")
Back to Journals » Cancer Management and Research » Volume 13
Silencing of LncRNA SNHG16 Downregulates Cyclin D1 (CCND1) to Abrogate Malignant Phenotypes in Oral Squamous Cell Carcinoma (OSCC) Through Upregulating miR-17-5p
Authors Wang Q, Han J, Xu P, Jian X, Huang X, Liu D
Received 22 December 2020
Accepted for publication 26 January 2021
Published 22 February 2021 Volume 2021:13 Pages 1831—1841
DOI https://doi.org/10.2147/CMAR.S298236
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Qiuling Wang,1,* Jingxin Han,1,* Pu Xu,1 Xinchun Jian,1,2 Xieshan Huang,1 Deyu Liu1
1Stomatology Center, Affiliated Haikou Hospital of Xiangya Medical College, Central South University, Haikou, Hainan, 570208, People’s Republic of China; 2Department of Oral and Maxillofacial Surgery, Xiangya Hospital, Central South University, Central South University, Changsha, Hunan, 410008, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Deyu Liu
Stomatology Center, Affiliated Haikou Hospital, Xiangya Medical College, Central South University, Renmin Road No. 43, Haikou, Hainan, 570208, People’s Republic of China
Email [email protected]
Background: Targeting the long non-coding RNAs (LncRNAs)-microRNAs (miRNAs)-mRNA competing endogenous RNA (ceRNA) networks has been proved as an effective strategy to treat multiple cancers, including oral squamous cell carcinoma (OSCC). Based on this, the present study identified a novel LncRNA SNHG16/miR-17-5p/CCND1 signaling pathway that played an important role in regulating the pathogenesis of OSCC.
Methods: The expression levels of cancer-associated genes were examined by Real-Time qPCR and Western Blot at transcriptional and translated levels, respectively. CCK-8 assay was performed to determine cell proliferation, and cell apoptosis ratio was measured by the Annexin V-FITC/PI double staining assay. Transwell assay was performed to examine cell migration, and dual-luciferase reporter gene system assay was used to validate the ceRNA networks.
Results: LncRNA SNHG16 and CCND1 were upregulated, while miR-17-5p was downregulated in OSCC tissues and cell lines, compared to their normal counterparts. Also, miR-17-5p negatively correlated with both LncRNA SNHG16 and CCND1 mRNA, but LncRNA SNHG16 was positively relevant to CCND1 mRNA in OSCC tissues. By performing the gain- and loss-of-function experiments, we noticed that LncRNA SNHG16 overexpression aggravated the malignant phenotypes, such as cell proliferation, viability, migration and epithelial-mesenchymal transition (EMT) in OSCC cells, while LncRNA SNHG16 knock-down had opposite effects. Furthermore, our dual-luciferase reporter gene system evidenced that LncRNA SNHG16 sponged miR-17-5p to upregulate CCND1 in OSCC cells, and the inhibiting effects of LncRNA SNHG16 ablation on OSCC progression were abrogated by both downregulating miR-17-5p and overexpressing CCND1. Finally, the xenograft tumor-bearing mice models were established, and our data validated that LncRNA SNHG16 served as an oncogene to promote tumorigenicity of OSCC cells in vivo.
Conclusion: Taken together, targeting the LncRNA SNHG16/miR-17-5p/CCND1 axis hindered the development of OSCC, and this study provided potential diagnostic and therapeutic biomarkers for OSCC in clinic.
Keywords: oral squamous cell carcinoma, competing endogenous RNA, LncRNA SNHG16, MiR-17-5p, cyclin D1
Background
Oral squamous cell carcinoma (OSCC) is the predominant subtype of oral cancer,1 which is characterized by high incidence and mortality, and brought huge health burden to human beings worldwide.2–4 Although the advances in the traditional therapies, such as surgery, chemotherapy and radiotherapy had been reached, the prognosis in OSCC patients is still very poor as the result of its unclear underlying mechanisms.5–7 Therefore, researchers recently focused on exploring the molecular characteristics of OSCC pathogenesis,4,8 and identified that multiple long non-coding RNAs (LncRNAs) participated in modulating OSCC development.9–11 Interestingly, emerging data suggested that manipulation of the cancer-associated LncRNAs was effective to hamper OSCC development.9–11 Among all the LncRNAs, LncRNA SNHG16 was identified as an oncogene to facilitate the development of pancreatic cancer,12 gastric cancer,13 and bladder cancer.14 Notably, Li et al noticed that upregulation of LncRNA SNHG16 enhanced progression and carcinogenesis in OSCC,15 however, the regulating mechanisms of LncRNA SNHG16 in OSCC development were still largely unknown.
MicroRNAs (miRNAs) are a group of endogenous small non-coding RNAs with approximately 22 nucleotides,16,17 and recent data suggested that miRNAs acted as “tumor suppressors” or “oncogenes” to inhibit or promote cancer progression in OSCC.18–20 Specifically, our team selected miR-17-5p for investigation, which had been reported to be closely associated with the radio-resistance and development of OSCC,21–23 but the detailed regulating mechanisms were still unclear. Based on the information from the previous publications,24,25 miR-17-5p exerted opposite effects in different cancer types. On the one hand, miR-17-5p facilitated cancer progression in cervical cancer,25 on the other, miR-17-5p functioned as a “tumor suppressor” in triple-negative breast cancer,24 hence, it was necessary and meaningful to investigate the role of miR-17-5p in regulating OSCC development. In addition, researchers agreed that LncRNAs served as “RNA sponges” for miRNAs,26,27 and previous publications suggested that miR-17-5p was the downstream target of LncRNA SNHG16 in bladder cancer,28 which was validated by our bioinformatics analysis through the online starBase software (http://starbase.sysu.edu.cn/), and rendered the possibility that LncRNA SNHG16 might sponge miR-17-5p in OSCC cells in a similar manner.
Cyclin D1 (CCND1) is crucial for sustaining cell proliferation, and mutations or deficiency of CCND1 may cause cell-cycle arrest and death.29–31 Of note, CCND1 served as an oncogene to promote the development of various cancers, such as colorectal cancer,32 breast cancer33 and OSCC.34,35 Interestingly, researchers noticed that silencing of CCND1 was proved to be an effective strategy to slow down OSCC progression.36 Of note, recent data indicated that miRNAs targeted the 3ʹ untranslated regions (3ʹUTRs) of CCND1 mRNA for CCND1 degradation and inhibition,37–39 and our preliminary work showed that there existed potential binding sites in miR-17-5p and 3ʹUTRs of CCND1. Given the fact that miR-17-5p was the downstream target of LncRNA SNHG16,28 it was reasonable to conjecture that miR-17-5p might serve as a “bridge” for LncRNA SNHG16 and CCND1, and investigating the regulating mechanisms of LncRNA SNHG16-miR-17-5p-CCND1 competing endogenous RNA (ceRNA) networks in OSCC became meaningful and necessary.
Based on the information from the existed literatures, the present study managed to investigate the involvement of LncRNA SNHG16/miR-17-5p/CCND1 signaling cascade in regulating the pathogenesis of OSCC, which broadened our knowledge in this field and provided potential biomarkers for OSCC diagnosis and therapy.
Materials and Methods
Clinical Specimen Collection
The 50 paired clinical tissues, including OSCC tissues and their corresponding adjacent normal epithelia, were collected from the primary OSCC patients from 2014 to 2018 in Affiliated Haikou Hospital of Xiangya Medical College, Central South University. We assured that all the patients had not accepted any other therapies before surgical resection, such as chemotherapy, radiotherapy, etc., and all the tissues were examined and judged by two pathologists in our hospital. The above tissues were immediately stored at the refrigerator with −70°C conditions for further analysis, and Real-Time qPCR was used to examine the expression levels of LncRNA SNHG16, miR-17-5p and CCND1 mRNA in the tissues, and their correlations were analyzed. In addition, 8 paired tissues were randomly selected, and Western Blot analysis was performed to examine the protein levels of CCND1. We had obtained the signed informed consent from all the participants, and all the clinical experiments were conducted in accordance with the Declaration of Helsinki, and were approved by the Ethics Committee of Affiliated Haikou Hospital of Xiangya Medical College, Central South University (No. 201712DA7619382193).
Cell Culture and Vectors Transfection
The human normal oral cell line (NOK) and OSCC cell lines (CAL27, TCA8113, OEC-M1 and TW2.6) were purchased from American Type Culture Collection (ATCC, USA) and Cell Bank of Chinese Academy of Sciences (Shanghai, China), respectively. All the cells were cultured in the Dulbecco’s Modified Eagle’s medium (DMEM, Life Technology, NY, USA) containing 10% fetal bovine serum (FBS, Gibco, USA) under the standard culture conditions with 5% CO2 humidified atmosphere at 37°C. The overexpression and downregulation vectors for LncRNA SNHG16 were designed and synthesized by Sangon Biotech (Shanghai, China), miR-17-5p mimic/inhibitor and CCND1 overexpression vectors were obtained from GenePharma (Shanghai, China). All the above vectors were delivered into the OSCC cells by using the Lipofectamine 2000 transfection reagent (Invitrogen, CA, USA) in keeping with the manufacturer’s protocol.
Real-Time qPCR Analysis
The OSCC tissues and cells were harvested and prepared, the total RNA was extracted by using the TRIzol reagent (Ambion, USA) in keeping with the manufacturer’s protocol. Next, the cDNA for LncRNA SNHG16, miR-17-5p, CCND1 mRNA, β-actin mRNA and U6 mRNA were synthesized by using different cDNA synthesis kits. For mRNA and LncRNA, the SuperScript III reverse transcriptase reagent (Invitrogen, USA) was used. For miRNA, the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, USA) was employed. After that, the Maestro GreenEvaGreen qPCR Master Mix (Maestrogen, USA) was purchased to quantify the expression levels of LncRNA and mRNA, while the QuantiTect SYBR Green PCR system (QIAGEN, Shanghai, China) was utilized to examine the levels of miR-17-5p. β-actin and U6 were used as internal control, and each experiment contained at least 3 repetitions. The primer sequences for LncRNA SNHG16, miR-17-5p, CCND1, β-actin and U6 are listed in Table 1.
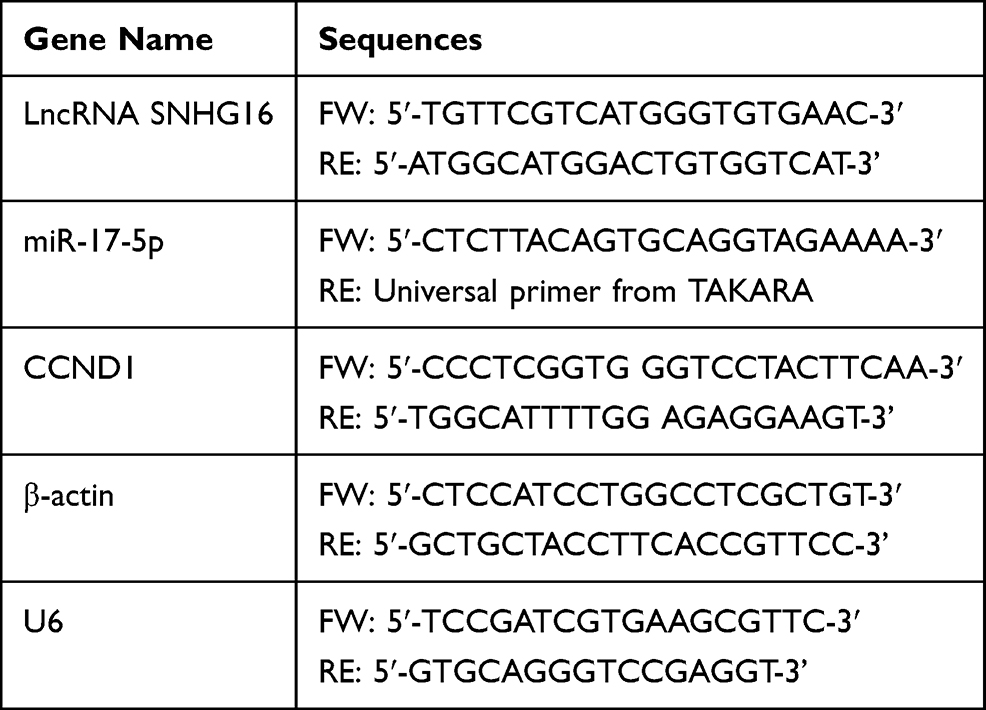 |
Table 1 The Primers Used for Real-Time qPCR |
Western Blot Analysis
The total proteins were extracted from the OSCC tissues and cells by using the commercial RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions, and the expression levels of CCND1, N-cadherin and Vimentin were examined by using the Western Blot analysis as previously described,34,35 which were normalized by the internal control β-actin. The detailed information for the primary antibodies against CCND1, N-cadherin, Vimentin and β-actin are listed in Table 2.
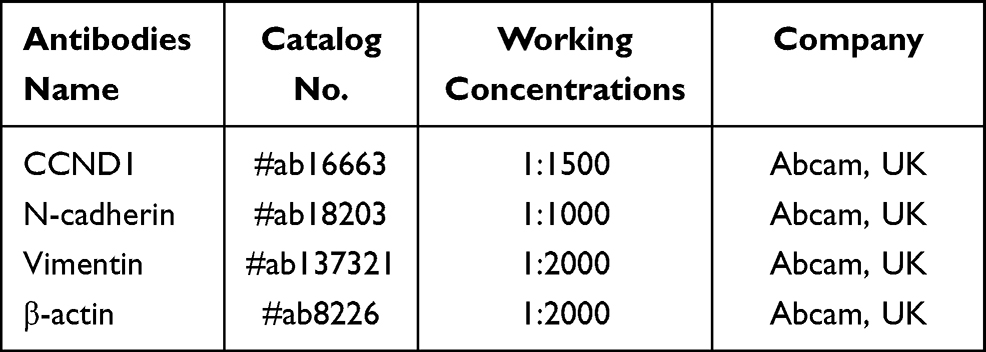 |
Table 2 The Antibodies Used for Western Blot Analysis |
Cell Counting Kit-8 (CCK-8) Assay
The OSCC cells were pre-transfected with different vectors, and were cultured in the 96-well plates under the standard culture conditions for 0 h, 24 h, 48 h and 72 h, respectively. After that, the commercial CCK-8 assay kit (YEASEN, Shanghai, China) was obtained to examine cell proliferation abilities based on the experimental procedures provided by the producer. Briefly, the cells were incubated with the CCK-8 solution for 1.5 h in the incubator, and the plates were shattered. After that, the optical density (OD) values were examined at the wavelength of 450 nm to reflect the relative proliferation abilities of the cells.
Transwell Assay
The OSCC cells were subjected to differential treatments, and the transwell assay was performed to evaluate cell migration abilities. In brief, the transwell plates were purchased from Corning Co-Star (USA), and the cells were detached and suspended in the serum-free DMEM medium (Life Technology, NY, USA). After that, the cells were seeded into the apical chamber, and the basolateral chambers were fulfilled with the DMEM medium containing 20% FBS (Gibco, USA). Subsequently, the transwell plates were placed in the incubator and the cells were cultured for 24 h, and the cells on the filters were stained with 0.1% crystal violet for 30 min at room temperature for visualization. Finally, the cells were photographed and counted under a light microscope to evaluate cell migration abilities.
Dual-Luciferase Reporter Gene System Assay
According to the previous publications and the data from the online starBase software (http://starbase.sysu.edu.cn/), we obtained the binding sites of miR-17-5p with the wild-type LncRNA SNHG16 and 3ʹ UTR of CCND1 mRNA, which were named as wild-type LncRNA SNHG16 (wt-SNHG16) and CCND1 (wt-CCND1). In addition, the targeting sites in the LncRNA SNHG16 and 3ʹ UTR of CCND1 mRNA were mutated, and were presented as mut-SNHG16 and mut-CCND1, respectively. The above vectors were cloned into the pmirGLO firefly luciferase-expressing vectors (Promega, USA), and were co-transfected with miR-17-5p mimic and inhibitor into the OSCC cells by using the Lipofectamine 2000 transfection reagent (Invitrogen, CA, USA) based on the protocol provided by the producer. At 48 h post-transfection, the Dual-luciferase Reporter Assay System (Promega, USA) was utilized to measure the relative luciferase activities in the above OSCC cells. Renilla luciferase was regarded as the control reporter for normalization.
Establishment of Xenograft Tumor-Bearing Mice Models
The male athymic Balb/c nude mice were purchased from Animal Center of Central South University, and all the mice aged 6 weeks and weighed 20–23 g. The OSCC cells were pre-transfected with LncRNA SNHG16 overexpression and downregulation vectors, and were subcutaneously implanted into the dorsal flank regions at the density of 2 × 10 7 per mouse. After that, the tumor volume was monitored every 5 days, and the volumes were calculated by using the following formula: (π × length × width2)/6. At the 5 days post-injection, the mice were anesthetized by intravenously injecting Barbiturate at the concentration of 100 mg/kg, and the standards for successful euthanasia included: (1) no cardiac arrest; (2) no spontaneous breath for at least 3 min; and (3) no blinking reflex in mice. Subsequently, the mice were sacrificed and the tumors were obtained by surgical resection. The tumors were photographed and weighed to reflect tumorigenesis of OSCC cells in vivo. All the animal experiments were conducted in accordance with the Laboratory Animal Welfare Guidelines of Research Animal Center of Xiangya Medical College, and were approved by the Ethics committee of Affiliated Haikou Hospital of Xiangya Medical College, Central South University (No. 201803DA76328123).
Data Analysis
The data were shown as Means ± Standard Deviation (SD), and SPSS 18.0 software and Graph Pad Prism 8.0 software were used to analyze the data. The means from two groups were compared by using the Student’s t-test, and one-way ANOVA analysis was conducted to compare the means from multiple groups. In addition, Pearson Correlation analysis was used to analyze the correlations among LncRNA SNHG16, miR-17-5p and CCND1 mRNA in OSCC clinical tissues. Each experiment had at least 3 repetitions, and P < 0.05 was regarded as statistical significance.
Results
LncRNA SNHG16, miR-17-5p and CCND1 Were Aberrantly Expressed in OSCC Tissues and Cells
The OSCC tissues (N = 50) and their paired adjacent normal tissues were collected and the expression levels of LncRNA SNHG16, miR-17-5p and CCND1 mRNA were determined by conducting Real-Time qPCR assay (Figure 1A–C). As shown in Figure 1A–C, LncRNA SNHG16 (Figure 1A) and CCND1 mRNA (Figure 1C) tended to be enriched, while miR-17-5p (Figure 1B) was downregulated in OSCC tissues, in contrast with their normal counterparts. Next, by performing the Pearson Correlation Analysis, we noticed that miR-17-5p negatively correlated with LncRNA SNHG16 (Figure 1D) and CCND1 mRNA (Figure 1E), while LncRNA SNHG16 was positively relevant to CCND1 mRNA in the OSCC tissues (Figure 1F). Finally, the human normal oral cell line (NOK) and OSCC cell lines (CAL27, TCA8113, OEC-M1 and TW2.6) were obtained, and we found that LncRNA SNHG16 (Figure 2A) and CCND1 (Figure 2C–E) were high-expressed, while miR-17-5p was low-expressed in OSCC cells (Figure 2B), compared to the normal NOK cells. Given that LncRNA SNHG16 tended to be enriched in CAL27 and TCA8113 cells (Figure 2A), the above two cell lines were chosen for further analysis.
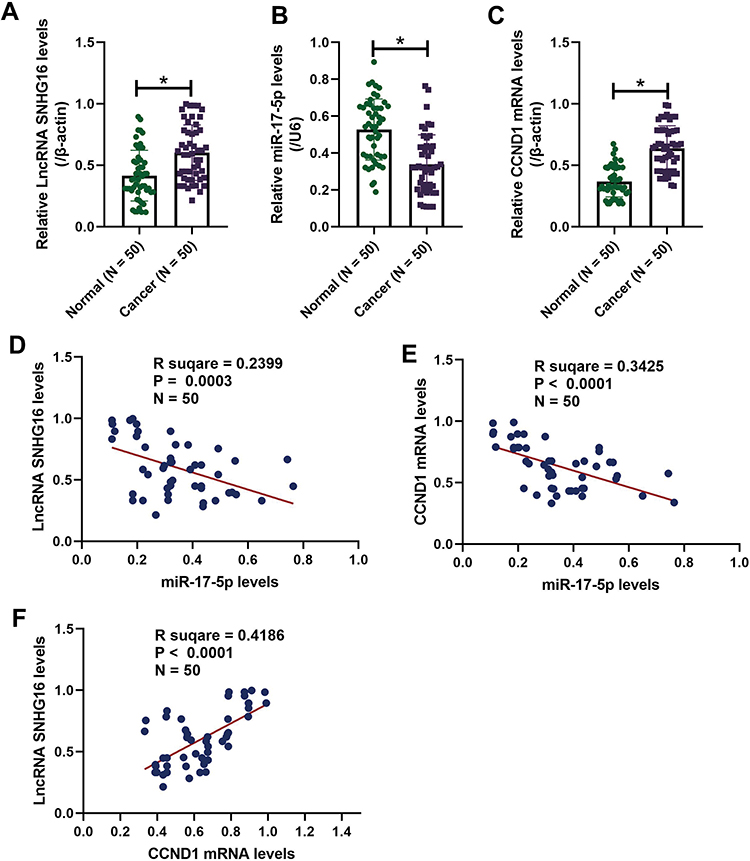 |
Figure 1 LncRNA SNHG16, miR-17-5p and CCND1 were aberrantly expressed in the clinical tissues collected from patients with oral squamous cell carcinoma (OSCC). The 50 paired cancer and adjacent normal tissues were obtained from OSCC patients, and Real-Time qPCR analysis was conducted to examine the expression levels of (A) LncRNA SNHG16, (B) miR-17-5p and (C) CCND1 mRNA. The Pearson correlation analysis was next conducted, and the results showed that miR-17-5p negatively correlated with (D) LncRNA SNHG16 and (E) CCND1 mRNA, but (F) LncRNA SNHG16 was positively relevant to CCND1 mRNA in OSCC tissues. *P < 0.05. |
 |
Figure 2 The expression status of LncRNA SNHG16, miR-17-5p and CCND1 in OSCC cells. The human normal oral cell line (NOK) and OSCC cell lines (CAL27, TCA8113, OEC-M1 and TW2.6) were obtained, and Real-Time qPCR was employed to determine the expression levels of (A) LncRNA SNHG16, (B) miR-17-5p and (C) CCND1 mRNA in the above cells. (D, E) Western Blot analysis was conducted to examine the protein levels of CCND1 in the OSCC cells. Each experiment repeated at least 3 times, and *P < 0.05. |
LncRNA SNHG16 Acted as an Oncogene to Promote OSCC Development in vitro and in vivo
Previous data suggested that LncRNA SNHG16 facilitated the development of OSCC,15 which were validated by our work. Initially, the LncRNA SNHG16 overexpression and downregulation vectors were successfully delivered into CAL27 and TCA8113 cells (Figure 3A), and further gain- and loss-function experiments evidenced that LncRNA SNHG16 positively regulated malignant phenotypes of OSCC cells in vitro and in vivo. Mechanistically, the CCK-8 assay results showed that OSCC cell proliferation abilities were enhanced by LncRNA SNHG16 overexpression, but were inhibited by LncRNA SNHG16 ablation (Figure 3B and C). Consistently, through Annexin V-FITC/PI double staining assay, we found that silencing of LncRNA SNHG16 triggered apoptotic cell death in OSCC cells (Figure 3D). Also, the xenograft tumor-bearing mice models were established, and the results evidenced that silencing of LncRNA SNHG16 slowed down tumor growth in vivo, which were promoted by upregulating LncRNA SNHG16 (Figure 3E). Furthermore, the transwell assay was utilized to examine migration, and the results showed that LncRNA SNHG16 positively regulated cell mobility in OSCC cells (Figure 4A and B). Finally, we noticed that LncRNA SNHG16 positively regulated N-cadherin and Vimentin to promote EMT in OSCC cells (Figure 4C–F).
 |
Figure 3 LncRNA SNHG16 promoted cell proliferation and growth, and inhibited cell apoptosis in OSCC cells (CAL27 and TCA8113) in vitro and in vivo. (A) The overexpression and downregulation vectors for LncRNA SNHG16 were delivered into the OSCC cells. (B, C) CCK-8 assay was performed to examine cell proliferation abilities in OSCC cells. (D) Knock-down of LncRNA SNHG16 triggered apoptotic cell death in OSCC cells. (E) The xenograft tumor-bearing mice models were established, and tumor weight was used to evaluate tumorigenesis of the OSCC cells. (Note: “Con” indicated “Control”, “OE” represented “LncRNA SNHG16 overexpression” and “KD” suggested “knock-down of LncRNA SNHG16”). Each experiment repeated at least 3 times, and *P < 0.05. |
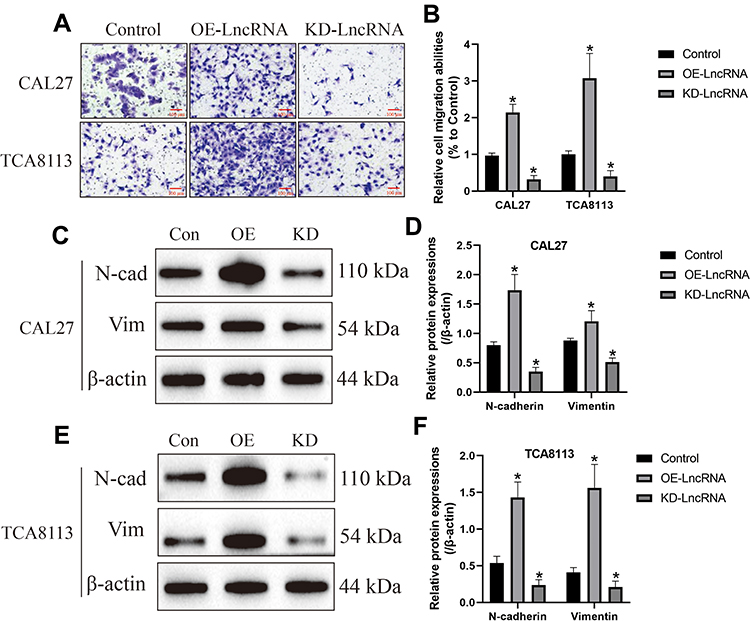 |
Figure 4 LncRNA SNHG16 positively regulated cell migration and EMT in OSCC cells. (A, B) Transwell assay was conducted to examine cell migration abilities. (C–F) Western Blot analysis was conducted to examine the expression levels of EMT-associated proteins (N-cadherin and Vimentin) in OSCC cells. (Note: “Con” indicated “Control”, “OE” represented “LncRNA SNHG16 overexpression” and “KD” suggested “knock-down of LncRNA SNHG16”). Each experiment repeated at least 3 times, and *P < 0.05. |
LncRNA SNHG16 Positively Regulated CCND1 by Sponging miR-17-5p in OSCC Cells
Based on the information from the previous publications and the online starBase software (http://starbase.sysu.edu.cn/), we noticed that miR-17-5p potentially bound to LncRNA SNHG16 (Figure 5A) and 3ʹ untranslated region (3ʹUTR) of CCND1 mRNA (Figure 5D). Therefore, we conjectured that miR-17-5p might serve as a “bridge” to combine LncRNA SNHG16 and CCND1 in OSCC. To validate this hypothesis, the binding sites in LncRNA SNHG16 and CCND1 were mutated, and were cloned into the luciferase reporter vectors by a third-party company (Sangon, Shanghai, China). Subsequently, the above vectors were co-transfected with miR-17-5p mimic and inhibitor into CAL27 and TCA8113 cells, and the results suggested that miR-17-5p mimic decreased luciferase activities in the OSCC cells co-transfected with wild-type LncRNA SNHG16 and CCND1, instead of their mutant counterparts, while miR-17-5p inhibitor had opposite effects (Figure 5B and C; Figure 5E and F). Next, we noticed that knock-down of LncRNA SNHG16 decreased the expression levels of CCND1 at both transcriptional (Figure 5G) and translated (Figure 5H–I) levels, which were reversed by silencing miR-17-5p (Figure 5G–I), indicating that LncRNA SNHG16 sponged miR-17-5p to upregulate CCND1 in OSCC cells.
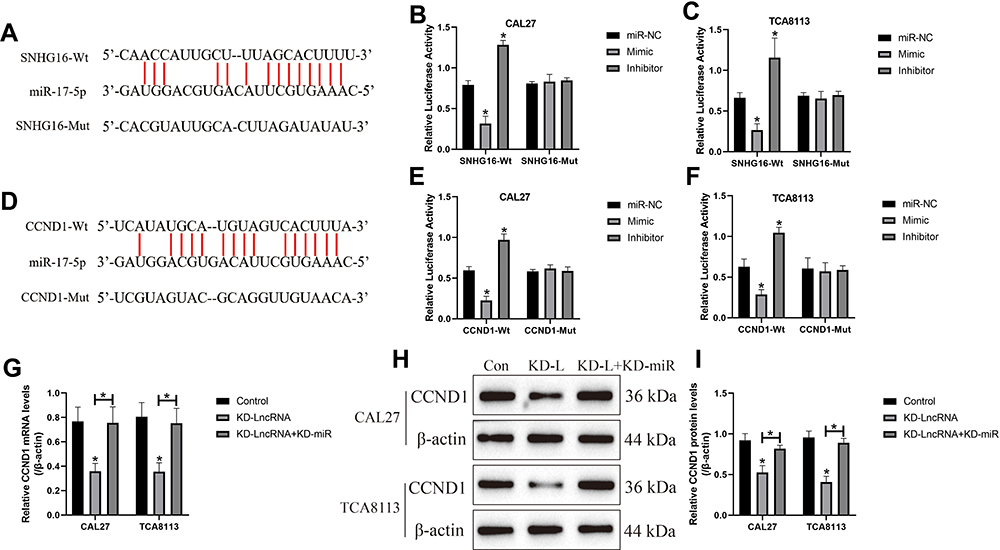 |
Figure 5 LncRNA SNHG16 sponged miR-17-5p to upregulate CCND1 in OSCC cells. The binding sites of miR-17-5p with (A) LncRNA SNHG16 and (D) 3ʹUTR of CCND1 mRNA were predicted by using the online starBase software (http://starbase.sysu.edu.cn/). The targeting sites of miR-17-5p with (B, C) LncRNA SNHG16 and (E, F) CCND1 mRNA were validated by using the dual-luciferase reporter gene system assay. (G) The mRNA and (H, I) protein levels of CCND1 were determined by Real-Time qPCR and Western Blot analysis, respectively. (Note: “Con” indicated “Control”, “KD-L” indicated “Knock-down of LncRNA SNHG16” and “KD-L+KD-miR” suggested “both LncRNA SNHG16 and miR-17-5p ablation”). Each experiment repeated at least 3 times, and *P < 0.05. |
Knock-Down of LncRNA SNHG16 Inhibited OSCC Progression Through the miR-17-5p/CCND1 Axis
We next investigated whether LncRNA SNHG16 regulated OSCC development by targeting the miR-17-5p/CCND1 axis. To achieve this, the LncRNA SNHG16 silencing vectors, miR-17-5p inhibitor and CCND1 overexpression vectors were delivered into OSCC cells (Figure 6A and B), which were divided into four groups, including Control, KD-SNHG16, KD-SNHG16+miR-17-5p inhibitor, and KD-SNHG16+OE-CCND1. As expected, the data in Figure 6C and D showed that knock-down of LncRNA SNHG16 inhibited cell proliferation in OSCC cells, which were restored by silencing miR-17-5p and upregulating CCND1. Consistently, the promoting effects of LncRNA SNHG16 ablation on cell apoptosis were also abrogated by miR-17-5p downregulation and CCND1 overexpression (Figure 6E and F). Next, by performing Western Blot analysis, we noticed that both miR-17-5p ablation and CCND1 overexpression restored the expressions of N-cadherin and Vimentin in LncRNA SNHG16-deficient OSCC cells, resulting in the promotion of EMT (Figure 7A–D).
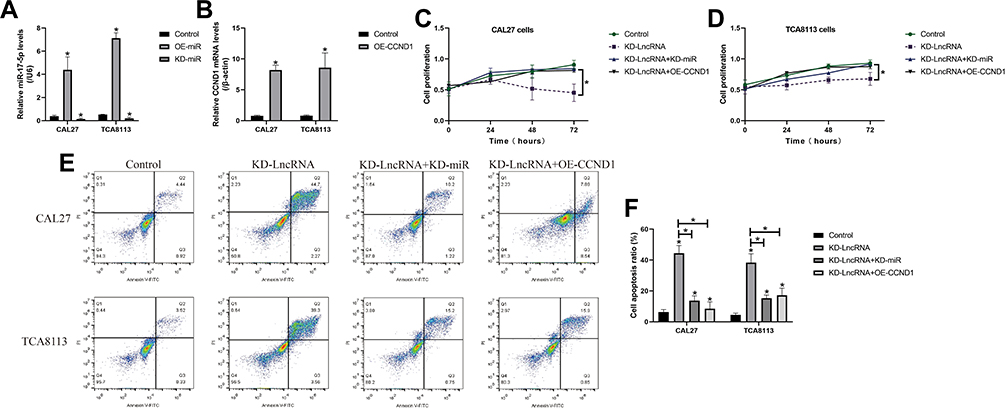 |
Figure 6 The role of LncRNA SNHG16/miR-17-5p/CCND1 axis in regulating OSCC cell proliferation and apoptosis. (A) The miR-17-5p mimic and inhibitor were transfected into OSCC cells for its overexpression and silencing. (B) CCND1 was overexpressed in OSCC cells. (C, D) CCK-8 assay was performed to examine cell proliferation abilities in OSCC cells. (E, F) Annexin V-FITC/PI double staining assay was used to determine cell apoptosis. Each experiment repeated at least 3 times, and *P < 0.05. |
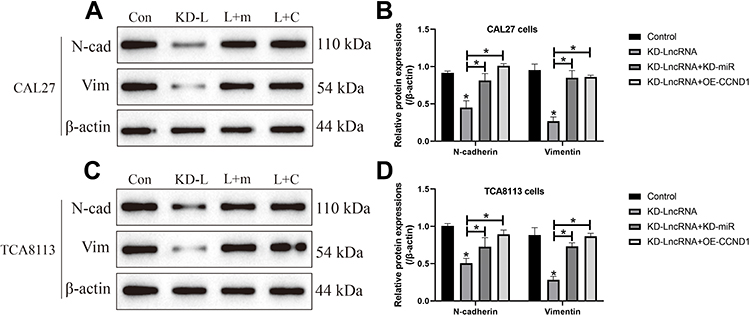 |
Figure 7 LncRNA SNHG16 regulated EMT in OSCC cells through targeting miR-17-5p/CCND1 axis. The expression levels of N-cadherin and Vimentin in (A, B) CAL27 and (C, D) TCA8113 cells were determined by using the Western Blot analysis. (Note: “Con” indicated “Control”, “KD-L” indicated “Knock-down of LncRNA SNHG16”, “L+M” indicated “Knock-down of LncRNA SNHG16 plus miR-17-5p silencing”, and “L+C” represented “LncRNA SNHG16 ablation plus CCND1 overexpression”). Each experiment repeated at least 3 times, and *P < 0.05. |
Discussion
Emerging evidences evidenced that targeting the long non-coding RNAs (LncRNAs)-microRNAs (miRNAs)-mRNA competing endogenous RNA (ceRNA) networks were effective therapy strategies to slow down the development of multiple cancers,40,41 including oral squamous cell carcinoma (OSCC).9–11 Based on this, the present study first identified a novel LncRNA SNHG16/miR-17-5p/CCND1 signaling cascade that involved in regulating OSCC pathogenesis. Specifically, according to the previous publication,15 LncRNA SNHG16 functioned as an oncogene to promote the development of OSCC, which were validated by our work, and the results showed that LncRNA SNHG16 tended to be enriched in OSCC tissues and cells, compared to their normal counterparts. In addition, LncRNA SNHG16 positively regulated the malignant phenotypes, including cell proliferation, invasion, migration, epithelial-mesenchymal transition (EMT) and tumorgenicity in vitro and in vivo, and knock-down of LncRNA SNHG16 triggered apoptotic cell death in OSCC cells, implying that silencing of LncRNA SNHG16 was effective to hamper OSCC progression.
MiRNAs also regulated the development of OSCC,18–20 which had widely been reported as the downstream target of LncRNAs.26,27 Specifically, previous literatures reported that miR-17-5p was relevant to OSCC pathogenesis,21–23 but the underlying mechanisms are still unclear. By analyzing the clinical tissues, we noticed that miR-17-5p was downregulated in cancer tissues. In addition, the expression levels of miR-17-5p and LncRNA SNHG16 showed negative correlations in OSCC tissues. Given the fact that there existed potential binding sites in the two genes,28 our further experiments validated that LncRNA SNHG16 sponged miR-17-5p in OSCC cells. Next, by performing the gain- and loss-function experiments, we found that the inhibiting effects of LncRNA SNHG16 ablation on the malignant phenotypes in OSCC were abrogated by silencing miR-17-5p, indicating that LncRNA SNHG16 involved in regulating OSCC progression by targeting miR-17-5p. According to the previous publications, the role of miR-17-5p in modulating cancer progression was controversial based on cancer types,24,25 and our data evidenced that miR-17-5p functioned as a tumor suppressor to repress OSCC development.
CCND1 was a key regulator for cell cycle29–31 and was often aberrantly overexpressed in OSCC, contributing to cancer development and deterioration.34,35 Interestingly, as the post-transcriptional regulators, multiple miRNAs had been identified to negatively regulate CCND1 through targeting its 3ʹ untranslated regions (3ʹ UTRs).37–39 Interestingly, our team validated that miR-17-5p bound the 3ʹUTR of CCND1 mRNA for degradation and inhibition, and miR-17-5p negatively correlated with CCND1 mRNA in OSCC clinical tissues. Of note, the ceRNA mechanisms theory depicted that miRNAs served as a “bridge” to combine the upstream LncRNAs and downstream mRNA targets,40,41 and we validated that LncRNA SNHG16 positively regulated CCND1 in OSCC cells through targeting miR-17-5p. Consistently, our clinical data also evidenced that LncRNA SNHG16 was positively relevant to CCND1 in OSCC tissues. Finally, we proved that silencing of LncRNA SNHG16 inhibited OSCC progression by downregulating CCND1. Moreover, in addition to CCND1, other anti-apoptotic proteins, such as nuclear Survivin,42 were closely associated with OSCC development. As the pathogenesis of OSCC was very complicated, our future work will focus on investigating the synergistic roles of CCND1 and other tumor-promoting proteins in regulating OSCC progression.
Conclusions
Taken together, we evidenced that knock-down of LncRNA SNHG16 inhibited CCND1 expressions through sponging miR-17-5p in a ceRNA-dependent manner, resulting in the inhibition of OSCC development in vitro and in vivo. The present study first identified a novel LncRNA SNHG16/miR-17-5p/CCND1 axis in regulating OSCC progression, and provided potential diagnostic and therapeutic agents for OSCC in clinic.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Thomson PJ. Perspectives on oral squamous cell carcinoma prevention-proliferation, position, progression and prediction. J Oral Pathol Med. 2018;47(9):803–807.
2. Eguchi T, Basugi A, Kanai I, Miyata Y, Suzuki T, Hamada Y. Adenosquamous carcinoma development as a recurrence of squamous cell carcinoma in the oral floor: a case report. Medicine. 2019;98(43):e17688.
3. Gawas NP, Navarange SS, Chovatiya GL, Chaturvedi P, Waghmare SK. Establishment and characterization of novel human oral squamous cell carcinoma cell lines from advanced‑stage tumors of buccal mucosa. Oncol Rep. 2019;41(4):2289–2298.
4. Panarese I, Aquino G, Ronchi A, et al. Oral and Oropharyngeal squamous cell carcinoma: prognostic and predictive parameters in the etiopathogenetic route. Expert Rev Anticancer Ther. 2019;19(2):105–119.
5. Bharadwaj R, Sahu BP, Haloi J, et al. Combinatorial therapeutic approach for treatment of oral squamous cell carcinoma. Artif Cells Nanomed Biotechnol. 2019;47(1):572–585.
6. Liu T, David M, Ellis O, et al. Treatment for oral squamous cell carcinoma: impact of surgeon volume on survival. Oral Oncol. 2019;96:60–65.
7. Mroueh R, Haapaniemi A, Saarto T, et al. Non-curative treatment of patients with oral tongue squamous-cell carcinoma. Eur Arch Otorhinolaryngol. 2019;276(7):2039–2045.
8. Xu Y, Liu Y, Xiao W, et al. MicroRNA-299-3p/FOXP4 axis regulates the proliferation and migration of oral squamous cell carcinoma. Technol Cancer Res Treat. 2019;18:1533033819874803.
9. Wang J, Huo F, Wang XR, Xu YY. LncRNA LACAT1 promotes proliferation of oral squamous cell carcinoma cells by inhibiting microRNA-4301. Eur Rev Med Pharmacol Sci. 2019;23(6):2427–2435.
10. Zhang L, Meng X, Zhu XW, et al. Long non-coding RNAs in oral squamous cell carcinoma: biologic function, mechanisms and clinical implications. Mol Cancer. 2019;18(1):102.
11. Zhang X, Guo B, Zhu Y, Xu W, Ning S, Liu L. Up-regulation of plasma lncRNA CACS15 distinguished early-stage oral squamous cell carcinoma patient. Oral Dis. 2019;1:2145.
12. Yu Y, Dong JT, He B, et al. LncRNA SNHG16 induces the SREBP2 to promote lipogenesis and enhance the progression of pancreatic cancer. Future Oncol. 2019;15(33):3831–3844.
13. Zhou C, Zhao J, Liu J, et al. LncRNA SNHG16 promotes epithelial-mesenchymal transition via down-regulation of DKK3 in gastric cancer. Cancer Biomark. 2019;26(4):393–401.
14. Cao X, Xu J, Yue D. LncRNA-SNHG16 predicts poor prognosis and promotes tumor proliferation through epigenetically silencing p21 in bladder cancer. Cancer Gene Ther. 2018;25(1–2):10–17.
15. Li S, Zhang S, Chen J. c-Myc induced upregulation of long non-coding RNA SNHG16 enhances progression and carcinogenesis in oral squamous cell carcinoma. Cancer Gene Ther. 2019;26(11–12):400–410.
16. Correia De Sousa M, Gjorgjieva M, Dolicka D. Deciphering miRNAs’ action through miRNA editing. Int J Mol Sci. 2019;20:24.
17. Sharma N, Baruah MM. The microRNA signatures: aberrantly expressed miRNAs in prostate cancer. Clin Transl Oncol. 2019;21(2):126–144.
18. Chamorro Petronacci CM, Pérez-Sayáns M, Padín Iruegas ME, Suárez Peñaranda JM, Lorenzo Pouso AI, Blanco Carrión A. and García García A, miRNAs expression of oral squamous cell carcinoma patients: validation of two putative biomarkers. Medicine. 2019;98(13):e14922.
19. Lu Z, He Q, Liang J, et al. miR-31-5p is a potential circulating biomarker and therapeutic target for oral cancer. Mol Ther Nucleic Acids. 2019;16:471–480.
20. Zhao J, Fang Z, Zha Z, et al. Quercetin inhibits cell viability, migration and invasion by regulating miR-16/HOXA10 axis in oral cancer. Eur J Pharmacol. 2019;847:11–18.
21. Coutinho-Camillo CM, Lourenço SV, de Araújo Lima L, Kowalski LP, Soares FA. Expression of apoptosis-regulating miRNAs and target mRNAs in oral squamous cell carcinoma. Cancer Genet. 2015;208(7–8):382–389.
22. Wu SY, Lin KC, Chiou JF, et al. MicroRNA-17-5p post-transcriptionally regulates p21 expression in irradiated betel quid chewing-related oral squamous cell carcinoma cells. Strahlenther Onkol. 2013;189(8):675–683.
23. Wu SY, Wu AT, Liu SH. MicroRNA-17-5p regulated apoptosis-related protein expression and radiosensitivity in oral squamous cell carcinoma caused by betel nut chewing. Oncotarget. 2016;7(32):51482–51493.
24. Li J, Lai Y, Ma J, et al. miR-17-5p suppresses cell proliferation and invasion by targeting ETV1 in triple-negative breast cancer. BMC Cancer. 2017;17(1):745.
25. Cai N, Hu L, Xie Y, et al. MiR-17-5p promotes cervical cancer cell proliferation and metastasis by targeting transforming growth factor-β receptor 2. Eur Rev Med Pharmacol Sci. 2018;22(7):1899–1906.
26. Tam C, Wong JH, Tsui SKW, Zuo T, Chan TF, Ng TB. LncRNAs with miRNAs in regulation of gastric, liver, and colorectal cancers: updates in recent years. Appl Microbiol Biotechnol. 2019;103(12):4649–4677.
27. Tang F, Lu Z, Wang J, et al. Competitive endogenous RNA (ceRNA) regulation network of lncRNAs, miRNAs, and mRNAs in Wilms tumour. BMC Med Genomics. 2019;12(1):194.
28. Peng H, Li H. The encouraging role of long noncoding RNA small nuclear RNA host gene 16 in epithelial-mesenchymal transition of bladder cancer via directly acting on miR-17-5p/metalloproteinases 3 axis. Mol Carcinog. 2019;58(8):1465–1480.
29. Chen G, Ding XF, Bouamar H, Pressley K, Sun LZ. Everolimus induces G(1) cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells. Am J Physiol Cell Physiol. 2019;317(2):C244–c252.
30. Sun F, Li N, Tong X, et al. Ara-c induces cell cycle G1/S arrest by inducing upregulation of the INK4 family gene or directly inhibiting the formation of the cell cycle-dependent complex CDK4/cyclin D1. Cell Cycle. 2019;18(18):2293–2306.
31. Xu S, Zhang H, Liu T, et al. 6-Gingerol induces cell-cycle G1-phase arrest through AKT-GSK 3β-cyclin D1 pathway in renal-cell carcinoma. Cancer Chemother Pharmacol. 2020;85(2):379–390.
32. Albasri AM, Elkablawy MA, Ansari IA, Alhujaily AS. Prognostic significance of Cyclin D1 over-expression in colorectal cancer: an experience from Madinah, Saudi Arabia. Asian Pac J Cancer Prev. 2019;20(8):2471–2476.
33. Choi C, Park S, Cho WK, Choi DH. Cyclin D1 is associated with radiosensitivity of triple-negative breast cancer cells to proton beam irradiation. Int J Mol Sci. 2019;20:19.
34. Kujan O, Huang G, Ravindran A, Vijayan M, Farah CS. CDK4, CDK6, cyclin D1 and Notch1 immunocytochemical expression of oral brush liquid-based cytology for the diagnosis of oral leukoplakia and oral cancer. J Oral Pathol Med. 2019;48(7):566–573.
35. Ramos-García P, González-Moles M, González-Ruiz L, et al. Clinicopathological significance of tumor cyclin D1 expression in oral cancer. Arch Oral Biol. 2019;99:177–182.
36. Gan CP, Sam KK, Yee PS, et al. IFITM3 knockdown reduces the expression of CCND1 and CDK4 and suppresses the growth of oral squamous cell carcinoma cells. Cell Oncol. 2019;42(4):477–490.
37. Liu L, Li J, Wang R, Wang Y, Wang G. MicroRNA-298 exacerbates myocardial ischemic injury via targeting cyclin D1. Pharmazie. 2019;74(6):369–373.
38. Sun W, Lv J, Duan L, et al. Long noncoding RNA H19 promotes vascular remodeling by sponging let-7a to upregulate the expression of cyclin D1. Biochem Biophys Res Commun. 2019;508(4):1038–1042.
39. Wang F, Mao A, Tang J, et al. microRNA-16-5p enhances radiosensitivity through modulating Cyclin D1/E1-pRb-E2F1 pathway in prostate cancer cells. J Cell Physiol. 2019;234(8):13182–13190.
40. Liang W, Sun F. Identification of pivotal lncRNAs in papillary thyroid cancer using lncRNA-mRNA-miRNA ceRNA network analysis. PeerJ. 2019;7:e7441.
41. Tang XJ, Wang W, Hann SS. Interactions among lncRNAs, miRNAs and mRNA in colorectal cancer. Biochimie. 2019;163:58–72.
42. Santarelli A, Mascitti M, Rubini C, et al. Nuclear survivin as a prognostic factor in squamous-cell carcinoma of the oral cavity. Appl Immunohistochem Mol Morphol. 2017;25(8):566–570.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.