovic D, Liang X, Zeng H, Lin L, Xu F, Luo Y. Single-cell RNA sequencing technologies and applications: a brief overview. Clin transl med. 2022;12(3):e694. doi:10.1002/ctm2.694
24. Tan W, Chen J, Wang Y, et al. Single-cell RNA sequencing in diabetic kidney disease: a literature review. Renal Failure. 2024;46(2):2387428. doi:10.1080/0886022X.2024.2387428
25. Yang J, Li S, Li Z, et al. Targeting YAP1-regulated glycolysis in fibroblast-like synoviocytes impairs macrophage infiltration to ameliorate diabetic osteoarthritis progression. Adv Sci. 2024;11(5):e2304617. doi:10.1002/advs.202304617
26. Xia H, Ning J, Guo X, Song H, Li X, Wang X. TMSB10 drives prostate cancer aggressiveness via immune microenvironment regulation. Molecular med. 2025;31(1):160. doi:10.1186/s10020-025-01211-8
27. Yang J, Yang X, Guo T, et al. Can thymosin beta 10 function both as a non-invasive biomarker and chemotherapeutic target in human colorectal cancer? Transl Oncol. 2024;46:102026. doi:10.1016/j.tranon.2024.102026
28. Wu Y, Yao M, Wu Z, Ma L, Liu C. A new prognostic model based on gamma-delta T cells for predicting the risk and aiding in the treatment of clear cell renal cell carcinoma. Discover Oncol. 2024;15(1):185. doi:10.1007/s12672-024-01057-2
29. Li Z, Li Y, Tian Y, Li N, Shen L, Zhao Y. Pan-cancer analysis identifies the correlations of Thymosin Beta 10 with predicting prognosis and immunotherapy response. Front Immunol. 2023;14:1170539. doi:10.3389/fimmu.2023.1170539
30. Xu X, Huang X, Zhang C, et al. METTL3 promotes podocyte pyroptosis in diabetic nephropathy through N(6)-methyladenosine modification of TRIM29 mRNA. Renal Failure. 2025;47(1):2497492. doi:10.1080/0886022X.2025.2497492
31. Sembach FE, Østergaard MV, Vrang N, et al. Rodent models of diabetic kidney disease: human translatability and preclinical validity. Drug Discovery Today. 2021;26(1):200–217. doi:10.1016/j.drudis.2020.05.004
32. Liu TT, Sun HF, Tang MZ, et al. Bicyclol attenuates pulmonary fibrosis with silicosis via both canonical and non-canonical TGF-β1 signaling pathways. J Transl Med. 2024;22(1):682. doi:10.1186/s12967-024-05399-x
33. Liu HL, Huang Z, Li QZ, et al. Schisandrin A alleviates renal fibrosis by inhibiting PKCβ and oxidative stress. Phytomedicine. 2024;126:155372. doi:10.1016/j.phymed.2024.155372
34. Giglio RV, Patti AM, Rizvi AA, et al. Advances in the pharmacological management of diabetic nephropathy: a 2022 international update. Biomedicines. 2023;11(2):291. doi:10.3390/biomedicines11020291
35. Qiu D, Song S, Chen N, et al. NQO1 alleviates renal fibrosis by inhibiting the TLR4/NF-κB and TGF-β/Smad signaling pathways in diabetic nephropathy. Cell. Signalling. 2023;108:110712. doi:10.1016/j.cellsig.2023.110712
36. Zhang Y, Ren L, Tian Y, Guo X, Wei F, Zhang Y. Signaling pathways that activate hepatic stellate cells during liver fibrosis. Front Med. 2024;11:1454980. doi:10.3389/fmed.2024.1454980
37. Jin Z, Jin Z, Liu Z, et al. Pantothenic acid ameliorates hepatic fibrosis by targeting IGFBP6 to regulate the TGF-β/SMADs pathway. Commun Biol. 2025;8(1):1127. doi:10.1038/s42003-025-08527-5
38. Ghafouri-Fard S, Askari A, Shoorei H, et al. Antioxidant therapy against TGF-β/SMAD pathway involved in organ fibrosis. J Cell & Mol Med. 2024;28(2):e18052. doi:10.1111/jcmm.18052
39. Lan HY. Transforming growth factor-β/Smad signalling in diabetic nephropathy. Clin Exp Pharmacol Physiol. 2012;39(8):731–738. doi:10.1111/j.1440-1681.2011.05663.x
40. Aragón E, Wang Q, Zou Y, et al. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-β signaling. Genes Dev. 2019;33(21–22):1506–1524. doi:10.1101/gad.330837.119
41. Yu XY, Sun Q, Zhang YM, Zou L, Zhao YY. TGF-β/Smad signaling pathway in tubulointerstitial fibrosis. Front Pharmacol. 2022;13:860588. doi:10.3389/fphar.2022.860588
42. Ye Z, Cheng M, Lian W, et al. GPX4 deficiency-induced ferroptosis drives endometrial epithelial fibrosis in polycystic ovary syndrome. Redox Biol. 2025;83:103615. doi:10.1016/j.redox.2025.103615
43. Li J, Zou Y, Kantapan J, Su H, Wang L, Dechsupa N. TGF‑β/Smad signaling in chronic kidney disease: exploring post‑translational regulatory perspectives (Review). Mol med rep. 2024;30(2). doi:10.3892/mmr.2024.13267
44. Chen JY, Yiu WH, Tang PM, Tang SC. New insights into fibrotic signaling in renal cell carcinoma. Front Cell Develop Biol. 2023;11:1056964. doi:10.3389/fcell.2023.1056964
45. Zhao X, Psarianos P, Ghoraie LS, et al. Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat Metab. 2019;1(1):147–157. doi:10.1038/s42255-018-0008-5
46. Li M, Cui H, Deng H, et al. Urolithin A promotes the degradation of TMSB10 to deformation F-actin in non-small-cell lung cancer. Phytomedicine. 2024;135:156109. doi:10.1016/j.phymed.2024.156109
47. Huang Q, Li F, Liu L, et al. Construction of EMT related prognostic signature for kidney renal clear cell carcinoma, through integrating bulk and single-cell gene expression profiles. Front Pharmacol. 2023;14:1302142. doi:10.3389/fphar.2023.1302142
48. Li W, Chen J, Xiang C, Long Y, Wu K, Li J. The clinical relevance and functional implications of thymosin beta-10 in glioma. Genetics Research. 2023;2023:5517445. doi:10.1155/2023/5517445
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.