Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Simvastatin pretreatment ameliorates t-PA–induced hemorrhage transformation and MMP-9/TIMP-1 imbalance in thromboembolic cerebral ischemic rats
Authors Yin B ![]() , Li DD
, Li DD ![]() , Xu SY, Huang H, Lin J, Sheng HS, Fang JH, Song JN, Zhang M
, Xu SY, Huang H, Lin J, Sheng HS, Fang JH, Song JN, Zhang M
Received 29 December 2018
Accepted for publication 1 May 2019
Published 16 July 2019 Volume 2019:15 Pages 1993—2002
DOI https://doi.org/10.2147/NDT.S199371
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jun Chen
Bo Yin,*,1 Dan-Dong Li,*,2,3 Shang-Yu Xu,3 Huan Huang,4 Jian Lin,3 Han-Song Sheng,3 Jun-Hao Fang,3 Jin-Ning Song,2 Ming Zhang1
1Department of Medical Imaging, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, People’s Republic of China; 2Department of Neurosurgery, The First Affiliated Hospital, Medical School of Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China; 3Department of Neurosurgery, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 4Department of Radiology, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Background: The use of thrombolysis with tissue-plasminogen activator (t-PA) in patients with acute ischemic stroke (AIS) is limited by increased levels of matrix metalloproteinase-9 (MMP-9) and by the increased risk of hemorrhagic transformation (HT). In this study, we investigated the effects of simvastatin pretreatment on t-PA-induced MMP-9/tissue inhibitor of metalloproteinase-1 (TIMP-1) imbalance and HT aggravation in a rat AIS model.
Methods: The rat AIS model was established by autologous blood emboli. Two weeks before surgery, rats were pretreated with simvastatin (60 mg/kg/d), and three hours after surgery, t-PA (10 mg/kg) was administered. MMP-9 and TIMP-1 levels in the infarcted zone and plasma were evaluated by Western blot analysis and ELISA; the level of HT was quantified by determining the hemoglobin content. RhoA activation was determined to clarify the potential effect.
Results: The results suggested that pretreatment with simvastatin suppressed the increase in t-PA-induced MMP-9 levels and neutralized the elevated MMP-9/TIMP-1 ratio, but had no effect on TIMP-1 levels. Thrombolysis with t-PA after ischemia improved neurological outcome, but increased intracranial hemorrhage. Moreover, t-PA-induced HT aggravation was reduced by simvastatin pretreatment. In addition, we showed that t-PA-induced activation of RhoA was suppressed by simvastatin, and that t-PA-induced MMP-9/TIMP-1 imbalance and hemorrhage was reduced by Rho kinases (ROCK) inhibitor Y-27632.
Conclusion: In this study, we showed that simvastatin pretreatment ameliorated t-PA-induced HT and MMP-9/TIMP-1 imbalance, and demonstrated that the RhoA/ROCK pathway was implicated.
Keywords: simvastatin, acute ischemic stroke, t-PA, MMP-9/TIMP-1, RhoA/ROCK pathway, hemorrhagic transformation
Introduction
In previous studies, thrombolysis with tissue-plasminogen activator (t-PA) has shown to improve the outcome for acute ischemic stroke (AIS) patients who were treated within 3 hrs of symptom onset,1 the fear of hemorrhagic transformation (HT) frequently limits the use of thrombolysis in clinical practice. In fact, t-PA is associated with a 5–6% risk of symptomatic intracerebral hemorrhage when given within 3 hrs of stroke onset.1,2 Currently, t-PA therapy is the only approved treatment for AIS,2 hence, identifying methods that may reduce the risk of hemorrhage is of utmost importance.
Matrix metalloproteinase-9 (MMP-9), a member of the matrix metalloproteinases family, degrades components of the extracellular matrix (ECM). An abnormal ECM regulation has been implicated as a causal factor of HT.3,4 Moreover, experimental and clinical studies5,6 have confirmed that after AIS, the level of MMP-9 significantly increased in both peripheral blood and brain tissue. Indeed, MMP-9 has been demonstrated a predictor of t-PA-induced HT after stroke.7 In general, MMP-9 is secreted together with variable amounts of its specific inhibitor, tissue inhibitor of metalloproteinase 1 (TIMP-1), which controls its proteolytic activity. Disturbances in the balance between MMP-9 and TIMP-1 may result in excessive ECM degradation, a condition that is often associated with vascular diseases. In our previous study, we have shown that the MMP-9/TIMP-1 ratio closely correlated with brain edema in a rat AIS model.8 In previous studies, it has been suggested that measures that aim to prevent MMP-9 upregulation and MMP-9/TIMP-1 imbalance may provide a novel approach for stroke therapy.
Statin, a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor, has been used to control serum cholesterol levels by inhibiting HMG-CoA reductase. Increasing evidence has shown that statins decrease MMPs levels and downregulate MMP-9/TIMP-1 ratio in vitro;9 however, little is known about the effects of statins on the MMP-9/TIMP-1 ratio in AIS models, especially in models in which the animals received rt-PA thrombolysis. Statins reduce MMP-9 secretion by inhibiting the RhoA/ROCK pathway and by decreasing MMP-9 mRNA levels independent of effects on the signal transduction pathways that are required for MMP-9 expression.10 Treatment with stains that are initiated in the acute phase of ischemic stroke improved neurologic outcome.11 Statin’s mechanism of action on the inhibition of MMP-9 expression is known;10 however, little is known about its effect on t-PA thrombolytic individuals. Hence, additional studies are needed to clarify its effect on MMP-9 expression and the MMP-9/TIMP-1 ratio, especially in patients in which acute stroke was treated with t-PA.
In the present study, we aimed to investigate the effect of pretreatment with simvastatin (SIM) on t-PA-induced MMP-9/TIMP-1 imbalance and HT aggravation in a rat AIS model, and to reveal the potential pathway involved.
Materials and methods
Animals and experimental groups
This study was approved by the Biomedical Ethics Committee of the Medical College of Xi’an Jiaotong University (Xi’an, China), conform the NIH Guide for the Care and Use of Laboratory Animals. Adult (9–10 weeks) male Sprague–Dawley rats (300±20 g) were purchased from the Laboratory Animal Center of Wenzhou Medical University (Wenzhou, China). Animals were randomly divided into four groups: a sham group (n=12, operation (-), SIM-/t-PA-), one group treated with both SIM and t-PA (n=12, operation (+), SIM+/t-PA+), rats treated with t-PA only (n=12, operation (+) SIM-/t-PA+), and a saline group, treated with normal saline (n=12, operation (+) SIM-/t-PA-). Rats in the SIM-/t-PA+ group and the saline group received normal saline (1 mL/d) and rats in the SIM+/t-PA+ group received SIM (60 mg/kg/d suspended in 1 mL water, Merck Sharp & Dohme, Hangzhou, China) via gavage for 2 weeks until surgery. Gavage involved the passage of a tube via the mouth down into the stomach followed by gentle administration. In the current study, the dose of SIM used was comparable to the dose used in previous studies.12,13 Rats in the SIM-/t-PA+ group and SIM+/t-PA+ group received t-PA (10 mg/kg, Boehringer Ingelheim, Ingelheim, Germany), and rats in the saline group received the same volume of normal saline via femoral venous catheterization 3 hrs after surgery, 20% of the total volume as a bolus injection given over 1 min, followed by the remainder infused over 30 mins. This dosage regimen of recombinant t-PA has commonly been used for investigating the effect of fibrinolysis in rodents.14,15 At 30 mins before surgery, Y27632, a specific inhibitor of ROCK, was given via intracerebroventricular injection, and the stereotactic coordinates were as follows: 1.0 mm caudal to the bregma, 1.5 mm lateral to the midline, and 4.0 mm deep from the dura. At the end of SIM treatment, physiological and biochemical indicators were evaluated as follows: blood pressure was measured by tail-cuff method (bp-98a, Softron, Japan), and lipid indicators were measured by commercial ELISA kits (Jianglaibio, Shanghai, China).
Surgery and specimen collection
AIS was induced in rats by focal embolic occlusion of the right middle cerebral artery (MCA) according to the method described by Busch16 with minor changes as follows. Anesthesia induction was carried out using 5% isoflurane and maintained with 2.5% isoflurane. Ten clots (each 0.35×2.0 mm size) were prepared before surgery. After exposure of the right common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA), and ligation of extracranial branches of the ICA including the pterygopalatine artery, the clots were transferred to a PE50 catheter. The catheter was inserted into the ECA until the tip reached the bifurcation. Then, the clots were injected over a period of 30 s, during which the CCA was temporarily clamped. Rats in the sham group underwent the same surgery but were only injected with an equal amount of phosphate-buffered saline (PBS). All animals were euthanized 24 hrs after surgery using an overdose of anesthetic chloral hydrate (10%, 1 mL/100 g). Per group, 6 rats were perfused with 250 mL saline, and brains were collected and divided into an infarct hemisphere and contralateral hemisphere and were homogenized. Per group, another 6 rats were perfused with 250 mL saline and brains were collected and divided as described earlier. After homogenization and centrifugation, supernatants were harvested for quantification of hemoglobin. From six rats per group, blood samples were collected from the abdominal aorta prior to perfusion.
ELISA
The plasma levels of secreted MMP-9 and TIMP-1 protein were determined using commercially available quantitative sandwich ELISA kits obtained from R&D Systems, USA. The rat hemoglobin ELISA kit for quantification of hemoglobin in cerebral homogenates was purchased from Cusabio (Wuhan, China). Blood samples were centrifuged at 3000× g for 10 mins, and cerebral homogenates were centrifuged at 15,000×g for 15 mins, the supernatant was removed and stored at −20°C until processing. The optical density (OD) in the supernatants was determined at 450 nm using an automatic immunoenzymatic analytic system (TECAN, Männedorf, Switzerland).
Western blot analysis
Total protein was extracted from brain homogenate using RIPA lysis buffer (Sigma, St. Louis, MO, USA). Equal protein samples (30 μg) were separated on denaturing 10% polyacrylamide gels electrophoresis and transferred to a polyvinylidene difluoride membrane. MMP-9 and β-actin proteins were determined by probing the membranes with the following primary antibodies, rabbit monoclonal antibody to MMP-9 (ab76003, Abcam, UK, 1:10000), rabbit polyclonal antibody to TIMP-1 (sc-5538, Santa Cruz, USA, 1:1000), mouse monoclonal to β-actin (sc-81178, 1:1000), and secondary antibody mouse anti-rabbit IgG-HRP, goat anti-mouse IgG-horseradish peroxidase (HRP) (sc-2357 and sc-2005, 1:5000). Quantitative analysis was performed with Gel-Pro Analyzer 3.1 software.
Real-time PCR
Total blood RNA was isolated using an RNAfast2000 kit (Fastagen, Shanghai, China). cDNA was prepared using the reverse transcriptase method as described by the manufacturer (PrimeScript RT reagent Kit, TaKaRa, Dalian, China) and subjected to quantitative real-time PCR using the primers presented in Table 1 and SYBR Green dye (TaKaRa, Dalian, China). The PCR program was as follows: 95°C for 1 min, followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 30 mins, and a final elongation of 10 mins at 72°C. Amplification and melting curves were prepared using an ABI 7500 system (Bio-Rad, Gibbstown, NJ, USA). A relative quantification (RQ) method was employed to calculate the relative levels of target gene mRNA, using β-actin as an internal reference gene. cDNA from normal rat blood extract served as a control reference sample. The equation was as follows: RQ=2−ΔΔCT, where ΔΔCT=(CT target gene−CT β-actin)embolism group−(CT target gene−CT β-actin)sham group.
 |
Table 1 The forward and reverse primers used |
Rho pull-down assay
The RhoA activity was determined using a rhotekin Rho-binding domain (RBD) affinity precipitation assay, followed by immunoblotting using a RhoA antibody. Active RhoA was affinity purified with GST bead-bound Rhotekin, which only binds RhoA in its active GTP-bound form. Then, Western blot analysis was performed to detect both active levels and total levels using a RhoA-specific antibody. A NewEast Biosciences RhoA Activation Assay Kit (Malvern, PA, USA) was used to detect the levels. Briefly, ice-cold lysis buffer (25 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 1% Igepal CA-630, 10 mmol/L MgCl2, 1 mmol/L EDTA, 10% glycerol, 1 Ag/mL aprotinin, 10 Ag/mL leupeptin, and 1 mmol/L Na3VO4) was added to brain homogenate (2:1 v:v), and incubated on ice for 30 mins. The lysates were centrifuged for 10 mins (12,000×g at 4°C). Lysates were incubated with agarose-conjugated rhotekin-RBD for 1 hr at 4°C and were then washed three times using lysis buffer. Agarose beads were boiled in SDS-PAGE sample buffer to release active Rho, and samples were resolved on a 12.5% polyacrylamide gel followed by immunoblotting using a RhoA antibody.
Neurological outcome
The modified neurological severity score (mNSS) is a composite of motor, sensory, reflex, and balance tests.17 Prior to euthanizing the animals, their neurologic function was graded using mNSS by three observers who were blinded to the group settings.
Statistical analysis
For quantitation of Western blots, the background was set for each blot, and each lane was sequentially analyzed. Data were expressed as the mean±standard deviation (SD) and analyzed using SPSS 19.0 software. Values of p<0.05 were considered statistically significant. When the data distribution was skewed, the Wilcoxon rank sum test was used (real-time PCR for MMP-9 and TIMP-1 and mNSS). The significance level in these tests was adjusted by Bonferroni adjustment. In all other measures (ELISA, Western blot analysis, and hemoglobin content), a one-way ANOVA followed by Dunnett t-tests for comparisons between the embolism groups and the sham group, Tukey’s tests for comparisons between the three embolism groups (SIM+/t-PA+ group, SIM-/t-PA+ group, and saline group). Data involving physiological and biochemical indicators were analyzed using a paired t-test.
Results
Physiological and biochemical indicators
Previous studies have revealed that low cholesterol levels are associated with increased stroke severity, symptomatic hemorrhagic transformation, and poorer functional outcome in patients with and without pre-stroke statin use.18,19 To prevent the lipid-lowering effect from interfering with our data, the dosage was selected strictly. Before treatment was initiated, six rats were randomly selected, and physiological indicators (blood pressure and body weight) and biochemical indicators (triglyceride, TG; total cholesterol, T-cho; high-density lipoprotein cholesterol, HDL-cho; low-density lipoprotein cholesterol, LDL-cho) were evaluated, and these values were used as baseline values. Blood pressure (BP) and body weight were not different among the groups during treatment. However, SIM significantly decreased the triglyceride level (p<0.05 versus water) but did not affect cholesterol levels (Table 2). These data indicated that SIM (60 mg/kg/d) treatment did not significantly alter rat blood pressure and cholesterol levels, therefore, the data were independent of lipid-lowering effects.
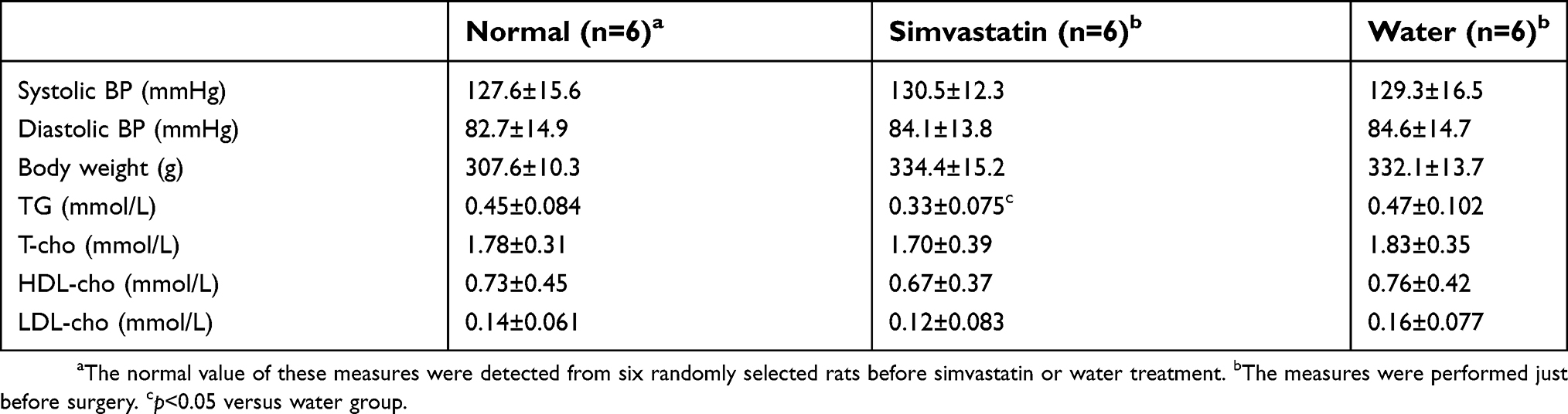 |
Table 2 Physiological and biochemical indicators |
Blood MMP-9 and TIMP-1 levels and MMP-9/TIMP-1 ratio
To investigate the effect of SIM on blood MMP-9 and TIMP-1 levels, ELISA and real-time PCR were performed. After 2 weeks of SIM treatment, levels of MMP-9 in the SIM+/t-PA+ group were significantly lower when compared to that in the SIM-/t-PA+ group (p<0.01). In addition, the MMP-9 level in the SIM-/t-PA+ group was higher compared to that in the saline group (p<0.01). After embolus injection, the TIMP-1 level in SIM-/t-PA+, SIM+/t-PA+ as well as in the saline group was higher compared to that in the sham group (p<0.01). However, no significant differences were observed between the SIM+/t-PA+ group, saline group, and SIM-/t-PA+ group (p>0.05) (Figure 1A). In addition, the blood MMP-9/TIMP-1 ratio was calculated, and t-PA significantly upregulated the MMP-9/TIMP-1 ratio (p<0.01 versus saline group). However, in the presence of SIM, the increased ratio was significantly neutralized (0.31±0.072 versus 0.20±0.056, p<0.01) (Figure 1C).
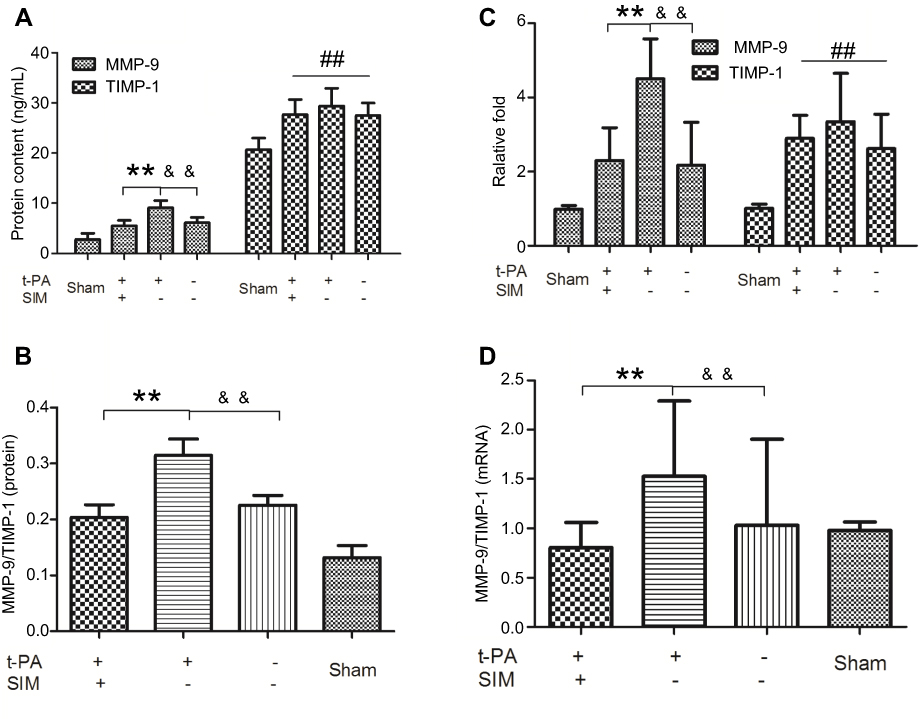 |
Figure 1 Simvastatin ameliorates t-PA-induced blood MMP-9/TIMP-1 ratio imbalance. Rats were pretreated with SIM (60 mg/kg/d) for the SIM+ group or water for the SIM- group for two weeks, then rats were received t-PA (10 mg/kg) for the t-PA+ group or normal saline for the t-PA- group 3 hrs after operation. Blood samples were collected via abdominal aorta puncture before sacrificed. (A) ELISA analysis of MMP-9 and TIMP-1 protein expression. (B) Real-time PCR analysis of MMP-9 and TIMP-1 mRNA levels. (C) The blood MMP-9/TIMP-1 protein ratio. (D) The blood MMP-9/TIMP-1 mRNA ratio. Bars express the mean ± SD of duplicates (n=6). **p<0.01 t-PA alone versus t-PA plus SIM; &&p<0.01 t-PA alone versus t-PA-/SIM-; ##p<0.01 versus sham group. Abbreviation: SIM, simvastatin. |
Leukocytes are the most important source of endogenous MMP-9 and TIMP-1;5 therefore, we investigated whether SIM might inhibit t-PA-induced MMP-9 and TIMP-1 mRNA. Data showed that mRNA levels of MMP-9 were increased in the SIM-/t-PA+ group (p<0.01 versus saline group); however, the t-PA-induced upregulated expression of MMP-9 mRNA was significantly inhibited by pretreatment with SIM (p<0.01). Furthermore, there was a marked increase in TIMP-1 mRNA level in the three embolism groups (all p<0.01 versus sham group); however, no significant difference was observed between these three embolism groups (Figure 1B). The MMP-9/TIMP-1 mRNA ratio was increased by t-PA treatment (p<0.01 versus saline group), and the effect was suppressed by treatment with SIM (1.53±0.76 versus 0.81±0.25, p<0.01) (Figure 1D).
Brain MMP-9 and TIMP-1 levels and ratio
To reveal MMP-9 and TIMP-1 expression, Western blot analysis was performed on ischemic brain tissue homogenates. The ischemic surgery significantly elevated the MMP-9 level in brain tissues (p<0.01, versus saline group). The MMP-9 level, which was increased by t-PA (p<0.01, SIM+/t-PA+ group versus saline group), was significantly suppressed by SIM treatment (p<0.01) (Figure 2A and B). In contrast, TIMP-1 expression increased after ischemic stroke; however, the difference between the SIM-/t-PA+ group and SIM+/t-PA+ group did not reach statistical significance (Figure 2A–C). Consistently, t-PA increased the MMP-9/TIMP-1 ratio (p<0.05 versus saline group), and SIM pretreatment neutralized the t-PA-induced increase (0.62±0.19 versus 1.15±0.33, p<0.01) (Figure 2D).
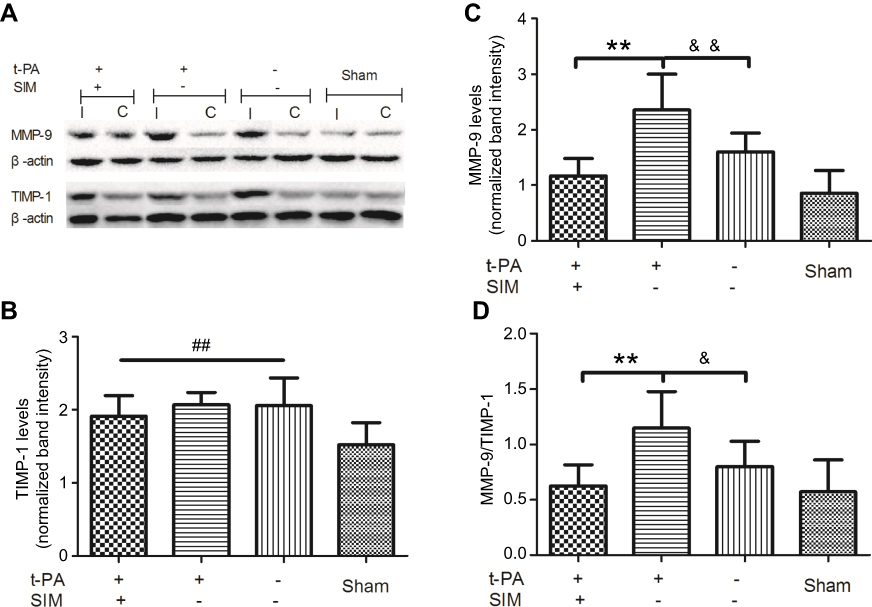 |
Figure 2 Simvastatin ameliorates t-PA-induced brain MMP-9/TIMP-1 ratio imbalance. Rats were pretreated with SIM (60 mg/kg/d) for the SIM+ group or water for the SIM- group for two weeks, then rats were received t-PA (10 mg/kg) for the t-PA+ group or normal saline for the t-PA- group 3 hrs after operation. (A) Representative images of gel electrophoresis results. (B) Quantitative analysis of MMP-9 level. (C) Quantitative analysis of TIMP-1 level. The normalized band intensity was calculated according to the formula: (MMP-9 or TIMP-1/β-actin)I/(MMP-9 or TIMP-1/β-actin)C. Bars express the mean±SD of duplicates (n=6). (D) The result of brain MMP-9/TIMP-1 protein ratio. Bars express the mean±SD of duplicates (n=6). **p<0.01 t-PA alone versus t-PA plus SIM; &p<0.05, &&p<0.01 t-PA alone versus t-PA-/SIM-; ##p<0.01 versus sham group. Abbreviations: I, infarct hemisphere; C, contralateral hemisphere; SIM, simvastatin. |
Neurological outcome and hemorrhagic transformation
The mNSS tests were used to assess the short-term functional outcome of ischemic injury during the first 24 hrs after surgery. The animals that received SIM treatment had significantly better neurologic function prior to saline (p<0.01), but showed no differences with the SIM-/t-PA+ group (Figure 3A). HT was analyzed as an index of vascular damage, and t-PA thrombolysis caused more severe vascular damage compared to the saline group as shown by a higher hemoglobin level (p<0.01) (Figure 3B). Hemorrhage severity in the SIM+/t-PA+ group was significantly lower compared to that in the SIM-/t-PA+ group (p<0.01), and no significant differences were observed when compared with the saline group.
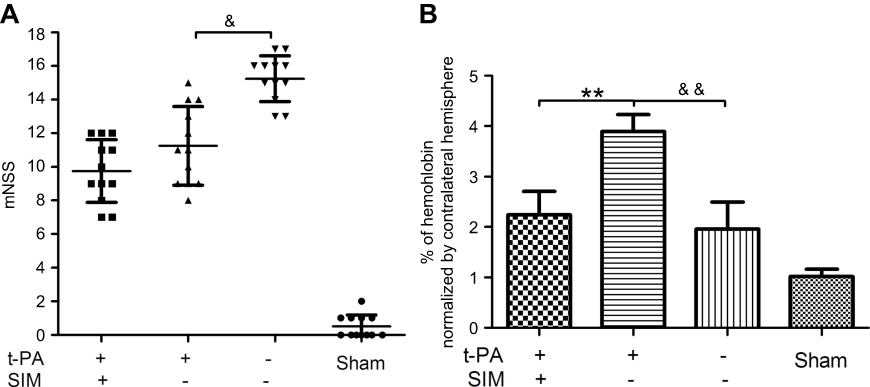 |
Figure 3 Pretreatment with simvastatin ameliorates t-PA-induced hemorrhage transformation in cerebral ischemic stroke without a significant change in neurological outcome in rats. (A) The result of mNSS tests. The mNSS tests were performed just before sacrificed (n=12/group). (B) The simvastatin-pretreated group showed lower intracranial hemorrhage than that of the t-PA thrombolysis-alone group. **p<0.01 t-PA alone versus t-PA plus SIM; &p<0.05, &&p<0.01 t-PA alone versus t-PA-/SIM. Abbreviation: mNSS, modified neurological severity score. |
Activity of RhoA and the effect of ROCK inhibitor
Little is known about the effect of t-PA thrombolysis on the activity of RhoA. To clarify this issue, we determined the changes in RhoA activity after different treatments. Twenty-four hours after infarction, the RhoA activity in the SIM-/t-PA+ group increased more than 1.5-fold compared with the saline group. However, the increased activity was suppressed by SIM pretreatment, which in the SIM+/t-PA+ group was significantly lower than in the SIM-/t-PA+ group. These findings suggested that the RhoA/ROCK pathway was involved in the inhibitory effect of SIM on t-PA-induced MMP-9/TIMP-1 imbalance (Figure 4A and B).
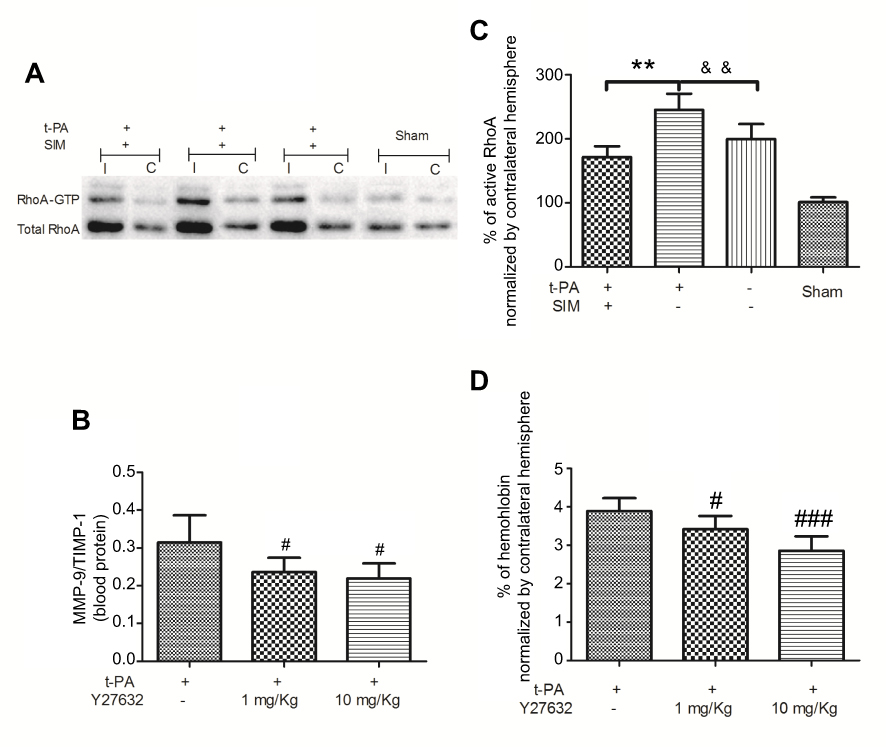 |
Figure 4 Simvastatin regulates the t-PA-induced RhoA activity and the t-PA-induced MMP-9/TIMP-1 imbalance and hemorrhage are ameliorated by Y27632. (A) Representative images of Rho pull-down assay. (B) Quantitative analysis of RhoA activity. The result is normalized as the formula: (active RhoA/total RhoA)I/(active RhoA/total RhoA)C. Rats were received t-PA (10 mg/kg) plus Y27632 (1 mg/kg or 10 mg/kg) or DMSO. (C) The t-PA-induced MMP-9/TIMP-1 imbalance is ameliorated by Y27632 with different concentrations. (D) The t-PA-induced hemorrhage is ameliorated by Y27632 with different concentrations. **p<0.01 t-PA alone versus t-PA plus SIM; &&p<0.01 t-PA alone versus t-PA-/SIM-; #p<0.05, ###p<0.001 versus t-PA alone. Abbreviations: I, infarct hemisphere; C, contralateral hemisphere; SIM, simvastatin. |
Hence, the role of the RhoA/ROCK pathway was further investigated by administration of t-PA with or without Y27632 in 36 rats. Utilizing the autologous blood emboli model, we compared the MMP-9/TIMP-1 ratio and hemoglobin level in Y-27632-treated rats versus other treatment groups. Detection of the MMP-9/TIMP-1 ratio and hemoglobin level was statistically different between Y-27632-absence and Y-27632-treated rats, thereby suggesting that RhoA/ROCK signaling is essential for the t-PA-induced MMP-9/TIMP-1 imbalance (Figure 4C) and hemorrhage (Figure 4D). Interestingly, both the MMP-9/TIMP-1 ratio and hemoglobin level in SIM-pretreated rats were lower when compared to that in Y-27632-treated rats (data not shown).
Discussion
Statins are established to prevent cardiac-cerebral vascular diseases. However, it has been revealed that the pharmacology properties of statins are not bound to the lipid-lowering effect.20 Moreover, the use of statins may be beneficial in ischemic stroke through their pleiotropic effects, such as anti-inflammatory activity,21 upregulation of endothelial nitric oxide synthase (eNOS), and t-PA.22,23 However, other beneficial activities are believed to be included, such as amelioration of t-PA-induced MMP-9.23 Kilic et al found that statins reduced t-PA induced reperfusion injury in a mouse AIS model,24 and Wang et al found that the effect may be performed through SIM by directly suppressing MMP-9 expression.23 However, the focus of the study by Kilic et al was to show that statins reversed the effects of t-PA by a mechanism that was independent from eNOS, and Wang et al only investigated the effect in astrocytes.23 Here, in addition to the pleiotropic effects of statins in the nervous system, we studied the action of statin on the suppression of t-PA-induced MMP-9/TIMP-1 imbalance in a rat AIS model. Moreover, we also observed the effect of statin on the reduction in t-PA-induced hemorrhagic transformation.
Previous studies have focused on the effects of combining statins with t-PA on experimental stroke;25–27 however, the results were contradictory. Statins were administrated 15 mins and 1 hr after ischemia in the studies performed by Campos25 and Guluma,26 respectively. Moreover, SIM did not effect neurological deficits or HT in these two studies. The results in a study by Lapchak et al, on the contrary, showed a salutary effect, which was verified in the study. The critical conflict was the different time points of SIM administrated, prior or post embolization. In the Lapchak study,27 SIM was pretreated to achieve a loading dose immediately after stroke to shorten the onset time. In contrast, in our study, SIM was pretreated 2 weeks prior to thrombolytic therapy to achieve a basic statinized condition. Because statins were used as one of the secondary prevention drugs for stroke and many AIS patients had a history of statin use, to simulate the conditions of these patients was a key objective.
It is known that t-PA treatment increases MMP-9 expression both in vivo and ex vivo.28,29 Our results are consistent with these previously published studies, the blood MMP-9 content and mRNA expression in the SIM-/t-PA+ group were significantly higher compared to the saline group. On the contrary, both the blood TIMP-1 content and mRNA expression were not significantly increased in t-PA-treated rats when compared with SIM-/t-PA-treated rats. We also revealed that SIM pretreatment could significantly reverse the t-PA-induced MMP-9 mRNA increase and therefore prevented the increase in MMP-9 levels. Furthermore, we demonstrated that in AIS, thrombosis with t-PA was associated with an abnormal expression of MMP-9 and imbalance of MMP-9/TIMP-1. The extent of remodeling the ECM depends upon the critical equilibrium between MMP-9 and TIMP-1. For ischemic stroke, an imbalance between MMP-9 and TIMP-1 is a key event in degradation of the blood–brain barrier (BBB) and therefore contributes to intracranial hemorrhage and vasogenic edema.8 In the present study, we investigated the changes in MMP-9/TIMP-1 ratio in brain tissue and serum, and both showed the same trends regarding changes. The higher MMP-9/TIMP-1 ratio potentially reflected increased MMP-9 activity, combined with elevated mRNA levels of MMP-9 in peripheral blood leukocytes after t-PA stimulation, may suggest a pathogenic link between elevated fibrinolysis and enhanced MMP-9 activity, potentially contributing to the increased hemorrhagic risk.
HT can be categorized based on its association with acute clinical symptoms, and only the parenchymal hematoma with substantial space-occupying effect influenced long-term outcome.1,30 Contrarily, some types of asymptomatic hemorrhage may be a marker of revascularization and may be helpful to resume neurologic outcome.31 In the present study, the hemoglobin level in rats in the SIM-/t-PA+ group was higher compared to that in the saline group as shown in Figure 3B. In addition, rats in the SIM-/t-PA+ group had a worse neurologic outcome (p<0.05, versus saline group), suggesting suggested that the type of HT in the animals was mostly symptomatic (Figure 3A). Consistent with clinical practice, symptomatic hemorrhage is usually attributed to thrombolytics if it occurs within 24 to 36 hrs of treatment.1 In fact, the consequences of symptomatic hemorrhage in the clinic, with a mortality rate of 45–83%,1 were more serious than were revealed in this study. Therefore, identifying a treatment modality that can ameliorate t-PA-induced hemorrhage is critical for stroke care. In the present study, SIM pretreatment significantly ameliorated t-PA-induced hemorrhage. A neurological improvement was found in the SIM pretreatment group, although, no statistically significant reduction in mNSS when compared with the t-PA alone group. This may attribute to the limited simvastatin pretreatment time. The pathogenesis of most patients who received thrombolysis was atherosclerosis, and statin administration was applicable to them for years before stroke onset. Therefore, the beneficial effect observed in our study has real potential for practical application.
Our results showed that the RhoA/ROCK pathway was involved in the inhibitory effect of SIM on t-PA-induced MMP-9/TIMP-1 imbalance. In previous studies, it has been demonstrated that SIM inhibited geranylgeranylation of RhoA, thereby preventing its translocation to the plasma membrane and subsequent activation of downstream activators, including ROCK.32 The effects of SIM were mimicked by the ROCK inhibitor Y27632, suggesting that the effects of SIM were mediated via inhibition of the RhoA/ROCK pathway, which was independent from cholesterol synthesis. In addition, we revealed that SIM had a greater inhibitory effect than Y-27362.
There were some limitations to our study. First, although SIM pretreatment could reverse t-PA-induced MMP-9/TIMP-1 imbalance both in brain and blood, it was still too cursory to conclude that SIM could reduce brain MMP-9 expression and activity. In addition, because SIM is lipophilic, it is expected to more likely penetrate the BBB when compared to hydrophilic statin (pravastatin, for example).33 Izidoro-Toledo and colleagues9 have demonstrated that lipophilic statins downregulated the MMP-9/TIMP-1 ratio in vitro, and no reputable study has ever conclusively demonstrated its BBB penetrability. Another reason may be that the source of MMP-9 in infarcted brain remains controversial. Recently, evidence has shown that the blood MMP-9 level has an intimate connection with the genesis and prognosis of HT,34,35 but in no study it is clarified where is the brain MMP-9 from. Second, although statins have shown vasoprotective effects, different statins may have different noncholesterol effects.36 Whether statins other than SIM will have the same effects observed in our study remains to be elucidated.
In conclusion, in our study, we simulated AIS patients on a SIM regimen and showed that pretreatment with statins ameliorated t-PA–induced hemorrhage transformation and MMP-9/TIMP-1 imbalance. We also demonstrated that the RhoA/ROCK pathway was involved.
Acknowledgment
This study was supported by the National Natural Science Foundation of China, No. 81501011.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Khatri P, Wechsler LR, Broderick JP. Intracranial hemorrhage associated with revascularization therapies. Stroke. 2007;38(2):431–440. doi:10.1161/01.STR.0000254524.23708.c9
2. Graham GD. Tissue plasminogen activator for acute ischemic stroke in clinical practice: a meta-analysis of safety data. Stroke. 2003;34(12):2847–2850. doi:10.1161/01.STR.0000101752.23813.C3
3. Montaner J, Alvarez-Sabin J, Molina CA, et al. Matrix metalloproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke. 2001;32(12):2762–2767.
4. Latour LL, Kang DW, Ezzeddine MA, Chalela JA, Warach S. Early blood-brain barrier disruption in human focal brain ischemia. Ann Neurol. 2004;56(4):468–477. doi:10.1002/ana.20199
5. Rosell A, Ortega-Aznar A, Alvarez-Sabin J, et al. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37(6):1399–1406. doi:10.1161/01.STR.0000223001.06264.af
6. Castellanos M, Sobrino T, Millan M, et al. Serum cellular fibronectin and matrix metalloproteinase-9 as screening biomarkers for the prediction of parenchymal hematoma after thrombolytic therapy in acute ischemic stroke: a multicenter confirmatory study. Stroke. 2007;38(6):1855–1859. doi:10.1161/STROKEAHA.106.481556
7. Dong X, Song YN, Liu WG, Guo XL. MMP-9, a potential target for cerebral ischemic treatment. Curr Neuropharmacol. 2009;7(4):269–275. doi:10.2174/157015909790031157
8. Li DD, Song JN, Huang H, et al. The roles of MMP-9/TIMP-1 in cerebral edema following experimental acute cerebral infarction in rats. Neurosci Lett. 2013;550:168–172. doi:10.1016/j.neulet.2013.06.034
9. Izidoro-Toledo TC, Guimaraes DA, Belo VA, Gerlach RF, Tanus-Santos JE. Effects of statins on matrix metalloproteinases and their endogenous inhibitors in human endothelial cells. Naunyn Schmiedebergs Arch Pharmacol. 2011;383(6):547–554. doi:10.1007/s00210-011-0623-0
10. Turner NA, O’Regan DJ, Ball SG, Porter KE. Simvastatin inhibits MMP-9 secretion from human saphenous vein smooth muscle cells by inhibiting the RhoA/ROCK pathway and reducing MMP-9 mRNA levels. Faseb J. 2005;19(7):804–806. doi:10.1096/fj.04-2852fje
11. Montaner J. Treatment with statins in the acute phase of ischemic stroke. Expert Rev Neurother. 2005;5(2):211–221. doi:10.1586/14737175.5.2.211
12. Delbosc S, Cristol JP, Descomps B, Mimran A, Jover B. Simvastatin prevents angiotensin II-induced cardiac alteration and oxidative stress. Hypertension. 2002;40(2):142–147.
13. Kalyanasundaram A, Elmore JR, Manazer JR, et al. Simvastatin suppresses experimental aortic aneurysm expansion. J Vasc Surg. 2006;43(1):117–124. doi:10.1016/j.jvs.2005.08.007
14. Niessen F, Hilger T, Hoehn M, Hossmann KA. Thrombolytic treatment of clot embolism in rat: comparison of intra-arterial and intravenous application of recombinant tissue plasminogen activator. Stroke. 2002;33(12):2999–3005.
15. Zhang L, Chopp M, Jia L, Cui Y, Lu M, Zhang ZG. Atorvastatin extends the therapeutic window for tPA to 6 h after the onset of embolic stroke in rats. J Cereb Blood Flow Metab. 2009;29(11):1816–1824. doi:10.1038/jcbfm.2009.105
16. Busch E, Kruger K, Hossmann KA. Improved model of thromboembolic stroke and rt-PA induced reperfusion in the rat. Brain Res. 1997;778(1):16–24.
17. Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–1011.
18. Koton S, Molshatzki N, Bornstein NM, Tanne D. Low cholesterol, statins and outcomes in patients with first-ever acute ischemic stroke. Cerebrovasc Dis. 2012;34(3):213–220. doi:10.1159/000342302
19. Tanaka T, Okamura T. Blood cholesterol level and risk of stroke in community-based or worksite cohort studies: a review of Japanese cohort studies in the past 20 years. Keio J Med. 2012;61(3):79–88.
20. Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14(1):37–44. doi:10.1016/j.molmed.2007.11.004
21. Menge T, Hartung HP, Stuve O. Statins–a cure-all for the brain? Nat Rev Neurosci. 2005;6(4):325–331. doi:10.1038/nrn1652
22. Asahi M, Huang Z, Thomas S, et al. Protective effects of statins involving both eNOS and tPA in focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25(6):722–729. doi:10.1038/sj.jcbfm.9600070
23. Wang S, Lee SR, Guo SZ, et al. Reduction of tissue plasminogen activator-induced matrix metalloproteinase-9 by simvastatin in astrocytes. Stroke. 2006;37(7):1910–1912. doi:10.1161/01.STR.0000226923.48905.39
24. Kilic E, Kilic U, Matter CM, Luscher TF, Bassetti CL, Hermann DM. Aggravation of focal cerebral ischemia by tissue plasminogen activator is reversed by 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor but does not depend on endothelial NO synthase. Stroke. 2005;36(2):332–336. doi:10.1161/01.STR.0000152273.24063.f7
25. Campos M, García-Bonilla L, Hernández-Guillamon M, et al. Combining statins with tissue plasminogen activator treatment after experimental and human stroke: a safety study on hemorrhagic transformation. CNS Neurosci Ther. 2013;19(11):863–870. doi:10.1111/cns.12181
26. Guluma KZ, Lapchak PA. Comparison of the post-embolization effects of tissue-plasminogen activator and simvastatin on neurological outcome in a clinically relevant rat model of acute ischemic stroke. Brain Res. 2010;1354:206–216. doi:10.1016/j.brainres.2010.07.050
27. Lapchak PA, Han MK. The 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor simvastatin reduces thrombolytic-induced intracerebral hemorrhage in embolized rabbits. Brain Res. 2009;1303:144–150. doi:10.1016/j.brainres.2009.09.064
28. Cuadrado E, Ortega L, Hernandez-Guillamon M, et al. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol. 2008;84(1):207–214. doi:10.1189/jlb.0907606
29. Ning M, Furie KL, Koroshetz WJ, et al. Association between tPA therapy and raised early matrix metalloproteinase-9 in acute stroke. Neurology. 2006;66(10):1550–1555. doi:10.1212/01.wnl.0000216133.98416.b4
30. Berger C, Fiorelli M, Steiner T, et al. Hemorrhagic transformation of ischemic brain tissue: asymptomatic or symptomatic? Stroke. 2001;32(6):1330–1335.
31. Molina CA, Alvarez-Sabin J, Montaner J, et al. Thrombolysis-related hemorrhagic infarction: a marker of early reperfusion, reduced infarct size, and improved outcome in patients with proximal middle cerebral artery occlusion. Stroke. 2002;33(6):1551–1556.
32. Turner NA, Aley PK, Hall KT, et al. Simvastatin inhibits TNFalpha-induced invasion of human cardiac myofibroblasts via both MMP-9-dependent and -independent mechanisms. J Mol Cell Cardiol. 2007;43(2):168–176. doi:10.1016/j.yjmcc.2007.05.006
33. Schweitzer M, Mitmaker B, Obrand D, et al. Atorvastatin modulates matrix metalloproteinase expression, activity, and signaling in abdominal aortic aneurysms. Vasc Endovascular Surg. 2010;44(2):116–122. doi:10.1177/1538574409348352
34. Barr TL, Latour LL, Lee KY, et al. Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke. 2010;41(3):e123–e128. doi:10.1161/STROKEAHA.109.570515
35. Kelly MA, Shuaib A, Todd KG. Matrix metalloproteinase activation and blood-brain barrier breakdown following thrombolysis. Exp Neurol. 2006;200(1):38–49. doi:10.1016/j.expneurol.2006.01.032
36. Turner NA, Midgley L, O’Regan DJ, Porter KE. Comparison of the efficacies of five different statins on inhibition of human saphenous vein smooth muscle cell proliferation and invasion. J Cardiovasc Pharmacol. 2007;50(4):458–461. doi:10.1097/FJC.0b013e318123767f
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.