Back to Journals » Vascular Health and Risk Management » Volume 17
Single-Cell RNA Sequencing (scRNA-seq) in Cardiac Tissue: Applications and Limitations
Authors Wang M ![]() , Gu M, Liu L, Liu Y
, Gu M, Liu L, Liu Y ![]() , Tian L
, Tian L ![]()
Received 8 May 2021
Accepted for publication 14 September 2021
Published 2 October 2021 Volume 2021:17 Pages 641—657
DOI https://doi.org/10.2147/VHRM.S288090
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Harry Struijker-Boudier
Mingqiang Wang,1 Mingxia Gu,2,3 Ling Liu,4 Yu Liu,1 Lei Tian1
1Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, CA, 94305, USA; 2Perinatal Institute, Division of Pulmonary Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, 45229, USA; 3Center for Stem Cell and Organoid Medicine, CuSTOM, Division of Developmental Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, 45229, USA; 4Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, CA, 94305, USA
Correspondence: Mingqiang Wang; Lei Tian
Stanford University School of Medicine, Stanford Cardiovascular Institute, 1291 Welch Road, Biomedical Innovations Building, Stanford, CA, 94305-5454, USA
Tel +1 650 723-5386
Fax +1 650 736-7925
Email [email protected]; [email protected]
Abstract: Cardiovascular diseases (CVDs) are a group of disorders of the blood vessels and heart, which are considered as the leading causes of death worldwide. The pathology of CVDs could be related to the functional abnormalities of multiple cell types in the heart. Single-cell RNA sequencing (scRNA-seq) technology is a powerful method for characterizing individual cells and elucidating the molecular mechanisms by providing a high resolution of transcriptomic changes at the single-cell level. Specifically, scRNA-seq has provided novel insights into CVDs by identifying rare cardiac cell types, inferring the trajectory tree, estimating RNA velocity, elucidating the cell–cell communication, and comparing healthy and pathological heart samples. In this review, we summarize the different scRNA-seq platforms and published single-cell datasets in the cardiovascular field, and describe the utilities and limitations of this technology. Lastly, we discuss the future perspective of the application of scRNA-seq technology into cardiovascular research.
Keywords: cardiovascular diseases, clustering, trajectory inference, RNA velocity, cell–cell communication, spatial genomics
Introduction
Cardiovascular diseases (CVDs) are the leading cause of death globally, taking an estimated 17.9 million (32.1%) lives in 2015, up from 12.3 million (25.8%) in 1990.1,2 CVDs are highly heterogeneous diseases involving a group of disorders of the heart and blood vessels, which include cardiomyopathy, hypertensive heart disease, heart failure, coronary artery disease, cerebrovascular disease, rheumatic heart disease and others.3 CVDs are complex in nature, stemming from molecular alternations at the genetic, epigenetic, transcriptomic, and even proteomic levels in various cardiac cell types.4,5 Accurate elucidation of cellular heterogeneities is necessary for decoding the pathogenic mechanisms of CVDs, identifying novel therapeutic targets, and developing effective treatment strategies.6
The profiling of cellular heterogeneity at the transcriptomic level in cardiac tissues has been considered as a promising direction for measuring the global transcriptional activity dynamics, which underlie the phenotypic diversity of multiple cardiac cell types.7,8 Over the years, next-generation sequencing (NGS) technologies have led to many discoveries in biomedical sciences, including the phenotypic consequences of molecular variation in cardiovascular research.9–11
Until recently, bulk RNA sequencing (RNA-seq) had been primarily used to profile the averaged gene expression from tissues that consist of various cell types.12 Bulk profiling hence ignores the stochasticity of gene expression in each cell type and indicates average values from the heterogeneous population of cells, which are affected by the relative cell-type abundance and the states of each cell type within a sequencing sample.13 Molecular differences at the transcriptional level between distinct sub-cell types are also missing. In order to measure the transcriptome of each cell, several high-throughput single-cell RNA-sequencing (scRNA-seq) technologies have been developed and commercialized (Table 1).14–24 scRNA-seq enables the characterization and identification of transcriptionally different subpopulations at the single-cell level. This approach has the potential to identify novel directions to develop therapeutic strategies.25
 |
Table 1 scRNA-Seq Sequencing Methods Comparison |
The number of scRNA-seq studies in cardiovascular research has rapidly increased in recent years. A recent search with the keyword “(scRNA-seq or single-cell transcript*)[TIAB] AND (heart or cardiac or cardio*)[TIAB] in NCBI’s PubMed database of scientific publications returned 1238 articles (Sept. 5, 2021), 257 of which were published in 2020 and 254 of which published in 2021, at the time this review was written (Figure 1). The application of scRNA-seq has transformed how we understand CVDs, with a growing recognition that cardiac cell populations are far more heterogeneous than previously expected, and that bulk population analysis is inadequate for fully characterizing the biological complexity of these various cell types (Table 2). In terms of computational processing, each particular scRNA-seq protocol, platform, and technology may require different pipelines of preprocessing of sequencing reads, quality control (QC), normalization, dimension reduction, clustering, and differentially expressed gene (DEGs) calling.26 Along with the development of various methodologies in single-cell capture, a paradigm has been occurring for the computational methodologies for the applications in biomedical research.27 In this review, we highlight the utilities of scRNA-seq in various analyses for cardiovascular research (Figure 2), including 1) unsupervised clustering of scRNA-seq data to identify cardiac cell types and states within both healthy and diseased conditions, 2) characterization of the dynamics of transcriptional states by trajectory inference, 3) prediction of the future transcriptional dynamic state by the estimation of RNA velocity, 4) inferring of cell–cell communication from the expression of genes encoding receptors and ligands, 5) single-cell integration of multiple datasets to identify rare cell types, 6) detecting genetic variants from scRNA-seq datasets, and 7) construction of the spatial genomic map of cardiac tissues. In the near future, advances in scRNA-seq research will provide further insights in better understanding the mechanisms of CVDs and in improving the diagnosis, treatment and prognosis of a broad range of CVDs.
 |
 |
 |
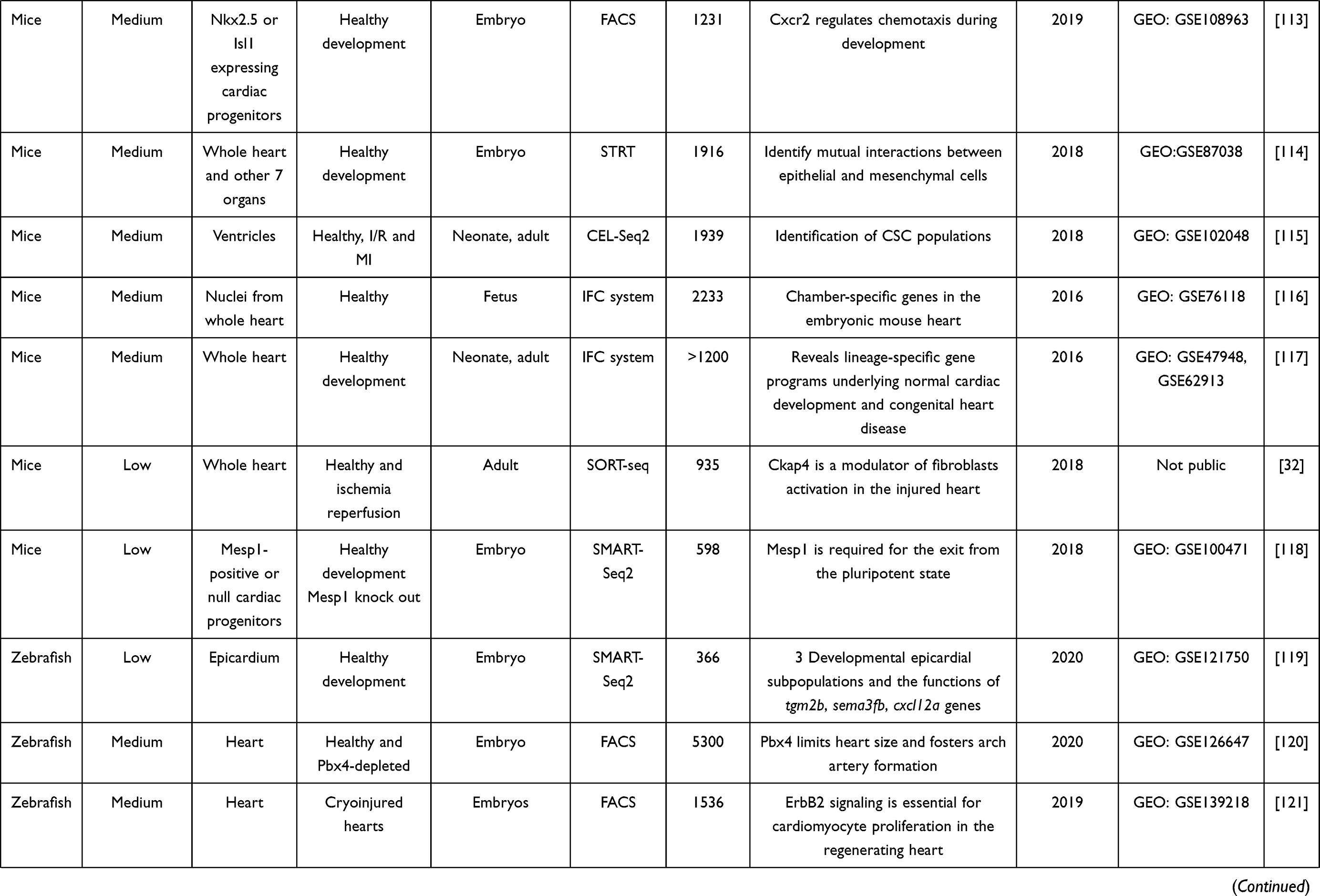 |
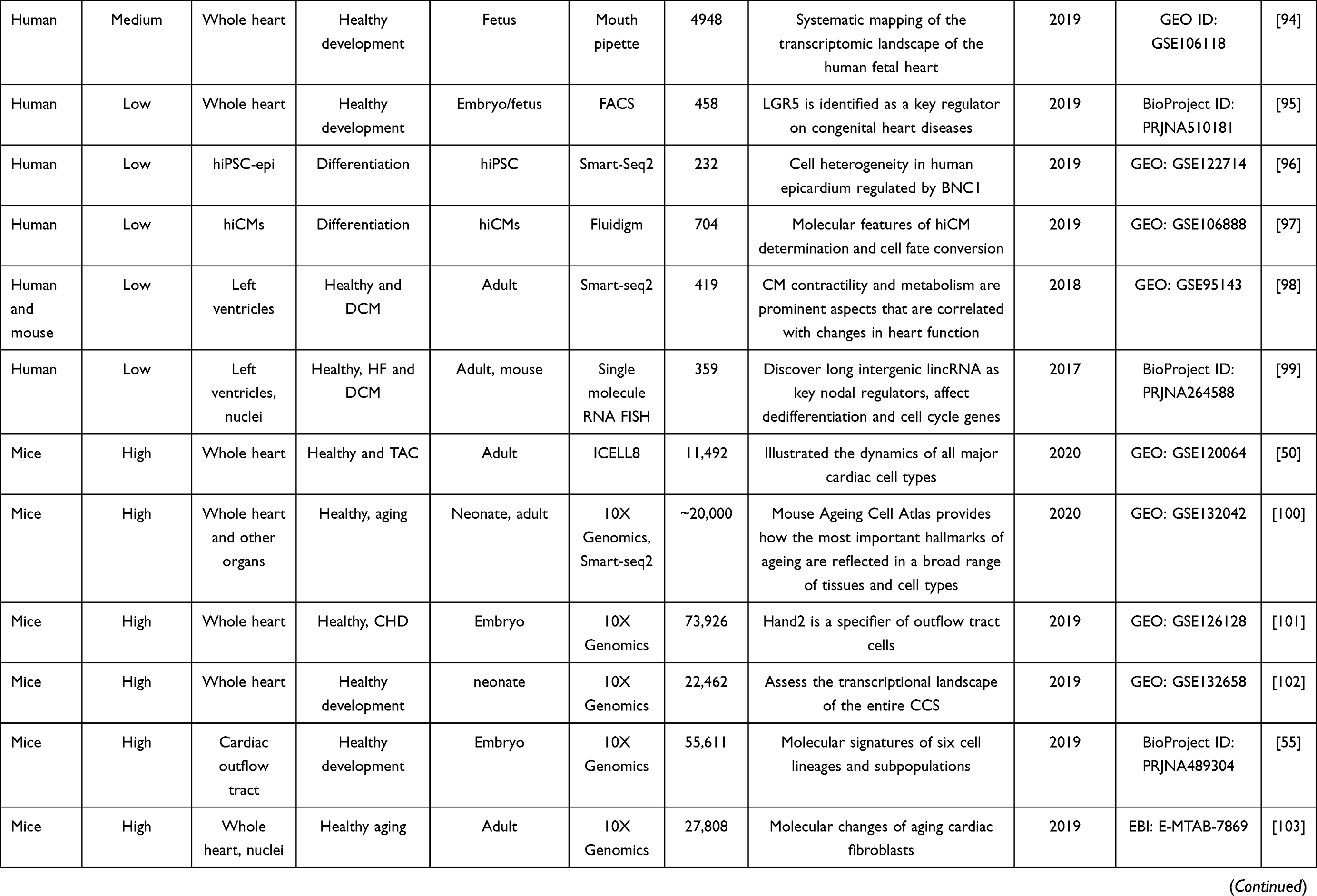
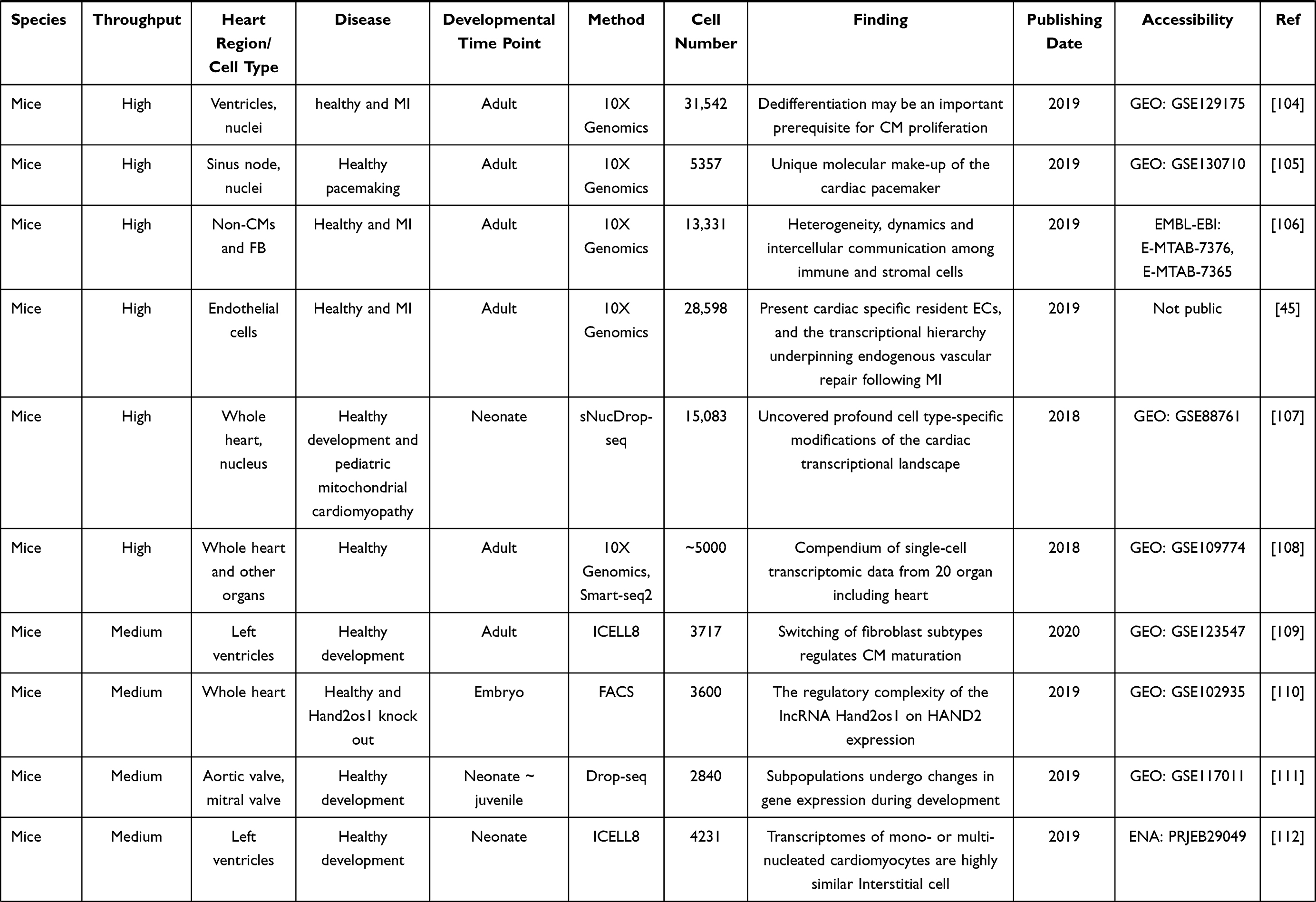
Table 2 Summary of Single-Cell RNA-Seq Studies on Cardiovascular Research |
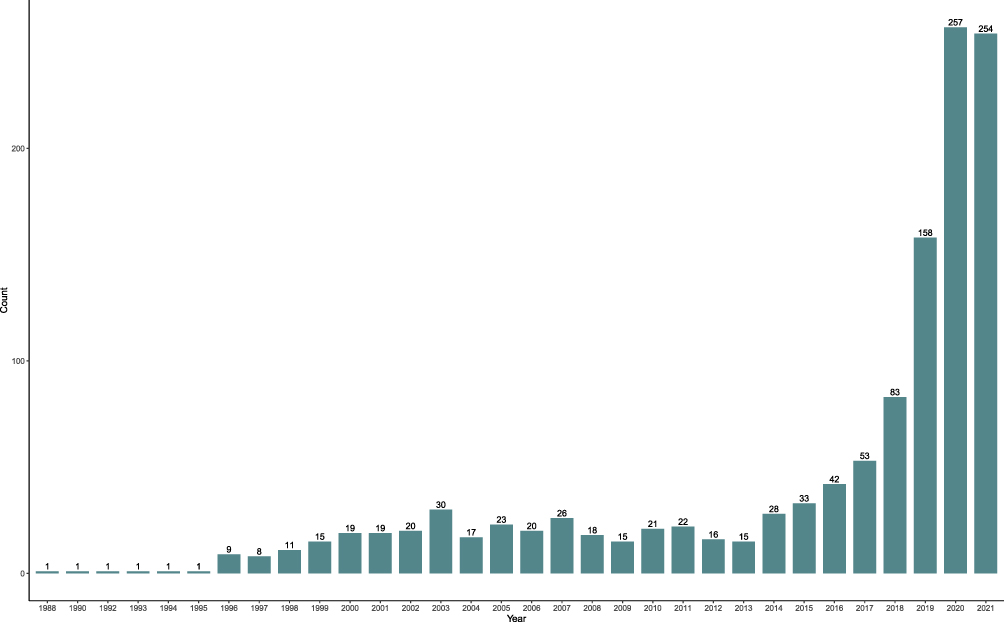 |
Figure 1 The number of papers published in the application of scRNA-seq in cardiovascular research in the past decades. Publications with the keyword “(scRNA-seq or single-cell transcript*)[TIAB] AND (heart or cardiac or cardio*)[TIAB]” in the NCBI PubMed database as of Aug 2021. Note the exponential growth in the number of published articles, in particular in the last 30 years. |
 |
Figure 2 Experimental workflow of single-cell RNA-seq. The general experimental workflow of single-cell RNA- study begins with sample preparation. Prepared cells are captured by various single-cell methods. Reverse transcription of single-cell RNA is performed, followed by PCR amplification and library preparation of the resulting cDNA. Next-generation sequencing is performed to generate the raw reads. |
The Workflow of scRNA-seq
A number of scRNA-seq techniques have been developed in the past decade. Different approaches of cell capture and transcript amplification result in differences in transcript length, target cell number, and read depth.28 Despite the differences, the scRNA-seq experimental techniques have a common workflow: sample preparation, dissociation, single-cell capture, cell lysis, reverse transcription (RT) and cDNA amplification, library preparation and RNA sequencing (Figure 2).29
Proper sample preparation is the key to generate high-quality single-cell transcriptome data. Considering the different properties of each cell type, the protocol should be optimized based on cell size, cell viability, and culture conditions.30 Single-cell suspensions are often achieved by a combination of enzymatic and physical dissociation. Subsequently, single cells are captured using different techniques including plate-based fluorescence-activated cell sorting (FACS) and droplet-based approaches. A broadly used droplet-based platform is the Chromium (10X Genomics) system, a microfluid device that allows rapid profiling of thousands of cells in droplets simultaneously. The Chromium system restricts the size of cells to be less than 30μm in diameter.31 Alternatively, plate-based FACS with a larger nozzle size (up to 130μm) can be used to capture cells of large size such as adult cardiomyocytes (CMs).32 In addition, single-nuclei RNA sequencing (snRNA-seq), a method for profiling gene expression in cells that are difficult to be dissociated, is an alternative to scRNA-seq to capture adult and large cardiomyocytes. To isolate RNA inside the nucleus, snRNA-seq uses a nuclear dissociation method that allows for minimization of technical issues.33
After individual cells are captured, they are lysed and processed for the first-strand cDNA synthesis by reverse transcription, followed by the second-strand synthesis and polymerase chain reaction (PCR) amplification. Single-cell system such as the Fluidigm C1 requires multiple PCR amplification, whereas most of the droplet-based techniques including the Chromium system allow pooled PCR using cell barcoding techniques, which significantly improves throughput.34 The sequencing libraries of cDNA fragments are then constructed and sequenced by high-throughput next-generation sequencers such as NextSeq 500. Sequencing libraries constructed with 3ʹ end enrichment is more cost-effective and produces less sequencing noise, whereas libraries retained full-length transcripts often obtain increased sequencing depth.35
Subsequently, raw data generated from sequencers are processed to obtain the gene expression matrix based on the unique molecular identifiers (UMIs). Raw data processing software such as the Cell Ranger (10X Genomics) is capable of performing QC, assignment of reads to the corresponding barcodes, demultiplexing, genome alignment, and read count quantification.21 The resulting count matrices, usually in the form of sparse matrices, are represented in dimensions with the row defined as the gene, and column defined as each cell. Reads assigned to unique barcodes, however, may not always originate from a single cell, as the barcodes may appear doublets, triplets, or may not tag any cells.36
The single-cell sparse matrix is used to normalize variance and to identify overdispersion genes. Principal component analysis (PCA) is applied to detect principal components (PCs) that capture the greatest variance among all cells. The resulting data is subjected to the graph-based Louvain clustering in high-dimensional PC space to identify cell clusters. Finally, these data are projected into 2D/3D space using dimensionality reduction methods such as t-distributed stochastic neighbor embedding (tSNE) or uniform manifold approximation and projection (UMAP) for visualization (Figure 3).36
 |
Figure 3 Application of scRNA-seq computational approach. Preprocessing steps convert the raw reads to sparse expression matrix. Downstream data analysis includes clustering, differentially expressed gene calling, cell trajectory analysis, RNA velocity, cell–cell interactions, identify mutations, integration (Reprinted from Cell, 177(7), Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. 1888-1902 e182, Copyright 2019, with permission from Elsevier)64 and spatial genomics. |
Comparisons of Different scRNA-seq Platforms
Among numerous single-cell platforms varied in captured cell number and read depth per cell, the plate-based Smart-seq2 method and droplet-based 10X Genomics Chromium approach are the two frequently used scRNA-seq platforms (Table 1). The plate-based Smart-seq2 platform has a high sensitivity for gene detection, especially for transcripts with low abundance. Depending on the method used for library construction and sequencing depth, the plate-based platform can simultaneously capture the full-length transcripts and reliably quantify more than 10,000 genes in each cell.37 The capacity of capturing full-length transcripts has the added advantage of facilitating the identification of splicing isoforms in single cells.38 This approach also allows for the profiling of more cell types with a wide range of cell size, permitting the analysis of large cells such as adult cardiomyocytes that is currently impossible to be profiled in droplet-based methods. However, reverse transcription performed in individual wells in the plate-based method prolongs the working process, limits throughput and increases noise in downstream steps.39
Droplet-based method such as the Chromium system from 10X Genomics enables 3′-end or 5′-end sequencing of single cells with higher throughput compared to the plate-based Smart-seq2 platform. Droplet-based methods encapsulate the single cells in oil droplets using DNA barcoding technology, substantially reducing the time and cost.40 Meanwhile, massive parallelization profiles up to 10,000 cells per sample for a given run.41 However, this method has a higher noise in accurately detecting the transcripts, especially for transcripts with low expression levels. The sensitivity of the protocols is expected to improve with continued protocol optimization and cost reductions. Despite the poly(A) enrichment, approximately 10–30% of all detected transcripts by both platforms are from non-coding genes, with lncRNA accounting for a higher proportion using the Chromium system.42
Applications of scRNA-seq in Cardiac Tissue
Unsupervised Clustering to Annotate Cardiac Cell Subtypes
scRNA-Seq can be used to annotate multiple cell types within the heart tissues based on the transcriptomic data of thousands of individual cells. In cardiovascular research, the clustered cell populations often include cardiomyocytes, fibroblasts, vascular smooth muscle cells, endothelial cells (ECs), epicardial adipocytes, immune cells and neural cells. Those identified cardiac cell clusters can also represent distinctive functional states in the different chambers such as ventricular and atrial chambers. Thus, performing unsupervised clustering to annotate cell populations represents cell types with biological relevance. Recently, scRNA-seq has been widely used to study heart development and disease. These studies utilized samples from various heart regions with or without disease, and identified multiple cardiac cell types.11,43–46 In addition, snRNA-seq also can assess the cellular and transcriptional diversity of the human heart. For example, Tucker et al sequenced the transcriptomes of 287,269 single cardiac nuclei, which have been clustered into a total of 9 major and 20 subclusters of cardiac cell types within the human heart.10 Cell types that were subclustered include two distinct groups of resident macrophages, four endothelial subtypes, and two fibroblasts subsets. They also identified strong enrichment for the role of cell subtypes in cardiac traits and diseases by using genetic association data.10 The newly defined subtypes transform our understanding of human heart and may pave the way for developing new therapeutics for CVDs.
Trajectory Inference Discovers Transition States in Heart Development
Trajectory inference is a computational technique used in single-cell transcriptomic analysis to determine the pattern of the dynamic cell transitional states based on the gene expression profiles of cells in varying states. It characterizes the expression pattern of the cells and places them along a pseudotime axis, which is a time-like variable demonstrating the relative position a cell takes in a lineage representing the evolution of the process rather than placing the cells in discrete clusters.47 By calculating a temporal dimension from the static scRNA-seq gene expression matrix, trajectory inference allows the probing of individual genes’ expression dynamics along with continuous cell-state changes. If the mean expression level of a gene can be changed along pseudotime, the gene is indicated as differentially expressed which can be crucial for the underlying cellular process that generated the pseudotime.47,48 Trajectory inference can thus illuminate the underlying biological processes by identifying key genes that play important roles in the development of particular lineages and genes differentially expressed between different lineages. Recently, Phansalkar et al applied the trajectory inference methods in developing human coronary arteries to illustrate coronary blood vessels from distinct origins can converge to equivalent states. The trajectory analysis result also suggested that artery ECs are formed by capillary ECs differentiation.49 Ren et al used trajectory inference to reconstruct the progression trajectory to reveal intervention principles in pathological cardiac hypertrophy. The trajectory analysis also showed that activation of proinflammatory macrophages was a key event for the transition from normal to reduced ejection fraction.50 Zhang et al used trajectory inference to illuminate the cell fate decisions and developmental origins of organ-specific cell types such as endothelial, muscle and cranial pharyngeal cell types in mesodermal progenitor cells. And they also uncovered intraembryonic progenitor from the lateral plate mesoderm (LPM) and cardiac progenitor from the late extraembryonic mesoderm can contribute to the development of cardiomyocytes.51 Collectively, these studies have utilized trajectory inference tools to better understand cellular transitions and intercellular communication in the early stages of human cardiac development.
RNA Velocity Predicts the Future Transcriptional Dynamic State
RNA velocity, a high-dimensional vector estimated by the ratio of unspliced and spliced mRNA reads in scRNA-seq data, is defined as the time derivative of the gene expression state.52 RNA velocity predicts the future transcriptional dynamic state of individual cells on a timescale and enables the identification of novel cell states in a systematic and quantitative manner. It has greatly aided the analysis of developmental lineages and cellular dynamics in the human heart.53 Recently, Wolfien et al utilized RNA velocity analysis to study the transcription kinetics and to visualize the dynamics of the transitions between mature and nascent cellular states of the cell types in the mammalian heart.54 They found that different subgroups of mammalian cardiomyocytes have distinct marker profiles, especially for the profile of RNA velocity in cardiomyocytes. Meanwhile, via RNA velocity analysis, they identified a cell population that expressed the canonical endothelial markers that are also associated with cardiac contractile function. Thus, the RNA velocity results generated in the mammalian hearts support the hypothesis that this population is in a trans-differentiation process from an ECs-like phenotype towards a cardiomyocyte-like phenotype. In addition, Liu et al used RNA velocity analysis to identify the convergent development of the vascular smooth muscle cell (vSMC) lineage and to infer the direction and rate of the changes in vSMC state changes during heart development.55 They found that the convergent development of vSMC lineage cell is involved in mesenchymal-to-vSMC transition or myocardial-to-vSMC transdifferentiation. Taken together, RNA velocity tools have paved new ways of studying heart development using scRNA-seq.
Identification of Unique Ligand–Receptor Interactions During Cardiac Cell–Cell Communication
Cell–cell communication is the essence of complex multicellular behaviors, of which cells communicate with one another via the binding of ligands and receptors that regulate cellular function, structure, and maintenance. The complex network of cell–cell communications among various cell types in the heart is essential to maintain the regular heart, whose disruption can lead to CVDs. These interactions underlying an intercellular network can be inferred from scRNA-seq data. Wang et al studied the cell–cell interaction networks in the human heart. The authors showed that cardiomyocytes and ECs are major cell-communication hubs and that cardiomyocytes’ contractility and metabolism are the most prominent aspects that are correlated with changes in heart function.46 For instance, Paik et al predicted the intercellular communication between ECs and other cell types in 12 major adult murine organs. This study reveals the existence of unique angiocrine ligand–receptor pairings between ECs and parenchymal cells in each major organs including heart and brain.8 Recent studies demonstrated the unrecognized functions of the immune cell during cardiac function and diseases. In the mouse heart, macrophages were found to facilitate electrical conduction and have crucial roles in myocardial infarction and aging. These discoveries emphasize the deeper investigation of the interplay among different cardiac cell types.56–58 Several statistical frameworks based on ligand–receptor interaction, such as CellPhoneDB, CellChat, SingleCellSignalR, have been developed to predict the enriched cellular interactions between two cell types from single-cell transcriptomic data.59–61 Discoveries from the study of cell–cell communication have the potential to identify novel therapeutic targets for treatments of CVD patients.
scRNA-Seq Integration Discovers a Novel Subset of Cardiomyocytes Population
The broad application of scRNA-seq technologies generated an unprecedented amount of data for cardiovascular research listed in Table 2. Integrated datasets from separate studies have the potential to provide biological insights that will not be possible from analyzing individual datasets. For instance, the integration of multiple scRNA-seq datasets derived from subpopulations of cells of a particular tissue can aid in characterizing heterogeneity in these tissues under different conditions. Many powerful methods have been developed to integrate individual scRNA-seq datasets such as Seurat v3, Harmony, SIMLR, SC3.62–65 Recently, Galow et al used the single-cell integration method to discover a minor population of cardiomyocytes characterized by proliferation markers that could not be identified by analyzing the datasets individually. The integration analysis also gave evidence that the renewal of the cardiomyocyte pool is driven by cytokinesis of resident cardiomyocytes rather than the differentiation of progenitor cells.66 Kuppe et al used scRNA-seq, scATAC-seq and spatial transcriptomic to profile the various physiological timepoints and zones of human health myocardium and myocardial infarction to build an integrative high-resolution map of cardiac remodeling. This integrated method increases cell-type composition spatial resolution and identifies the distinct injury, repair and remodeling cellular spatial zones.67 In summary, single-cell integration genomics has played a fundamental role in our understanding of tissue heterogeneity and cross-species analyses may yield similar insights toward our understanding of cardiac cell diversity.
Detection of Genetic Variants from scRNAs-Seq Data
Genetic variants are generally identified from whole-genome sequencing (WGS) and whole-exon sequencing (WES) studies.61 Detecting genetic variants from scRNA-seq data is rarely reported because of the inherited limitations of scRNA-seq platform such as low transcript abundance, allelic dropout, and incomplete transcript coverage. To overcome these limitations, SCmut has been developed to identify the cell-level recurrent variants in many single cells by controlling the false discoveries using the 2D local false discovery rate (FDR). The variants detected from scRNA-seq data can facilitate the investigation of cell-to-cell heterogeneity.68 Compared to variants identified from WGS/WES dataset, the cell-level mutations can only be found in the exonic regions and are affected by stochastic monoallelic expression. Although the methods for detecting genetic variants from scRNA-seq data have been reported in studies of the area of cancer biology, to date, no scRNA-seq studies in cardiovascular research reported mutation analysis. However, detecting genetic variants from scRNA-seq dataset has the potential to reveal the precise mechanisms of the pathogenesis in CVDs.
Construction of Spatial Subcellular Map During Heart Development
Spatial transcriptomics has been developed to characterize the gene expression profiles simultaneously retaining spatial information in various biological contexts.69 These methods aim to elucidate the function of individual cells in the context of their spatial organization in the tissue.70 The methodologies in spatial transcriptomics provide important insights in cardiovascular research to explore the process of cardiac morphogenesis in humans. For example, fluorescence in situ hybridization (FISH)–based method has been developed to directly label in tissue sections to visualize each single cell, even in subcellular location.71–73 In the latest work reporting the application of spatial genomic in the cardiovascular field, Asp et al used spatial transcriptomic method to study the transcriptional landscape of cardiac cells during the development of the embryonic heart and mapped the specific genes to the corresponding anatomical domains. They characterized the unique gene profiles in distinct anatomical regions and constructed a spatial subcellular map for the three developmental phases.43 In a separate study, Mohenska et al used spatial transcriptomics to reconstruct a 3D gene expression pattern in the mouse adult heart. They revealed specific gene lists that displayed complex spatial expression in organ sub-compartments, and deciphered gene expression profiles of the atria and the transcriptional complexity within the ventricles, and predicted the localization of non-myocytes within the heart.74 These spatial transcriptomics methods have greatly facilitated future studies on cardiogenesis with unprecedented resolution.
Limitations of scRNA-seq in Cardiac Tissue
Current scRNA-seq technologies are still confronting many challenges and limitations to profile the transcriptomic panorama of individual cells.75 For instance, scRNA-seq is unable to reliably detect low-abundance transcripts. It has been reported that only approximately 10% of the transcript could be detected from a single cell and the percentage of the lost RNA content reached to 60%. Both contribute to a higher difficulty in detecting the low abundant transcripts.76,77 The low amount of transcripts often resulted from library preparation leads to high levels of computational noise, which disturbs data analysis and may mask underlying biological variation. For example, long non-coding RNAs (lncRNAs), which have critical roles in regulatory functions, typically are presented in several copies in a cell but often could not be detected.78 Thus, it is necessary to improve the sensitivity of scRNA-seq to detect low copy transcripts in one cell to gain a full understanding of many regulatory processes.
Furthermore, some cell types such as cardiomyocytes may not be compatible with the processing steps of popular scRNA-seq techniques. The droplet-based Chromium platform is suitable for scRNA-seq studies with cells smaller than 30μm.31 However, adult CMs in mice and humans are relatively larger than 100μm in diameter, which deterred the use of this single-cell system.31 Therefore, snRNA-seq, which extracted nuclei rather than intact CMs, or plate-based FACS platform could be considered as alternative methods to profile CMs or cells from frozen specimens.79 Future improvement in these single-cell protocols using intact cells or nuclei will help to overcome the limitations and biases.
Unsupervised clustering of scRNA-seq data is crucial for the downstream data analysis, as it annotates the cell types. In clustering, the hypothesis is that each cluster will represent one cell type. However, there is no golden standard for defining the cluster as a specific cell type.75,80 This is partly due to the expression matrix exhibited more zero values (known as dropouts). The dropouts cause higher levels of noise to annotate the state and identity of the cells accurately.81 The technical noise is also generated in the preparation of sequencing samples such as single-cell digestion resulted disproportionately enriches for one cell type over another.80
Trajectory inference can use single-cell sequencing data to infer the cells along the developmental trajectories and facilitate mapping clonal relationships onto these landscapes.47 However, these sequencing-based lineage-tracing methods are still in their infancy. Compared with the DNA barcodes and clone analysis to reconstruct lineage relationships, sequencing-based lineage-tracing methods are sensitive to the choice of experimental platform to perform the scRNA-seq, which could affect the conclusions.82 However, it can anticipate that lineage-tracing methods might be integrated with clonal analysis and DNA barcoding methods, which will significantly track the number of clones and establish clonal composition without requiring prior knowledge of the marker genes.82 To accurately decipher the spatial gene expression, it is important to capture gene identity along with quantitative data. And efforts have been made to achieve higher spatial resolution due to the integrated single-cell imaging techniques.83
Perspective
Over the past decade, there has been an increasing interest in using scRNA-seq technologies to study cardiovascular development and disease. Imaging technologies such as FISH have been proposed to combine with scRNA-seq to study the spatial single-cell transcriptomic profiles in the cardiovascular environment.84 In addition, long-read sequencing technology such as PacBio and Oxford Nanopore platforms can be combined with high-throughput droplet-based scRNA-Seq workflows to capture gene-expression profiles with targeted full-length mRNA sequences from a large number of cells. Such combination has the potential to achieve both high sensitivity and accuracy in capturing full-length transcripts, which could be further used for the identification of somatic mutations and the inference of clonal evolution of distinct cell types.85 In addition, the integration of scRNA-seq and chromatin accessibility data can provide more comprehensive insights into gene regulation and cellular dynamics. As the numbers of scRNA-seq datasets are rapidly increasing due to collaborative efforts such as the Human Cell Atlas consortium,86 it is necessary to optimize the algorithm and develop sophisticated computational methods for data analysis. Collectively, scRNA-seq technologies will greatly expand our knowledge in cardiac cell heterogeneity, CVD pathogenesis and microenvironmental interactions, and ultimately lay a foundation for precision medicine in cardiovascular diseases.
Abbreviations
CF, cardiac fibroblast; CMs, cardiomyocytes; CVDs, cardiovascular diseases; DE, differential expressed; ECs, endothelial cells; FACS, fluorescence-activated cell sorting; FDR, false discovery rate; FISH, fluorescence in situ hybridization; hiPSC, human-induced pluripotent stem cell; lncRNAs, long non-coding RNAs; LPM, lateral plate mesoderm; NGS, next-generation sequencing; PCs, principal components; PCA, principal component analysis; PCR, polymerase chain reaction; QC, quality control; RNA-seq, RNA sequencing; RT, reverse transcription; scRNA-seq, single-cell RNA sequencing; snRNA-seq, single-nucleus RNA sequencing; tSNE, t-distributed stochastic neighbor embedding; UMAP, uniform manifold approximation and projection; UMIs, unique molecular identifiers; vSMC, vascular smooth muscle cell; WES, whole-exon sequencing; WGS, whole-genome sequencing.
Acknowledgments
We thank Dr David Paik for the constructive suggestions for the manuscript.
Author Contributions
All authors made a significant contribution to the manuscript, whether that is in the conception, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
We are grateful for funding support from the American Heart Association Postdoctoral Fellowship 20POST35210924 (Dr Tian) and the National Heart, Lung, and Blood Institute R00HL135258 (Dr Gu).
Disclosure
The authors have no conflicts of interest to declare in this work.
References
1. Mortality GBD, Causes of Death C. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117–171. doi:10.1016/S0140-6736(14)61682-2
2. Mortality GBD, Causes of Death C. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1459–1544.
3. Thomas H, Diamond J, Vieco A, et al. Global atlas of cardiovascular disease 2000–2016: the path to prevention and control. Glob Heart. 2018;13(3):143–163. doi:10.1016/j.gheart.2018.09.511
4. Leon-Mimila P, Wang J, Huertas-Vazquez A. Relevance of multi-omics studies in cardiovascular diseases. Front Cardiovasc Med. 2019;6:91. doi:10.3389/fcvm.2019.00091
5. Leopold JA, Loscalzo J. Emerging role of precision medicine in cardiovascular disease. Circ Res. 2018;122(9):1302–1315. doi:10.1161/CIRCRESAHA.117.310782
6. Neeland IJ, Poirier P, Despres JP. Cardiovascular and metabolic heterogeneity of obesity: clinical challenges and implications for management. Circulation. 2018;137(13):1391–1406.
7. McCormick ME, Manduchi E, Witschey WR, et al. Integrated Regional Cardiac Hemodynamic Imaging and RNA Sequencing Reveal Corresponding Heterogeneity of Ventricular Wall Shear Stress and Endocardial Transcriptome. J Am Heart Assoc. 2016;5(4):e003170. doi:10.1161/JAHA.115.003170
8. Paik DT, Tian L, Williams IM, et al. Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. 2020;142(19):1848–1862. doi:10.1161/CIRCULATIONAHA.119.041433
9. Ruan H, Liao Y, Ren Z, et al. Single-cell reconstruction of differentiation trajectory reveals a critical role of ETS1 in human cardiac lineage commitment. BMC Biol. 2019;17(1):89. doi:10.1186/s12915-019-0709-6
10. Tucker NR, Chaffin M, Fleming SJ, et al. Transcriptional and cellular diversity of the human heart. Circulation. 2020;142(5):466–482. doi:10.1161/CIRCULATIONAHA.119.045401
11. Litvinukova M, Talavera-Lopez C, Maatz H, et al. Cells of the adult human heart. Nature. 2020;588(7838):466–472. doi:10.1038/s41586-020-2797-4
12. Kamdar F, Das S, Gong W, et al. Stem cell-derived cardiomyocytes and beta-adrenergic receptor blockade in Duchenne muscular dystrophy cardiomyopathy. J Am Coll Cardiol. 2020;75(10):1159–1174. doi:10.1016/j.jacc.2019.12.066
13. Kulkarni A, Anderson AG, Merullo DP, Konopka G. Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr Opin Biotechnol. 2019;58:129–136. doi:10.1016/j.copbio.2019.03.001
14. Pollen AA, Nowakowski TJ, Shuga J, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol. 2014;32(10):1053–1058. doi:10.1038/nbt.2967
15. Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10(11):1096–1098. doi:10.1038/nmeth.2639
16. Sheng K, Cao W, Niu Y, Deng Q, Zong C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat Methods. 2017;14(3):267–270. doi:10.1038/nmeth.4145
17. Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343(6172):776–779. doi:10.1126/science.1247651
18. Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2(3):666–673. doi:10.1016/j.celrep.2012.08.003
19. Macosko EZ, Basu A, Satija R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–1214. doi:10.1016/j.cell.2015.05.002
20. Bagnoli JW, Ziegenhain C, Janjic A, et al. Sensitive and powerful single-cell RNA sequencing using mcSCRB-seq. Nat Commun. 2018;9(1):2937. doi:10.1038/s41467-018-05347-6
21. Zheng GX, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi:10.1038/ncomms14049
22. Gierahn TM, Wadsworth MH
23. Rosenberg AB, Roco CM, Muscat RA, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018;360(6385):176–182. doi:10.1126/science.aam8999
24. Goldstein LD, Chen YJ, Dunne J, et al. Massively parallel nanowell-based single-cell gene expression profiling. BMC Genomics. 2017;18(1):519. doi:10.1186/s12864-017-3893-1
25. Chaudhry F, Isherwood J, Bawa T, et al. Single-cell RNA sequencing of the cardiovascular system: new looks for old diseases. Front Cardiovasc Med. 2019;6:173. doi:10.3389/fcvm.2019.00173
26. Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 2019;15(6):e8746. doi:10.15252/msb.20188746
27. Andrews TS, Kiselev VY, McCarthy D, Hemberg M. Tutorial: guidelines for the computational analysis of single-cell RNA sequencing data. Nat Protoc. 2021;16(1):1–9. doi:10.1038/s41596-020-00409-w
28. Svensson V, Vento-Tormo R, Teichmann SA. Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc. 2018;13(4):599–604. doi:10.1038/nprot.2017.149
29. Potter SS. Single-cell RNA sequencing for the study of development, physiology and disease. Nat Rev Nephrol. 2018;14(8):479–492. doi:10.1038/s41581-018-0021-7
30. Santoro F, Chien KR, Sahara M. Isolation of human ESC-derived cardiac derivatives and embryonic heart cells for population and single-cell RNA-seq analysis. STAR Protoc. 2021;2(1):100339. doi:10.1016/j.xpro.2021.100339
31. Yamada S, Nomura S. Review of single-cell RNA sequencing in the heart. Int J Mol Sci. 2020;21:21. doi:10.3390/ijms21218345
32. Gladka MM, Molenaar B, de Ruiter H, et al. Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation. 2018;138(2):166–180. doi:10.1161/CIRCULATIONAHA.117.030742
33. Lacar B, Linker SB, Jaeger BN, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun. 2016;7:11022. doi:10.1038/ncomms11022
34. Xin Y, Kim J, Ni M, et al. Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proc Natl Acad Sci U S A. 2016;113(12):3293–3298. doi:10.1073/pnas.1602306113
35. Nayak R, Hasija Y. A hitchhiker’s guide to single-cell transcriptomics and data analysis pipelines. Genomics. 2021;113(2):606–619. doi:10.1016/j.ygeno.2021.01.007
36. McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8(4):329–337 e324. doi:10.1016/j.cels.2019.03.003
37. Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9(1):171–181. doi:10.1038/nprot.2014.006
38. Picelli S. Full-Length single-cell RNA sequencing with Smart-seq2. Methods Mol Biol. 2019;1979:25–44.
39. Ziegenhain C, Vieth B, Parekh S, et al. Comparative Analysis of single-cell RNA sequencing methods. Mol Cell. 2017;65(4):631–643 e634. doi:10.1016/j.molcel.2017.01.023
40. Salomon R, Kaczorowski D, Valdes-Mora F, et al. Droplet-based single cell RNAseq tools: a practical guide. Lab Chip. 2019;19(10):1706–1727. doi:10.1039/C8LC01239C
41. Lareau CA, Ma S, Duarte FM, Buenrostro JD. Inference and effects of barcode multiplets in droplet-based single-cell assays. Nat Commun. 2020;11(1):866. doi:10.1038/s41467-020-14667-5
42. Liu SJ, Nowakowski TJ, Pollen AA, et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016;17:67. doi:10.1186/s13059-016-0932-1
43. Asp M, Giacomello S, Larsson L, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179(7):1647–1660 e1619. doi:10.1016/j.cell.2019.11.025
44. Xu D, Ma M, Xu Y, et al. Single-cell transcriptome analysis indicates new potential regulation mechanism of ACE2 and NPs signaling among heart failure patients infected with SARS-CoV-2. medRxiv. 2020;8:103.
45. Li Z, Solomonidis EG, Meloni M, et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur Heart J. 2019;40(30):2507–2520. doi:10.1093/eurheartj/ehz305
46. Wang L, Yu P, Zhou B, et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol. 2020;22(1):108–119. doi:10.1038/s41556-019-0446-7
47. Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single-cell trajectory inference methods. Nat Biotechnol. 2019;37(5):547–554. doi:10.1038/s41587-019-0071-9
48. Song D, Li JJ. PseudotimeDE: inference of differential gene expression along cell pseudotime with well-calibrated p-values from single-cell RNA sequencing data. Genome Biol. 2021;22(1):124. doi:10.1186/s13059-021-02341-y
49. Phansalkar RS, Krieger J, Zhao M, et al. Coronary blood vessels from distinct origins converge to equivalent states during mouse and human development. bioRxiv. 2021;2:584.
50. Ren Z, Yu P, Li D, et al. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation. 2020;141(21):1704–1719. doi:10.1161/CIRCULATIONAHA.119.043053
51. Zhang Q, Carlin D, Zhu F, et al. Unveiling complexity and multipotentiality of early heart fields. Circ Res. 2021;129(4):474–487. doi:10.1161/CIRCRESAHA.121.318943
52. La Manno G, Soldatov R, Zeisel A, et al. RNA velocity of single cells. Nature. 2018;560(7719):494–498. doi:10.1038/s41586-018-0414-6
53. Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38(12):1408–1414. doi:10.1038/s41587-020-0591-3
54. Wolfien M, Galow AM, Muller P, et al. Single-nucleus sequencing of an entire mammalian heart: cell type composition and velocity. Cells. 2020;9:2.
55. Liu X, Chen W, Li W, et al. Single-cell RNA-Seq of the developing cardiac outflow tract reveals convergent development of the vascular smooth muscle cells. Cell Rep. 2019;28(5):1346–1361 e1344. doi:10.1016/j.celrep.2019.06.092
56. Hulsmans M, Clauss S, Xiao L, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169(3):510–522 e520. doi:10.1016/j.cell.2017.03.050
57. King KR, Aguirre AD, Ye YX, et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23(12):1481–1487. doi:10.1038/nm.4428
58. Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res. 2018;191:15–28. doi:10.1016/j.trsl.2017.10.001
59. Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. 2020;15(4):1484–1506. doi:10.1038/s41596-020-0292-x
60. Jin S, Guerrero-Juarez CF, Zhang L, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12(1):1088. doi:10.1038/s41467-021-21246-9
61. Cabello-Aguilar S, Alame M, Kon-Sun-Tack F, Fau C, Lacroix M, Colinge J. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020;48(10):e55. doi:10.1093/nar/gkaa183
62. Wang B, Zhu J, Pierson E, Ramazzotti D, Batzoglou S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat Methods. 2017;14(4):414–416. doi:10.1038/nmeth.4207
63. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. doi:10.1038/s41592-019-0619-0
64. Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–1902 e1821. doi:10.1016/j.cell.2019.05.031
65. Kiselev VY, Kirschner K, Schaub MT, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods. 2017;14(5):483–486. doi:10.1038/nmeth.4236
66. Galow AM, Wolfien M, Muller P, et al. Integrative cluster analysis of whole hearts reveals proliferative cardiomyocytes in adult mice. Cells. 2020;9:5. doi:10.3390/cells9051144
67. Kuppe C, Flores RO, Li Z, et al. Spatial multi-omic map of human myocardial infarction. BioRxiv. 2020. doi:10.1101/2020.12.08.411686
68. Vu TN, Nguyen HN, Calza S, Kalari KR, Wang L, Pawitan Y. Cell-level somatic mutation detection from single-cell RNA sequencing. Bioinformatics. 2019;35(22):4679–4687. doi:10.1093/bioinformatics/btz288
69. Linnarsson S, Teichmann SA. Single-cell genomics: coming of age. Genome Biol. 2016;17:97. doi:10.1186/s13059-016-0960-x
70. Asp M, Bergenstrahle J, Lundeberg J. Spatially resolved transcriptomes-next generation tools for tissue exploration. Bioessays. 2020;42(10):e1900221. doi:10.1002/bies.201900221
71. Moffitt JR, Zhuang X. RNA imaging with multiplexed error-robust fluorescence in situ hybridization (MERFISH). Methods Enzymol. 2016;572:1–49.
72. Gelali E, Custodio J, Girelli G, Wernersson E, Crosetto N, Bienko M. An application-directed, versatile DNA FISH Platform for Research and Diagnostics. Methods Mol Biol. 2018;1766:303–333.
73. Gelali E, Girelli G, Matsumoto M, et al. iFISH is a publically available resource enabling versatile DNA FISH to study genome architecture. Nat Commun. 2019;10(1):1636. doi:10.1038/s41467-019-09616-w
74. Mohenska M, Tan NM, Tokolyi A, et al. 3D-Cardiomics: a spatial transcriptional atlas of the mammalian heart. bioRxiv. 2019:792002. doi:10.1101/792002
75. Lahnemann D, Koster J, Szczurek E, et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020;21(1):31.
76. Islam S, Zeisel A, Joost S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11(2):163–166. doi:10.1038/nmeth.2772
77. Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343(6167):193–196. doi:10.1126/science.1245316
78. Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. doi:10.1101/gr.132159.111
79. Sun W, Dong H, Balaz M, et al. snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature. 2020;587(7832):98–102. doi:10.1038/s41586-020-2856-x
80. Kiselev VY, Andrews TS, Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet. 2019;20(5):273–282. doi:10.1038/s41576-018-0088-9
81. Cui Y, Zhang S, Liang Y, Wang X, Ferraro TN, Chen Y. Consensus clustering of single-cell RNA-seq data by enhancing network affinity. Brief Bioinform. 2021. doi:10.1093/bib/bbab236
82. Wagner DE, Klein AM. Lineage tracing meets single-cell omics: opportunities and challenges. Nat Rev Genet. 2020;21(7):410–427. doi:10.1038/s41576-020-0223-2
83. Waylen LN, Nim HT, Martelotto LG, Ramialison M. From whole-mount to single-cell spatial assessment of gene expression in 3D. Commun Biol. 2020;3(1):602. doi:10.1038/s42003-020-01341-1
84. Roth R, Kim S, Kim J, Rhee S. Single-cell and spatial transcriptomics approaches of cardiovascular development and disease. BMB Rep. 2020;53(8):393–399. doi:10.5483/BMBRep.2020.53.8.130
85. Singh M, Al-Eryani G, Carswell S, et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat Commun. 2019;10(1):3120. doi:10.1038/s41467-019-11049-4
86. Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA. The Human Cell Atlas: from vision to reality. Nature. 2017;550(7677):451–453. doi:10.1038/550451a
87. Suryawanshi H, Clancy R, Morozov P, Halushka MK, Buyon JP, Tuschl T. Cell atlas of the foetal human heart and implications for autoimmune-mediated congenital heart block. Cardiovasc Res. 2020;116(8):1446–1457. doi:10.1093/cvr/cvz257
88. Hu Z, Liu W, Hua X, et al. Single-cell transcriptomic atlas of different human cardiac arteries identifies cell types associated with vascular physiology. Arterioscler Thromb Vasc Biol. 2021;41(4):1408–1427. doi:10.1161/ATVBAHA.120.315373
89. Miao Y, Tian L, Martin M, et al. Intrinsic endocardial defects contribute to hypoplastic left heart syndrome. Cell Stem Cell. 2020;27(4):574–589 e578. doi:10.1016/j.stem.2020.07.015
90. Cao J, O’Day DR, Pliner HA, et al. A human cell atlas of fetal gene expression. Science. 2020;370:6518. doi:10.1126/science.aba7721
91. Han X, Zhou Z, Fei L, et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581(7808):303–309. doi:10.1038/s41586-020-2157-4
92. Friedman CE, Nguyen Q, Lukowski SW, et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell. 2018;23(4):586–598 e588. doi:10.1016/j.stem.2018.09.009
93. Churko JM, Garg P, Treutlein B, et al. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat Commun. 2018;9(1):4906. doi:10.1038/s41467-018-07333-4
94. Cui Y, Zheng Y, Liu X, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019;26(7):1934–1950 e1935. doi:10.1016/j.celrep.2019.01.079
95. Sahara M, Santoro F, Sohlmer J, et al. Population and single-cell analysis of human cardiogenesis reveals unique lgr5 ventricular progenitors in embryonic outflow tract. Dev Cell. 2019;48(4):475–490 e477. doi:10.1016/j.devcel.2019.01.005
96. Gambardella L, McManus SA, Moignard V, et al. BNC1 regulates cell heterogeneity in human pluripotent stem cell-derived epicardium. Development. 2019;146:24.
97. Zhou Y, Liu Z, Welch JD, et al. Single-cell transcriptomic analyses of cell fate transitions during human cardiac reprogramming. Cell Stem Cell. 2019;25(1):149–164 e149. doi:10.1016/j.stem.2019.05.020
98. Nomura S, Satoh M, Fujita T, et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat Commun. 2018;9(1):4435. doi:10.1038/s41467-018-06639-7
99. See K, Tan WLW, Lim EH, et al. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat Commun. 2017;8(1):225. doi:10.1038/s41467-017-00319-8
100. Tabula Muris C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. 2020;583(7817):590–595. doi:10.1038/s41586-020-2496-1
101. de Soysa TY, Ranade SS, Okawa S, et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature. 2019;572(7767):120–124. doi:10.1038/s41586-019-1414-x
102. Goodyer WR, Beyersdorf BM, Paik DT, et al. Transcriptomic profiling of the developing cardiac conduction system at single-cell resolution. Circ Res. 2019;125(4):379–397. doi:10.1161/CIRCRESAHA.118.314578
103. Vidal R, Wagner JUG, Braeuning C, et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight. 2019;4:22. doi:10.1172/jci.insight.131092
104. Zhang Y, Gago-Lopez N, Li N, et al. Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discov. 2019;5:30. doi:10.1038/s41421-019-0095-9
105. Linscheid N, Logantha S, Poulsen PC, et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat Commun. 2019;10(1):2889. doi:10.1038/s41467-019-10709-9
106. Farbehi N, Patrick R, Dorison A, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;1:8.
107. Hu P, Liu J, Zhao J, et al. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018;32(19–20):1344–1357. doi:10.1101/gad.316802.118
108. Tabula Muris C, Overall C, Logistical C, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562(7727):367–372.
109. Wang Y, Yao F, Wang L, et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat Commun. 2020;11(1):2585. doi:10.1038/s41467-020-16204-w
110. Han X, Zhang J, Liu Y, et al. The lncRNA Hand2os1/Uph locus orchestrates heart development through regulation of precise expression of Hand2. Development. 2019;146:13. doi:10.1242/dev.176198
111. Hulin A, Hortells L, Gomez-Stallons MV, et al. Maturation of heart valve cell populations during postnatal remodeling. Development. 2019;146:12.
112. Yekelchyk M, Guenther S, Preussner J, Braun T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res Cardiol. 2019;114(5):36. doi:10.1007/s00395-019-0744-z
113. Xiong H, Luo Y, Yue Y, et al. Single-cell transcriptomics reveals chemotaxis-mediated intraorgan crosstalk during cardiogenesis. Circ Res. 2019;125(4):398–410. doi:10.1161/CIRCRESAHA.119.315243
114. Dong J, Hu Y, Fan X, et al. Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 2018;19(1):31. doi:10.1186/s13059-018-1416-2
115. Kretzschmar K, Post Y, Bannier-Helaouet M, et al. Profiling proliferative cells and their progeny in damaged murine hearts. Proc Natl Acad Sci U S A. 2018;115(52):E12245–E12254. doi:10.1073/pnas.1805829115
116. Li G, Xu A, Sim S, et al. Transcriptomic Profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev Cell. 2016;39(4):491–507. doi:10.1016/j.devcel.2016.10.014
117. DeLaughter DM, Bick AG, Wakimoto H, et al. Single-cell resolution of temporal gene expression during heart development. Dev Cell. 2016;39(4):480–490. doi:10.1016/j.devcel.2016.10.001
118. Lescroart F, Wang X, Lin X, et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science. 2018;359(6380):1177–1181. doi:10.1126/science.aao4174
119. Weinberger M, Simoes FC, Patient R, Sauka-Spengler T, Riley PR. Functional heterogeneity within the developing Zebrafish Epicardium. Dev Cell. 2020;52(5):574–590 e576. doi:10.1016/j.devcel.2020.01.023
120. Holowiecki A, Linstrum K, Ravisankar P, Chetal K, Salomonis N, Waxman JS. Pbx4 limits heart size and fosters arch artery formation by partitioning second heart field progenitors and restricting proliferation. Development. 2020;147:5.
121. Honkoop H, de Bakker DE, Aharonov A, et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife. 2019;1:8.
122. Chestnut B, Casie Chetty S, Koenig AL, Sumanas S. Single-cell transcriptomic analysis identifies the conversion of zebrafish Etv2-deficient vascular progenitors into skeletal muscle. Nat Commun. 2020;11(1):2796. doi:10.1038/s41467-020-16515-y
123. Zhang W, Zhang S, Yan P, et al. A single-cell transcriptomic landscape of primate arterial aging. Nat Commun. 2020;11(1):2202. doi:10.1038/s41467-020-15997-0
124. Ma S, Sun S, Li J, et al. Single-cell transcriptomic atlas of primate cardiopulmonary aging. Cell Res. 2021;31(4):415–432. doi:10.1038/s41422-020-00412-6
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.