Back to Journals » Journal of Inflammation Research » Volume 15
Shifting the Biosynthesis of Leukotrienes Toward Specialized Pro-Resolving Mediators by the 5-Lipoxygenase-Activating Protein (FLAP) Antagonist BRP-201
Authors Kretzer C, Jordan PM, Bilancia R, Rossi A, Gür Maz T ![]() , Banoglu E
, Banoglu E ![]() , Schubert US, Werz O
, Schubert US, Werz O ![]()
Received 21 October 2021
Accepted for publication 28 December 2021
Published 9 February 2022 Volume 2022:15 Pages 911—925
DOI https://doi.org/10.2147/JIR.S345510
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Christian Kretzer,1 Paul M Jordan,1 Rossella Bilancia,2 Antonietta Rossi,2 Tuğçe Gür Maz,3 Erden Banoglu,3 Ulrich S Schubert,4,5 Oliver Werz1,5
1Department of Pharmaceutical/Medicinal Chemistry, Institute of Pharmacy, Friedrich Schiller University Jena, Jena, 07743, Germany; 2Department of Pharmacy, School of Medicine and Surgery, University of Naples Federico II, Naples, I-80131, Italy; 3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Gazi University, Yenimahalle, 06560, Ankara, Turkey; 4Laboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Jena, 07743, Germany; 5Jena Center for Soft Matter (JCSM) Friedrich Schiller University Jena, Jena, 07743, Germany
Correspondence: Oliver Werz, Email [email protected]
Background and Purpose: Lipid mediators (LM) play crucial roles in the complex inflammation process with respect to initiation, maintenance, and resolution. Proinflammatory leukotrienes (LTs), generated by 5-lipoxygenase (LOX) and the 5-LOX-activating protein (FLAP), initiate and maintain inflammation while specialized pro-resolving mediators (SPMs) formed by various LOXs as key enzymes promote inflammation resolution and the return to homeostasis. Since 5-LOX also contributes to SPM biosynthesis, smart pharmacological manipulation of the 5-LOX pathway and accompanied activation of 12-/15-LOXs may accomplish suppression of LT formation but maintain or even elevate SPM formation. Here, we demonstrated that the FLAP antagonist BRP-201 possesses such pharmacological profile and causes a switch from LT toward SPM formation.
Methods and Results: Comprehensive LM metabololipidomics with activated human monocyte-derived macrophages (MDM) of M1 or M2 phenotype showed that BRP-201 strongly inhibits LT formation induced by bacterial exotoxins. In parallel, SPM levels and 12/15-LOX-derived products were markedly elevated, in particular in M2-MDM. Intriguingly, in unstimulated MDM, BRP-201 induced formation of 12/15-LOX products including SPM and caused 15-LOX-1 subcellular redistribution without affecting 5-LOX. Experiments with HEK293 cells stably expressing either 5-LOX with or without FLAP, 15-LOX-1 or 15-LOX-2 confirmed suppression of 5-LOX product formation due to FLAP antagonism by BRP-201 but activated 15-LOX-1 in the absence of FLAP. Finally, in zymosan-induced murine peritonitis, BRP-201 (2 mg/kg, ip) lowered LT levels but elevated 12/15-LOX products including SPMs.
Conclusion: BRP-201 acts as FLAP antagonist but also as 12/15-LOX activator switching formation of pro-inflammatory LTs toward inflammation-resolving SPM, which reflects a beneficial pharmacological profile for intervention in inflammation.
Keywords: lipoxygenase, specialized pro-resolving mediators, leukotrienes, lipid mediators
Introduction
Lipid mediators (LM) orchestrate inflammatory responses by modulation of the innate immune system and thereby determine the body’s reaction to harmful stimuli like microbial infections or tissue damage.1,2 Upon cell activation during inflammation, phospholipase (PL)A2 enzymes release polyunsaturated fatty acids (PUFA) like arachidonic acid (AA), eicosapentanoic acid (EPA), and docosahexaenoic acid (DHA) that are transformed into LM in complex interconnected networks.3–5 Within these complex LM networks (Figure 1), enzymatic pathways leading to leukotriene (LT) and prostaglandin (PG) formation from AA predominate at the onset of inflammation.2,6 In contrast, the specialized pro-resolving mediators (SPM) that encompass lipoxins (LX), resolvins (RV), protectins (PD), and maresins (MaR) are produced from these PUFAs in a delayed manner in order to terminate and resolve inflammation leading to tissue repair and regeneration.2,4
 |
Figure 1 Biochemical pathways of lipid mediator formation and the influence of BRP-201. Schematic overview of LM-biosynthetic pathways during the acute phase of inflammation (red) and the inflammation resolution phase (blue). COX-1/2 and 5-LOX/FLAP generate PGE2 and LTB4, respectively, which are the major pro-inflammatory LM, while 12- and 15-LOX, partially in conjunction with 5-LOX, biosynthesize the SPMs namely lipoxins, E- and D-series resolvins, protectins and maresins. The FLAP antagonist BRP-201 suppresses LTB4 formation but elevates generation of SPM, and may thereby promote inflammation resolution. |
For PG formation, cyclooxygenase (COX)-1 and -2 produce the intermediate PGH2 from AA, which is further metabolized by different synthases to PG and thromboxane (TX), where PGE2 is massively formed in inflammation by microsomal prostaglandin E2 synthase-1 (mPGES-1) with vasodilatory, tissue permeabilizing, pain sensitizing, and fever inducing effects.7 The formation of LT is accomplished by conversion of AA by 5-lipoxygenase (5-LOX) aided by the nuclear membrane-bound 5-LOX-activating protein (FLAP) to 5(S)-hydroperoxyeicosatetraenoic acid (5-HPETE) that is further dehydrated to the epoxide LTA4 or reduced to 5(S)-hydroxyeicosatetraenoic acid (5-HETE).8 Conversion of LTA4 by LTA4 hydrolase (LTA4H) yields LTB4 that exacerbates inflammation and recruits neutrophils while LTC4 synthase converts LTA4 to LTC4, D4 and E4 that increase vasopermeability and constrict small vessels and bronchi.9,10 For FLAP, no enzymatic activity has yet been shown and it is believed that FLAP facilitates the access of 5-LOX to AA that is provided by cPLA2 at the nuclear membrane.8 FLAP is mandatory for LT formation in vivo and in intact cells from endogenously provided AA, and genetic or pharmacological interference with FLAP efficiently blocks LT formation, conferring FLAP as promising drug target for LT-related disorders.11,12 In contrast, transformations of PUFAs by the 12/15-LOXs are apparently independent of FLAP.13,14
The biosynthesis of SPMs is mainly driven by 12/15-LOXs, partially in conjunction with 5-LOX at least for LX and RV biosynthesis, while MaR and PD formation is 5-LOX-independent.4 Furthermore, CYP enzymes or acetylated COX-2 (by aspirin) may act together with 5-LOX to generate EPA-derived RVs via 18-HEPE, and epimers of LX and RV via 15R-HETE or 17R-HDHA, respectively. 12/15-LOX, CYP and Ac-COX-2 confer the first step in the conversion of PUFAs and then 5-LOX acts on the de-novo-biosynthesized precursors 15-HETE, 15-HEPE, 18-HEPE and 17-HDHA as substrates for SPM formation. Whether FLAP assists 5-LOX in the production of SPM from those mono-hydroxylated precursors is still a matter of debate15 but accumulating evidence indicates that SPM formation is FLAP-independent.13,14,16
Anti-inflammatory drugs like COX or 5-LOX inhibitors suppress formation of all PG or LT, respectively, but are essentially inefficient to resolve inflammation and cause adverse side effects in clinical therapy.17–19 Novel smart inhibitors that promote the switch from pro-inflammatory to pro-resolving LM might have potential as new pharmacological strategy not only to dampen inflammation but also to push its resolution, tissue regeneration and return to homeostasis.20 Here, we studied the pharmacological profile of the recently identified FLAP antagonist BRP-201 (5-{1-[(2-chlorophenyl)methyl]-2-{1-[4-(2-methylpropyl)phenyl]ethyl}-1H-benzimidazole-5-yl}-2,3-dihydro-1,3,4-oxadiazole-2-thione)21 for modulation of broad LM networks (Figure 1) in human pro-inflammatory M1 and anti-inflammatory M2 monocyte-derived macrophages (MDM) and in the peritoneum of zymosan-challenged mice in vivo. Our data show that in both experimental models BRP-201 efficiently blocked LT formation but elevated concomitant generation of SPM and their 12/15-LOX-derived precursors.
Materials and Methods
Materials
BRP-201 was synthesized as reported by us before.21 Deuterium-labeled and non-labeled LM standards for ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS-MS) quantification were obtained from Cayman Chemical/Biomol (Hamburg, Germany). All other chemicals were obtained at Sigma-Aldrich (Taufkirchen, Germany) unless stated otherwise.
Cell Isolation and Cell Culture
Leukocyte concentrates obtained from the Institute of Transfusion Medicine of the University Hospital Jena were prepared from peripheral blood from healthy human adult donors that had not taken any anti-inflammatory drugs for the last 10 days prior to blood donation. Informed consent was provided by the donors. The ethical committee of the University Hospital approved the protocol, and all performed methods were in accordance with the relevant regulations and guidelines. To isolate monocytes, the erythrocytes were sedimented by mixing the leucocyte concentrates with dextran (from leuconostoc spp. MW ~40,000, Sigma Aldrich, Taufkirchen, Germany). The supernatant was covered with lymphocyte separation medium (Histopaque®-1077, Sigma Aldrich, Taufkirchen, Germany) and centrifuged (2000 g, 10 min, 4°C). The peripheral blood mononuclear cells (PBMC) on the top of the lymphocyte separation medium were washed twice with ice-cold PBS and seeded in cell culture flasks for 1 h (37°C, 5% CO2) in PBS with Ca2+/Mg2+ to isolate the adherent monocytes. Differentiation and polarization into M1 and M2 macrophages was performed as described.14 In brief, to obtain M1 macrophages, adherent monocytes were treated with 20 ng mL−1 granulocyte macrophage-colony stimulating factor (GM-CSF, Peprotech, Hamburg, Germany) for six days in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 2 mmol L−1 L-glutamine, penicillin (100 U mL−1) and streptomycin (100 µg mL−1) and subsequently incubated for another 48 h with 100 ng mL−1 lipopolysaccharide (LPS) and 20 ng mL−1 interferon-γ (IFN-γ, Peprotech). To obtain M2 macrophages, 20 ng mL−1 M-CSF (Peprotech) was added to monocytes for six days, followed by 20 ng mL−1 IL-4 (Peprotech) for 48 h. Correct polarization and purity of macrophages were routinely checked by flow cytometry (FACS Canto Plus flow cytometer, BD Biosciences, Heidelberg, Germany) as reported14 using the following antibodies: FITC anti-human CD14 (2 µg/test, clone M5E2, BD Biosciences), PE anti-human CD54 (1 µg/test, clone HA58, BD Biosciences), APC-H7 anti-human CD80 (0.25 µg/test, clone L307.4, BD Biosciences), PE-Cy7 anti-human CD163 (2 µg/test, clone RM3/1, Biolegend, San Diego, CA, USA), PerCP-eFluor710 anti-human CD206 (0.06 µg/test, clone 19.2, BD Biosciences, San Diego, CA, USA).
HEK293 cells (purchased commercially from ATCC) stably expressing human 5-LOX, 5-LOX plus FLAP, 15-LOX-1 and 15-LOX-222 were cultured in monolayers (37°C, 5% CO2) in DMEM containing 10% FCS, penicillin (100 U/mL) and streptomycin (100 µg/mL). HEK293 cells expressing 5-LOX or 5-LOX/FLAP were selected by 200 µg mL−1 hygromycin B and/or 400 µg mL−1 geneticin, respectively; cells expressing 15-LOX-1 or 15-LOX-2 were obtained by using the plasmids pCMV6_15-LOX-1 (Origene, NM_001140) and pcDNA3.1/neom (+) 15-LOX-2 and selected with 400 µg mL−1 geneticin.
Immunofluorescence Microscopy
M2-MDM (1 × 106 cells) were seeded onto glass coverslips in a 12-well plate and cultured for 48 h. BRP-201, vehicle (0.1% DMSO) or 1% Staphylococcus aureus 6850-conditioned medium (SACM) was added for 180 min at 37°C. The process was stopped by fixation with 4% paraformaldehyde solution. Acetone (3 min, 4°C) and 0.25% triton X-100 (10 min, RT) were used for permeabilization before blocking with normal goat serum 10% (50062Z, ThermoFisher). Coverslips were incubated with mouse monoclonal anti-15-LOX-1 antibody, 1:100 (ab119774, Abcam, Cambridge, UK) and rabbit anti-5-LOX antibody, 1:100 (1550 AK6, kindly provided by Dr. Olof Radmark, Karolinska Institutet, Stockholm, Sweden) at 4°C overnight. 15-LOX-1 was stained with the fluorophore-labeled secondary antibodies; Alexa Fluor 488 goat anti-rabbit IgG (H + L), 1:500 (A11034, ThermoFisher) and Alexa Fluor 555 goat anti-mouse IgG (H + L); 1:500 (A21424, ThermoFisher). Nuclear DNA was stained with ProLong Gold Antifade Mountant with DAPI (15395816, ThermoFisher). Samples were analyzed by a Zeiss Axiovert 200M microscope, and a Plan Neofluar ×40/1.30 Oil (DIC III) objective (Carl Zeiss, Jena, Germany). An AxioCam MR camera (Carl Zeiss) was used for image acquisition.
Evaluation of Lipoxygenase Product Formation in HEK293 Cells
For evaluation of the effects on LOX product formation in stably transfected HEK293 cells, 2 × 106 cells per mL were preincubated with BRP-201 or vehicle (0.1% DMSO) in PBS pH 7.4 containing 0.1% glucose and 1 mM CaCl2 for 15 min. LM biosynthesis was initiated by addition of 2.5 µM A23187 plus 1 µM AA at 37°C and terminated after 15 min. Alternatively, to determine the stimulatory effects of BRP-201, cells were incubated with BRP-201 or vehicle for 180 min at 37°C. The reactions were stopped by addition of 2 mL ice-cold methanol containing 10 µL of deuterium-labeled internal standards (200 nM d8-5S-HETE, d4-LTB4, d5-LXA4, d5-RvD2, d4-PGE2 and 10 µM d8-AA) to facilitate LM quantification. Samples were kept at –20°C for one day to allow protein precipitation. After centrifugation (2000 g, 4°C, 10 min), the supernatants were subjected to solid phase extraction of formed LM that were then separated and analyzed by UPLC MS/MS as reported13 and described in the following sections.
Determination of Lipid Mediator Profile in Human Monocyte-Derived Macrophages
Human monocyte-derived M1 and M2 macrophages (2 × 106 cells) were seeded in 6-well-plates and incubated with BRP-201 or vehicle at 37°C with or without subsequent (after 10 min) addition of SACM, (from 24 h culture with OD = 0.05) for 180 min. The reaction was stopped with 2 mL ice-cold methanol containing deuterium-labeled internal standards (200nM, d8-5S-HETE, d4-LTB4, d5-LXA4, d5-RvD2, d4-PGE2 and 10 µM d8-AA). Samples were kept at –20°C for one day to allow protein precipitation. After centrifugation (2000 × g, 4°C, 10 min), supernatants were subjected to solid phase extraction and LM analysis by UPLC-MS-MS exactly as reported before13 and described in the following paragraph.
Lipid Mediator Metabololipidomics by UPLC-MS-MS
Analysis of LM by UPLC-MS-MS was performed as reported by us before.13 In brief, to 2 mL aliquots of supernatants obtained from incubated HEK293 cells and MDM as described previously, 8 mL acidified H2O (final pH = 3.5) was added and the samples were subjected to solid phase cartridges (Sep-Pak® Vac 6cc 500 mg/6 mL C18; Waters, Milford, MA). The columns had been equilibrated with 6 mL methanol and 2 mL H2O prior to sample loading. After washing with 6 mL H2O and subsequently with 6 mL n-hexane, LM were eluted with 6 mL methyl formate. The samples were dried using a TurboVap LV evaporation system (Biotage, Uppsala, Sweden) and resuspended in 100 µL methanol/water (50/50, v/v) for UPLC-MS-MS analysis. LM analysis was conducted with an Acquity™ UPLC system (Waters, Milford, MA, USA) and a QTRAP 5500 Mass Spectrometer (ABSciex, Darmstadt, Germany) equipped with a Turbo V™ Source and electrospray ionization. The LM were separated on an ACQUITY UPLC® BEH C18 column (1.7 µm, 2.1×100 mm; Waters, Eschborn, Germany) at 50°C at a flow rate of 0.3 mL/min and a mobile phase consisting of methanol-water-acetic acid of 42:58:0.01 (v/v/v) that was ramped to 86:14:0.01 (v/v/v) over 12.5 min and then to 98:2:0.01 (v/v/v) for 3 min. The QTRAP 5500 was operated in the negative ionization mode using scheduled multiple reaction monitoring (MRM) coupled with information-dependent acquisition. The scheduled MRM window was 60 sec, optimized LM parameters were adopted,13 and the curtain gas pressure was set to 35 psi. The retention time and at least six diagnostic ions for each LM were confirmed by means of an external standard (Cayman Chemical/Biomol GmbH, Hamburg, Germany). Quantification was achieved by calibration curves for each LM. Linear calibration curves were obtained for each LM and gave r2 values of 0.998 or higher. Additionally, the limit of detection for each targeted LM was determined.13
Zymosan-Induced Peritonitis Mouse Model
Adult (8 weeks) male CD1 mice (Charles River, Calco, Italy) were housed at the animal care facility of the Department of Pharmacy of the University of Naples “Federico II” and kept under controlled environment (ie, temperature 21 ± 2°C and humidity 60 ± 10%) and provided with normal chow and water ad libitum. Mice were allowed to acclimatize for four days prior to experiments and were subjected to 12 h light/dark schedule. Experiments were conducted during the light phase. The experimental procedures were approved by the Italian Ministry and carried out in accordance with the EU Directive 2010/63/EU and the Italian DL 26/2014 for animal experiments and in compliance with the ARRIVE guidelines and Basel declaration including the 3R concept. Mice (n=6/group) received BRP-201 (2 mg/kg) or vehicle (2% DMSO in saline) by intraperitoneal (ip) injection of 0.5 mL/mouse, given 30 min prior to peritonitis induction by zymosan (1 mg/mouse in 0.5 mL saline, ip). After 2 h mice were euthanized in a saturated CO2 atmosphere and peritoneal lavage was obtained by washing the peritoneal cavity with 3 mL ice-cold PBS and subsequent centrifugation (18,000 × g, 5 min, 4°C). Samples were immediately frozen for further analysis of LMs via UPLC-MS-MS as described previously.
Statistics
Results are expressed as mean + S.E.M. of independent experiments, where n represents the indicated numbers from separate donors performed on different days which is given in the figure legends for each and every figure panel. For animal experiments n=6 mice in each group were examined. Statistical analysis and graphs were made using GraphPad Prism 8 software (San Diego, CA). Unpaired t-test was used to analyze experiments for comparison of two groups; while for multiple comparisons, ANOVA with Bonferroni or Dunnett multiple comparison tests were applied as indicated. p-value ≤ 0.05 is a criterion for statistical significance.
Results
BRP-201 Suppresses Formation of Pro-Inflammatory 5-LOX Products and Elevates 12/15-LOX Products in Activated Macrophages
Previous results showed that BRP-201 is able to effectively suppress LT formation in human primary neutrophils21 but whether or not other branches of the LM network are affected by this compound remains unknown. To study the influence of BRP-201 on LM networks in a broader context, we employed human M1- and M2-MDM that generate multiple LM upon exposure to pathogenic bacteria14 due to cell activation by exotoxins like α-hemolysin.23 M1- and M2-MDM were preincubated with BRP-201 (0.1 to 3 µM) and then LM biosynthesis was elicited by Staphylococcus aureus-conditioned medium (SACM, containing exotoxins) within 180 min, which are appropriate and biologically relevant experimental settings to induce LM biosynthesis in human MDM.23
In M1-MDM, BRP-201 reduced the formation of the 5-LOX products LTB4 and its trans-isomers, 5-HETE, 5-HEPE and 5S,6R-diHETE in a concentration-dependent manner, starting at 0.1 µM, where especially LTB4 was significantly impaired by more than 50% (Figure 2A and B). PGE2, another major pro-inflammatory LM typically derived from M1-MDM, as well as other PG were hardly reduced (approx 20% at 3 µM BRP-201) or not altered (Figure 2A), indicating that BRP-201 mainly acts as inhibitor of the 5-LOX pathway devoid of substrate shunting effects toward COX products observed for certain other 5-LOX inhibitors.14,24,25 Notably, 12/15-LOX-derived products formed from AA (12-HETE and 15-HETE), EPA (12-HEPE and 15-HETE) and DHA (17-HDHA, 14-HDHA) were elevated after BRP-201 treatment, while 5,15-diHETE and 7-HDHA that are proposed to be partially produced by 5-LOX,14 were rather impaired (Figure 2A and B). Because in particular 15-lipoxygenation was elevated, this may hint to LM shunting toward 15-LOX-2 which in contrast to 15-LOX-1 is constitutively expressed in M1 macrophages23 and specifically oxygenates C15 but not C12.26 Note that formation of SPM is low in M1-MDM, as reported before,13,14 and even though some SPM such as RvD1 and MaR1 were somewhat increased, the other members were inconsistently modulated, and also the release of PUFAs was not markedly affected by BRP-201 (Figure 2A).
 |
Figure 2 Modulation of exotoxin-induced LM formation in M1- and M2-MDM by BRP-201. M1- and M2-MDM (2 × 106) were resuspended in 1 mL PBS containing 1 mM CaCl2, pre-incubated with BRP-201 (0.1, 0.3, 1 or 3 µM, as indicated) or vehicle (0.1% DMSO) for 10 min at 37°C, and stimulated with 1% SACM (from 6850 strain) for 180 min at 37°C. Then, the supernatants were collected, formed LMs were extracted by SPE and analyzed by UPLC-MS/MS. (A and C) Results are presented in pg/2 × 106 M1-MDM (A) and M2-MDM (C) for vehicle control (100%) given as mean ± SEM, and as percentage ± SEM of BRP-201-treated cells versus vehicle control (100%) in a heatmap; n=3–6. (B and D) Effects of BRP-201 on the AA-, DHA-, and EPA-derived monohydroxylated products and bioactive PGs, LTB4 and SPM produced in M1-MDM (B) or M2-MDM (D). Results are shown as percentage, given as mean ± SEM, of BRP-201-treated cells versus vehicle control (100%), n=3–6. (E) Amounts of formed LM in pg/2 × 106 MDM are shown in a spider web graph indicating the impact of BRP-201 at 1 µM on LM signature profiles, n=3–6. (F) Principal component analysis of the LMs (PGD2, PGE2, PGF2α, TXB2, tr-LTB4, LTB4, 5-HEPE, 5-HETE, 5S,6R-diHETE, PDX, RvD5, MaR1, 4-HDHA, 7-HDHA, 18-HEPE, 14-HDHA, 17-HDHA, 12-HEPE, 15-HEPE, 12-HETE, 15-HETE, 5.15-diHETE) organized by LM classes in vehicle- and BRP-201-treated M1- and M2-MDM (corresponding data in (A) and (C) at 1 µM BRP-201), n=3–6. Statistical analysis was performed via ratio-paired t-test, and principal component analysis was performed using R Studio (version 1.4) with implemented R packages FactoMineR and factoextra. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. |
A similar pattern of modulation of LM biosynthesis by BRP-201 was evident in SACM-activated M2-MDM. Thus, BRP-201 at 0.1 µM suppressed formation of 5-LOX products by approx. 60%, accompanied by slightly elevated levels of COX products at higher BRP-201 concentrations (Figure 2C and D). Of interest, BRP-201 markedly elevated formation of all detectable SPM, namely PD1, PDX, RvD1, RvD2, RvD5, and MaR1 with most pronounced effects at 3 µM for RvD1 and RvD2 (> 3-fold increase) (Figure 2C and D). In line with these findings, the formation of 17-HDHA and 14-HDHA, the precursors of these SPM, as well as 18-HEPE and the 12/15-LOX-derived 12-HETE, 12-HEPE, 15-HETE and 15-HEPE were increased, while 4-HDHA was not altered. Like in M1-MDM, the release of PUFAs was not markedly affected by BRP-201 in the M2 phenotype (Figure 2C). In Figure 2E and D, LM class switch in M1- and M2-MDM due to BRP-201 is visualized by comparison of selected representative bioactive LM and their precursors. Principal component analysis of the LMs formed in vehicle- and BRP-201-treated M1- and M2-MDM revealed different clusters that are separated, especially for M2-MDM, supporting the LM class switch due to BRP-201 (Figure 2F).
Modulation of 5-LOX/FLAP and 15-LOX Pathways in Stably Transfected HEK293 Cells
To gain more insight into the modulation of different LOX pathways by BRP-201, we took advantage of HEK293 cells that neither express any LOX nor FLAP per se but are convenient cell-based LOX expression models for studying select LOX pathways.22,27 We employed stably transfected HEK293 cells with either 5-LOX alone, with 5-LOX and FLAP, with 15-LOX-1 or with 15-LOX-2, respectively, as described previously.22 The cells were pretreated with BRP-201 (3 µM) for 15 min, stimulated with 2.5 µM A23187 and 1 µM AA for 15 min, and LOX products were analyzed by UPLC-MS-MS. As expected from our previous studies on FLAP and FLAP antagonists,27 in HEK293 cells expressing only 5-LOX (no FLAP), the formation of 5-LOX products was low and not significantly suppressed by BRP-201. However, upon co-expression of 5-LOX with FLAP, BRP-201 efficiently inhibited the FLAP-dependent generation of LTA4 hydrolysis products LTB4 and trans-LTB4 (Figure 3A and B). In parallel, BRP-201 strongly elevated 15-HETE formation but only in cells that express both 5-LOX and FLAP (Figure 3A). Of interest, in HEK293 cells expressing 15-LOX-1, BRP-201 markedly elevated (up to 3-fold) the formation of 15-HEPE and of 17-HDHA and 14-HDHA from endogenous EPA and DHA, respectively, but also 15-HETE biosynthesis from endogenous and/or exogenous AA was increased by about twofold (Figure 3). In contrast, BRP-201 failed to enhance product formation by 15-LOX-2 (Figure 3). Taken together, BRP-201 inhibits the biosynthesis of pro-inflammatory 5-LOX products in activated M1- and M2-MDM as well as in stimulated HE293 cells, apparently by acting at FLAP, but stimulates SPM and 15-LOX product formation, especially in M2-MDM.
 |
Figure 3 Modulation of LM biosynthesis in HEK293 cells transfected with LOX isoforms by BRP-201; impact of co-expression of FLAP with 5-LOX. HEK293 cells (2 ×106) transfected with human recombinant 5-LOX, 5-LOX and FLAP, 15-LOX-1 or 15-LOX-2 were resuspended in PBS containing 1 mM CaCl2 and 0.1% glucose, incubated with vehicle (0.1% DMSO) or BRP-201 (3 µM) for 15 min at 37°C and then stimulated with A23187 (2.5 µM) plus AA (1 µM) at 37°C for 15 min. Afterwards, the formed LMs were extracted from the supernatants using SPE and analyzed by UPLC-MS/MS. Data, given as mean ± SEM, are shown as (A) absolute values in pg/2×106 cells, and (B) as percentage of BRP-201-treated cells versus vehicle control (100%), n=3. Statistical analysis was performed via logarithmic paired t-test, * p<0.05, ** p<0.01. |
BRP-201 Activates Macrophages for Formation of SPM and Related 12/15-LOX Products
To assess whether active induction of 15-LOXs is the reason for elevated SPMs and related LM we exposed M1- and M2-MDM to BRP-201 (0.3, 1 or 3 µM) without additional stimulus for 180 min and determined the LM profiles. Interestingly, BRP-201 concentration-dependently induced the formation of 12/15-LOX products in both M1- and M2-MDM, with comparable efficiencies (about 2.5- to 3-fold) for 12-lipoxygenated (12-HETE, 12-HEPE and 14-HDHA) and 15-lipoxygenated products (15-HETE, 15-HEPE, and 17-HDHA) (Figure 4A–D). Along these lines, BRP-201 elevated SPM levels, in particular PD1, RvD5 and MaR1 in M2-MDM (Figure 4B and D). In contrast, formation of COX- and 5-LOX-derived products were not or only marginally elevated by BRP-201 and also 4-HDHA and 7-HDHA were not or less increased. Moreover, BRP-201 failed to elevate the levels of free PUFAs in both MDM phenotypes (Figure 4A and B), implying that elevated 12/15-LOX product formation is not simply due to larger amounts of substrate.
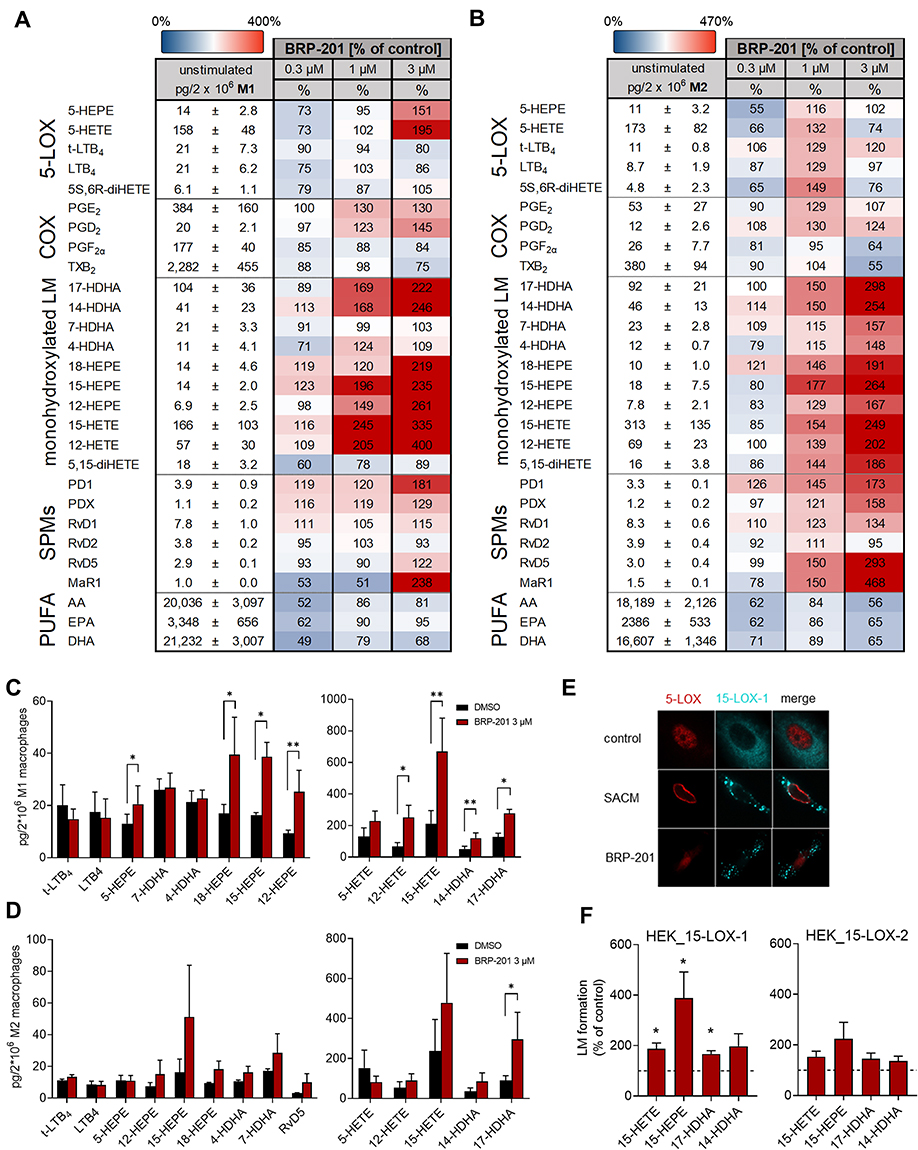 |
Figure 4 Induction of LM biosynthesis and 15-LOX-1 activation by BRP-201 in MDM and HEK293 cells. (A–D) Induction of LM biosynthesis by BRP-201 in MDM. M1- and M2-MDM (2 × 106) were resuspended in PBS containing 1 mM CaCl2 and incubated with vehicle (0.1% DMSO) or BRP-201 (0.3, 1, or 3 µM as indicated) for 180 min at 37°C. Then, formed LMs were extracted from the supernatants using SPE and analyzed by UPLC-MS/MS. Results are presented in pg/2 × 106 M1-MDM (A) and M2-MDM (B) for vehicle control (100%), given as mean ± SEM, and as percentage ± SEM of BRP-201-treated cells versus vehicle control (100%) in a heatmap; n=3–6. Induction of the most abundant LM in M1-MDM (C) and M2-MDM (D) by 3 µM BRP-201; results, given as mean ± SEM, are presented in pg/2 × 106 cells; n=3–6. (E) Subcellular redistribution of 5-LOX and 15-LOX-1 in M2-MDM. Cells were resuspended in PBS containing 1 mM CaCl2 and 5 mM MgCl2 and incubated with 1 µM BRP-201, 1% SACM (from strain 6850) or vehicle (0.1% DMSO). After 180 min, cells were fixed, permeabilized, and incubated with antibodies against 5-LOX (red) and 15-LOX-1 (cyan-blue), and analyzed by immunofluorescence microscopy; scale bars = 10 µm. Results shown for one single cell are representative of approximately 100 individual cells analyzed in n=3 independent experiments with separate donors, each. (F) HEK293 cells (2 × 106) transfected with human recombinant 15-LOX-1 or 15-LOX-2 were resuspended in PBS containing 1 mM CaCl2 and 0.1% glucose and incubated with vehicle (0.1% DMSO) or BRP-201 (3 µM) for 180 min at 37°C. Then, formed LMs were extracted from supernatants by SPE and analyzed by UPLC-MS/MS. Results, given as mean ± SEM, are presented as percentage of BRP-201-treated cells versus vehicle control (100%), n=3. Statistical analysis was performed with a ratio-paired t-test or logarithmic paired t-test, * p<0.05, ** p<0.01. |
Subcellular redistribution of 5-LOX and 15-LOX-1 from a soluble locale to a membrane compartment in (SACM- or bacteria-) activated human MDM is a determinant for their activation, access to substrate, and eventually for LM formation.14,23 We studied whether BRP-201 is able to induce such subcellular redistribution and, thus, activation of 5-LOX and 15-LOX-1 in M2-MDM by using immunofluorescence microscopy. In analogy to SACM, 1 µM BRP-201 caused 15-LOX-1 translocation from the cytosol to a membrane compartment within 180 min (Figure 4E). However, in contrast to SACM that caused 5-LOX nuclear membrane translocation, BRP-201 failed in this respect with 5-LOX remaining in the nucleosol (Figure 4E), confirming that BRP-201 may not activate 5-LOX.
To further support activation of 15-LOXs by BRP-201, we exposed HEK293 cells expressing 15-LOX-1 or 15-LOX-2 to 3 µM BRP-201 for 180 min and measured LM formation. For HEK cells expressing 15-LOX-1, an about 2- to 3-fold elevation of 15-HETE, 15-HEPE and 14-HDHA was observed (Figure 4F). Also, in HEK293 cells expressing the isoform 15-LOX-2, a moderate elevation of these LOX products in response to BRP-201 was evident. In conclusion, BRP-201 is able to activate 15-LOX-1 in MDM and in HEK293 cells to generate related LM.
BRP-201 Suppresses Formation of 5-LOX Products and Elevates 12/15-LOX Products in Zymosan-Induced Murine Peritonitis in vivo
In order to study if BRP-201 induces an LM class switch also in vivo, we pre-treated CD1 mice ip with 2 mg/kg BRP-201 for 30 min before peritonitis induction; due to the poor solubility of BRP-201, application of higher doses was not feasible. We then injected zymosan (ip) into the peritoneum in order to elicit broad spectrum LM formation within 2 hrs28 and analyzed the LM profile in the peritoneal lavage (exudates). As shown in Figure 5A and C, LTB4 formation was lowered upon BRP-201 treatment from 337±37 to 224±75 pg/mL exudate, and also the 5-LOX products trans-LTB4, 5S,6R-diHETE and 5-HETE were decreased by about 28 to 38%. In contrast, the sum of DHA- and EPA-derived 12/15-LOX products was significantly elevated (Figure 5B), with most pronounced effects for 12-HEPE and 14-HDHA (Figure 5A and C). Also, the sum of SPM was slightly increased (Figure 5B), in particular for PDX, but did not reach statistical significance (Figure 5A and C). AA-derived 12/15-LOX products (eg, 15-HETE), COX-derived prostanoids and LM formed by other pathways (eg, 4-HDHA and 7-HDHA), as well as PUFAs were not markedly changed by BRP-201 (Figure 5A and B). These data further support the hypothesis that BRP-201 induces a shift in the biosynthesis from pro-inflammatory 5-LOX- toward pro-resolving 12/15-LOX-derived LM.
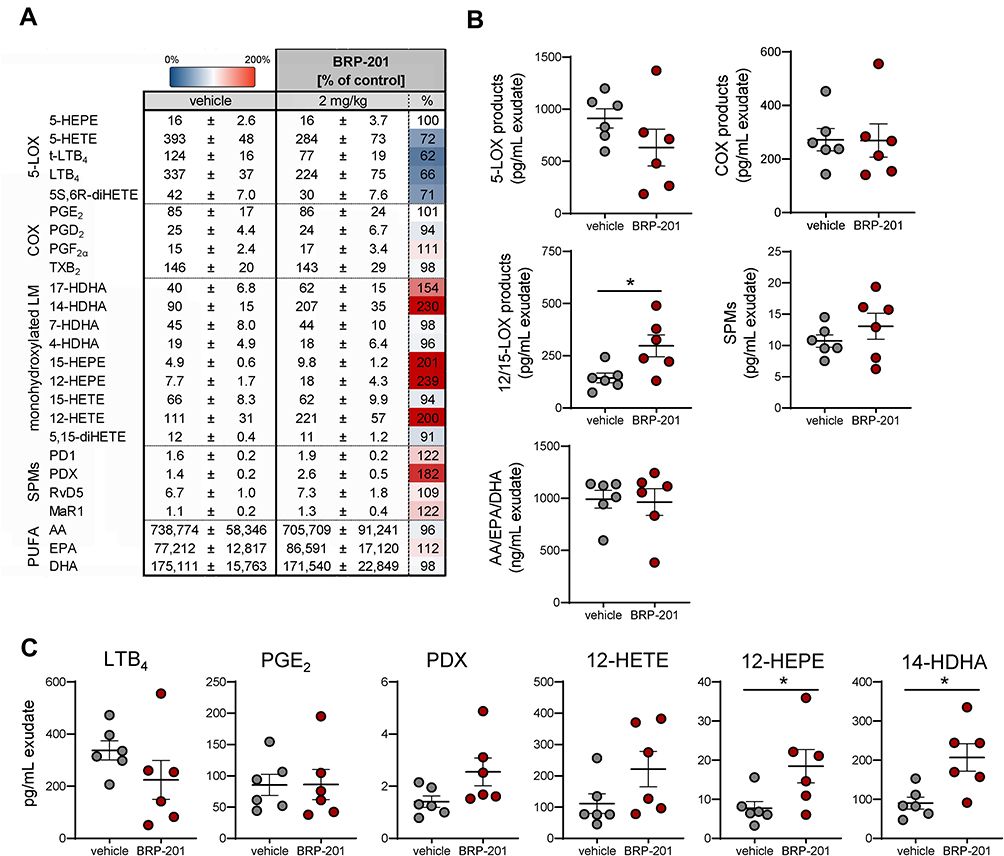 |
Figure 5 Effects of BRP-201 on LM biosynthesis during zymosan-induced murine peritonitis. Mice received BRP-201 (2 mg/kg) i.p. 30 min prior to i.p. injection of zymosan (1 mg/mouse). After 2 h, animals were sacrificed and peritoneal exudates were collected by lavage with 3 mL cold PBS. Formed LMs were extracted from the exudates using SPE and analyzed by UPLC-MS-MS. Results, given as means ± SEM, are presented in pg/mL exudate and as percentage of BRP-201-treated mice versus vehicle-treated controls (100%) in a heatmap (A), and as scatter dot plots (B) for LM grouped in different classes such as 5-LOX products (sum of LTB4, t-LTB4, 5S,6R-diHETE, 5-HETE and 5-HEPE), COX products (sum of PGE2, PGD2, PGF2α, TXB2), 12/15-LOX products (sum of 14-HDHA, 12-HEPE, 17-HDHA and 15-HEPE) and SPMs (sum of PD1, PDX, RvD5, and MaR1) in pg/mL exudate and PUFAs (sum of AA, EPA, DHA) in ng/mL exudate. Data are shown as single values (circles) and as means (black lines) ± SEM. (C) Selected bioactive and predominantly produced LMs are presented; given in pg/mL exudate; n = 6. Statistical analysis was performed by unpaired t-test; * p < 0.05. |
Discussion
Current anti-inflammatory pharmacotherapy related to intervention with LM relies on the interference with single enzymatic pathways in a complex LM network, that is, inhibition of COX-1/2-dependent PG formation by NSAIDs like ibuprofen, diclofenac or celecoxib, or suppression of 5-LOX-dependent LT biosynthesis by zileuton.29,30 NSAID-mediated reduction of the levels of PGs with homeostatic functions is frequently afflicted with severe side effects in the gastrointestinal tract, the kidneys and the cardiovascular system.31 Moreover, inhibition of PG formation by NSAIDs favors elevated LT levels by substrate redirection and cross talk-mediated shunting phenomena further promoting unwanted effects.13,18,24 Novel alternative strategies for intervention with inflammatory disorders focus on the development of dual inhibitors that block formation of both pro-inflammatory PGs (ie PGE2) and LTs, such as COX/5-LOX or mPGES-1/5-LOX inhibitors, aiming at circumventing these side effects.30,32 The discovery of SPMs and their favorable functions in inflammation resolution2 shifted the paradigm from anti-inflammatory toward resolution pharmacology.33 Thus, new concepts for inflammation pharmacotherapy were proposed that pursue the switch from pro-inflammatory eicosanoids toward inflammation-resolving SPMs, potentially accomplished by agents that dually block pro-inflammatory PGE2 and LT formation but stimulate SPM biosynthesis.28,34–37 Such pharmacological LM class switch strategies may bear a significant potential to effectively relieve chronic inflammation devoid of side effects of classical anti-inflammatory drugs.
Here, we showed that the FLAP antagonist BRP-201 causes an LM class switch in human macrophages, and to a minor extent also in murine peritoneum in vivo, by shifting the biosynthesis of LTs toward SPMs. BRP-201 was recently revealed as FLAP antagonist21 that efficiently inhibited LT formation in a convenient and well-recognized screening assay based on ionophore-activated human neutrophils38 with IC50 in the two-digit nanomolar range. To better estimate its pharmacological profile and potential, we analyzed the effects of BRP-201 within more complex LM networks using a more biologically relevant cellular system that allows better analysis of modulation of other LM branches. Thus, we employed human MDM with M1- and M2-like phenotype that when stimulated with bacterial exotoxins, produce a broad range of different types of LM, including PGs, LTs and SPM that can be analyzed by UPLC-MS-MS.13,14,23
The M1-phenotype generated substantial PG and LT but only moderate amounts of SPMs and their precursors upon exotoxin-challenge, which is in agreement with strong expression of COX-2 and FLAP but lack of 15-LOX-1, respectively.14,23 Accordingly, in M1-MDM, FLAP antagonism by BRP-201 caused potent suppression of 5-LOX products including LTB4, while the detectable SPMs were hardly and inconsistently affected. But human M1-MDM express low levels of 15-LOX-223 that in contrast to the 12/15-lipoxygenating 15-LOX-1 isoform, selectively oxygenate solely carbon 15 and 17 in AA/EPA and DHA, respectively, but not carbon 12 and 14.39 Indeed, BRP-201 elevated 15-HETE, 15-HEPE and 17-HDHA in M1-MDM but also 12-HETE, 12-HEPE and 14-HDHA. Shifts from 5- to 12/15-lipoxygenation in M1-MDM were observed also with 3-O-acetyl-11-keto-β-boswellic acid (AKBA) that alters the regiospecificity of 5-LOX via binding to an allosteric site of the enzyme.22 Whether BRP-201 acts in analogy to AKBA at this site of 5-LOX is unknown but conceivable, as discussed in the following paragraphs.
The pronounced abundance of 15-LOX-1 in M2-MDM enables formation of high amounts of SPMs and their precursors.14 In these cells, BRP-201 caused substantial elevation of SPM formation as well as the respective precursors 17-HDHA and 14-HDHA, suggesting that BRP-201 effectively redirects LM formation from 5-LOX/FLAP to 15-LOX-1. Among the mono-hydroxylated products in M2-MDM, only those generated by 15-LOX-1, namely 17-HDHA, 14-HDHA, 15-HEPE, 15-HETE, 12-HEPE and 12-HETE were concentration-dependently elevated by BRP-201 but not 7-HDHA, 4-HDHA, 18-HEPE and 5,15-diHETE that are formed independent of 15-LOX-1 or in conjunction with 5-LOX.13
The hypothesis that BRP-201 elevates 12/15-LOX product formation in two ways, that is, i) via a FLAP-dependent shift of the 5-LOX regiospecificity to a 12/15-lipoxygenating enzyme, and ii) by FLAP-independent activation of 15-LOX-1, is supported by our results obtained with LOX isoform-transfected HEK293 cells that are suitable as defined model systems to study selective LOX isoforms22 and the role of FLAP.27,40 Thus, BRP-201 redirected ionophore-induced LM formation only in 5-LOX-expressing HEK293 when FLAP was co-expressed, where classical FLAP-dependent 5-LOX products (ie, LTB4 and tr-LTB4) were suppressed but 12/15-LOX products were increased. Intriguingly however, BRP-201 elevated 12/15-LOX product formation also in HEK293 cells expressing 15-LOX-1 that are devoid of 5-LOX and FLAP, but not so in cells expressing the 15-LOX-2 isoform.
5-LOX plays a dual role in the formation of pro-inflammatory LTs on one hand and of anti-inflammatory/pro-resolving SPM on the other, which raises the question how the biosynthesis of these disparate LM is orchestrated in the cellular context.20 Moreover, it is questionable if pharmacological concepts can be pursued that allow favorable modulation of LM biosynthesis by interference with the 5-LOX pathway, that is, suppression of LTs without lowering SPM but rather stimulating the formation of the latter. As potential mechanisms, allosteric modulation/inhibition of 5-LOX leading to a shift of the regiospecificity toward a 12/15-lipoxygenating enzyme, but also antagonism of FLAP are both conceivable. The pentacyclic triterpene acids 3-O-acetyl-11-keto-β-boswellic acid (AKBA)22 and celastrol28 promote SPM formation in human neutrophils and macrophages as well as in inflamed murine peritoneum, apparently by allosteric modulation of 5-LOX shifting the regiospecificity from 5- to 12/15-lipoxygenation. Although BRP-201 clearly differs in structure form AKBA and celastrol, modulation of the 5-LOX regiospecificity by BRP-201 at this allosteric site is conceivable and would explain the elevated formation of 12/15-LOX products in M1-MDM and 5-LOX/FLAP-expressing HEK293 cells that are both devoid of 15-LOX-1.
The balance of LTs and SPM in leukocytes might be achieved through differential subcellular localization of 5-LOX where nuclear 5-LOX in proximity to FLAP favors LT generation while cytoplasmic 5-LOX distant from FLAP favors LXA4 formation.16 Thus, FLAP antagonists should prevent nuclear membrane-bound 5-LOX from generating LT but still permit activated cytosolic 5-LOX to oxygenate SPM precursors such as 15-HETE, 17-HDHA, and 18-HEPE to SPM. In fact, in human E. coli-activated M2-MDM, the FLAP antagonist MK886 blocked formation of classical 5-LOX-derived products while the SPM RvD5 and MaR1 and other 12/15-LOX products were elevated.13,14 Also, in murine peritonitis, MK886 markedly reduced the levels of LTB4, tr-LTB4 and 5-HETE in the peritoneal exudates while the amounts of LXA4, PD1, RvD4 and RvD5 remained elevated.35 Similarly, the FLAP antagonist BAY X-1005 blocked cysteinyl-LT formation but elevated SPM levels during murine liver injury.41 In agreement with these actions of FLAP antagonists in vivo, BRP-201 caused a trend toward lowered levels of LTB4 and of other classical 5-LOX products in the exudates of zymosan-challenged mice, while the levels of all detectable SPM, their precursors and most other 12/15-LOX-derived products were rather increased. Due to the poor water solubility of BRP-201, we could apply only a low dose of 2 mg/kg by ip injection which might be suboptimal, explaining why statistical significance could not be reached for single LM, except for 12-HEPE and 14-HDHA. Moreover, since the biology of murine 15-LOX-1 differs from that of the human ortholog in various aspects, in particular in the reaction specificity,26,39 it is also plausible that BRP-201 might be more active toward the human than to the mouse enzyme. Note that COX-derived prostanoids and LM formed by other pathways (eg, 4-HDHA and 7-HDHA) were not markedly affected in these murine peritoneal exudates, again supporting a concrete LM switch toward 12/15-LOX products. These data also imply that simple substrate redirection24 is unlikely as reason for elevated SPM and 12/15-LOX products. Rather, BRP-201, besides antagonizing FLAP, may cause stimulation of 15-LOX-1 activity in resident cells of the peritoneum thereby elevating formation of SPM and other 12/15-lipoxygenated products, supported by the induction of 12/15-LOX product formation in 15-LOX-1-expressing M2-MDM and HEK293 cells in the absence of a stimulus.
In general, the induction of cellular formation of LOX products requires the supply of free PUFA as substrates and the activation of LOXs that travel within the cell to access the substrate and to convert it.3,8,14,26 Our results suggest that elevation of 12/15-LOX products by BRP-201 in the absence of a stimulus (like A23187 or S. aureus) is primarily caused by stimulation of 15-LOX-1 and by facilitating the access of the LOX to its substrates, without marked increase of free PUFA supply. Thus, BRP-201 did not elevate PUFA levels in MDM but clearly induced 15-LOX-1 redistribution from the cytosol to a membrane compartment, along with 12/15-LOX product formation. Notably, BRP-201 failed to evoke translocation and product formation of 5-LOX in MDM, implying selectivity for modulation of LOX isoforms. Direct activators of 15-LOX in cell free-assays were reported42 and proposed to shift the AA metabolic network toward inflammation resolution. Allosteric activation of 15-LOX at a second AA binding site based on molecular dynamics simulations may be causative43 but experimental data confirming such shift in cells or in vivo are still missing. Interaction of BRP-201 with such allosteric AA binding site at 15-LOX-1 is conceivable, especially in view of the fact that BRP-201 acts as AA mimetic at FLAP competing with AA.21
Conclusion
Using human macrophages and mouse peritonitis as experimental models, we showed that BRP-201 induces a switch in the formation of pro-inflammatory 5-LOX-derived LT toward inflammation-resolving 12/15-LOX-derived SPM which reflects a beneficial pharmacological profile for intervention in inflammation. While the suppression of LT formation is obviously due to antagonism of FLAP, the stimulation of SPM and 12/15-LOX product formation by BRP-201 might be due to FLAP-dependent redirection of 5-LOX subcellular redistribution, allosteric modulation of 5-LOX, and by activation of 15-LOX-1. Therefore, BRP-201 is an interesting tool and lead for development of novel pharmacological strategies that pursue fostering of SPMs as immunoresolvents to promote inflammation resolution.
Data Sharing Statement
The datasets generated and analyzed in this study will be available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank Heidi Traber, Petra Wiecha, Katrin Fischer and Alrun Schumann for expert technical assistance.
Author Contributions
All authors significantly contributed to the reported work related to the conception, study design, execution, acquisition of data, analysis and interpretation; took part in drafting, revising or critically reviewing the article; gave final approval of the manuscript; have agreed on the journal to which the article has been submitted; agree to be accountable for all aspects of the work.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG), Collaborative Research Center SFB 1278 “PolyTarget” (project number 316213987, projects A04 and Z01). The study of the chemical development of BRP-201 was supported by The Scientific and Technological Research Council of Turkey (TÜBİTAK Grant No: 112S596).
Disclosure
Professor Erden Banoglu reports grants from TUBITAK, during the conduct of the study. The authors declare no conflicts of interest in this work.
References
1. Calder PC, Harwood J, Lloyd-Evans E. Eicosanoids. Essays Biochem. 2020;64(3):423–441. doi:10.1042/EBC20190083
2. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. doi:10.1038/nature13479
3. Astudillo AM, Balboa MA, Balsinde J. Selectivity of phospholipid hydrolysis by phospholipase A2 enzymes in activated cells leading to polyunsaturated fatty acid mobilization. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(6):772–783. doi:10.1016/j.bbalip.2018.07.002
4. Chiang N, Serhan CN, Harwood J, Lloyd-Evans E. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem. 2020;64(3):443–462. doi:10.1042/EBC20200018
5. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15(8):511–523. doi:10.1038/nri3859
6. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi:10.1126/science.294.5548.1871
7. Smith WL, Urade Y, Jakobsson PJ. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem Rev. 2011;111(10):5821–5865. doi:10.1021/cr2002992
8. Radmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim Biophys Acta. 2015;1851(4):331–339. doi:10.1016/j.bbalip.2014.08.012
9. Haeggstrom JZ. Leukotriene biosynthetic enzymes as therapeutic targets. J Clin Invest. 2018;128(7):2680–2690. doi:10.1172/JCI97945
10. Peters-Golden M, Henderson WR
11. Gur ZT, Caliskan B, Banoglu E. Drug discovery approaches targeting 5-lipoxygenase-activating protein (FLAP) for inhibition of cellular leukotriene biosynthesis. Eur J Med Chem. 2018;153:34–48. doi:10.1016/j.ejmech.2017.07.019
12. Pettersen D, Davidsson O, Whatling C. Recent advances for FLAP inhibitors. Bioorg Med Chem Lett. 2015;25(13):2607–2612. doi:10.1016/j.bmcl.2015.04.090
13. Werner M, Jordan PM, Romp E, et al. Targeting biosynthetic networks of the proinflammatory and proresolving lipid metabolome. FASEB J. 2019;33(5):6140-6153. doi: 10.1096/fj.201802509R
14. Werz O, Gerstmeier J, Libreros S, et al. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun. 2018;9(1):59. doi:10.1038/s41467-017-02538-5
15. Lehmann C, Homann J, Ball AK, et al. Lipoxin and resolvin biosynthesis is dependent on 5-lipoxygenase activating protein. FASEB J. 2015;29(12):5029–5043. doi:10.1096/fj.15-275487
16. Fredman G, Ozcan L, Spolitu S, et al. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc Natl Acad Sci USA. 2014;111(40):14530–14535. doi:10.1073/pnas.1410851111
17. Mahesh G, Anil Kumar K, Reddanna P. Overview on the discovery and development of anti-inflammatory drugs: should the focus be on synthesis or degradation of PGE2? J Inflamm Res. 2021;14:253–263. doi:10.2147/JIR.S278514
18. Rainsford KD. Anti-inflammatory drugs in the 21st century. Subcell Biochem. 2007;42:3–27.
19. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128(7):2657–2669. doi:10.1172/JCI97943
20. Gilbert NC, Newcomer ME, Werz O. Untangling the web of 5-lipoxygenase-derived products from a molecular and structural perspective: the battle between pro- and anti-inflammatory lipid mediators. Biochem Pharmacol. 2021;193:114759. doi:10.1016/j.bcp.2021.114759
21. Gur ZT, Caliskan B, Garscha U, et al. Identification of multi-target inhibitors of leukotriene and prostaglandin E2 biosynthesis by structural tuning of the FLAP inhibitor BRP-7. Eur J Med Chem. 2018;150:876–899. doi:10.1016/j.ejmech.2018.03.045
22. Gilbert NC, Gerstmeier J, Schexnaydre EE, et al. Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nat Chem Biol. 2020;16(7):783–790. doi:10.1038/s41589-020-0544-7
23. Jordan PM, Gerstmeier J, Pace S, et al. Staphylococcus aureus-derived alpha-hemolysin evokes generation of specialized pro-resolving mediators promoting inflammation resolution. Cell Rep. 2020;33(2):108247. doi:10.1016/j.celrep.2020.108247
24. He C, Wu Y, Lai Y, Cai Z, Liu Y, Lai L. Dynamic eicosanoid responses upon different inhibitor and combination treatments on the arachidonic acid metabolic network. Mol Biosyst. 2012;8(5):1585–1594. doi:10.1039/c2mb05503a
25. Werz O, Gerstmeier J, Garscha U. Novel leukotriene biosynthesis inhibitors (2012-2016) as anti-inflammatory agents. Expert Opin Ther Pat. 2017;27(5):607–620. doi:10.1080/13543776.2017.1276568
26. Ivanov I, Kuhn H, Heydeck D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene. 2015;573(1):1–32. doi:10.1016/j.gene.2015.07.073
27. Gerstmeier J, Weinigel C, Barz D, Werz O, Garscha U. An experimental cell-based model for studying the cell biology and molecular pharmacology of 5-lipoxygenase-activating protein in leukotriene biosynthesis. Biochim Biophys Acta. 2014;1840(9):2961–2969. doi:10.1016/j.bbagen.2014.05.016
28. Pace S, Zhang K, Jordan PM, et al. Anti-inflammatory celastrol promotes a switch from leukotriene biosynthesis to formation of specialized pro-resolving lipid mediators. Pharmacol Res. 2021;167:105556. doi:10.1016/j.phrs.2021.105556
29. Patrono C, Baigent C. Coxibs, traditional NSAIDs, and cardiovascular safety post-PRECISION: what we thought we knew then and what we think we know now. Clin Pharmacol Ther. 2017;102(2):238–245. doi:10.1002/cpt.696
30. Sala A, Proschak E, Steinhilber D, Rovati GE. Two-pronged approach to anti-inflammatory therapy through the modulation of the arachidonic acid cascade. Biochem Pharmacol. 2018;158:161–173. doi:10.1016/j.bcp.2018.10.007
31. Patrignani P, Patrono C. Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochim Biophys Acta. 2015;1851(4):422–432. doi:10.1016/j.bbalip.2014.09.016
32. Koeberle A, Werz O. Natural products as inhibitors of prostaglandin E2 and pro-inflammatory 5-lipoxygenase-derived lipid mediator biosynthesis. Biotechnol Adv. 2018;36(6):1709–1723. doi:10.1016/j.biotechadv.2018.02.010
33. Dalli J. Does promoting resolution instead of inhibiting inflammation represent the new paradigm in treating infections? Mol Aspects Med. 2017;58:12–20. doi:10.1016/j.mam.2017.03.007
34. Cheung SY, Werner M, Esposito L, et al. Discovery of a benzenesulfonamide-based dual inhibitor of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase that favorably modulates lipid mediator biosynthesis in inflammation. Eur J Med Chem. 2018;156:815–830. doi:10.1016/j.ejmech.2018.07.031
35. Gerstmeier J, Kretzer C, Di Micco S, et al. Novel benzoxanthene lignans that favorably modulate lipid mediator biosynthesis: a promising pharmacological strategy for anti-inflammatory therapy. Biochem Pharmacol. 2019;165:263–274. doi:10.1016/j.bcp.2019.03.003
36. Van Anh TT, Mostafa A, Rao Z, et al. From Vietnamese plants to a biflavonoid that relieves inflammation by triggering the lipid mediator class switch to resolution. Acta Pharm Sin B. 2021;11(6):1629–1647. doi:10.1016/j.apsb.2021.04.011
37. Zhang K, Pace S, Jordan PM, et al. Beneficial modulation of lipid mediator biosynthesis in innate immune cells by antirheumatic Tripterygium wilfordii glycosides. Biomolecules. 2021;11(5):746. doi:10.3390/biom11050746
38. Werz O, Steinhilber D. Development of 5-lipoxygenase inhibitors–lessons from cellular enzyme regulation. Biochem Pharmacol. 2005;70(3):327–333. doi:10.1016/j.bcp.2005.04.018
39. Kuhn H, Humeniuk L, Kozlov N, Roigas S, Adel S, Heydeck D. The evolutionary hypothesis of reaction specificity of mammalian ALOX15 orthologs. Prog Lipid Res. 2018;72:55–74. doi:10.1016/j.plipres.2018.09.002
40. Gerstmeier J, Weinigel C, Rummler S, Radmark O, Werz O, Garscha U. Time-resolved in situ assembly of the leukotriene-synthetic 5-lipoxygenase/5-lipoxygenase-activating protein complex in blood leukocytes. FASEB J. 2016;30(1):276–285. doi:10.1096/fj.15-278010
41. Titos E, Claria J, Planaguma A, et al. Inhibition of 5-lipoxygenase-activating protein abrogates experimental liver injury: role of Kupffer cells. J Leukoc Biol. 2005;78(4):871–878. doi:10.1189/jlb.1204747
42. Meng H, McClendon CL, Dai Z, et al. Discovery of novel 15-Lipoxygenase activators to shift the human arachidonic acid metabolic network toward inflammation resolution. J Med Chem. 2016;59(9):4202–4209. doi:10.1021/acs.jmedchem.5b01011
43. Meng H, Dai Z, Zhang W, Liu Y, Lai L. Molecular mechanism of 15-lipoxygenase allosteric activation and inhibition. Phys Chem Chem Phys. 2018;20(21):14785–14795. doi:10.1039/C7CP08586A
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.