Back to Journals » Cancer Management and Research » Volume 13
Silencing UHRF1 Enhances Radiosensitivity of Esophageal Squamous Cell Carcinoma by Inhibiting the PI3K/Akt/mTOR Signaling Pathway
Authors Hui B, Pan S, Che S, Sun Y, Yan Y, Guo J, Gong T, Ren J ![]() , Zhang X
, Zhang X
Received 17 March 2021
Accepted for publication 29 April 2021
Published 21 June 2021 Volume 2021:13 Pages 4841—4852
DOI https://doi.org/10.2147/CMAR.S311192
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bilikere Dwarakanath
Beina Hui,1 Shupei Pan,2 Shaomin Che,1 Yuchen Sun,1 Yanli Yan,1 Jia Guo,1 Tuotuo Gong,1 Juan Ren,1 Xiaozhi Zhang1
1Department of Radiation Oncology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi Province, 710061, People’s Republic of China; 2Department of Radiation Oncology, The Second Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi Province, 710004, People’s Republic of China
Correspondence: Juan Ren; Xiaozhi Zhang Tel +86 2985324029
; +86 2985324621
Email [email protected]; [email protected]
Purpose: Resistance to radiotherapy results in a high treatment failure rate for locally advanced esophageal squamous cell carcinoma (ESCC). Ubiquitin-like with plant homeodomain and ring-finger domains 1 (UHRF1), is associated with poor prognosis in ESCC. The present study aims to characterize the effect of UHRF1 silencing on the radiosensitivity of ESCC and its potential mechanism.
Methods: Both in vitro and in vivo experiments were conducted to observe the effects of UHRF1 silencing on the radiosensitivity of ESCC. The effects of UHRF1 silencing on the apoptosis of ESCC cells were assessed by flow cytometry. The expression of apoptosis-related factors (caspase-3 and Bcl-2), PI3K/Akt/mTOR signaling pathway-related factors (PTEN, p-Akt and Akt, p-mTOR and mTOR), and DNMT1 were measured via Western blot, and the status of PTEN methylation was detected by methylation-specific PCR. Immunohistochemistry was used to detect the expressions of PTEN, p-AKT, and p-mTOR in xenograft tumor tissues.
Results: In vitro and in vivo experiments showed that UHRF1 knock-down inhibited ESCC cell growth and enhanced their radiosensitivity. shUHRF1 combined with radiation significantly increased ESCC cell apoptosis. Meanwhile, it activated the expression of caspase-3 and inhibited the expression of Bcl-2. shUHRF1 inhibited the expression of DNMT1 and reduced the methylation of PTEN, and then upregulated the expression of PTEN to inhibit the PI3K/Akt/mTOR signaling pathway. On the contrary, the PI3K/Akt/mTOR signaling pathway can be activated by upregulation of UHRF1.
Conclusion: Our findings provide a theoretical basis for UHRF1 as a target to improve the radiosensitivity of ESCC.
Keywords: radiosensitivity, UHRF1, esophageal squamous carcinoma, shRNA, PI3K/Akt/mTOR
Introduction
Esophageal cancer currently ranks 9th in incidence and 6th in mortality among all cancers worldwide.1 China accounts for approximately 55% of new cases and deaths,1,2 of which more than 90% are esophageal squamous cell carcinoma (ESCC). Radiotherapy plays an important role in the comprehensive treatment of esophageal cancer. For patients with operable esophageal cancer, surgical resection after neoadjuvant chemoradiotherapy is the standard treatment. For patients with the non-operable disease, radical chemoradiotherapy is the only available treatment. Postoperative radiotherapy can improve the local control rate and survival rate of select ESCC cases. Due to advancements in radiotherapy and optimized dosage regimens, the quality of life for patients with esophageal cancer has improved over the last decade. However, the 5-year survival rate remains dismal at less than 20%.3 Acquired radio-resistance is associated with increased local tumor recurrence rates. Therefore, enhancing the radiosensitivity of esophageal cancer is an important task to improve its prognosis.
The ubiquitin-like with PHD and ring finger domains 1 (UHRF1), also known as ICBP90, is a nuclear protein involved in cell growth, which is an important connection between DNA methylation and histone modification.4–6 UHRF1 is also involved in important biological processes, such as cell proliferation, cell cycle regulation, and apoptosis.7–9 UHRF1 is overexpressed in various human tumor tissues and cells, such as hepatoblastoma, lung cancer, bladder cancer, and ESCC.10–14 The inhibition of UHRF1 can decrease tumor growth and alter the chemo- and radio-sensitivity of tumor cells.15,16 In ESCC, UHRF1 expression is closely related to poorer prognoses and regulates global DNA hypomethylation.13,14 However, the role of UHRF1 in the radiotherapy response of ESCC has not been fully investigated.
In this study, we investigated the function of UHRF1 on the radiosensitivity of ESCC by showing that UHRF1 inhibition increases the radiosensitivity of ESCC both in vitro and in vivo. The molecular mechanism was related to the induction of apoptosis, inhibition of PTEN methylation, and further inhibition of the PI3K/Akt/mTOR signaling pathway.
Materials and Methods
Cells Lines and Culture
The human ESCC, Eca109, and TE-1 cell lines were purchased from the Cell Bank of the Typical Culture Collection Committee of the Chinese Academy of Sciences (Shanghai, China). The 293T cells were purchased from Gima Gene Co., Ltd. (GenePharma, Jiangsu, China). The cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 µg/mL) in a humidified incubator at 37°C with 5% CO2.
shRNA Adenovirus Infection
RNA interference lentiviral recombinant vectors for targeting the UHRF1 gene were synthesized by Gima Gene Co., Ltd. (GenePharma, Jiangsu, China). The Eca109 and TE-1 cells were inoculated in a 24-well culture plate at 1×105 cells/well and cultured overnight. Next, the cells were infected with the lentiviral supernatant containing pGC-UHRF1-shRNA-LV-GFP or pGC-NC-LV-GFP according to the manufacturer’s instructions. After 48 h, the medium containing puromycin was used to select stable clones.
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted following the instructions of the Trizol reagent (Invitrogen, Carlsbad, CA, USA), reverse transcribed into cDNA, and amplified using the SYBR Green RT-PCR kit (Vazyme, Nanjing, China). The ABI7000 fluorescent quantitative PCR instrument (Applied Biosystems, Foster City, CA, USA) was used for amplification. The reaction procedure included 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 30 sec, and 60 °C for 30 sec. The melting curve was graphed, and the final data analyzed using the formula 2−ΔΔCt. Primer sequences were 5ʹ-GGGTTTCTCGTGTGGCGCTACCTTC-3ʹ (forward) and 5ʹ-TGCCGACTTCCGCTTCCACTTGC-3ʹ (reverse) for UHRF1.
Clonogenic Survival Assay for Cell Radiosensitivity
The cells were first cultured in six-well plates. Next, the cells were irradiated with a 4MV X-ray at a distance of 100 cm and dosages of 0, 2, 4, 6, and 8 Gy by a medical linear accelerator (Elekta Instruments, Inc., Stockholm, Sweden). At 14 post-irradiation, the cells were fixed and stained, and the clones were counted. The survival fraction (SF) was calculated, according to which survival lines were fitted using the single-hit multitarget model [Survival fraction (SF) = 1-(1-e^[-kD])^N] by GraphPad Prism 5.0 software. The radiobiological parameters (D0, Dq, N, and SF2) were calculated.
Western Blot
Cells were lysed using radioimmunoprecipitation assay lysis and extraction buffer (Pioneer Technology, Xi’an, China), and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Next, the proteins were transferred to polyvinylidene difluoride membranes (Millipore, MA, USA). The membranes were incubated at 4 °C overnight with the following primary antibodies: anti-UHRF1 (1:1000), anti-caspase-3 (1:2000), anti-Bcl-2 (1:1000), anti-PTEN (1:1000), anti-AKT (1:2000), anti-p-AKT (Ser473) (1:2000), anti-mTOR (1:1000), anti-mTOR (1:1000), and anti-GAPDH (1:1000). After washing the membranes with tris-buffered saline, 0.1% Tween 20 (TBST), an HRP-labeled goat anti-rabbit secondary antibody (1:8000) was added and incubated for 2 h at 37°C with gentle shaking. The signal was detected with an ECL kit (Millipore, Burlington, MA, USA), and the final scanned images were analyzed using Image Lab Imaging System software. The antibodies used for Western blot were all purchased from Cell Signaling Technology (Beverly, MA, USA).
Flow Cytometry Assay
ESCC cells were seeded in 6-well plates and divided into four groups: NC group (NC), shUHRF1 group (shUHRF1), NC + irradiation (NC + IR) group and shUHRF1+ irradiation group (shUHRF1 + IR). The IR groups received 6 Gy X-ray irradiation 24 h before detection. The apoptosis rate was measured following the instructions of the Annexin V-APC/7-AAD apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA). Briefly, the cells were resuspended in 500 μL binding buffer, mixed well after the addition of 5 μL Annexin V-APC, mixed well after the further addition of 5 μL 7-AAD, incubated at room temperature in the dark for 5–15 min, and analyzed by the FASC Calibur MT flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Subcutaneous Xenograft Mouse Model
Twelve four-week-old specific-pathogen-free (SPF) grade female BALB/c-nu nude mice, weighing 16 to 18 g [license number SCXK (Xiang) 2016–0002], were obtained from Hunan Slake Jingda Experimental Animal Co. Ltd. (Changsha, China), and housed in the Medical Experimental Animal Center (Xi’an Jiaotong University, China). To establish the ESCC xenograft model, Eca-109 cells (1 × 107) were injected subcutaneously into the right armpit of each BALB/c nude mouse. To explore the role of UHRF1 in radiosensitivity in vivo, four groups were designed: NC group (NC), shUHRF1 group (shUHRF1), NC + irradiation (NC + IR) group, and shUHRF1+ irradiation group (shUHRF1 + IR). The IR groups received 2 Gy X-ray IR for five consecutive days. The IR treatment was initiated 22 days after transplantation. The transplanted tumors were monitored from the first day after tumor formation. Tumor volume changes were monitored every 3 days by recording the (a) maximum diameter and (b) shortest diameter. Tumor volume was calculated as [tumor volume = ab2/2]. After 15 days, the mice were sacrificed, and the tumors were excised and weighed. The transplanted tumors were excised and photographed, and tumor volume was recorded. The tumor specimens were aliquoted and processed according to immunohistochemical methodologies. All mouse experiments were conducted according to the Public Health Service Policy on Humane Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Xi’an Jiaotong University.
UHRF1 Expression Vector Construction and Transfection
PCR primers were designed based on the UHRF1 gene number in GenBank (NM_013282) and the restriction map of the eukaryotic expression vector pIRES2-ZsGreen1 (Takara Bio USA, Inc., Mountain View, CA, USA). The plasmid pIRES2-ZsGreen1 and the target fragments of homo UHRF1 were digested with EcoR I and Xho I, recovered and purified from a 1% agarose gel, and ligated. The ligated product was transformed into DH5α competent cells. Single colonies were picked and identified by agarose gel electrophoresis, and the correct colonies were sequenced. The clone with the correct sequence was named pIRES2-ZsGreen1-Homo UHRF1. The recombinant pIRES2-ZsGreen1-Homo UHRF1 vector and empty pIRES2-ZsGreen1-NC vector were transfected into Eca109 cells using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
Methylation-Specific PCR (MSP)
Using methylation modified DNA as a template, PCR amplification was performed using methylated and un-methylated primer pairs separately. The ammonium sulfite modification kit was purchased from Qiagen (Hilden, Germany). The primers were synthesized by Shanghai Shenggong Biotechnology Co. (Japan). The methylated primer sequences of PTEN were 5ʹ-GGTGTTGATGTTTATTTTTTTTAAGC-3ʹ (forward) and 5ʹ-TATCAAATCTATTTACAACCCCGAT-3ʹ (reverse). The un-methylated primer sequences of PTEN were 5ʹ-TGGTGTTGATGTTTATTTTTTTTAAGT-3ʹ (forward) and 5ʹ-TATCAAATCTATTTACAACCCCAAT-3ʹ (reverse). The amplification product was 161 bp in length. Based on the instructions of the PCR kit (Invitrogen, Waltham, MA, USA), the amplification conditions were 95 °C for 5 min, 35 cycles at 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s, 72 °C for 10 min, and 4 °C for 10 min.
Immunohistochemical Staining
Tumor tissues were fixed with 10% methanol and embedded in paraffin for immunohistochemical staining. The tissue sections were heated in 100 °C citrate buffer (pH = 6.0) for 15 min to facilitate antigen retrieval. Endogenous peroxidase was blocked with 3% hydrogen peroxide, and a diluted primary antibody was added and incubated overnight in a wet box at 4 °C. Next, a horseradish peroxidase (HRP)-labeled goat anti-rabbit/mouse secondary antibody was added and incubated at room temperature for 20 min. A 3,3ʹ-diaminobenzidine (DAB) solution was dripped onto the sections and allowed to incubate for 5 min to produce a visible color, which was followed by counterstaining with hematoxylin and dehydration with ethanol. After adding xylene, the sections were sealed with neutral gum. The primary antibodies included anti-UHRF1 (1:50, SanYing, Wuhan, China), anti-PTEN (1:50, Abcam, Cambridge, UK), anti-pmTOR (1:20, Bioss, Beijing, China), and anti-pAKT (1:100, CST, Beverly, MA, USA). Image-pro Plus 6.0 software (Version 7, Media Cybernetics, Rockville, MD, USA) was used to quantitatively analyze the immunohistochemical staining of mouse tissues.
Statistical Analysis
All data were analyzed using SPSS 23.0 software (IBM, Chicago, IL, USA). The numerical data were reported as the mean ± standard deviation (SD). The SNK-q test was used for pairwise comparison. A p-value of <0.05 was considered statistically significant.
Results
UHRF1 Knockdown Sensitizes ESCC Cells to Irradiation in vitro and in vivo
The PGC-UHRF1-LV-GFP #1 - #3 and the pGC-NC-LV-GFP negative control lentiviral vectors were transduced into Eca109 and TE-1 cells separately. pGC-UHRF1-LV-GFP #2 demonstrated the clearest interference of UHRF1, as determined by qRT-PCR (Figure 1A). Hence, this vector was selected for subsequent experiments. The interference sequences of pGC-UHRF1-LV-GFP: 5ʹ-AGACGGAAUUGGGGCUGUATT-3ʹ (sense sequence) and 5ʹ-UACAGCCCCAAUUCCGUCUTT-3ʹ (antisense sequence). The negative control sequences of pGC-NC-LV-GFP: 5ʹ-UUCUUCGAACGUGUCACGUTT-3ʹ and antisense sequence: 5ʹ-AGCUGACACGUUCGGAGAATT-3ʹ. Western blot analysis further confirmed that shRNA successfully inhibited the expression of the UHRF1 protein in the two cell lines (Figure 1B). Colony formation assays showed that the radiosensitivity of ESCC cells increased after inhibition of UHRF1 expression compared with the NC group (P < 0.05) (Figure 2A and B). The specific radiobiological parameters are shown in Table 1.
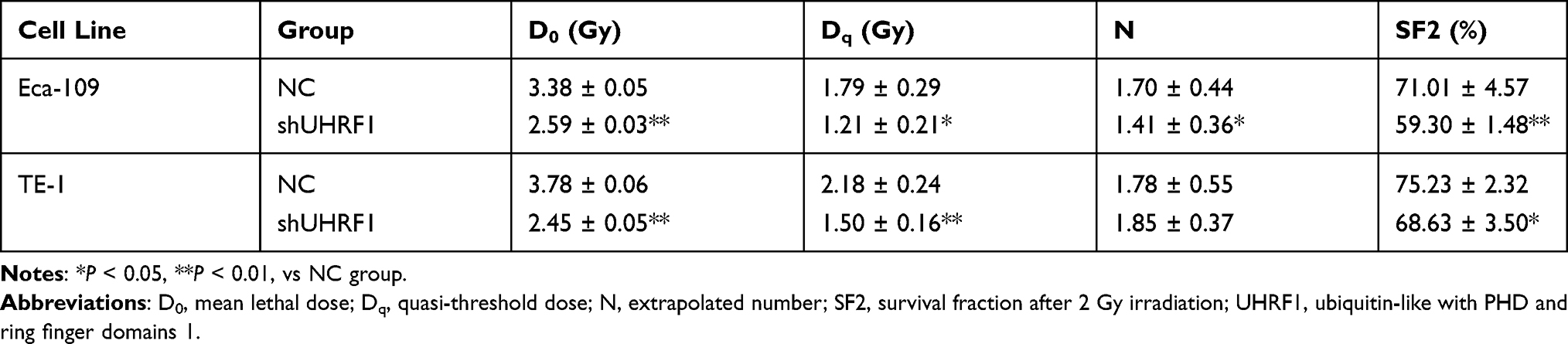 |
Table 1 Biological Radiation Parameters of ESCC Cells in the Different Groups |
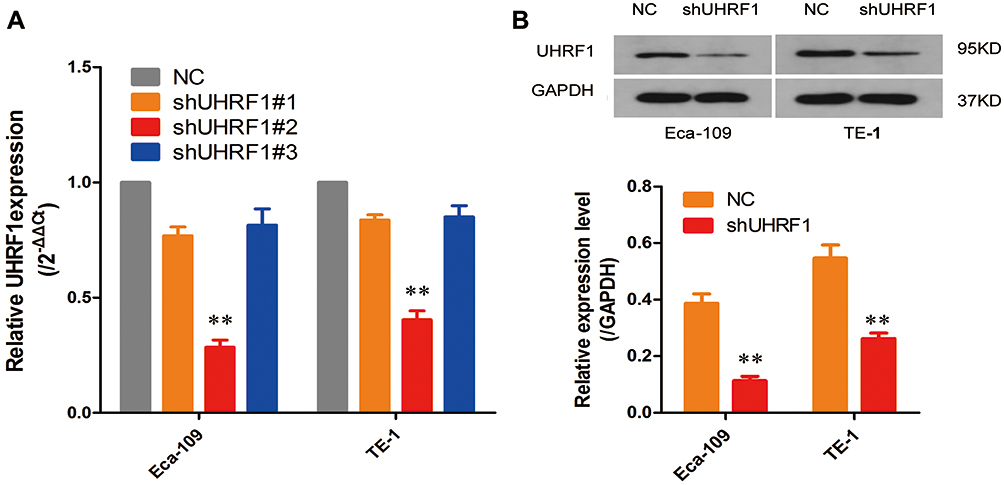 |
Figure 1 (A) The knockdown efficiency of the three UHRF1 shRNAs was confirmed by quantitative real-time polymerase chain reaction, qRT-PCR. (B) The knockdown efficiency of UHRF1 shRNA was confirmed by Western blot analysis in ECA109 and TE-1 cells. GAPDH was used for normalization (**P < 0.01 vs NC group). |
 |
Figure 2 UHRF1 knockdown sensitized ESCC cells to irradiation in vitro and in vivo. (A) Clonogenic survival assays were performed to measure radiosensitivity in Eca109 and TE-1 cells. (B) Survival curves in NC and shUHRF1 cells treated with irradiation are shown. (C) Tumor volumes of xenograft mouse tumors. (D) Tumor weight of xenograft mouse tumors. (E) Representative data of tumors in nude mice bearing Eca109 cells (*P < 0.05, **P < 0.01 vs NC group). |
To investigate the effects of the UHRF1 gene on the growth of ESCC in vivo, we constructed a nude mouse xenograft model using Eca109 cells with the UHRF1 gene stably downregulated. The effects of UHRF1 silencing on the growth of xenograft tumors in nude mice revealed a reduction in tumor growth (P < 0.05). This phenomenon was more obvious after combined with x-ray irradiation (P < 0.01) (Figure 2C). The weight and volume of the transplanted tumors were the largest in the NC group and the smallest in the shUHRF1 + IR group (Figure 2D and E). These results indicate that the tumorigenicity of ESCC cells in nude mice was significantly inhibited with the downregulation of UHRF1, while the radiosensitivity of the resulting xenografts increased substantially.
Depletion of UHRF1 Enhances Irradiation-Induced Apoptosis in ESCC Cells
Flow cytometry results showed that both the downregulation of UHRF1 expression and X-ray irradiation increased the apoptosis of ESCC cells (shUHRF1, NC+IR group vs NC group, P < 0.05). In addition, the combination of irradiation with UHRF1 down-regulation further increases the apoptosis rate (shUHRF1+IR group vs NC group, P < 0.01). The apoptosis rates of Eca109 cells were as follows: NC: 5.90 ± 0.76%, shUHRF1: 9.74 ± 0.96%, NC+IR: 14.33 ± 0.36%, shUHRF1+IR: 19.20 ± 0.68%. The apoptosis rates of TE-1 cells were as follows: NC:6.30 ± 0.71%, shUHRF1: 9.07 ± 0.11%, NC+IR: 12.28 ± 0.77%, shUHRF1+IR: 17.18 ± 1.75% (Figure 3A). To explore the molecular mechanism of how shUHRF1 enhanced radiation-induced apoptosis of ESCC cells, the expression levels of apoptosis-related proteins Bcl-2 and caspase-3 were examined. Western blot results demonstrated an increase in active caspase-3 protein expression, along with a decrease in Bcl-2 protein expression in both the shUHRF1 group and in NC+IR group. These changes were even more apparent in the shUHRF1+ IR group (Figure 3B). This phenomenon indicated that the inhibition of UHRF1 could enhance radiosensitivity by increasing apoptosis in esophageal cancer cells.
 |
Figure 3 Knockdown of UHRF1 induced apoptosis in ESCC cells before and after 6 Gy irradiation. (A) Apoptotic changes in ESCC cells in different groups were examined with FCM. (B) Western blot analysis of apoptosis-related markers in ESCC cells in different groups (*P < 0.05, **P < 0.01 vs NC group). |
Depletion of UHRF1 Enhances the Radiosensitivity of ESCC Cells by Changing the Methylation Status of PTEN and Weakening the Activity of the PI3K/Akt/mTOR Signaling Pathway
The PI3K/Akt/mTOR signaling pathway is a major anti-apoptotic pathway in tumor cells. After activation, Akt promotes cell survival and apoptotic resistance by regulating the expression of several apoptosis-related proteins, such as caspase-3 and Bcl-2. Inhibition of the PI3K/Akt/mTOR signaling pathway has been found to regulates radiosensitivity of tumor cells. In this study, the expression of key proteins involved in the PI3K/Akt/mTOR pathway, such as Akt, p-Akt (Ser473), mTOR, and p-mTOR (ser2448), along with the negative regulator of the signaling pathway, such as PTEN, were detected by Western blot. As shown in Figure 4, shUHRF1 could significantly decrease the expression of p-Akt and p-mTOR while increasing the expression of PTEN in Eca-109 and TE-1 cell lines (P < 0.05). This change was more pronounced in the shUHRF1+ IR group (P < 0.01). The expression of p-Akt and p-mTOR in the shUHRF1 group and NC+IR group showed no differences (P > 0.05). The expression levels of Akt and mTOR proteins also showed no differences between the groups (P > 0.05). These findings suggest that the enhanced ESCC radiosensitivity resulting from the downregulation of UHRF1 is associated with increased expression of PTEN and decreased PI3K/Akt/mTOR signaling.
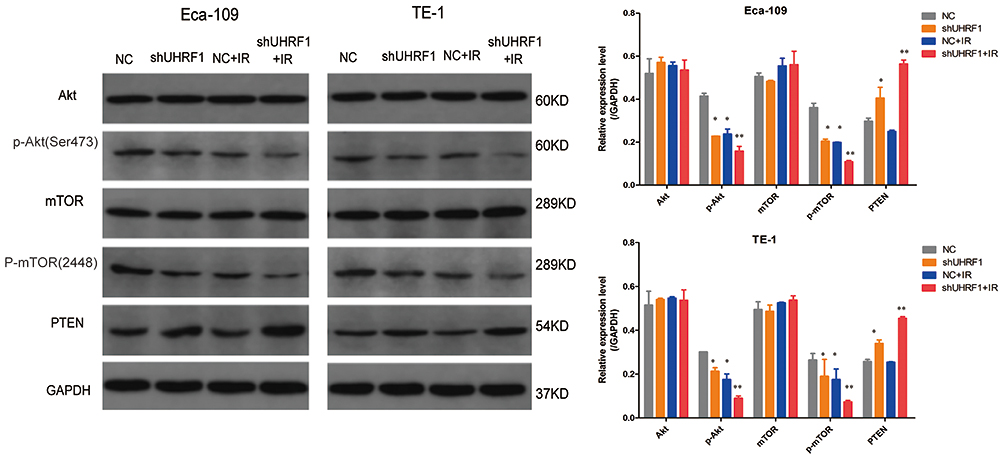 |
Figure 4 The effect of UHRF1 knockdown on the expression of key proteins in the PI3K/Akt/mTOR signaling pathway in ESCC cells before and after 6 Gy irradiation (*P < 0.05, **P < 0.01 vs NC group). |
To further verify the regulatory effect of UHRF1 on the PI3K/Akt/mTOR signaling pathway in ESCC, we constructed a eukaryotic UHRF1 expression vector (pIRES2-ZsGreen1-Homo UHRF1) and transfected it into Eca109 cells using liposomes (Eca109 UHRF1 cells). Western blot results showed an increase in p-Akt and p-mTOR protein expression, along with a decrease in PTEN protein expression (P < 0.05). However, the expression of total AKT and mTOR showed no significant alterations (Figure 5). These results indicate that UHRF1 regulates the expression of PTEN, thereby affecting the PI3K/Akt/mTOR signaling pathway.
 |
Figure 5 The effect of UHRF1 overexpression on the expression of key proteins in the PI3K/Akt/mTOR signaling pathway in Eca109 cells (*P < 0.05, **P < 0.01 vs NC group). |
UHRF1 is the “core protein” of epigenetic regulation and has been shown to bind DNMT1 to hemi-methylated DNA to maintain the stability of methylated DNA (7). PTEN is known to be methylated in esophageal cancer and other malignant tumor tissues. We hypothesized that the high expression of PTEN resulting from the downregulation of UHRF1 in ESCC cells might be related to PTEN demethylation. Western blot results showed that down-regulation of UHRF1 expression in Eca109 and TE-1 cells leads to significant reductions in DNMT1 expression, as compared with the NC group (P < 0.05) (Figure 6A). The MSP method was used to detect changes in PTEN methylation before and after the downregulation of UHRF1. PTEN methylation is weakened after UHRF1 is downregulated, while PTEN mRNA expression is increased (Figure 6B and C). Therefore, we speculate that UHRF affects the PI3K/Akt/mTOR signaling pathway activity by regulating PTEN methylation.
 |
Figure 6 Effect of UHRF1 knockdown on PTEN methylation. (A) Western blot analysis of the expression of DNMT1 in the NC and shUHRF1 groups (**P < 0.01 vs NC group). (B) Methylation of the PTEN gene was measured by MSP; M: amplified using primers for methylated DNA, U: amplified using primers for unmethylated DNA. (C) PTEN mRNA was measured by RT-PCR. (**P < 0.01 vs NC group). |
Depletion of UHRF1 Inhibited PI3K/Akt/mTOR Signaling Pathway Activity in vivo
To confirm the effects of UHRF1 on the PI3K/Akt/mTOR signaling pathway in vivo, we determined the expression of PI3K/Akt/mTOR signaling pathway-related proteins in the nude mice. The immunohistochemical analysis showed that the expressions of p-Akt and p-mTOR were all downregulated in the shUHRF1 group, NC + IR group, and shUHRF1 + IR group, when compared with the NC group. This decrease was more obvious in the shUHRF1+IR group than in the other two groups (P < 0.05). PTEN expression was higher in the shUHRF1 group, NC+IR group, and shUHRF1 + IR group, when compared with the NC group. The increase was more obvious in the shUHRF1 + IR group (Figure 7). These findings were consistent with the results in vitro.
 |
Figure 7 Representative images of immunohistochemical (IHC) staining of the PTEN, p-Akt, and p-mTOR proteins in xenograft mouse tumor samples (*P < 0.05, **P < 0.01 vs NC group). |
Discussion
Radiosensitivity can be influenced by hypoxia, cell cycle distribution, DNA damage repair and cytotoxicity, and the presence of tumor stem cells.17 Various genetic variations associated with the biological effects of radiation can cause differences in radiosensitivity. UHRF1 is overexpressed in many malignant tumors and is associated with several epigenetic alterations, such as DNA methylation and histone modification. It has been shown that inhibition of UHRF1 can inhibit tumor growth and change the sensitivity of cancer cells to chemotherapy and radiation. It was previously reported by Li et al16 that HeLa cells transfected with the UHRF1 sense sequence demonstrated lower radiosensitivity than non-transfected cells. Similarly, Muto et al18 found that UHRF1-inactivated homozygous mouse embryonic stem cells were more sensitive to x-rays and ultraviolet rays than embryonic stem cells containing wild-type UHRF1 or the heterozygous-inactivated UHRF1 gene. UHRF1 is overexpressed and acts as an independent prognostic factor in ESCC.14 In this study, we silenced the expression of UHRF1 in ESCC cells using shRNA and found that inhibition of UHRF1 expression could improve the radiosensitivity of ESCC. Radiobiological parameters showed that D0 and Dq and N values of shUHRF1 group were significantly reduced, and the cell survival curve was downward in the shUHRF1 group, suggesting that the interference with UHRF1 expression could be a potential target to improve the radiosensitivity of esophageal cancer.
Irradiation-induced apoptosis is the primary form of tumor cell death following radiotherapy. The Bcl-2 family plays a key role in the molecular mechanism of apoptosis induction, in which Bcl-2 and Bax proteins are directly related to apoptosis. The increase of Bax promotes apoptosis, as Bcl-2 is an apoptosis suppressor gene.19 The Caspase family is a group of cysteine proteases in the process of apoptosis, among which Caspase-3 is the most critical apoptotic protease in the apoptosis cascade, and once activated, a downstream cascade will occur, making apoptosis inevitable.20 We detected by flow cytometry and found that UHRF1 silencing, both alone and in combination with irradiation, could increase caspase-3 levels and the apoptosis of cells, while reducing the levels of the apoptosis inhibitory protein Bcl-2. These findings demonstrate that silencing UHRF1 can enhance the radiosensitivity by increasing the apoptosis of ESCC cells. The tumor growth rate depends on the proliferation rate and the degree of apoptosis. The inhibition of tumor growth is based on strategies to promote apoptosis while inhibiting proliferation. Nude mouse xenograft experiments showed that after silencing UHRF1 expression, tumor volumes and weights were lower than in the NC group. After combining UHRF1 silencing with radiotherapy, the growth inhibition of transplanted tumors was more obvious. Hence, the inhibition of UHRF1 expression using RNA interference in vivo significantly inhibits the growth of esophageal cancer xenograft tumors in nude mice. When compared with irradiation alone, the tumor inhibition effects become more apparent.
The PI3K/Akt/mTOR signaling pathway is an important signal transduction pathway in cells, as its activation is associated with angiogenesis, proliferation, differentiation, and apoptosis of tumor cells. The activation of components in this pathway contribute to the poor prognosis of many tumors.21,22 Inhibition of the PI3K/Akt/mTOR signaling pathway has been found to enhance radiosensitivity in nasopharyngeal and prostate cancers.23,24 Recent studies have shown that UHRF1 regulates the survival and differentiation of iNKT cells through the Akt-mTOR pathway.25 Knockdown of UHRF1 enhances the autophagy of chondrocytes through the PI3K/AKT/mTOR signaling pathway.26 Therefore, we speculated that the increased radiosensitivity resulting from the downregulation of UHRF1 in esophageal cancer might be related to the inhibition of the PI3K/Akt/mTOR signaling activity. In the current study, both in vivo and in vitro experiments showed that UHRF1 silencing in ESCC cells decreases the expression of p-Akt and p-mTOR, while increasing UHRF1 expression elevates the expression of pAkt and p-mTOR. This suggests that UHRF1 silencing may induce apoptosis by inhibiting the PI3K/Akt/mTOR signaling pathway, thereby increasing the radiosensitivity of ESCC cells.
The molecular structure of UHRF1 contains several functional domains, and UHRF1 can form a complex with histone deacetylase 1 (HDAC1) and DNA methyltransferase 1 (DNMT1) through its unique SET and RING finger-associated (SRA) domain. UHRF1 inhibits the expression of several tumor suppressor genes (TSG) through the regulation of histone modification and DNA methylation.27,28 For example, UHRF1 overexpression in breast cancer is related to the hypermethylation of the BRCA1,29 while UHRF1 overexpression in lung cancer is significantly associated with the hypermethylation of the promoter regions of CDKN2A and RASSF1. In addition, the silencing of UHRF1 expression weakens the methylation of RASSF1, CYGB, and CDH13.30 As such, PTEN is a negative regulator of the PI3K/Akt/mTOR signaling pathway. In the current study, we found that PTEN is negatively correlated with UHRF1, p-Akt, and p-mTOR. As a tumor suppressor gene, the abnormal methylation of PTEN can directly cause its silencing and loss of function as a tumor suppressor gene. Therefore, we examined the methylation status of PTEN before and after silencing UHRF1 and found that UHRF1 participates in the maintenance of PTEN methylation. After UHRF1 silencing, PTEN methylation was reduced, and PTEN mRNA expression levels were increased. In addition, DNMT1 expression decreased after UHRF1 silencing, indicating that UHRF1 indirectly affects the PI3K/Akt/mTOR signaling pathway through the regulation of PTEN methylation.
Conclusions
In summary, we demonstrate that silencing of UHRF1 can enhance the radiosensitivity of ESCC cells by inducing cell apoptosis and reducing the methylation level of PTEN. In return, this leads to the inhibition of the PI3K/Akt/mTOR signaling pathway. We hope that UHRF1 inhibitors may be developed as a combination therapy to improve the efficacy of radiotherapy for patients with esophageal cancer in the future.
Abbreviations
UHRF1, Ubiquitin-like with plant homeodomain and ring-finger domains 1; ESCC, esophageal squamous cell carcinoma; IR, irradiation; SF2, survival fraction after 2 Gy irradiation; D0, mean lethal dose; Dq, quasi-threshold dose; DNMT1, DNA methyltransferase 1.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author Xiaozhi Zhang upon reasonable request.
Funding
This work was supported by the Natural Science Basic Research Program of Shaanxi (Beina Hui, 2016JM8110) and the National Natural Science Foundation of China (Xiaozhi Zhang, 81773239). The funding bodies had no role in the design of the study, the collection, analysis, or interpretation of the data, or writing the manuscript.
Disclosure
The authors reported no conflicts of interest for this work and declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492
2. Liang H, Fan JH, Qiao YL. Epidemiology, etiology, and prevention of esophageal squamous cell carcinoma in China. Cancer Biol Med. 2017;14:33–41. doi:10.20892/j.issn.2095-3941.2016.0093
3. Zhang Y. Epidemiology of esophageal cancer. World J Gastroenterol. 2013;19:5598–5606. doi:10.3748/wjg.v19.i34.5598
4. Bronner C, Fuhrmann G, Chedin FL, Macaluso M, Dhe-Paganon S. UHRF1 links the histone code and DNA methylation to ensure faithful epigenetic memory inheritance. Genet Epigenet. 2010;2009:29–36.
5. Nishiyama A, Yamaguchi L, Sharif J, et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013;502:249–253. doi:10.1038/nature12488
6. Sharif J, Koseki H. Recruitment of Dnmt1 roles of the SRA protein Np95 (Uhrf1) and other factors. Prog Mol Biol Transl Sci. 2011;101:289–310.
7. Lorenzato M, Caudroy S, Bronner C, et al. Cell cycle and/or proliferation markers: what is the best method to discriminate cervical high-grade lesions? Hum Pathol. 2005;36:1101–1107. doi:10.1016/j.humpath.2005.07.016
8. Zhang H, Liu H, Chen Y, et al. A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat Commun. 2016;7:10201. doi:10.1038/ncomms10201
9. Zhang Q, Qiao L, Wang X, Ding C, Chen JJ. UHRF1 epigenetically down-regulates UbcH8 to inhibit apoptosis in cervical cancer cells. Cell Cycle. 2018;17:300–308. doi:10.1080/15384101.2017.1403686
10. Beck A, Trippel F, Wagner A, et al. Overexpression of UHRF1 promotes silencing of tumor suppressor genes and predicts outcome in hepatoblastoma. Clin Epigenetics. 2018;10:27. doi:10.1186/s13148-018-0462-7
11. Unoki M, Daigo Y, Koinuma J, Tsuchiya E, Hamamoto R, Nakamura Y. UHRF1 is a novel diagnostic marker of lung cancer. Br J Cancer. 2010;103:217–222. doi:10.1038/sj.bjc.6605717
12. Unoki M, Kelly JD, Neal DE, Ponder BA, Nakamura Y, Hamamoto R. UHRF1 is a novel molecular marker for diagnosis and the prognosis of bladder cancer. Br J Cancer. 2009;101:98–105. doi:10.1038/sj.bjc.6605123
13. Nakamura K, Baba Y, Kosumi K, et al. UHRF1 regulates global DNA hypomethylation and is associated with poor prognosis in esophageal squamous cell carcinoma. Oncotarget. 2016;7:57821–57831. doi:10.18632/oncotarget.11067
14. Ye J, Zhang Y, Liang W, Huang J, Wang L, Zhong X. UHRF1 is an independent prognostic factor and a potential therapeutic target of esophageal squamous cell carcinoma. J Cancer. 2017;8:4027–4039. doi:10.7150/jca.21256
15. Fang L, Shanqu L, Ping G, et al. Gene therapy with RNAi targeting UHRF1 driven by tumor-specific promoter inhibits tumor growth and enhances the sensitivity of chemotherapeutic drug in breast cancer in vitro and in vivo. Cancer Chemother Pharmacol. 2012;69:1079–1087. doi:10.1007/s00280-011-1801-y
16. Li XL, Meng QH, Fan SJ. Adenovirus-mediated expression of UHRF1 reduces the radiosensitivity of cervical cancer HeLa cells to gamma-irradiation. Acta Pharmacol Sin. 2009;30:458–466. doi:10.1038/aps.2009.18
17. Schulz A, Meyer F, Dubrovska A. Cancer stem cells and radioresistance: DNA repair and beyond. Cancers (Basel). 2019;11:862. doi:10.3390/cancers11060862
18. Muto M, Kanari Y, Kubo E, et al. Targeted disruption of Np95 gene renders murine embryonic stem cells hypersensitive to DNA damaging agents and DNA replication blocks. J Biol Chem. 2002;277:34549–34555. doi:10.1074/jbc.M205189200
19. Xie Z, Koyama T, Suzuki J, et al. Coronary reperfusion following ischemia: different expression of bcl-2 and bax proteins, and cardiomyocyte apoptosis. Jpn Heart J. 2001;42:759–770. doi:10.1536/jhj.42.759
20. Atkinson EA, Barry M, Darmon AJ, et al. Cytotoxic T lymphocyte-assisted suicide. Caspase 3 activation is primarily the result of the direct action of granzyme B. J Biol Chem. 1998;273:21261–21266. doi:10.1074/jbc.273.33.21261
21. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi:10.1038/nrc2664
22. Maity A, Bernhard EJ. Modulating tumor vasculature through signaling inhibition to improve cytotoxic therapy. Cancer Res. 2010;70:2141–2145. doi:10.1158/0008-5472.CAN-09-3615
23. Chang L, Graham PH, Hao J, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014;5:e1437. doi:10.1038/cddis.2014.415
24. Wang W, Li Q, Liu T, Chen L. Dual phosphoinositide 3-kinase/Mammalian target of rapamycin inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radiosensitivity in nasopharyngeal carcinoma. Am Assoc Cancer Res. 2014;74:3951.
25. Cui Y, Chen X, Zhang J, et al. Uhrf1 controls iNKT cell survival and differentiation through the Akt-mTOR axis. Cell Rep. 2016;15:256–263. doi:10.1016/j.celrep.2016.03.016
26. Shi X, Han L, Sun T, et al. Silencing UHRF1 enhances cell autophagy to prevent articular chondrocytes from apoptosis in osteoarthritis through PI3K/AKT/mTOR signaling pathway. Biochem Biophys Res Commun. 2020;529:1018–1024. doi:10.1016/j.bbrc.2020.06.032
27. Alhosin M, Omran Z, Zamzami MA, et al. Signalling pathways in UHRF1-dependent regulation of tumor suppressor genes in cancer. J Exp Clin Cancer Res. 2016;35:174. doi:10.1186/s13046-016-0453-5
28. Sidhu H, Capalash N. UHRF1: the key regulator of epigenetics and molecular target for cancer therapeutics. Tumour Biol. 2017;39:1010428317692205. doi:10.1177/1010428317692205
29. Jin W, Chen L, Chen Y, et al. UHRF1 is associated with epigenetic silencing of BRCA1 in sporadic breast cancer. Breast Cancer Res Treat. 2010;123:359–373. doi:10.1007/s10549-009-0652-2
30. Daskalos A, Oleksiewicz U, Filia A, et al. UHRF1-mediated tumor suppressor gene inactivation in nonsmall cell lung cancer. Cancer. 2011;117:1027–1037. doi:10.1002/cncr.25531
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.