Back to Journals » Infection and Drug Resistance » Volume 15
Short- and Long-Term Effects of Different Antibiotics on the Gut Microbiota and Cytokines Level in Mice
Authors Wang J, Xiang Q, Gu S, Gu Y, Yao M, Huang W ![]() , Gao W, Tang LL
, Gao W, Tang LL
Received 7 September 2022
Accepted for publication 14 November 2022
Published 23 November 2022 Volume 2022:15 Pages 6785—6797
DOI https://doi.org/10.2147/IDR.S388687
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Jingxia Wang,1,2,* Qiangqiang Xiang,1,2,* Silan Gu,1,* Yudan Gu,3 Mingfei Yao,1 Weixin Huang,4,5 Wang Gao,4 Ling-Ling Tang1,2
1State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 2Shulan (Hangzhou) Hospital Affiliated to Zhejiang Shuren University, Shulan International Medical College, Hangzhou, People’s Republic of China; 3Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 4Jinan Microecological Biomedicine Shandong Laboratory, Jinan, People’s Republic of China; 5Shaoxing Tongchuang Biotechnology Co., Ltd, Shaoxing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ling-Ling Tang, Shulan (Hangzhou) Hospital Affiliated to Zhejiang Shuren University, Shulan International Medical College, Hangzhou, People’s Republic of China, Email [email protected]
Background: Antibiotics are the first line of treatment for infectious diseases. However, their overuse can increase the spread of drug-resistant bacteria. The present study analyzed the impact of different types of antibiotics on the gut microbiome and cytokines level of mice.
Methods: A total of five groups of 8-week-old male BALB/c mice (n = 35) were treated with piperacillin-tazobactam (TZP), ceftriaxone (CRO), tigecycline (TGC), levofloxacin (LEV) or normal saline (Ctrl), respectively, for up to 4 weeks. Fecal samples were analyzed by bacterial 16S rRNA gene sequencing for bacterial identification. Blood samples were used for the determination of 23 serum cytokines using multiplex immunoassay.
Results: Exposure to antibiotics was shown to affect the normal weight gain of mice. Significant changes in gut composition caused by TZP, CRO and TGC treatment included the decreased abundance of Bacteroidetes (p < 0.01), Muribaculaceae (p < 0.01) and Lachnospiraceae (p < 0.01), and the increased abundance of Proteobacteria (p < 0.05), Enterobacteriaceae (including Klebsiella and Enterobacter) (p < 0.01) and Enterococcaceae (including Enterococcus) (p < 0.01). After 4-week treatment, the TZP, CRO and LEV groups had significantly lower concentrations of several serum cytokines. Correlation analysis of the top 30 bacterial genera and cytokines showed that Enterococcus and Klebsiella were strongly positively correlated with tumor necrosis factor-α (TNF-α), interleukins (IL) IL-12p70 and IL-1β. Desulfovibrio, Candidatus Saccharimonas, norank_f__norank_o__Clostridia_UCG-014, Lactobacillus, and Roseburia were strongly negatively correlated with these cytokines.
Conclusion: This study demonstrates the effects of various antibiotics on the intestinal microflora and immune status of mice. Compared with TZP, CRO and TGC, LEV had minimal impact on the gut microbiota. In addition to TGC, long-term TZP, CRO and LEV intervention can lead to a decrease in serum cytokine levels, which may depend on the intestinal microflora, antibiotic used and the duration of treatment.
Keywords: gut microbiota, piperacillin-tazobactam, ceftriaxone, tigecycline, levofloxacin, cytokine, immunity
Introduction
Since the discovery of penicillin by Alexander Fleming in 1928, antibiotics have been widely used to save millions of lives.1 However, the overuse and misuse of antibiotics have become a worldwide problem. There is increasing concern that antibiotic exposure may result in short- and/or long-term non-beneficial consequences, including an increased risk of necrotizing enterocolitis, bronchial allergy and asthma, obesity and autoimmune diseases.2–6 In recent years, disruption of the intestinal microbiota by antibiotics has received increasing attention. Some studies have demonstrated a direct link between antibiotic use and alterations in the intestinal microbiota.7–10 Moreover, it was found that antibiotic inducing changes in the gut microbiota may affect normal immune function or immune status by disrupting metabolic homeostasis.11–13
Among various antibiotics, the side effect of broad-spectrum antibiotics is easy to be depreciated as they are widely used in human and animal disease prevention and treatment. However, long-term high-dose application of broad-spectrum antibiotics can lead to intestinal microecological imbalance, reduce the colonization ability of normal bacteria, increase the number of potential pathogenic bacteria and even cause endogenous infection in severe cases.14 The amount of disturbance is closely related to the spectrum, dosage, route of administration, pharmacokinetic and pharmacodynamic properties.15 For example, β-lactams are widely used to treat infectious diseases and disrupt cell wall and peptidoglycan synthesis, leading to cell death. Among them, penicillin/β-lactamase inhibitor combinations (piperacillin/tazobactam, TZP) and Third-generation cephalosporin (ceftriaxone, CRO) are broad-spectrum antimicrobials commonly used in clinical.16 They have very similar pharmacokinetic and pharmacodynamic characteristics and spectra of activity against medically important pathogens.17 Gamage et al found the administration of either of two broad-spectrum antibiotics TZP or CRO could result in significant changes fecal microbiota of ICU patients.18 Tigecycline is an important glycylcycline antibiotic, which was designed to circumvent common tetracycline resistance mechanisms that is administered intravenously, while Levofloxacin is a type of fluoroquinolone antibiotic that is used to treat a wide range of infections caused by Gram-negative and Gram-positive bacteria with a wide margin of safety and efficacy.19,20 However, the specific correlation and mechanism between these broad-spectrum antibiotics with intestinal microbiota composition still remain elusive, which may can ultimately induce the body’s immune status imbalance.
A better understanding of this aspect will help in the rational use of broad-spectrum antibiotics in clinical practice. Therefore in our work, we have studied the effects of continuous administration of four types of widely-used broad-spectrum antibiotics on the intestinal microbiota and cytokines level of mice and attempted to establish the correlation between them. The main purpose of the experiment was to compare the differences in intestinal microbiota composition and cytokine levels, which can reflect the body’s immune status caused by different antibiotics under short- and long-term intervention. The secondary purpose was to understand the correlation between serum cytokines and intestinal microbiota changes through correlation analysis.
Materials and Methods
Experimental Groups
Six-week-old male BALB/c mice (n = 35) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd., acclimated for 2 weeks to reduce the impact of environmental stimuli on experimental results and properly fed under specific pathogen-free conditions. The mice were randomly divided into five groups (n = 7 per group) according to treatment: piperacillin-tazobactam (TZP), ceftriaxone (CRO), tigecycline (TGC), levofloxacin (LEV) or normal saline (Ctrl). The drugs were administered subcutaneously once a day for 4 weeks at the following doses (mg/20 gm body weight): 40.55 (TZP), 3 (CRO), 0.6 (TGC) and 1.5 (LEV), where the doses are equivalent to those recommended for adult human patients.21 The study was approved by the Research Ethics Committee of the First Affiliated Hospital from the School of Medicine of Zhejiang University (2022–1503) and conformed to the 2011 National Institutes of Health Guide for the care and use of laboratory animals. The experimental design scheme is shown in Figure 1A.
 |
Figure 1 (A) Scheme of the research design. A total of five groups of 8-week-old male BALB/c mice (n = 35) were continuously treated with piperacillin-tazobactam (TZP, n=7), ceftriaxone (CRO, n=7), tigecycline (TGC, n=7), levofloxacin (LEV, n=7) or normal saline (Ctrl, n=7), respectively, for up to 4 weeks. The fecal samples and serum before (0W) and after treatment for 1 and 4 weeks were used for 16S rRNA gene sequencing. Blood was before (0W) and after treatment for 1 and 4 weeks for the determination of serum cytokines. (B) Effect of antibiotic treatment on mouse growth performance. Time curve of body weight ratio changes at different time points. 0: before treatment. 3, 7, 14, 21 and 28: 3 7, 14, 21 and 28 days after treatment. Abbreviations: TZP, piperacillin/tazobactam; CRO, ceftriaxone; TGC, tigecycline; LEV, levofloxacin; Ctrl, control. |
Sample Collection and Serum Cytokines Test
We collected fresh stool samples at 3 time points– day 0 (0W), one-week (1W) and four-week (4W) of treatment for 16S rRNA sequencing for bacterial identification. The blood samples were also collected at the 3 time points for the cytokines 23-plex assay.22 The blood samples were immediately frozen and stored at −80 C until analysis. Then before test, the blood samples were centrifuged at 2500 to 3000 revolutions per minute to obtain the serum in the supernatant. The cytokine levels in the serum were analyzed by Bio-PlexTM 200 System (Bio-Rad) with the Bio-Plex murine 23-Plex Panel Kit (Bio-Rad Laboratories, Inc.) according to the manufacturer’s instructions. The measured cytokines were as follows: IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-17, IFN-γ, eotaxin, G-CSF, GM-CSF, KC, MCP-1, MIP-1α, MIP-1β, RANTES, and TNF-α. Standard curves for each cytokine (in duplicate) were generated using the reference cytokine concentrations supplied in the kit and then used to calculate the cytokine concentrations in the serum.
16S rRNA Gene Sequence Analysis
The QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) was used to extract genomic DNA from 200 mg of frozen stool, which was subjected to 16S rRNA gene sequencing.23 The V3-V4 hypervariable region of the bacterial 16S rRNA gene was amplified by PCR with barcoded primers 338F and 806R. The quality of amplified products was checked on 1% agarose gel and purified using the GeneJET Gel Extraction Kit (Thermo Fisher Scientific, Waltham, MA, USA). The PCR cycling conditions were as follows: 95°C for 1 min; 30 cycles of 98°C for 10s, 50°C for 30s, and 72°C for 30s; and a final extension for 5 min at 72°C. The Illumina MiSeq platform (Majorbio Bio-pharm Technology Co., Ltd., Shanghai, China) was used for sequencing after identification and purification of the amplified products. The raw data were filtered for quality and trimmed of any sequencing adapters using Fastp v0.19.6 software,24 and the sequences were assembled and error-corrected using FLASH 1.2.11 software.25 All sequence data were deposited in the NCBI BioProject: PRJNA890946. Single-end reads were separated by barcode, and barcodes and primers were removed. High-quality reads were obtained using Cutadapt. Chimeric sequences were detected and removed using UCHIME.26 Sequences were analyzed using UPARSE software and the sequences with ≥ 97% similarity were assigned to the same operational taxonomic units (OTUs).27 The median OTU count per sample was 30,869. Classified, unfiltered OTU counts were used to calculate the relative abundance of bacterial groups (phylum, family and genus). Main compositional shifts in the microbiome were visualized using bar charts generated in QIIME. Diversity within samples as species richness (a-diversity) was measured by comparison of Shannon and Abundance-based coverage estimate (ACE) diversity indices calculated in Mothur.28 Principal-coordinate analysis (PCoA) was used to assess community similarity among samples (b-Diversity) using Bray-Curtis based on the generated phylogeny tree.
Statistical Analysis
For the test of two groups of data, Student’s T-test was used for normally distributed data, while the Wilcoxon test was used for non-normally distributed data. For data tests of three or more groups: one-way ANOVA was used for normal distribution and Kruskal–Wallis H-test was used for non-normal distribution. Correlations were performed using a non-parametric Spearman’s test. Data were analyzed using SPSS version 26.0 (SPSS Inc., Chicago IL, USA) and GraphPad version 9.0.
Results
Changes in Mouse Body Weight During Antibiotic Treatment
After antibiotic treatment, the weight gain of mice in TZP, CRO, TGC and LEV groups was delayed when compared to the Ctrl group (Figure 1B). The body weight of the mice was significantly lower (p < 0.05) than the Ctrl group on day 7, 14, and 21 for the TZP group, and only on day 14 and 21 for the mice in the CRO and LEV groups while the same as the group TGC on day 21.
Effect of Long-Term Antibiotic Treatment on Cytokine Levels
Treatment with antibiotics can induce immune responses in mice, which could be dependent on the antibiotic class and the timing of treatment. We compared the levels of all cytokines between 0W, 1W and 4W in the control mice (Figure 2). There were no significant differences in cytokine levels between different groups on 0W and 1W. However, after 4 weeks, the levels of IL-1α, IL-3, IL-9, IL-12(P40), IL-17, eotaxin, GM-CSF, IFN-γ, MCP-1, MIP-1α, MIP-1β, RANTES, TNF-α in the TZP and CRO groups were significantly lower than Ctrl group(p < 0.05). In addition, there was a significant decrease of IL-6 in TZP group (p < 0.01) and IL-4 in CRO group (p < 0.05). Serum IL-1β, IL-3, IL-17, eotaxin, GM-CSF, MCP-1 and RANTES cytokine concentrations were significantly decreased in LEV group (p < 0.05). There were no significant changes in serum cytokine concentrations in the TGC group. Only long-term treatment with TZP, CRO and LEV was found to have an effect on the levels of serum cytokines. TGC did not cause any drastic changes in the cytokine levels in the short- and long-term.
 |
Figure 2 Changes in levels of serum cytokines after antibiotics treatment for 4 weeks. (A) IL-1α, (B) IL-1β, (C) IL-3, (D) IL-4, (E) IL-6, (F) IL-9, (G) IL-12 (p40), (H) IL-17, (I) IFN-γ, (J) eotaxin, (K) GM-CSF, (L) MCP-1, (M) MIP-1α, (N) MIP-1β, (O) RANTES, (P) TNF-α. Significant differences between the two groups were marked, and *Indicates p < 0.05; **p < 0.01 and ***p < 0.001. Abbreviations: TZP, piperacillin/tazobactam; CRO, ceftriaxone; TGC, tigecycline; LEV, levofloxacin; Ctrl, control. |
Effect of Antibiotic Treatment on Gut Microbiome Structure
The diversity and richness of the mouse gut microbiota can be evaluated by calculating alpha diversity (Shannon and Ace indexes) (Figure 3A–D). Our data suggested that α-diversity of the antibiotic groups was dramatically reduced on 1W and 4W when compared to control group (p < 0.05). In the fourth week, the α-diversity of LEV was higher when compared to other treatment groups (p < 0.05). β-diversity can reflect the degree of similarity or difference in community composition in different samples. Principal coordinate analysis (PCoA) was subsequently performed using distance matrix analysis to compare microbiota structure. The results demonstrated that there were obvious differences among the different groups on 1W and 4W (Figure 3E and F). But it was observed that the mice treated with LEV resembled the control group more closely, compared to the other groups.
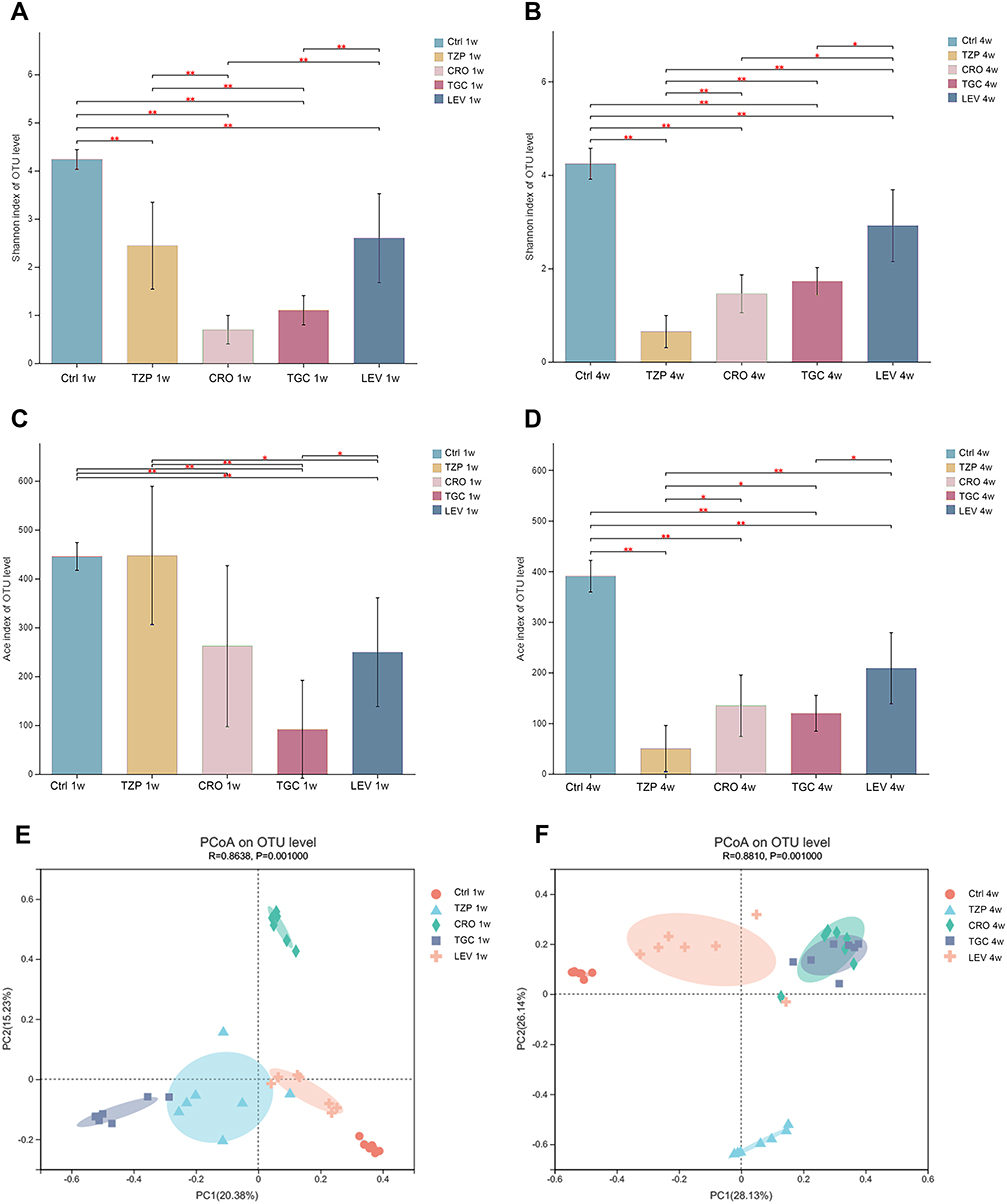 |
Figure 3 Microbial diversity measured by the Shannon index and Ace index was altered by antibiotic treatment across the treatment groups on 1W (A and B) and 4W (C and D). *P < 0.05, **P < 0.01 vs the control group. Analysis of microbial communities by principal coordinate analysis (PCoA) at different time points after antibiotic treatment on 1W and 4W (E and F). 1 W and 4 W: 1 W and 4W after treatment. Abbreviations: TZP, piperacillin/tazobactam; CRO, ceftriaxone; TGC, tigecycline; LEV, levofloxacin; Ctrl, control. |
The Impact of Different Antibiotics on the Gut Microbial Composition
During the experiment, antibiotic exposure greatly disturbed the microbial composition at the phylum, family and genus levels of distribution (Figure 4A–C). The composition of gut microbiota of mice in the control and antibiotic groups was similar before treatment. Firmicutes, Bacteroidetes, Desulfobacterota and Patescibacteria were the dominant bacterial phyla, accounting for more than 85% of the gut microbiota, of which Firmicutes and Bacteroidetes accounted for more than 75% (Figure 4A).
 |
Figure 4 Antibiotic-induced composition changes of gut microbiota. Distribution of the predominant bacteria with antibiotics-treated at (A) phylum level, (B) family level and (C) genus level on 0W, 1W and 4W. Species with less than 0.5% abundance in all samples were merged into the “Others” category. 0W: before treatment. 1 W and 4 W: 1 W and 4W after treatment. Abbreviations: TZP, piperacillin/tazobactam; CRO, ceftriaxone; TGC, tigecycline; LEV, levofloxacin; Ctrl, control. |
After short-term (1W) antibiotic exposure, Proteobacteria showed significant increase in TZP, CRO and TGC (p < 0.05), while Bacteroidetes, Patescibacteria, and Desulfobacterota decreased significantly (p < 0.05) (Figure 5A). The dominant bacterial phylum was composed of Firmicutes and Proteobacteria, accounting for more than 90%. A significant decrease in Desulfobacterota was observed in LEV (p < 0.001). At the family and genus levels, the concentrations of several beneficial symbionts from the families Muribaculaceae and Lachnospiraceae (p < 0.01) were dramatically decreased in the TZP, CRO and TGC groups. This was largely due to a significant reduction in norank_f__Muribaculaceae, Lachnospiraceae_NK4A136_group, and norank_f__Lachnospiraceae (p < 0.05). In addition, Roseburia was found to decrease in the CRO and TGC groups (p < 0.01), while Lachnospiraceae_NK4A136_group also showed a decrease in LEV group (p < 0.001). However, a significantly higher relative abundance of opportunistic pathogens in TZP, CRO and TGC groups. The population of Enterococcaceae and its genus Enterococcus (p < 0.01) was higher in TZP and TGC groups. The population of Enterobacteriaceae and its genus unclassified_f__Enterobacteriaceae (p < 0.001) was higher in TGC while the abundance of Bacillaceae and its genus Bacillus (p < 0.001), Enterobacteriaceae (p < 0.01) and its genus Enterobacter, and Klebsiella (p < 0.001) was higher in CRO (Figure 5B and C).
 |
Figure 5 Comparison of dominant bacteria at the (A) Phylum, (B) Family and (C) Genus levels among five groups on 1W. Comparison of dominant bacteria at the (D) Phylum, (E) Family and (F) Genus levels among five groups on 4W. 1 W and 4 W: 1 W and 4W after treatment. Abbreviations: TZP, piperacillin/tazobactam; CRO, ceftriaxone; TGC, tigecycline; LEV, levofloxacin; Ctrl, control. |
After long-term (4W) antibiotic intervention, compared to control group, a significant increase of Proteobacteria (p < 0.001) was observed in the TZP group, accompanied by a decrease in Firmicutes and Bacteroidetes(p < 0.001). Except, Proteobacteria (p < 0.05), Firmicutes (p < 0.01) also increased in CRO as well as TGC, and was accompanied by a significant decrease in Bacteroidetes (p < 0.01). In addition, Verrucomicrobiota was significantly increased in LEV (p < 0.01) (Figure 5D). At the family and genus levels, families Muribaculaceae and Saccharimonadaceae (p < 0.01) dramatically decreased in the TZP, CRO and TGC groups, which was largely due to a significant reduction in norank_f__Muribaculaceae and Candidatus_Saccharimonas (p < 0.01). Lachnospiraceae (p < 0.001) decreased only in TZP; however, its genus Lachnospiraceae_NK4A136_group (p < 0.05), unclassified_f__Lachnospiraceae (p < 0.05), norank_f__Lachnospiraceae (p < 0.01) was decreased in TZP, CRO and TGC groups. Enterococcaceae (including Enterococcus) and Enterobacteriaceae (p < 0.05) were higher in the TZP, CRO and TGC. Enterobacter (p < 0.01) and Blautia (p < 0.01) were higher in the CRO and TGC. Klebsiella was tremendous higher in the TZP (p < 0.0001). Akkermansiaceae and its genus Akkermansia (p < 0.001) increased in LEV, accompanied with the decrease of Saccharimonadaceae and its genus Candidatus_Saccharimonas (p < 0.001) (Figure 5E and F).
Correlation Between Gut Microbiota and Cytokines
To study the interaction between the intestinal microbiota and the host immune system, we calculated the correlations between microbiome compositions (at the genus level) and the 23 serum cytokine levels of host immune markers by Spearman correlation analysis (Figure 6A and B). The significance thresholds were absolute correlation coefficients higher than 0.1 and P values lower than 0.05. Enterococcus, Klebsiella, Enterobacter and Akkermansia were found to be positively correlated with most cytokines. Desulfovibrio, Candidatus_Saccharimonas, norank_f__norank_o__Clostridia_UCG-014, Roseburia, Lactobacillus and Helicobacter were negatively correlated with most cytokines. Next, the significance thresholds were defined as absolute correlation coefficients higher than 0.3. Enterococcus and Klebsiella were strongly positively correlated with IL-1β, IL-12 (p70) and TNF-α. Erysipelatoclostridium was strongly positively correlated with IL-1α, IL-6, IFN-γ and MIP-1β. Desulfovibrio, Candidatus_Saccharimonas, norank_f__norank_o__Clostridia_UCG-014, Roseburia, Lactobacillus and Helicobacter were strongly negatively correlated with IL-1β, IL-12 (p70) and TNF-α.
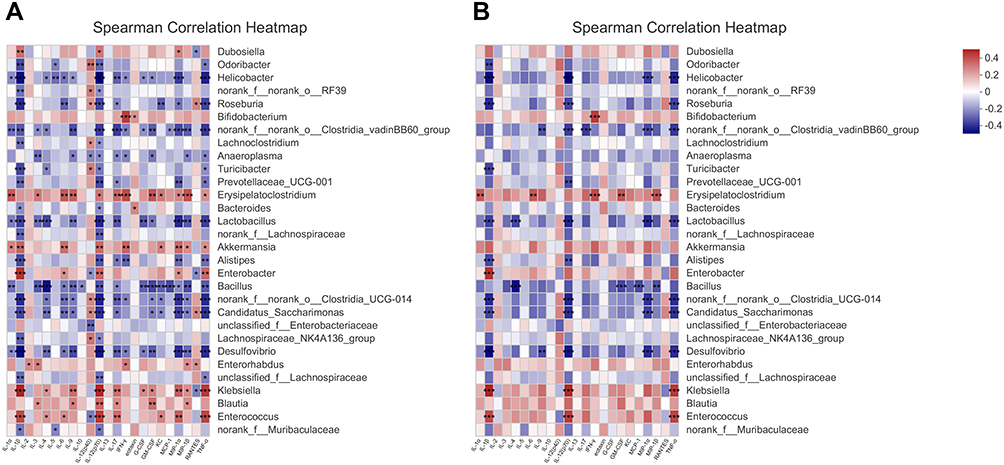 |
Figure 6 Correlations microbiome compositions (at the top 30 genus level) and the 23 serum cytokine levels of host immune markers on 0W, 1W and 4W. (A) Significance thresholds included absolute correlation coefficients higher than 0.1 and P < 0.05. (B) Significance thresholds included absolute correlation coefficients higher than 0.3 and P < 0.05. The number in color present Spearman correlation coefficient: red color indicates positive correlation; blue color indicates negative correlation. *P < 0.05, **P < 0.01, ***P < 0.001. |
Discussion
Antibiotics are still indispensable for the treatment of infectious diseases, which makes their rational use even more important.29,30 The various factors influencing the choice of the antibiotic include the administration, antibacterial spectrum, drug resistance, and organ functions of the patients. However, a lot of research in recent years has focused on the impact of antibiotics on gut microbiota and host immunity. In spite of the interspecies differences as the human gut microbiome is prone to fluctuations due to various factors including diet and environment, mouse models remain valuable for studying the human microbiome.17 In our study, four types of antibiotics were continuously administered to mice for 4 weeks. Subsequent studies analyzed the impact of different antibiotics on the intestinal microbiota and host immunity after 1 and 4 weeks of treatment, and attempted to find the correlation between the gut microbiome and immune response.
Consistent with previous studies, four broad-spectrum antibiotics significantly reduced the α- and β-diversity of gut microbiota.31 During the experiment, the fluctuating dominant bacteria were similar in the TGC, CRO and TZP groups. There was a decreased abundance of Bacteroidetes, Muribaculaceae and Lachnospiraceae, and an increased abundance of Firmicutes, Proteobacteria, Enterobacteriaceae (including Klebsiella and Enterobacter) and Enterococcaceae (including Enterococcus).
Continuous intervention with TZP, CRO and TGC significantly decreased the α-diversity of the gut microbiota and Bacteroidetes in short- and long-term exposure. Previous studies have demonstrated that substantial reductions in the abundance of Bacteroidetes, an important producer of short-chain fatty acids, have been associated with intestinal inflammation and various other pathological states.32,33 On the contrary, a normal gut microbiome dominated by Firmicutes and Bacteroidetes is unfavorable for Gram-negative bacterial colonization and can reduce the risk of corresponding bloodstream infections.34 During antibiotic intervention, the microbial community shifted from a normal microbiota structure dominated by Firmicutes and Bacteroidetes to Firmicutes and Proteobacteria, which includes many potent pathogenic microorganisms. The significant increase in the abundance of Proteobacteria may be due to the antibiotic treatment disrupting the anaerobic environment in the gut.35 Current research suggests that a high relative abundance of Proteobacteria is not beneficial to human health. For instance, patients with obesity, diabetes or nonalcoholic fatty liver disease often show a higher abundance of Proteobacteria.36–38
When the intervention time was the same, the impact of different antibiotics on the gut flora of mice showed specific changes. Under the short-term effects of TZP, CRO and TGC, the abundance of Muribaculaceae and Lachnospiraceae (a family of butyrate-producing bacteria) significantly reduced compared with the control group.39 The reduction of normal flora can provide a breeding ground for the growth of opportunistic pathogens in the gut.40 However, a significantly higher relative abundance of opportunistic pathogens (p < 0.01) was observed in the antibiotic groups except for LEV. Enterococcus was significantly increased in the TZP and TGC groups. Although Enterococcus constitute only a small proportion (<0.1%) of the intestinal microbiota of healthy individuals, they are also common pathogens. Excess Enterococcus can lead to abdominal and pelvic infections and can cause intestinal inflammation by stimulating antigen-presenting cells and CD4+RORγ+ T cell infiltration.41 In addition, in our study, the abundance of bacilli such as Bacillus, Enterobacter and Klebsiella significantly increased in CRO. Enterobacter and Klebsiella are representative pathogenic bacteria genera of the Enterobacteriaceae. Studies have shown that the presence of Klebsiella spp. in the gut is a serious risk factor for infection.42 Klebsiella also plays a key role in causing pathological damage in patients suffering from Crohn’s disease and diabetes.42–44
After long-term intervention with antibiotics, the abundance of Muribaculaceae was still decreased in the TZP, CRO and TGC groups. But Lachnospiraceae was not decreased in the CRO and TGC groups because of an increase of Blautia. Klebsiella was significantly increased and replaced Enterococcus as the main pathogen in the TZP group. Klebsiella spp. can develop resistance to β-lactam antibiotics by producing β-lactamase. Therefore, both TZP and CRO, both of which belong to β-lactam antibiotics, can cause Klebsiella to increase during the intervention process.45 Enterococcus and Enterobacter became the dominant pathogenic bacteria in the CRO and TGC groups. The dominant pathogenic bacteria were constantly changing during the course of treatment.
Compared with the control, LEV treatment did not induce any significant changes in the bacterial composition. The minor changes were observed as a decrease in Desulfobacterota and Lachnospiraceae_NK4A136_group in the first week, and a decrease in Saccharimonadaceae and Candidatus_Saccharimonas and an increase in Verrucomicrobiota Akkermansiaceae and Akkermansia in the fourth week. Akkermansia is a promising probiotic that can help patients with inflammatory bowel disease and diabetes by regulating intestinal homeostasis and basal metabolism.46 It has also recently been found to reduce intestinal injury and mortality in mice infected with Clostridium difficile. But whether a simple antibiotic-mediated Akkermansia increase is beneficial to host is not clear.47 Studies have also found that vancomycin-treated mice had a gut compositional shift to Verrucomicrobia and its species Akkermansia muciniphila, and Proteobacteria and its species Escherichia coli. The increased abundance of L-aspartate mediated by Akkermansiaceae could be associated with inflammation in the host, which was partially confirmed in our correlation results.48 Correlation results found that Akkermansia is positively correlated with IL-1α, IL-1β, IL-6, IL-12(p70), eotaxin, KC, MIP-1α, MIP-1β, and TNF-α, but the specific mechanism of action needs to be further explored.
Previous studies have found that antibiotic-induced microbiota perturbations are accompanied by modulation of host immune status. For example, antibiotic treatment for up to 4 weeks, especially with vancomycin, increases susceptibility to invasive candidiasis by reducing IL-17A and GM-CSF and promoting non-inflammatory escape of gut bacteria, leading to systemic coinfection and death.49 This immune response is associated with a decrease in some bacterial families such as Bacteroidales, Deferribacterales, Erysipelotrichales, and Clostridiales, and their short-chain fatty acid-producing species, such as Ruminococcus, Roseburia, and Butyricimonas.50 These findings were partially confirmed by our study results. Long-term treatment with TZP, CRO and LEV can decrease the level of serum IL-17 and GM-CSF. In addition, long-term effects of TZP and CRO, both belonging to the β-lactam class, include reducing the levels of IL-1α, IL-9, IL-12(P40), eotaxin, IFN-γ, MIP-1α, MIP-1β, RANTES, and TNF-α. Serum IL-1β, IL-10, and RANTES cytokine concentrations were also significantly decreased with the quinolone levofloxacin (p < 0.05). However, in the case of TGC, there was no significant change in the level of cytokines. Immunodeficiency is not the only immune regulation disorder caused by antibiotics. A number of studies have shown that antibiotics cause excessive inflammation in the host. Zhang et al found that serum IFN-γ, IL-13, and IL-17A levels in vancomycin-treated mice and IL-13 and IL-17 levels in polymyxin B-treated mice were significantly increased compared with control mice after 8 weeks of treatment.12 Combining our own findings with previous reports, we postulate that immune function is not only related to the timing of antibiotic treatment, but also the class of antibiotic used.
In addition to the timing and type of antibiotic treatment, which may affect the host’s immune function, it is currently believed that the influence of host immunity is closely related to antibiotic-mediated changes in the microbiota. Through correlation analysis, it was found that some bacteria had a certain correlation with cytokines. Klebsiella and Enterobacter were strongly positively correlated with proinflammatory factors such as TNF-α, IL-12p70 and IL-1β. One mechanism underlying the microbiome–cytokine interaction may be mediated by the metabolites originating from gut microbiota. It is known that Klebsiella and Enterobacter belong to the Enterobacteriaceae family, and can increase the concentration of lipopolysaccharides (LPS) to promote the release of proinflammatory cytokines. Desulfovibrio, Lactobacillus, Roseburia, Candidatus_Saccharimonas were strongly negatively correlated with these cytokines. Almost all of them are butyrate-producing bacteria.49 Studies have demonstrated that short chain fatty acids (SCFAs), have a role in reducing inflammatory immune responses.51
Conclusion
Our study demonstrated that different antibiotics have different effects on the intestinal flora and immune response of mice. Compared to TZP, CRO and TGC, LEV had minimal impact on the gut microbiota, both in short-term and long-term interventions. In terms of the influence of intestinal microecology, LEV is more recommended clinically than β-lactam antibiotics (TZP and CRO) and tetracycline antibiotics (TGC). In addition to TGC, long-term CRO, TGC, and LEV treatment may compromise immune response through a decrease in serum cytokine levels, and this change may be related to gut microbiota, the class of antibiotic, and the timing of antibiotic treatment.
Funding
This study was supported by grants from the Research Project of Jinan Microecological Biomedicine Shandong Laboratory (JNL-2022014B), the National Science and Technology Major Project (No.2017ZX10204401), and the Academician Shusen Lanjuan Talent Foundation of Zhejiang University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rodrigues RR, Greer RL, Dong X, et al. Antibiotic-induced alterations in gut microbiota are associated with changes in glucose metabolism in healthy mice. Front Microbiol. 2017;8:2306. doi:10.3389/fmicb.2017.02306
2. Bailey LC, Forrest CB, Zhang P, et al. Association of antibiotics in infancy with early childhood obesity. JAMA Pediatr. 2014;168(11):1063–1069. doi:10.1001/jamapediatrics.2014.1539
3. Tilg H, Adolph TE, Gerner RR, et al. The intestinal microbiota in colorectal cancer. Cancer Cell. 2018;33(6):954–964. doi:10.1016/j.ccell.2018.03.004
4. Routy B, Gopalakrishnan V, Daillere R, et al. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol. 2018;15(6):382–396. doi:10.1038/s41571-018-0006-2
5. Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut. 2018;67(2):226–236. doi:10.1136/gutjnl-2017-314205
6. Ogino S, Nowak JA, Hamada T, et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut. 2018;67(6):1168–1180. doi:10.1136/gutjnl-2017-315537
7. Lavelle A, Hoffmann TW, Pham H-P, et al. Baseline microbiota composition modulates antibiotic-mediated effects on the gut microbiota and host. Microbiome. 2019;7(1):111. doi:10.1186/s40168-019-0725-3
8. Ferrer M, Mendez-Garcia C, Rojo D, et al. Antibiotic use and microbiome function. Biochem Pharmacol. 2017;134:114–126. doi:10.1016/j.bcp.2016.09.007
9. Palleja A, Mikkelsen KH, Forslund SK, et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat Microbiol. 2018;3(11):1255–1265. doi:10.1038/s41564-018-0257-9
10. Guo Y, Yang X, Qi Y, et al. Long-term use of ceftriaxone sodium induced changes in gut microbiota and immune system. Sci Rep. 2017;7(1):43035. doi:10.1038/srep43035
11. Sun L, Zhang X, Zhang Y, et al. Antibiotic-induced disruption of gut microbiota alters local metabolomes and immune responses. Front Cell Infect Microbiol. 2019;9:99. doi:10.3389/fcimb.2019.00099
12. Zhang N, Liu J, Chen Z, et al. Integrated analysis of the alterations in gut microbiota and metabolites of mice induced after long-term intervention with different antibiotics. Front Microbiol. 2022;13:832915. doi:10.3389/fmicb.2022.832915
13. Angoa-Perez M, Zagorac B, Francescutti DM, et al. Effects of gut microbiota remodeling on the dysbiosis induced by high fat diet in a mouse model of Gulf war illness. Life Sci. 2021;279:119675. doi:10.1016/j.lfs.2021.119675
14. Greenwood C, Morrow AL, Lagomarcino AJ, et al. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J Pediatr. 2014;165(1):23–29. doi:10.1016/j.jpeds.2014.01.010
15. Bhalodi AA, van Engelen TSR, Virk HS, et al. Impact of antimicrobial therapy on the gut microbiome. J Antimicrob Chemother. 2019;74(Suppl 1):i6–i15. doi:10.1093/jac/dky530
16. Magill SS, Edwards JR, Beldavs ZG, et al. Prevalence of antimicrobial use in US acute care hospitals, May–September 2011. JAMA. 2014;312(14):1438–1446. doi:10.1001/jama.2014.12923
17. Ng KM, Aranda-Diaz A, Tropini C, et al. Recovery of the gut microbiota after antibiotics depends on host diet, community context, and environmental reservoirs. Cell Host Microbe. 2020;28(4):628. doi:10.1016/j.chom.2020.09.001
18. Gamage H, Venturini C, Tetu SG, et al. Third generation cephalosporins and piperacillin/tazobactam have distinct impacts on the microbiota of critically ill patients. Sci Rep. 2021;11(1):7252. doi:10.1038/s41598-021-85946-4
19. Jump RLP, Kraft D, Hurless K, et al. Impact of tigecycline versus other antibiotics on the fecal metabolome and on colonization resistance to clostridium difficile in mice. Pathog Immun. 2017;2(1):1–20. doi:10.20411/pai.v2i1.159
20. Croom KF, Goa KL. Levofloxacin: a review of its use in the treatment of bacterial infections in the United States. Drugs. 2003;63(24):2769–2802. doi:10.2165/00003495-200363240-00008
21. Rice LB, Hutton-Thomas R, Lakticova V, et al. Beta-lactam antibiotics and gastrointestinal colonization with vancomycin-resistant enterococci. J Infect Dis. 2004;189(6):1113–1118. doi:10.1086/382086
22. Gu S, Chen Y, Zhang X, et al. Identification of key taxa that favor intestinal colonization of Clostridium difficile in an adult Chinese population. Microbes Infect. 2016;18(1):30–38. doi:10.1016/j.micinf.2015.09.008
23. Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4516–4522. doi:10.1073/pnas.1000080107
24. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
25. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi:10.1093/bioinformatics/btr507
26. Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi:10.1093/bioinformatics/btr381
27. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998. doi:10.1038/nmeth.2604
28. McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi:10.1371/journal.pone.0061217
29. Nicolaou KC, Rigol S. A brief history of antibiotics and select advances in their synthesis. J Antibiot. 2018;71(2):153–184.
30. Benko R, Gajdacs M, Matuz M, et al. Prevalence and antibiotic resistance of ESKAPE pathogens isolated in the emergency department of a tertiary care teaching hospital in Hungary: a 5-year retrospective survey. Antibiotics. 2020;9(9):624. doi:10.3390/antibiotics9090624
31. Jones-Nelson O, Tovchigrechko A, Glover MS, et al. Antibacterial monoclonal antibodies do not disrupt the intestinal microbiome or its function. Antimicrob Agents Chemother. 2020;64(5). doi:10.1128/AAC.02347-19
32. De Schepper S, Verheijden S, Aguilera-Lizarraga J, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. 2019;176(3):676. doi:10.1016/j.cell.2019.01.010
33. Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–662. doi:10.1038/s41586-019-1237-9
34. Kim S, Covington A, Pamer EG. The intestinal microbiota: antibiotics, colonization resistance, and enteric pathogens. Immunol Rev. 2017;279(1):90–105. doi:10.1111/imr.12563
35. Litvak Y, Byndloss MX, Tsolis RM, et al. Dysbiotic proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol. 2017;39:1–6. doi:10.1016/j.mib.2017.07.003
36. Vatanen T, Franzosa EA, Schwager R, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature. 2018;562(7728):589–594. doi:10.1038/s41586-018-0620-2
37. Crovesy L, Masterson D, Rosado EL. Profile of the gut microbiota of adults with obesity: a systematic review. Eur J Clin Nutr. 2020;74(9):1251–1262. doi:10.1038/s41430-020-0607-6
38. Fukui H. Role of gut dysbiosis in liver diseases: what have we learned so far? Diseases. 2019;7(4):58. doi:10.3390/diseases7040058
39. Shi Y, Kellingray L, Zhai Q, et al. Structural and functional alterations in the microbial community and immunological consequences in a mouse model of antibiotic-induced dysbiosis. Front Microbiol. 2018;9:1948. doi:10.3389/fmicb.2018.01948
40. Garrett WS, Gallini CA, Yatsunenko T, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8(3):292–300. doi:10.1016/j.chom.2010.08.004
41. Fiore E, Van Tyne D, Gilmore MS. Pathogenicity of Enterococci. Microbiol Spectr. 2019;7(4). doi:10.1128/microbiolspec.GPP3-0053-2018
42. Hassan MA, Abd El-Aziz S, Elbadry HM, et al. Prevalence, antimicrobial resistance profile, and characterization of multi-drug resistant bacteria from various infected wounds in North Egypt. Saudi J Biol Sci. 2022;29(4):2978–2988. doi:10.1016/j.sjbs.2022.01.015
43. Rashid T, Ebringer A, Wilson C. The role of Klebsiella in Crohn’s disease with a potential for the use of antimicrobial measures. Int J Rheumatol. 2013;2013:610393. doi:10.1155/2013/610393
44. Hassan MA, Tamer TM, Rageh AA, et al. Insight into multidrug-resistant microorganisms from microbial infected diabetic foot ulcers. Diabetes Metab Syndr. 2019;13(2):1261–1270. doi:10.1016/j.dsx.2019.01.044
45. Bouza E, Cercenado E. Klebsiella and Enterobacter: antibiotic resistance and treatment implications. Semin Respir Infect. 2002;17(3):215–230. doi:10.1053/srin.2002.34693
46. Png CW, Linden SK, Gilshenan KS, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105(11):2420–2428. doi:10.1038/ajg.2010.281
47. Wu Z, Xu Q, Gu S, et al. Akkermansia muciniphila Ameliorates Clostridioides difficile infection in mice by modulating the intestinal microbiome and metabolites. Front Microbiol. 2022;13:841920. doi:10.3389/fmicb.2022.841920
48. Rao Y, Kuang Z, Li C, et al. Gut Akkermansia muciniphila ameliorates metabolic dysfunction-associated fatty liver disease by regulating the metabolism of L-aspartate via gut-liver axis. Gut Microbes. 2021;13(1):1–19. doi:10.1080/19490976.2021.1927633
49. Khan I, Wei J, Li A, et al. Lactobacillus plantarum strains attenuated DSS-induced colitis in mice by modulating the gut microbiota and immune response. Int Microbiol. 2022;25(3):587–603.
50. Sencio V, Machelart A, Robil C, et al. Alteration of the gut microbiota following SARS-CoV-2 infection correlates with disease severity in hamsters. Gut Microbes. 2022;14(1):2018900. doi:10.1080/19490976.2021.2018900
51. Awoniyi M, Wang J, Ngo B, et al. Protective and aggressive bacterial subsets and metabolites modify hepatobiliary inflammation and fibrosis in a murine model of PSC. Gut. 2022:gutjnl-2021–326500. doi:10.1136/gutjnl-2021-326500
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.