Back to Journals » OncoTargets and Therapy » Volume 14
Single-Cell Sequencing, an Advanced Technology in Lung Cancer Research
Authors Wang H, Meng D, Guo H, Sun C, Chen P ![]() , Jiang M, Xu Y
, Jiang M, Xu Y ![]() , Yu J, Fang Q, Zhu J
, Yu J, Fang Q, Zhu J ![]() , Zhao W, Wu S
, Zhao W, Wu S ![]() , Zhao S, Li W, Chen B, Wang L, He Y
, Zhao S, Li W, Chen B, Wang L, He Y ![]()
Received 1 December 2020
Accepted for publication 16 February 2021
Published 16 March 2021 Volume 2021:14 Pages 1895—1909
DOI https://doi.org/10.2147/OTT.S295102
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Hao Wang,1,2 Die Meng,1,2 Haoyue Guo,1,2 Chenglong Sun,1,2 Peixin Chen,1,2 Minlin Jiang,1,2 Yi Xu,1,2 Jia Yu,1,2 Qiyu Fang,1,2 Jun Zhu,1,2 Wencheng Zhao,1,2 Shengyu Wu,1,2 Sha Zhao,1 Wei Li,1 Bin Chen,1 Lei Wang,1 Yayi He1
1Department of Medical Oncology, Shanghai Pulmonary Hospital, Tongji University Medical School Cancer Institute, Tongji University School of Medicine, Shanghai, 200433, People’s Republic of China; 2Tongji University, School of Medicine, Shanghai, 200433, People’s Republic of China
Correspondence: Yayi He
Department of Medical Oncology, Shanghai Pulmonary Hospital, Tongji University Medical School Cancer Institute, Tongji University School of Medicine, No 507 Zhengmin Road, Shanghai, 200433, People’s Republic of China
Tel +86 13818828623
Email [email protected]
Abstract: Single-cell sequencing (SCS) which has an unprecedentedly high resolution is an advanced technique for cancer research. Lung cancer still has a high mortality and morbidity. For further understanding the lung cancer, SCS is also been applied to lung cancer research to investigate its heterogeneity, metastasis, drug resistance, tumor microenvironment and many other issues. In this review, we summarized lung cancer research using SCS and their research achievements.
Keywords: single-cell sequencing, lung cancer, intratumoral heterogeneity, tumor microenvironment, lung cancer therapy
Introduction
As an individual cell is the most basic unit of life, single-cell molecular profiling can make us know more about life in different states. Single-cell sequencing (SCS) composed of single-cell isolation, nucleic acid amplification, sequencing and analysis (Figure 1) can conduct molecular profiling at single-cell resolution. Up to now, many techniques (Table 1) and bioinformatic methods have been developed. Through these techniques, researchers can detect single nucleotide variations (SNVs), copy number variations (CNVs), gene expression alteration, etc., at single-cell level.1
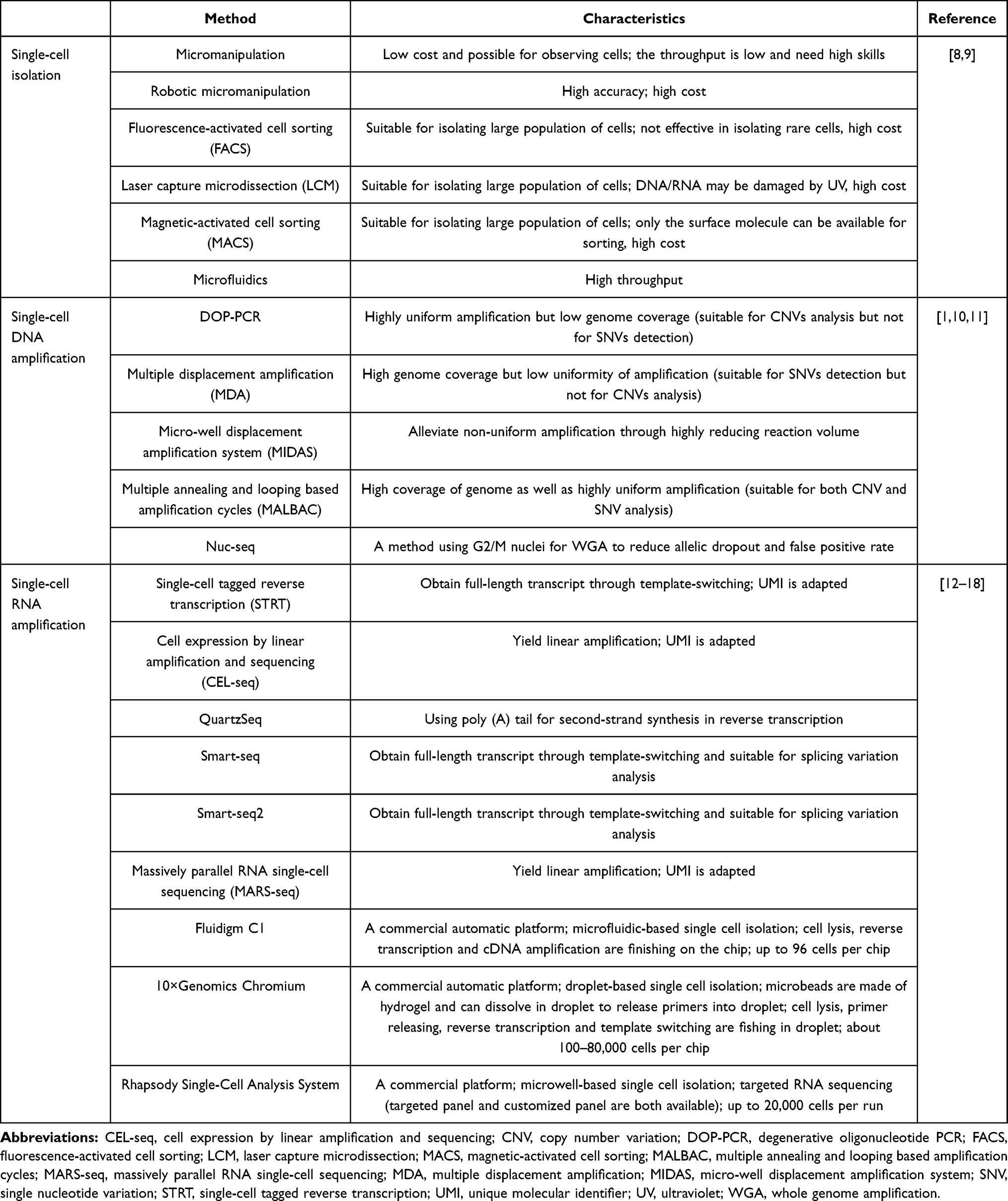 |
Table 1 Part of Methods Used in Single-Cell Sequencing |
 |
Figure 1 A brief schematic diagram of the process of single-cell sequencing. |
Tumor is still a non-negligible health issue. As a disease caused by genetic abnormalities, knowing more about genetic changes at different levels will give us a great insight into tumor and treatment strategies. We have got information about gene mutations that exist in tumor and found therapeutic targets through bulk sequencing. And some target therapies have worked effectively in cancer control.2 However, the material used in conventional bulk sequencing is a mixture of many different cells, and data obtained are average consequence. While intratumoral heterogeneity, a character of tumor, has been linked to drug resistance, and tumor showing higher invasiveness in pathology will have higher heterogeneity.3,4 Bulk sequencing cannot deal with intratumoral heterogeneity well, which will cause some rare information undetected. SCS can analyze the molecular profiling of individual cells which makes it possible to obtain more precise information of tumor.
Lung cancer is one of the tumors with high morbidity and mortality.5 Although at present many therapies including target therapies2 and immune therapies6 have shown a promising anti-tumor effect, many problems still require further exploration. Considering the high resolution of SCS, this method has been used in lung cancer research. This review focuses on this technique applied to lung cancer studies and their findings.
A Brief Overview of Single-Cell Sequencing
Sample acquirement is the first step for SCS. The sample can be surgically resected tissue, biopsy samples, tissue from mouse model and circulating tumor cells. Fresh tissue and rapidly frozen tissue are adaptive for SCS and the sample needs to >100 mg. Recently, researchers developed a technique that could use formalin-fixed paraffin-embedded (FFPE) samples to conduct whole-genome copy number profiling.7
After the acquirement of samples, single-cell isolation will be indispensable. At present, many single-cell isolation techniques have been developed, such as micromanipulation, robotic micromanipulation, fluorescence-activated cell sorting (FACS), laser capture microdissection (LCM), magnetic-activated cell sorting (MACS) and microfluidics.8 Micromanipulation is a useful method for isolating cultured cells. For FACS, MACS and microfluidics, samples need to be minced into approximately 1 mm3, digested by multiple enzymes and filtered to make cell suspension. Over 10,000 cells will be needed in suspension for FACS, which makes it unsuitable for isolating cells from suspension with a small population of cells.8 LCM does not need cell suspension preparation and it can isolate cells directly from tissue slide visually. A more detailed introduction about single-cell isolation techniques has been well reviewed in the literature.9
For genome analysis, DNA amplification is necessary because of scarce DNA in a single cell. Degenerative oligonucleotide PCR (DOP-PCR) can provide a highly uniform amplification but a low genome coverage, which makes it suitable for CNV analysis but not for SNV detection. Multiple displacement amplification (MDA), a method using the polymerase with strand displacement activity and high fidelity, can achieve high coverage of the genome, but it will yield non-uniform amplification. Thus, it can be used in SNV detection instead of CNV detection.1 Nuc-seq, a modified MDA protocol, uses flow-sorting to select G2/M nuclei for DNA amplification to reduce the false-positive rate and allelic dropout.10 Micro-well displacement amplification system (MIDAS) has an extremely small reaction volume than MDA through which the concentration of primer can be augmented and then eliminates non-uniform amplification.1 Multiple annealing and looping-based amplification cycles (MALBAC), which combines quasi-linear pre-amplification with strand displacement active polymerase, can achieve 93% genome coverage and the efficiency of SNV detection can be 76% which makes it suitable for both CNV and SNV detection. Meanwhile, its allelic drop rate is lower than 1% and the false-positive rate is less than 4×10−5.11 For latter quantification, DNA microarrays and next-generation sequencing (NGS) can be selected. Compared to the former method, NGS can detect all types of abnormalities.
For transcriptome analysis, many protocols have been developed, such as single-cell tagged reverse transcription (STRT), cell expression by linear amplification and sequencing (CEL-seq), CEL-seq2, QuartzSeq, droplet-based RNA-seq, Smart-seq, Smart-seq2 and massively parallel RNA single-cell sequencing (MARS-seq).12–15 Comparing to STRT, CEL-seq, which could achieve linear amplification, had higher sensitivity and lower technical noise.12 Droplet-based RNA-seq includes Drop-seq, inDrop and 10× Genomics Chromium. Among these systems, 10× Genomics Chromium has the highest sensitivity, accuracy and lowest technical noise, but its cost is also the highest. Thus, choosing 10× Genomics Chromium is safe for most applications but when the sample size is really large Drop-seq may be more cost-efficient, while if a custom protocol is wanted inDrop may be better.14 Different scRNA-seq methods have their own characteristics and can be preferable under specific circumstances. Drop-seq would be better when transcriptome of a large population of cells with low sequencing depth was needed. SCRB-seq and MARS-seq would be preferable when the sample size was small and Smart-seq2 would be preferable in analyzing splice variation.15 Nowadays, many commercial automatic platforms based on microfluidic, droplet or microwell have been established. Fluidigm C1, a representative platform based on microfluidic, isolates single cell with integrated fluidic circuits.16 One of its limitations was that the cell isolated might have a size bias because of its fixed size range of capture site for a certain chip.17 10× Genomics Chromium is a commercial platform based on droplet and has been discussed formerly. As for the microwell-based platform, Rhapsody Single-cell Analysis System has been commercial, and it is designed for targeted RNA sequencing, which makes it more sensitive for rare information.18 More platforms for SCS have been more detailed summarized.18
Uncovering the Heterogeneity of Lung Cancer Through SCS
Intratumoral heterogeneity has been found in many cancers. It correlates with clinical and pathological characteristics and is an important factor that drives drug resistance.3,4
Significant intratumoral heterogeneity also exists in lung cancer.3 High resolution of SCS makes it more effective to assess heterogeneity and detect scarce clone (Figure 2), thus SCS has been used in further research of this field (Table 2). In the mouse lung squamous cell carcinoma (LUSC) model, it was demonstrated different cancer cells harbor different non-silent somatic mutations of cancer genes.19 In the lung adenocarcinoma (LUAD) patient-derived xenograft (PDX) model, researchers found a heterogeneous expression pattern of cancer-specific SNVs including KRASG12D.20 Another study found heterogeneously expressed 64 genes among single cells from the LUAD PDX model and this heterogeneity was also detected in bulk sequencing. Patients with upregulated G64 expression would have a poor prognosis.21 But how different expression of G64 influences the biological behavior of individual cells still needs elucidate, which may be important for finding new therapeutic targets. Recently, Ma et al showed heterogeneous expression of antigen-presenting genes and neoantigens among individual cells,22 which gave us more information about tumor antigenicity and suggested that cancer vaccine targeted multiple neoantigens might have a better effect on cancer suppression. The alterations of driver genes were heterogeneous within non-small cell lung cancer (NSCLC) which might explain the incomplete response of target therapy.23 In small cell lung cancer (SCLC), CNV patterns of single cells from primary and different metastatic lesions also exhibit heterogeneity.24
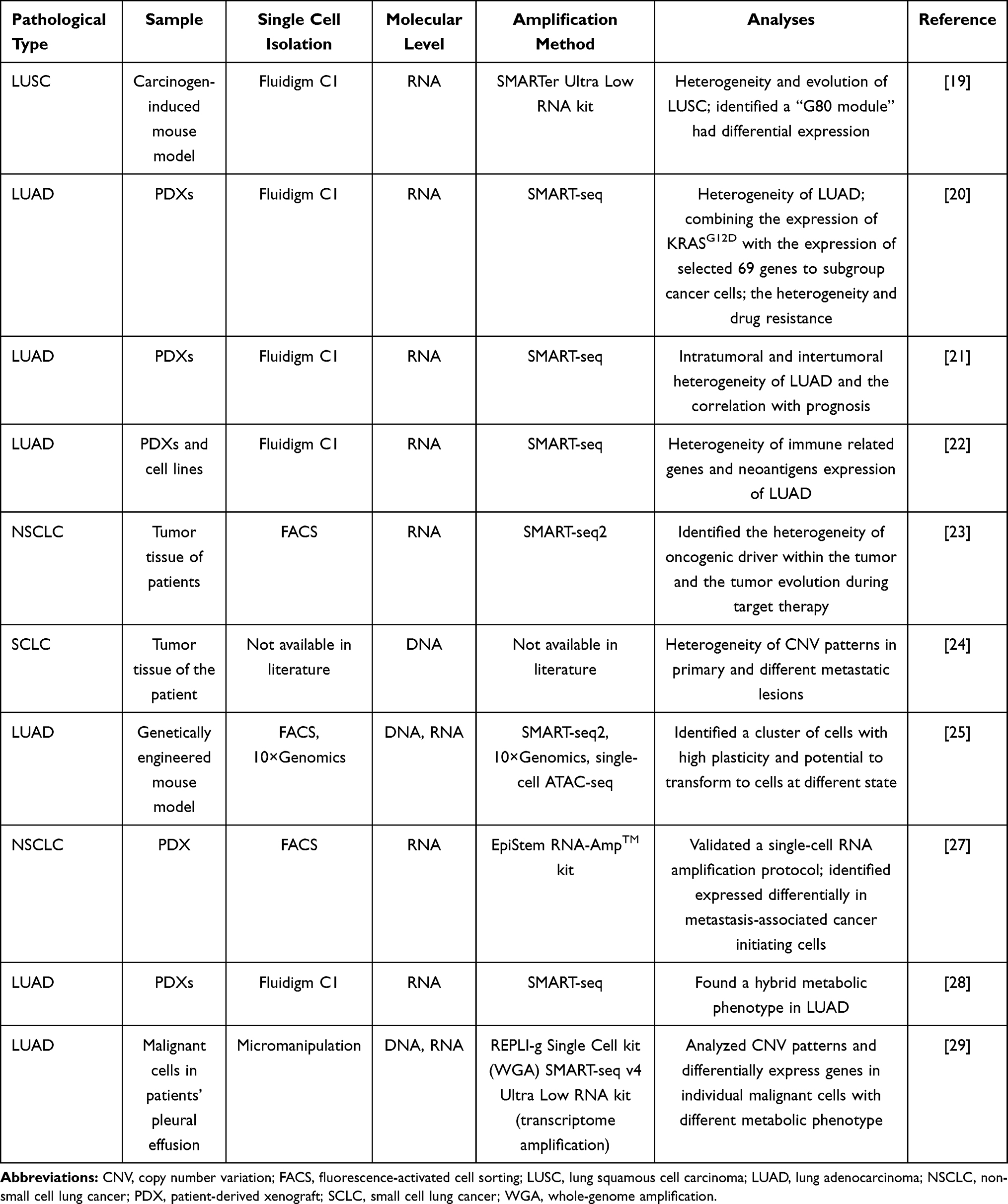 |
Table 2 SCS in Lung Cancer Heterogeneity, Metastasis and Metabolic |
 |
Figure 2 With the single-cell resolution of the technique, single-cell sequencing can assess heterogeneity better and be more sensitive for detecting rare mutations than conventional bulk sequencing. And these scarce mutations may be related to carcinogenesis, proliferative, and primary or acquired drug resistance. |
Investigation of Lung Cancer Evolution and Metastasis
Although significant heterogeneity of somatic mutation was detected among individual cells, Xiong et al also found all 28 single cells from one mouse LUSC model harbored the same mutation, IGFBP7R45P, which indicated these cells were from a common ancestor and it might be an important driver mutation.19 This study indicated IGFBP7 might be a new target for target therapy, but it still needed further investigation. In another study, high plastic cells with the potential of evolving to robust cell states were identified and marked by TIGIT. These cells might contribute to the heterogeneity of advanced LUAD.25 Metastasis, an important part of cancer progression, has not been fully understood. In SCLC, researchers found liver metastatic cells had a distinct CNV pattern, and the heterogeneity of CNV in the liver metastatic lesion was the lowest among all metastatic lesions.24 It suggested these cells tended to metastasize to the liver, but the mechanism was unknown yet. And further comprehensive studies should investigate whether the presence of the CNV pattern in tumor tissue can be an indicator to predict the tendency of liver metastasis in SCLC patients. It has been thought that a rare population of cells exists in cancer to initiate metastasis.26 However, these cells are scarce in tumor tissue. Taking advantage of high resolution, SCS could do well in analyzing these cells. Rothwell et al found metastasis-associated cancer-initiating cells in NSCLC PDX model and single-cell transcriptional profiling of these cells found increased expression of genes were related to ribosomal processing, cytoskeleton, glutathione transferase and stemness.27 This study demonstrated the existence of metastatic initiating cells and delineated the gene expression of these cells and their potential drug resistance.
We can also use SCS to get more information about lung cancer evolution including evolution during treatment. Through scRNA-seq, Maynard et al showed us the plasticity of the transcriptome during target therapy of NSCLC and transcriptional signature at different states of treatment. This gave insight into tumor changes during treatment and provided potentially targetable pathways to promote current therapy.23
SCS in Research on Lung Cancer Metabolism
Abnormal metabolism is a hallmark of cancer. SCS could be combined with other techniques to analyze cell metabolism and show molecular profiling in cells with different metabolic phenotypes (Table 2). Yu et al tested the expression of downstream targets of AMPK and HIF-1 in single cells through SCS, thus further demonstrated glycolysis and oxidative respiration could exist in a cancer cell simultaneously.28 In malignant pleural effusion (MPE), tumor cells exhibiting oxidative phosphorylation phenotype upregulated expression of genes enriched in E-Cadherin and integrin signaling. While cells with glycolytic phenotype upregulated expression of genes related to epithelial–mesenchymal transition (EMT) and they had higher expression of program cell death ligand. And patients with a larger proportion of glycolytic phenotypic cells in MPE would show poor prognosis.29
SCS in Investigating the Microenvironment of Lung Cancer
Tumor cells are surrounded by stroma, which has a cellular part and a non-cellular part. These compartments consist of a complex tumor microenvironment (TME) and interact with cancer cells.30 SCS can provide a more detailed profile of cancer TME (Figure 3), which will be powerful and necessary in lung cancer TME investigation (Table 3).
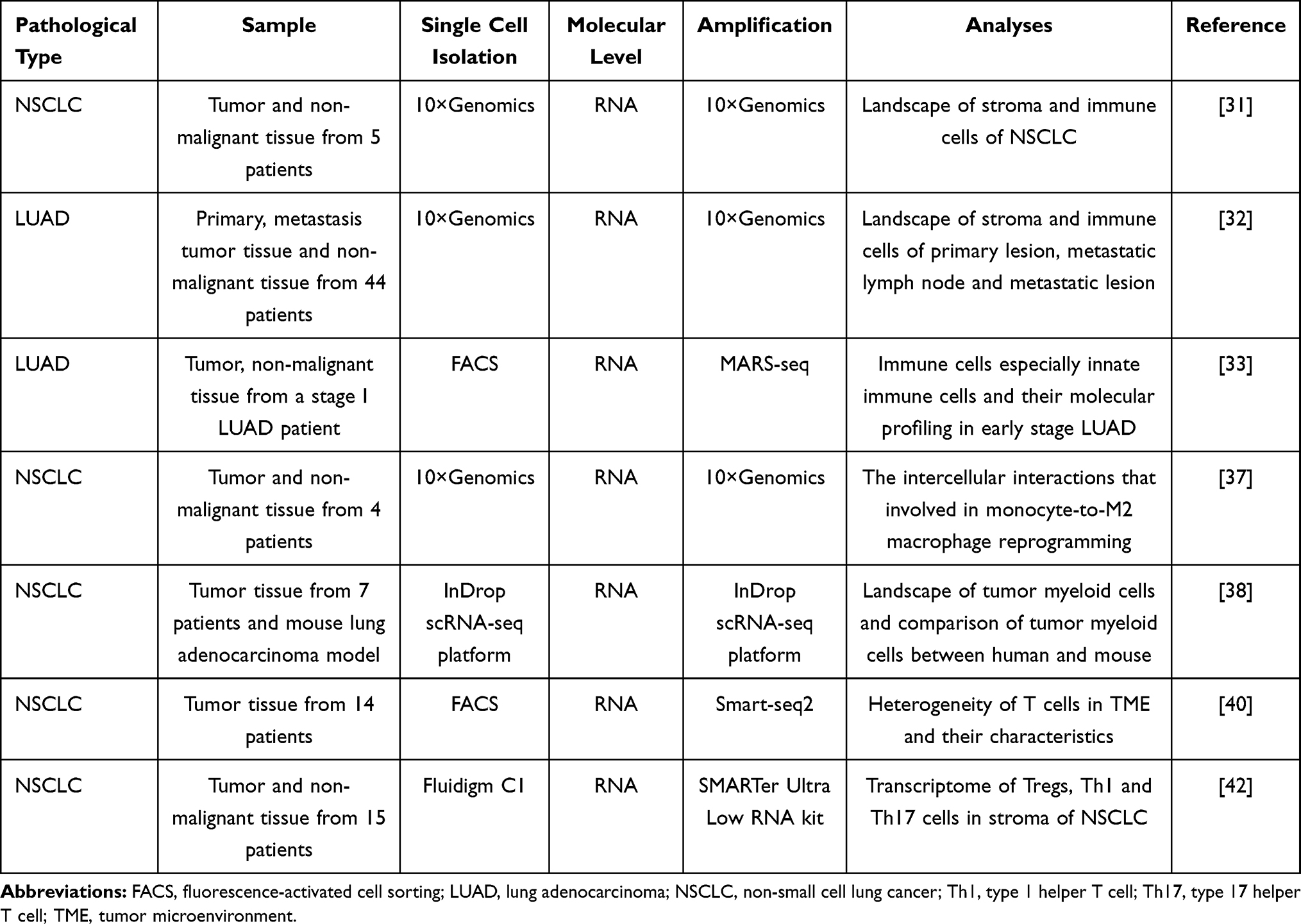 |
Table 3 SCS in Lung Cancer TME Analysis |
 |
Figure 3 Single-cell sequencing makes it accessible to analyze every single cell in the TME and it can delineate different cell types in the TME more minutely. It can even provide detailed information on subgroups of a group of cells in the TME. Abbreviation: Treg, regulatory T cell. |
Lambrechts et al found pathways related to nucleotide metabolism, oxidative phosphorylation and glycolysis were upregulated in tumor endothelial cells. However, genes involved in antigen presentation, chemotaxis and immune cell homing were downregulated.31 Downregulation of immune-related pathways was also confirmed in another study.32 These suggested tumor endothelial cells potentially gave tumor vessels the ability to prevent immune cell infiltration and it might be one reason for the generation of immune-desert tumor. More studies would be indispensable to investigate whether we could find methods to modulate these cells to enhance immune cell infiltration. Fibroblast is also an important part of TME which can influence cancer progression and prognosis of patients.30 Heterogeneity of fibroblasts also existed in NSCLC. Each subpopulation might have specialized functions in TME and myofibroblasts were found to replace fibroblasts in TME during tumor progression.31,32
Tumor-infiltrating immune cell is a compartment of TME and the progression of immune therapy depends on further understanding of these cells. Researchers had shown that not only in the advanced stage but also in the early stage LUAD lesion, nature killer (NK) cell infiltration was lower than that in normal tissue and the cytotoxic function of infiltrated NK cells was reduced,32,33 which indicated dysfunction of NK cells occurred at an early stage and reinvigorating NK cells might be a strategy for early LUAD treatment. Combining mass cytometry by time-of-flight (CyTOF) and scRNA-seq, researchers found that infiltration of CD14+ monocyte was comparable between tumor and normal tissue but monocyte expressed fewer proinflammatory cytokines in tumor site.33 Macrophages had two subsets and M2 macrophage secreted anti-inflammatory cytokines to facilitate tissue repair and sustain homeostasis. Tumor-associated macrophages (TAMs) showed M2 phenotype and limited tumor elimination.34 SCS showed that genes regulated by IRF2, IRF7, IRF9 and STAT2 transcription factors were upregulated while genes regulated by IRF8 and Fos/Jun transcription factors were downregulated in TAMs.31 Another research confirmed the higher level of PPAR-γ, triggering receptor expressed on myeloid cells-2 (TREM2) in TAMs than in macrophages from normal tissue.33 Recently, researchers demonstrated TREM2 as a vital molecule for the suppressive function of tumor-infiltrating myeloid cells, and combining a blockade of TREM2 with anti-PD-1 therapy could enhance treatment efficacy.35,36 The cellular and molecular interaction involved in reprogramming of monocyte to M2 macrophage has been revealed, which could give us more potential targets to modulate the TAM formation.37 Moreover, even in macrophage with M2 phenotype, the heterogeneity also exists.38 Whether these different subtypes of macrophages have distinct functions in TME still need further elucidation. Neutrophil is also a vital compartment in TME, and scRNA-seq has demonstrated that most neutrophils in TME of NSCLC would progress to pro-tumoral neutrophils gradually, but a distinct subtype with low abundance still maintained anti-tumor potential.38 Further investigations of how TME and tumor cells force neutrophils to become pro-tumoral and how the rare subtype is maintained are necessary, which will lead us to develop therapies to elicit neutrophils’ function and against the transformation of neutrophils. scRNA-seq also suggested the antigen presentation mediated by CD141+ dendritic cell (DC) was dysregulated in TME.33 Except for that, plasmacytoid DCs was identified in NSCLC tissues and might contribute to the formation of immunosuppressive TME.32,38 Researchers also pointed out that all innate immune changes shown in the advanced stage had presented in early-stage lesion.33 This finding suggests that dysfunction of innate immunity may play a critical role in tumor development and reinvigorate the innate immunity against tumor will be a potential way to block tumor formation and progression.
Adaptive immune cell, a non-negligible part of TME, can interact with tumor cells to impact their progression and they are major targets for immune therapies presently.6 But there still exist some problems, such as the exact mechanism of therapies, more appropriate markers for patient selection. SCS is an advanced technique to explain unsolved problems and find new immune therapies through uncovering the composition of tumor-infiltrating lymphocytes (TILs) and their molecular profiling more clearly. In hepatocellular carcinoma, significant infiltration of regulatory T cells (Tregs) in TME and new markers for exhausted CD8+T cells were found. LAYN encoded layilin, a cell surface protein, especially expressed on Tregs and exhausted CD8+T cells. It also had been confirmed Tregs with LAYN expression were those with higher repression and stability. Meanwhile, LAYN exclusively expressed on LAG-3− CD8+T cells which indicated LAYN might be a marker of a unique subset of CD8+T cells.39 Exhausted CD8+T cells with LAYN expression were also detected in NSCLC.40 It is necessary for analyzing the function of LAYN in TILs to investigate whether it could be a new target for NSCLC immune therapy. Guo et al showed the complexity of CD8+T cells in NSCLC through scRNA-seq. They found two clusters of pre-exhausted CD8+T cells correlating with the prognosis of patients.40 Another research also demonstrated that most CD8+T cells in TME had downregulated proliferation pathways. But two subpopulations of CD8+T cells had a highly proliferative and cytotoxic activity with high expression of immune checkpoints as well.31 It would be necessary to explore whether the proportion of these pre-exhausted cells could be an appropriate marker for selecting patients to use immune checkpoint blockades (ICBs). A study found some potential novel checkpoints, such as APOBEC3G which could regulate cytotoxic lymphocytes to adapt to hypoxia.31,41 In TME, Tregs will suppress anti-tumor immunity and they have been found enriched in tumor tissue.32 Moreover, researchers found the expression of 45 genes that were related to receptors of Tregs, signaling and enzyme activity, transcription factors, and cytokines was upregulated in Tregs that infiltrated in colorectal cancer and NSCLC. Some were also demonstrated at the protein level. And this expression pattern was consistent in primary and metastasis lesions.42 These upregulated molecules might be targets for immune therapy to prevent Tregs infiltrating or repress their immunosuppression activity. Recently, it was demonstrated that B cells also play an important role in anti-tumor immunity.43 scRNA-seq revealed that heterogeneous B cells were enriched in lung tumors. The proportion of different B cell subpopulation was different between tumor tissue and matched normal tissue.32 Meanwhile, the function of follicular B cells was also found to decrease in tumor.31 But the influence of this phenomenon is unclear. Although analysis of TME of SCLC was also intriguing, because of difficulty in sample acquisition, related research was limited. Analyzing the peripheral blood by SCS may be an alternative way to indirectly indicate the immune alterations in SCLC.
Although SCS is a powerful technique for analyzing TME, spatial information is sacrificed during sample preparation. When we are desire to obtain spatial information, other methods should be used. Monkman et al used Digital Spatial Profiling (DSP) system and found CD3, CD34 and ICOS expression in tumor regions were associated with better OS of NSCLC patients.44 Zugazagoitia et al used the same system and found CD4 and CD56 expression in the immune compartment were positively correlated with the efficacy of PD-1 blockade, while CD127 and VISTA expression in the tumor compartment was negatively correlated with the efficacy of PD-1 blockade.45 Liu et al used quantitative immunofluorescence and found macrophages were the predominant cells with PD-L1 expression in the stromal compartment instead of CD8+ or CD56+ cells. PD-L1+CD68+ macrophage was positively correlated with the outcome of PD-(L) 1 monotherapy.46 Recently, spatial transcriptome has been developed and it has been combined with SCS which dramatically improves the resolution of spatial analysis of TME of cancer.47 Combining spatial transcriptome and SCS can give us more precise information of individual cells at a different location in TME of lung cancer.
Research on Lung Cancer Treatment with SCS
Target therapy is an important and widely used treatment for lung cancer,2 but the drug resistance will be inevitable.48 SCS can analyze molecular characteristics of individual cells which will help explore drug resistance mechanisms and understand the evolution during therapy. In different cell lines, researchers found that compared to vandetanib sensitive cells, resistant cells had distinct expression pattern.49 Kim et al found that cells survived after docetaxel, phosphatidylinositide 3 kinase (PI3K) inhibitors or selumetinib treatment were cells with low-risk score, decreased total KRAS expression, downregulated RAS-MAPK pathway activation, downregulated cell cycle pathway and upregulated ion channel transport. And these cells could convert to high-risk score phenotype with the recovered signature of KRAS overexpression and MAKP pathway activation after withdrawing selumetinib.20 This research demonstrated cells in tumor had different sensitivity to therapies. The proportion of different cell groups may be a marker for evaluating the efficacy of the therapy for individual patient, and dynamic changes of the proportion may predict the occurrence of resistance. The distinct molecular phenotype might be used to infer mechanisms of drug resistance and find combination therapies. However, it was still not clear that drug-resistant cells existed at first or were generated during the evolution of cancer. Data obtained from an osimertinib resistant patient suggested that MET amplification might be a reason for osimertinib resistance, and the existence of a rare clone with KRASG12C would be a potential reason for osimertinib and c-MET inhibitor resistance.50 It also confirmed that SCS could be a powerful technique for detecting scarce clone. By applying scRNA-seq on samples during different states of target therapy, Maynard et al found that plasminogen activation pathway-related genes and some gap-junction genes were overexpressed in samples from progressive disease.23 This indicates that these may have a relationship with resistance to target therapy. To our limited knowledge, studies using SCS in finding novel resistant mechanisms of target therapies are still limited. As SCS has a high resolution, it will show us a more detailed mutation profile of patients and the evolution during therapies, which may give more basis for combination therapies and treatment after disease progression. And with the SCS technique progressing and more widely applying, more resistant mechanisms will be deciphered for precision medicine.
There are also some unsolved problems in immune therapy and SCS has been used in the relative fields. Yost et al found in basal or squamous cell carcinoma patients who had used anti-PD-1 agents, expanded clones were derived from novel clonotypes and pre-existing exhausted T cells did not proliferate or turn to non-exhausted phenotype.51 Miller et al found pre-exhausted CD8+T cells exhibited as Slamf6+Tim−. Compared to terminally exhausted T cells, they had different expression states and were maintained by different epigenetic states. Furthermore, these were the cells that reacted to anti-PD-1 therapy.52 Slamf6, a cell surface molecule, had the potential ability to count the pre-exhausted T cells in TME and be a biomarker for predicting patients’ response to anti-PD-1 therapy. Other studies found memory-precursor-like T cells instead of terminally exhausted T cells were the target for ICB. And Tcf1 would be important for maintaining this subpopulation and impact efficacy of ICB.53,54 In NSCLC, CXCR5+CD8+T cells with an intermediate expression of PD-1 and TIGIT had both memory-precursor features and cytotoxic potential.55 Thus, these cells might be the target of anti-PD-1 therapy. Thommen et al showed CD8+T cells with highly PD-1 expression in NSCLC had upregulated expression and secretion of chemoattractant CXCL13 that could bind to chemokine receptor CXCR5 to attract B cells, CD4+ follicular helper T cells which might be related to the generation of tertiary lymphoid structures and showed a predictive value for better response to anti-PD-1 therapy.56 Thus, considering CD8+T as a fully dysfunctional cell simply according to high PD-1 expression may be not precise. Another study also demonstrated this by identifying a new subset of tissue-resident memory T cells (TRM) characterized as high PD-1 and TIM-3 expression and cytotoxic feature simultaneously and these TRM cells enriched in patients who responded to PD-1 blockade.57 Changes of T cell in peripheral blood during anti-PD-1 therapy also have been detected through SCS. In NSCLC patients, Ki-67+PD-1+CD8+T cells would increase in peripheral blood after anti-PD-1 therapy and these cells were tumor-specific.58 Another study suggested that a reduced abundance of PD-1+CD4+T in peripheral blood might be a predictor of acquired resistance to anti-PD-1 therapy.59 These studies indicate that using SCS to investigate cellular and molecular changes of immune cells in peripheral blood during ICB treatment may also lead us to know mechanisms of drug resistance and monitor it. Moreover, as ICBs are administered systemically, knowing the systemic changes of immune cells may also make us understand and monitor the occurrence of immune-related adverse effects.
SCS for Studying Lung Cancer Circulating Tumor Cells (CTCs)
It had been confirmed patient-derived CTCs were tumorigenic, and tumor generated from CTCs would preserve most morphological and genetic characteristics as primary tumor.60 They were thought of being involved in metastasis and as a potential marker for drug responses.61,62 Pulmonary venous CTCs were also associated with disease-free survival of patients after surgery.63,64 SCS also has been applied to lung cancer CTC investigation (Table 4) in different aspects (Figure 4).
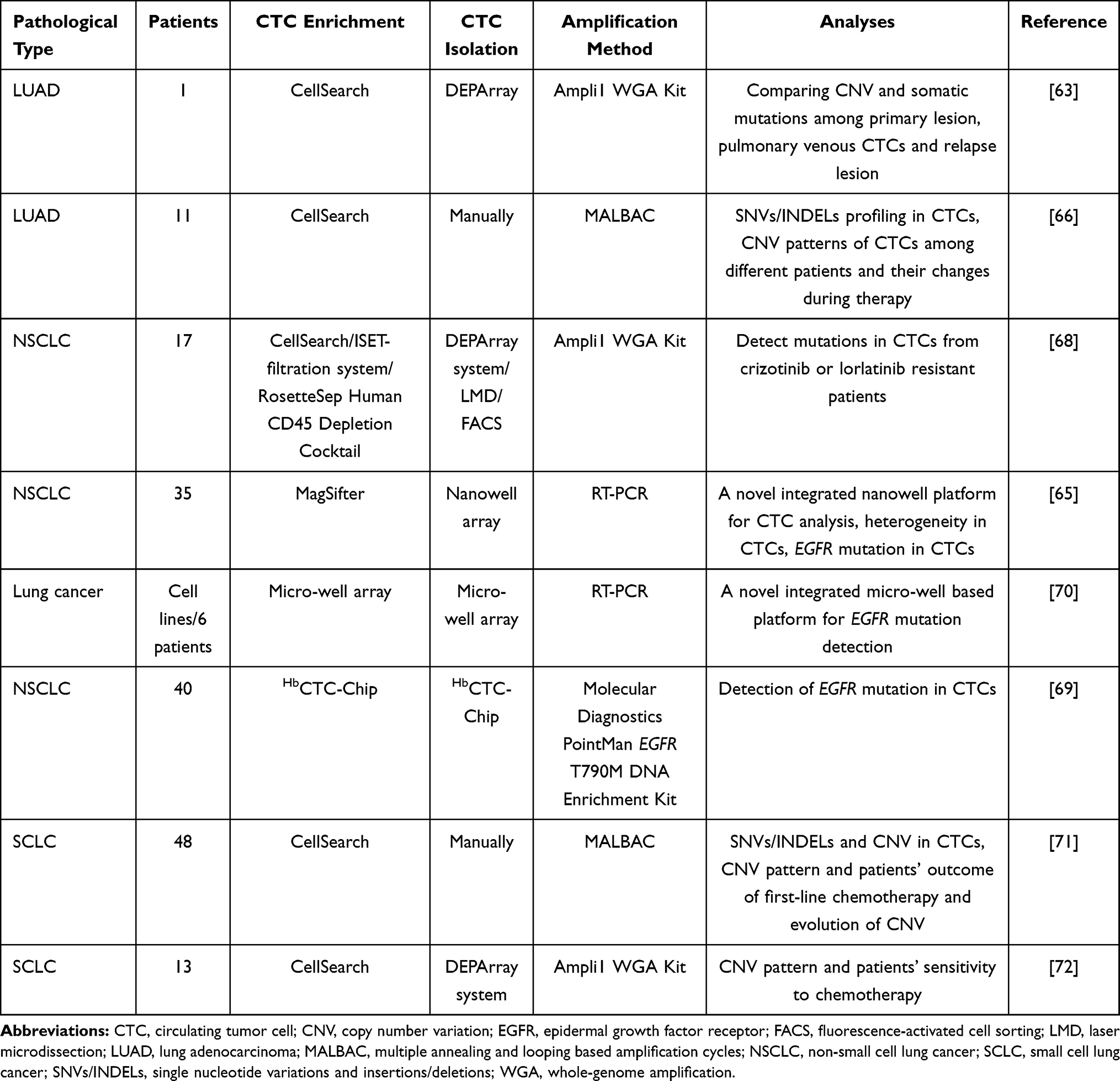 |
Table 4 Study on Lung Cancer Circulating Tumor Cells (CTCs) with SCS |
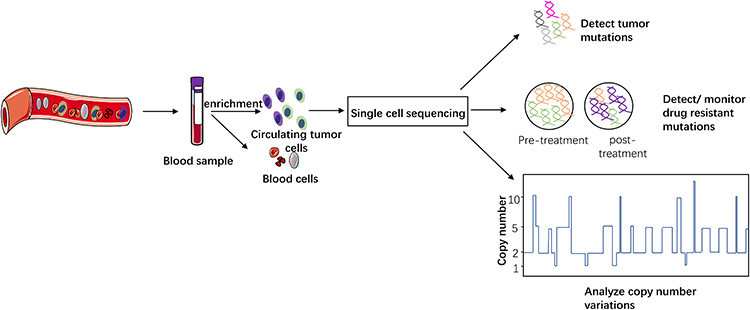 |
Figure 4 CTC has been used to detect tumor mutation, find or monitor resistant mutations and analyze the correlation between CNV pattern and clinical issues. With the development of this technology, it may become a less invasive approach for selecting proper patients, predicting outcome and monitoring drug resistance in clinical work. |
Park et al observed heterogeneous expression of vimentin and aldehyde dehydrogenase gene in CTCs from NSCLC patients and EGFR mutation was also detected.65 But the relationship between the heterogeneity and outcome of patients had not been elucidated. In a LUAD patient, most somatic mutations which had been related to cancer could be detected in primary/metastasis lesion and CTCs simultaneously. But phosphatidylinositol 3-kinase catalytic subunit α (PIK3CA), TP53 and RB1 mutations were only found in CTCs and liver metastatic lesion at which phenotypic transition from LUAD to SCLC occurred.66 Moreover, TP53 and RB1 mutations were correlated with the generation of SCLC.67 This result confirmed that CTCs could show the molecular characteristics of primary and metastatic lesions simultaneously. Phylogenetic analysis in another study suggested pulmonary venous CTCs and relapse lesion were close with each other but were distinct from the primary lesion. This finding further demonstrated the pre-existed CTCs before surgery was responsible for relapse.63 It also indicated we might use CTCs to partly represent the mutational characteristics of metastatic lesions when the sample obtained from the metastatic lesion was difficult. Moreover, finding out mutations only exist in CTCs and metastatic lesion might help us uncover mechanisms of metastasis. CNV patterns of CTCs were demonstrated to be reproducible and stable during treatment. And CTCs’ CNV patterns were similar among patients with the same pathological type, while would be different among patients with different pathological types.66 The formation of specific CNV patterns could be caused by intrinsic features of different pathological types and selection pressure in peripheral blood, but exact mechanisms remained unclear. Further investigation could lead us to understand how specific CNV patterns influence CTCs’ presence and survival. For clinical application, monitoring CNV patterns of CTCs might be a non-invasive method for detecting the pathological transition in LUAD patients. Drug resistance research also can be conducted in CTCs. In CTCs from crizotinib-resistant patients, mutations in downstream or parallel pathways which were defined as “off-target” mechanisms were detected. While in lorlatinib-resistant patients, “on-target” mechanisms instead of “off-target” mechanisms, such as ALKG1202R/F1147C and ALKG1202R/T1151M compound mutations, were detected in CTCs. And some mutations even had not been detected in bulk sequencing of single-site tumor biopsy sample.68 This study suggested that the resistant mechanisms of different ALK inhibitors were not the same. Through single CTCs sequencing, we would dig out information that was missed in bulk sequencing and find treatment targets after progression. For detecting EGFR T790M mutation, single CTCs sequencing would achieve a 70% detection rate.69 Recently, micro-well array-based integrated platform had been established to detect EGFR mutation rapidly at a low cost.65,70 In an analysis of CTCs from SCLC patients, Su et al grouped 10 CNV locations as a classifier, and SCLC patients with a low score would have a better outcome from etoposide plus platinum chemotherapy. Highly reproducible CNV was also confirmed in this study but heterogeneity was observed in some certain regions.71 Another classifier composed of 16 CNV regions was constructed to distinguish chemosensitive and chemorefractory SCLC patients.72 These data indicated that CTCs had the potential ability to predict chemotherapy response and monitor the sensitivity during chemotherapy in a non-invasive approach. But we had to reduce interference from the heterogeneity of CTCs and large trials were indispensable. Meanwhile, further analysis in these CNV regions might find some precise mechanisms of drug resistance in SCLC and get strategies to enhance the efficacy of chemotherapy. Recently, researchers designed a workflow in which CellSearch®-enriched CTCs could be stored in glycerol at -20°C before isolating a single CTC and analyzing.73 Furthermore, methods independent of surface marker to enrich CTCs were also developed and a novel platform, Parsortix, had been demonstrated as a system to isolate CTCs and cell-free DNA simultaneously.74–76 These efforts will promote the application of CTCs in clinical as liquid biopsy.
Conclusions
SCS with unprecedented high resolution has been widely used in lung cancer research. It is a powerful method for heterogeneity and evolution studies. In TME analysis, this technique will give us extensive information on molecular profiling of stromal cells which is necessary for us to understand TME deeply, guide current therapies and explore future therapies. As it can detect rare clones and delineate the landscape of mutations and gene expression in therapy-resistant cells, this technique is useful in uncovering mechanisms for drug resistance and finding predictors for patients’ response to drugs. SCS also promotes CTCs research and its clinical application. After further investigation, single CTC sequencing may become a convenient and non-invasive approach for monitoring metastasis, selecting patients for specific therapies and monitoring drug resistance of patients in clinical work. However, limitations do exist, such as its high cost, losing the spatial information of cancer cells.77 Although it has limitations, according to our current knowledge, we still think SCS will develop and be more and more widely used in the future instead of being replaced by current technology.
Abbreviations
CEL-seq, cell expression by linear amplification and sequencing; CyTOF, mass cytometry by time-of-flight; CNVs, copy number variations; CTCs, circulating tumor cells; DC, dendritic cell; DOP-PCR, degenerative oligonucleotide PCR; EMT, epithelial–mesenchymal transition; FACS, fluorescence-activated cell sorting; FFPE, formalin-fixed paraffin-embedded; ICBs, immune checkpoint blockades; LCM, laser capture microdissection; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MACS, magnetic-activated cell sorting; MALBAC, multiple annealing and looping based amplification cycles; MARS-seq, massively parallel RNA single-cell sequencing; MDA, multiple displacement amplification; MIDAS, micro-well displacement amplification system; MPE, malignant pleural effusion; NGS, next-generation sequencing; NK, natural killer; NSCLC, non-small cell lung cancer; PDX, patient-derived xenograft; PI3K, phosphatidylinositide 3 kinase; PIK3CA, phosphatidylinositol 3-kinase catalytic subunit α; SCLC, small cell lung cancer; SCS, single-cell sequencing; SNVs, single nucleotide variations; STRT, single-cell tagged reverse transcription; TRM, tissue-resident memory T cells; Tregs, regulatory T cells; TAMs, tumor-associated macrophages; TILs, tumor infiltrating lymphocytes; TME, tumor microenvironment; TREM2, triggering receptor expressed on myeloid cells-2.
Consent for Publication
All authors are in agreement with the content of the manuscript and agree to the submission.
Author Contributions
Hao Wang is the first author. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was supported in part by a grant of Young Talents in Shanghai, National Natural Science Foundation of China (81802255), Young Talents in Shanghai (2019QNBJ), Scientific research project of Shanghai Pulmonary Hospital (fkcx1903), “Dream Tutor” Outstanding Young Talents Program (fkyq1901), Clinical Research Project of Shanghai Pulmonary Hospital (fk18005), Key Discipline in 2019 (oncology), Project of Shanghai Municipal Science and Technology Commission (Project of Municipal Science and Technology Commission), Scientific research project of Shanghai Pulmonary Hospital (fkcx1903), Shanghai Municipal Commission of Health and Family Planning (2017YQ050), Innovation Training Project of SITP of Tongji University, and Key Projects of Leading Talent (19411950300). Youth Project of Hospital Management Research Fund of Shanghai Hospital Association (Q1902037).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Grun D, van Oudenaarden A. Design and analysis of single-cell sequencing experiments. Cell. 2015;163(4):799–810. doi:10.1016/j.cell.2015.10.039
2. Hida T, Nokihara H, Kondo M, et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised Phase 3 trial. Lancet. 2017;390(10089):29–39. doi:10.1016/S0140-6736(17)30565-2
3. Andor N, Graham TA, Jansen M, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22(1):105–113. doi:10.1038/nm.3984
4. Saunders NA, Simpson F, Thompson EW, et al. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol Med. 2012;4(8):675–684. doi:10.1002/emmm.201101131
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
6. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643
7. Martelotto LG, Baslan T, Kendall J, et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nature Med. 2017;23(3):376–385. doi:10.1038/nm.4279
8. Hu P, Zhang W, Xin H, Deng G. Single cell isolation and analysis. Front Cell Dev Biol. 2016;4:116. doi:10.3389/fcell.2016.00116
9. Gross A, Schoendube J, Zimmermann S, Steeb M, Zengerle R, Koltay P. Technologies for single-cell isolation. Int J Mol Sci. 2015;16(8):16897–16919. doi:10.3390/ijms160816897
10. Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–160. doi:10.1038/nature13600
11. Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338(6114):1622–1626. doi:10.1126/science.1229164
12. Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2(3):666–673. doi:10.1016/j.celrep.2012.08.003
13. Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343(6172):776–779. doi:10.1126/science.1247651
14. Zhang X, Li T, Liu F, et al. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-Seq systems. Mol Cell. 2019;73(1):130–142.e135. doi:10.1016/j.molcel.2018.10.020
15. Ziegenhain C, Vieth B, Parekh S, et al. Comparative analysis of single-cell RNA sequencing methods. Mol Cell. 2017;65(4):631–643.e634. doi:10.1016/j.molcel.2017.01.023
16. Xin Y, Kim J, Ni M, et al. Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proc Natl Acad Sci U S A. 2016;113(12):3293–3298. doi:10.1073/pnas.1602306113
17. Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58(4):610–620. doi:10.1016/j.molcel.2015.04.005
18. Valihrach L, Androvic P, Kubista M. Platforms for single-cell collection and analysis. Int J Mol Sci. 2018;19:3. doi:10.3390/ijms19030807
19. Xiong D, Pan J, Yin Y, et al. Novel mutational landscapes and expression signatures of lung squamous cell carcinoma. Oncotarget. 2018;9(7):7424–7441. doi:10.18632/oncotarget.23716
20. Kim KT, Lee HW, Lee HO, et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015;16:127. doi:10.1186/s13059-015-0692-3
21. Min JW, Kim WJ, Han JA, et al. Identification of distinct tumor subpopulations in lung adenocarcinoma via single-cell RNA-seq. PLoS One. 2015;10(8):e0135817. doi:10.1371/journal.pone.0135817
22. Ma KY, Schonnesen AA, Brock A, et al. Single-cell RNA sequencing of lung adenocarcinoma reveals heterogeneity of immune response-related genes. JCI Insight. 2019;4:4. doi:10.1172/jci.insight.121387
23. Maynard A, McCoach CE, Rotow JK, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182(5):1232–1251.e1222. doi:10.1016/j.cell.2020.07.017
24. Ferronika P, van den Bos H, Taudt A, et al. Copy number alterations assessed at the single-cell level revealed mono- and polyclonal seeding patterns of distant metastasis in a small-cell lung cancer patient. Ann Oncol. 2017;28(7):1668–1670. doi:10.1093/annonc/mdx182
25. Marjanovic ND, Hofree M, Chan JE, et al. Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell. 2020;38(2):229–246.e213. doi:10.1016/j.ccell.2020.06.012
26. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. doi:10.1038/nrc2499
27. Rothwell DG, Li Y, Ayub M, et al. Evaluation and validation of a robust single cell RNA-amplification protocol through transcriptional profiling of enriched lung cancer initiating cells. BMC Genomics. 2014;15:1129. doi:10.1186/1471-2164-15-1129
28. Yu L, Lu M, Jia D, et al. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. 2017;77(7):1564–1574. doi:10.1158/0008-5472.CAN-16-2074
29. Li Z, Wang Z, Tang Y, et al. Liquid biopsy-based single-cell metabolic phenotyping of lung cancer patients for informative diagnostics. Nat Commun. 2019;10(1):3856. doi:10.1038/s41467-019-11808-3
30. Bremnes RM, Donnem T, Al-Saad S, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thoracic Oncol. 2011;6(1):209–217. doi:10.1097/JTO.0b013e3181f8a1bd
31. Lambrechts D, Wauters E, Boeckx B, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24(8):1277–1289. doi:10.1038/s41591-018-0096-5
32. Kim N, Kim HK, Lee K, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020;11(1):2285. doi:10.1038/s41467-020-16164-1
33. Lavin Y, Kobayashi S, Leader A, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169(4):750–765.e717. doi:10.1016/j.cell.2017.04.014
34. Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi:10.1002/jcp.26429
35. Katzenelenbogen Y, Sheban F, Yalin A, et al. Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell. 2020;182(4):872–885.e819. doi:10.1016/j.cell.2020.06.032
36. Molgora M, Esaulova E, Vermi W, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing Anti-PD-1 immunotherapy. Cell. 2020;182(4):886–900.e817. doi:10.1016/j.cell.2020.07.013
37. Song Q, Hawkins GA, Wudel L, et al. Dissecting intratumoral myeloid cell plasticity by single cell RNA-seq. Cancer Med. 2019;8(6):3072–3085. doi:10.1002/cam4.2113
38. Zilionis R, Engblom C, Pfirschke C, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50(5):1317–1334.e1310. doi:10.1016/j.immuni.2019.03.009
39. Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342–1356.e1316. doi:10.1016/j.cell.2017.05.035
40. Guo X, Zhang Y, Zheng L, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24(7):978–985. doi:10.1038/s41591-018-0045-3
41. Sharma S, Wang J, Alqassim E, et al. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol. 2019;20(1):37. doi:10.1186/s13059-019-1651-1
42. De Simone M, Arrigoni A, Rossetti G, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016;45(5):1135–1147. doi:10.1016/j.immuni.2016.10.021
43. Petitprez F, De Reynies A, Keung EZ, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):1–5. doi:10.1038/s41586-019-1906-8
44. Monkman J, Taheri T, Ebrahimi Warkiani M, et al. High-plex and high-throughput digital spatial profiling of Non-Small-Cell Lung Cancer (NSCLC). Cancers. 2020;12(12):35551.
45. Zugazagoitia J, Gupta S, Liu Y, et al. Biomarkers associated with beneficial PD-1 checkpoint blockade in Non-Small Cell Lung Cancer (NSCLC) identified using high-plex digital spatial profiling. Clin Cancer Res. 2020;26(16):4360–4368. doi:10.1158/1078-0432.CCR-20-0175
46. Liu Y, Zugazagoitia J, Ahmed FS, et al. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res. 2020;26(4):970–977. doi:10.1158/1078-0432.CCR-19-1040
47. Moncada R, Barkley D, Wagner F, et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol. 2020;38(3):333–342. doi:10.1038/s41587-019-0392-8
48. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17(11):637–658.
49. Suzuki A, Matsushima K, Makinoshima H, et al. Single-cell analysis of lung adenocarcinoma cell lines reveals diverse expression patterns of individual cells invoked by a molecular target drug treatment. Genome Biol. 2015;16:66. doi:10.1186/s13059-015-0636-y
50. Martinez-Marti A, Felip E, Matito J, et al. Dual MET and ERBB inhibition overcomes intratumor plasticity in osimertinib-resistant-advanced non-small-cell lung cancer (NSCLC). Ann Oncol. 2017;28(10):2451–2457. doi:10.1093/annonc/mdx396
51. Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25(8):1251–1259. doi:10.1038/s41591-019-0522-3
52. Miller BC, Sen DR, Al Abosy R, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–336. doi:10.1038/s41590-019-0312-6
53. Siddiqui I, Schaeuble K, Chennupati V, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50(1):195–211.e110. doi:10.1016/j.immuni.2018.12.021
54. Kurtulus S, Madi A, Escobar G, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(-)CD8(+) tumor-infiltrating T cells. Immunity. 2019;50(1):181–194.e186. doi:10.1016/j.immuni.2018.11.014
55. Brummelman J, Mazza EMC, Alvisi G, et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J Exp Med. 2018;215(10):2520–2535. doi:10.1084/jem.20180684
56. Thommen DS, Koelzer VH, Herzig P, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. 2018;24(7):994–1004. doi:10.1038/s41591-018-0057-z
57. Clarke J, Panwar B, Madrigal A, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med. 2019;216(9):2128–2149.
58. Kamphorst AO, Pillai RN, Yang S, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017;114(19):4993–4998. doi:10.1073/pnas.1705327114
59. Zhang F, Bai H, Gao R, et al. Dynamics of peripheral T cell clones during PD-1 blockade in non-small cell lung cancer. Cancer Immunol Immunother. 2020.
60. Hodgkinson CL, Morrow CJ, Li Y, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014;20(8):897–903. doi:10.1038/nm.3600
61. Tanaka F, Yoneda K, Kondo N, et al. Circulating tumor cell as a diagnostic marker in primary lung cancer. Clin Cancer Res. 2009;15(22):6980–6986. doi:10.1158/1078-0432.CCR-09-1095
62. Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C. Molecular analysis of circulating tumour cells-biology and biomarkers. Nat Rev Clin Oncol. 2014;11(3):129–144. doi:10.1038/nrclinonc.2013.253
63. Chemi F, Rothwell DG, McGranahan N, et al. Pulmonary venous circulating tumor cell dissemination before tumor resection and disease relapse. Nat Med. 2019;25(10):1534–1539. doi:10.1038/s41591-019-0593-1
64. Crosbie PA, Shah R, Krysiak P, et al. Circulating tumor cells detected in the tumor-draining pulmonary vein are associated with disease recurrence after surgical resection of NSCLC. J Thoracic Oncol. 2016;11(10):1793–1797. doi:10.1016/j.jtho.2016.06.017
65. Park SM, Wong DJ, Ooi CC, et al. Molecular profiling of single circulating tumor cells from lung cancer patients. Proc Natl Acad Sci U S A. 2016;113(52):E8379–e8386. doi:10.1073/pnas.1608461113
66. Ni X, Zhuo M, Su Z, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci U S A. 2013;110(52):21083–21088. doi:10.1073/pnas.1320659110
67. Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19(6):754–764. doi:10.1016/j.ccr.2011.04.019
68. Pailler E, Faugeroux V, Oulhen M, et al. Acquired resistance mutations to alk inhibitors identified by single circulating tumor cell sequencing in ALK-rearranged non–small-cell lung cancer. Clin Cancer Res. 2019;25:6671–6682. doi:10.1158/1078-0432.CCR-19-1176
69. Sundaresan TK, Sequist LV, Heymach JV, et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin Cancer Res. 2016;22(5):1103–1110. doi:10.1158/1078-0432.CCR-15-1031
70. Gao W, Zhang X, Yuan H, et al. EGFR point mutation detection of single circulating tumor cells for lung cancer using a micro-well array. Biosens Bioelectron. 2019;139:111326. doi:10.1016/j.bios.2019.111326
71. Su Z, Wang Z, Ni X, et al. Inferring the evolution and progression of small-cell lung cancer by single-cell sequencing of circulating tumor cells. Clin Cancer Res. 2019;25(16):5049–5060. doi:10.1158/1078-0432.CCR-18-3571
72. Carter L, Rothwell DG, Mesquita B, et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med. 2017;23(1):114–119. doi:10.1038/nm.4239
73. Mesquita B, Rothwell DG, Burt DJ, et al. Molecular analysis of single circulating tumour cells following long-term storage of clinical samples. Mol Oncol. 2017;11(12):1687–1697. doi:10.1002/1878-0261.12113
74. Kulasinghe A, Kapeleris J, Cooper C, Warkiani ME, O’Byrne K, Punyadeera C. Phenotypic characterization of circulating lung cancer cells for clinically actionable targets. Cancers. 2019;11(3):380. doi:10.3390/cancers11030380
75. Chudziak J, Burt DJ, Mohan S, et al. Clinical evaluation of a novel microfluidic device for epitope-independent enrichment of circulating tumour cells in patients with small cell lung cancer. Analyst. 2016;141(2):669–678. doi:10.1039/C5AN02156A
76. Zhou J, Kulasinghe A, Bogseth A, O’Byrne K, Punyadeera C, Papautsky I. Isolation of circulating tumor cells in non-small-cell-lung-cancer patients using a multi-flow microfluidic channel. Microsyst Nanoeng. 2019;5:8. doi:10.1038/s41378-019-0045-6
77. Ren X, Kang B, Zhang Z. Understanding tumor ecosystems by single-cell sequencing: promises and limitations. Genome Biol. 2018;19(1):211. doi:10.1186/s13059-018-1593-z
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.