Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Serum IL-1β and IL-17 levels in patients with COPD: associations with clinical parameters
Authors Zou Y ![]() , Chen X, Liu J, Zhou DB, Kuang X, Xiao J, Yu Q, Lu XX, Li W, Xie B, Chen Q
, Chen X, Liu J, Zhou DB, Kuang X, Xiao J, Yu Q, Lu XX, Li W, Xie B, Chen Q
Received 7 January 2017
Accepted for publication 3 April 2017
Published 24 April 2017 Volume 2017:12 Pages 1247—1254
DOI https://doi.org/10.2147/COPD.S131877
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Yong Zou,1 Xi Chen,1 Jie Liu,2 Dong bo Zhou,1 Xiao Kuang,1 Jian Xiao,1 Qiao Yu,1 Xiaoxiao Lu,1 Wei Li,1 Bin Xie,1 Qiong Chen1
1Department of Geriatrics, Respiratory Medicine, Xiangya Hospital of Central South University, 2Department of Emergency, The First People’s Hospital of Changsha, Changsha, People’s Republic of China
Abstract: COPD is a chronic airway inflammatory disease characterized mainly by neutrophil airway infiltrations. Interleukin (IL)-1β and IL-17 are the key mediators of neutrophilic airway inflammation in COPD. This study was undertaken to evaluate the serum IL-1β and IL-17 levels and associations between these two key mediators with clinical parameters in COPD patients. Serum samples were collected from 60 COPD subjects during the acute exacerbation of COPD, 60 subjects with stable COPD and 40 healthy control subjects. Commercial enzyme-linked immunosorbent assay kits were used to measure the serum IL-1β and IL-17 concentrations. The association between serum IL-1β and IL-17 with FEV1% predicted, C-reactive protein, neutrophil percentage and smoking status (pack-years) was assessed in the COPD patients. We found that serum IL-1β and IL-17 levels in acute exacerbation of COPD subjects were significantly higher than that in stable COPD or control subjects and were positively correlated to serum C-reactive protein levels, neutrophil % and smoking status (pack-years) but negatively correlated with FEV1% predicted in COPD patients. More importantly, serum IL-1β levels were markedly positively associated with serum IL-17 levels in patients with COPD (P=0.741, P<0.001). In conclusion, elevated serum IL-1β and IL-17 levels may be used as a biomarker for indicating persistent neutrophilic airway inflammation and potential ongoing exacerbation of COPD.
Keywords: neutrophilic airway inflammation, cigarette smoking, exacerbation
Introduction
COPD, which features chronic airway inflammation and incompletely reversible airflow obstruction, is mainly caused by chronic cigarette smoking (CS) and exposure to various noxious gases or particles.1 Although the pathogenesis of COPD is not clear, as is known to all, cigarette smoke-induced chronic inflammatory reactions in the airways and lung parenchyma are the leading causes of COPD.2,3 Toxic chemicals in cigarette smoke give rise to abnormal airway inflammation, which triggers the release of chemokines, promoting the infiltration of neutrophils and other inflammatory cells into airways.4,5 Particularly, neutrophils are the key players in the development of COPD, as demonstrated by the prominent increase in neutrophil count and percentage observed in sputum from COPD patients.6 Accumulated neutrophils can produce and release a variety of proinflammatory mediators and enzymes including neutrophil elastase (NE) and matrix metalloproteinases, which work together to promote the development of chronic bronchitis and emphysema.7–9
Neutrophil airway inflammation, one of the prominent features of COPD, is mediated by proinflammatory cytokines. Interleukin (IL)-1β, a typical innate immune cytokine that has been correlated to COPD, plays an important role in initiating and maintaining airway inflammation.10,11 IL-1β secretion is increased in stable and exacerbating COPD.12,13 Many studies in the COPD model in mice demonstrated that IL-1β was pivotal in contributing to the development of airway inflammation and emphysema.12,14–16 IL-1β is a key driver of neutrophil airway inflammation in COPD, mainly involving the following mechanisms: on the one hand, IL-1β is a potent inducer of IL-8 and IL-6 in normal human bronchial epithelial cells, two key cytokines promoting neutrophil recruitment and activation in airway;17 on the other hand, IL-1β mediates increase in the number of T-lymphocytes and dendritic cells in bronchoalveolar lavage fluid (BLF) and lung tissues in CS-induced pulmonary inflammation model, and these two types of cells promote the release of IL-6 and IL-17, increasing the recruitment of neutrophils into airway.12,18 A recent study reported that IL-1β-driven neutrophilia was mediated by IL-17 in the early stage, rather than in the peak phase of viral replication, in a chronic lung inflammation model in mice induced by lipopolysaccharide and elastase.19
Another important cytokine implicated in the pathogenesis of COPD is IL-17 (previously known as IL-17A). Increased levels of IL-17 and enhanced number of IL-17+cells have been detected in the bronchial mucosa and sputum of COPD patients.20–23 IL-17, which is primarily produced by the T-lymphocytes, can mobilize the neutrophils to infiltrate into airways through the induction of release of IL-8 or rat macrophage inflammatory protein-2 (rMIP-2) in airway inflammation models in mice.24–26
Apart from ongoing local airway inflammation, it is increasingly recognized that systemic inflammation, which is possibly derived from a spillover from the lung into the systemic circulation, may be a key link between COPD and comorbidities.27,28 Thus, the aim of this study was to evaluate serum IL-1β and IL-17 levels and associations between these two cytokines and clinical parameters (C-reactive protein [CRP], FEV1% predicted, neutrophil percentage [Neu%] and smoking status [pack-years]) in patients with COPD.
Methods
Recruitment of subjects
In total, 60 subjects with stable COPD (S-COPD), 60 subjects with acute exacerbation of COPD (AE-COPD) and 40 healthy control subjects were recruited from Xiangya hospital, Central South University between April and November 2016. Lung function tests were performed by skilled technicians in all subjects. In order to meet the inclusion criteria, COPD subjects were recruited on the basis of clinical history of chronic respiratory symptoms (ie, coughing, sputum or both, dyspnea), physical examination, chest radiography and a post-bronchodilator FEV1/forced vital capacity (FVC) ratio <70% according to the Global Initiative for Chronic Obstructive Lung Disease guidelines.1 Adults with normal pulmonary function (FEV1/FVC >70% and FEV1 >80% of predicted) can be included as healthy control subjects. Exclusion criteria are listed as follows: asthma or atopy, a history of coronary heart disease or malignancy or a systemic infection or an inflammatory process that could be associated with abnormal IL-1β and IL-17 levels.
An acute exacerbation was defined as a change in the symptoms of cough, expectoration and dyspnea that was beyond the daily variation and required changes in therapy in patients with COPD. All exacerbation visits occurred within 7 days after its onset. S-COPD was defined as having no symptoms correlated with exacerbation over at least 6 weeks.29 Serum CRP was measured and routine blood examination conducted, and baseline characteristics, including sex, age, body mass index and smoking history (pack-years), were collected in all subjects.
Specimen collection
Blood samples were drawn in vacuum blood tube and centrifuged (1,500 × g for 20 minutes) at room temperature within 60 minutes of collection. The supernatants were frozen at −80°C until analysis. This study was approved by the Ethics Committee of Xiangya Hospital, Central South University, and all subjects signed informed consent to participate in this study.
Measurements of serum IL-1β and IL-17
Serum IL-1β and IL-17 concentrations were measured using commercial enzyme-linked immunosorbent assay kits in accordance with the manufacturer’s protocols (R&D Systems, Minneapolis, MN, USA). All samples were measured in duplicate.
Statistical analysis
All statistical analyses were performed with SPSS version 19.0 (IBM Corporation, Armonk, NY, USA). Data were presented as the mean ± standard deviation (SD). Baseline characteristics of the subjects were compared by the independent t-test for continuous variables and the chi-square test for categorical variables. Differences between subject groups were analyzed by one-way analysis of variance with post hoc analysis carried out using Tamhane’s test. Correlations between serum IL-1β and IL-17 levels and FEV1% predicted, CRP, Neu% and smoking status (pack-years) were performed using Spearman or Pearson methods, depending on the normality of the data distribution. A two-tailed P<0.05 was considered significant.
Results
Study population
The demographic, clinical and lung function characteristics of the study subjects are presented in Table 1. As expected, the COPD patients had higher smoking exposure (pack-years), obvious airflow obstruction and higher serum CRP and Neu% than the controls. Patients with COPD and healthy control subjects were of similar age, sex and body mass index (P>0.05).
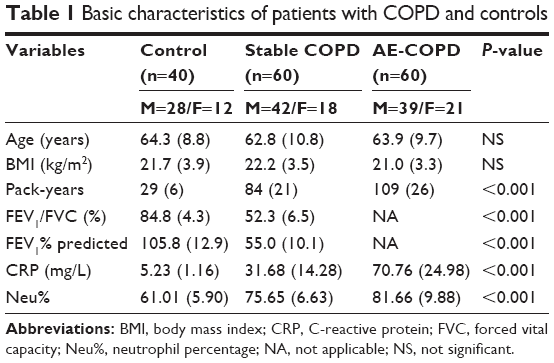 | Table 1 Basic characteristics of patients with COPD and controls |
IL-1β and IL-17 levels in serum
Serum IL-1β and IL-17 concentrations of the three subject groups are listed in Table 2. Figure 1 shows significantly higher serum IL-1β (12.73±3.16 vs 6.19±2.44 vs 2.66±0.52 pg/mL, P<0.001) and IL-17 (63.10±10.78 vs 50.47±8.94 vs 32.50±5.40 pg/mL, P<0.001) levels in the AE-COPD group than in the S-COPD or control group.
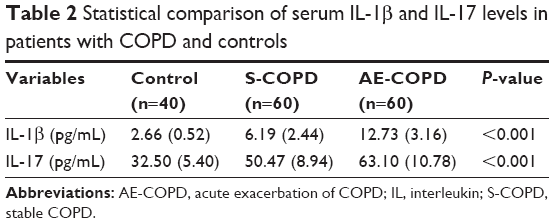 | Table 2 Statistical comparison of serum IL-1β and IL-17 levels in patients with COPD and controls |
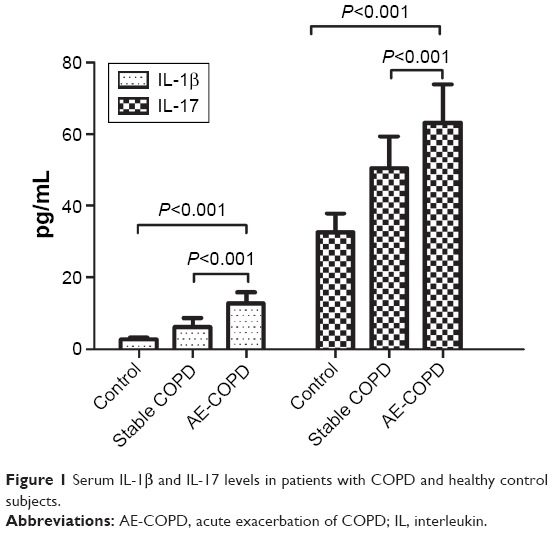 | Figure 1 Serum IL-1β and IL-17 levels in patients with COPD and healthy control subjects. |
Relationship between serum IL-1β and IL-17 levels in patients with COPD
In patients with COPD, serum IL-1β levels showed a notable positive correlation with the levels of IL-17 (r=0.741, P<0.001; Figure 2A). Further regression analysis showed that IL-1β might be an important factor contributing to elevation of IL-17 in patients with COPD (R2=0.55, regression coefficient =2.01; Figure 2B).
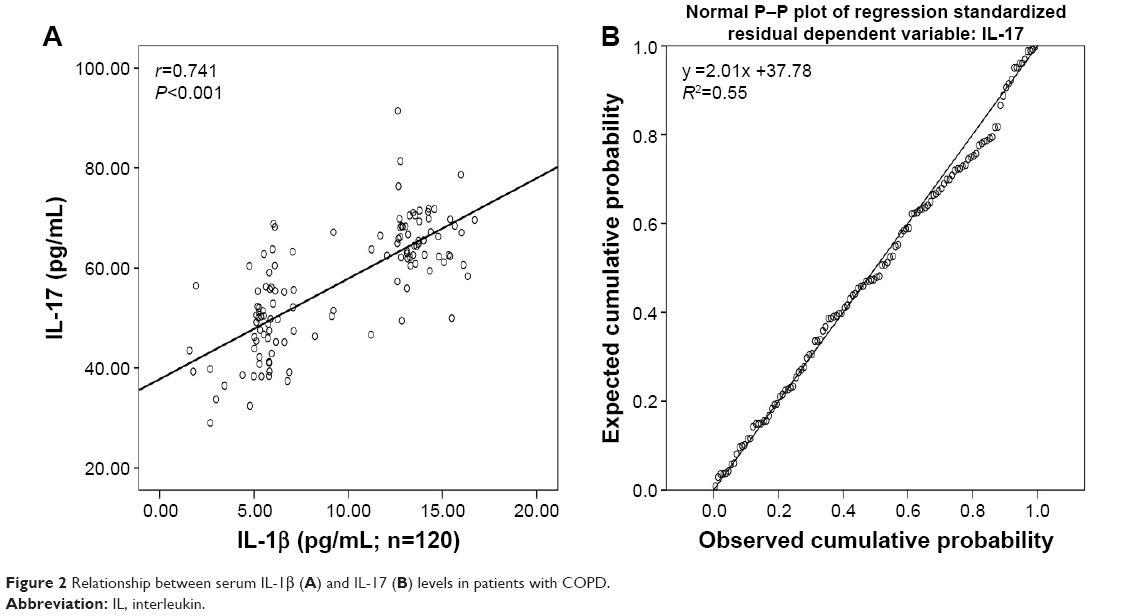 | Figure 2 Relationship between serum IL-1β (A) and IL-17 (B) levels in patients with COPD. |
Association of serum IL-1β and IL-17 levels with pulmonary function, CRP, Neu% and smoking status
There was significant inverse correlation between serum IL-1β (r=−0.634, P<0.001; Figure 3A) or IL-17 (r=−0.562, P<0.001; Figure 3B) levels and FEV1% predicted. Serum IL-1β levels showed a significant positive correlation with the levels of CRP (r=0.751, P<0.001; Figure 4A), Neu% (r=0.497, P<0.001; Figure 5A) and smoking status (r=0.628, P<0.001; Figure 6A). Serum IL-17 levels also showed a significant positive correlation with the levels of CRP (r=0.506, P<0.001; Figure 4B), Neu% (r=0.252, P=0.005; Figure 5B) and smoking status (r=0.403, P<0.001; Figure 6B).
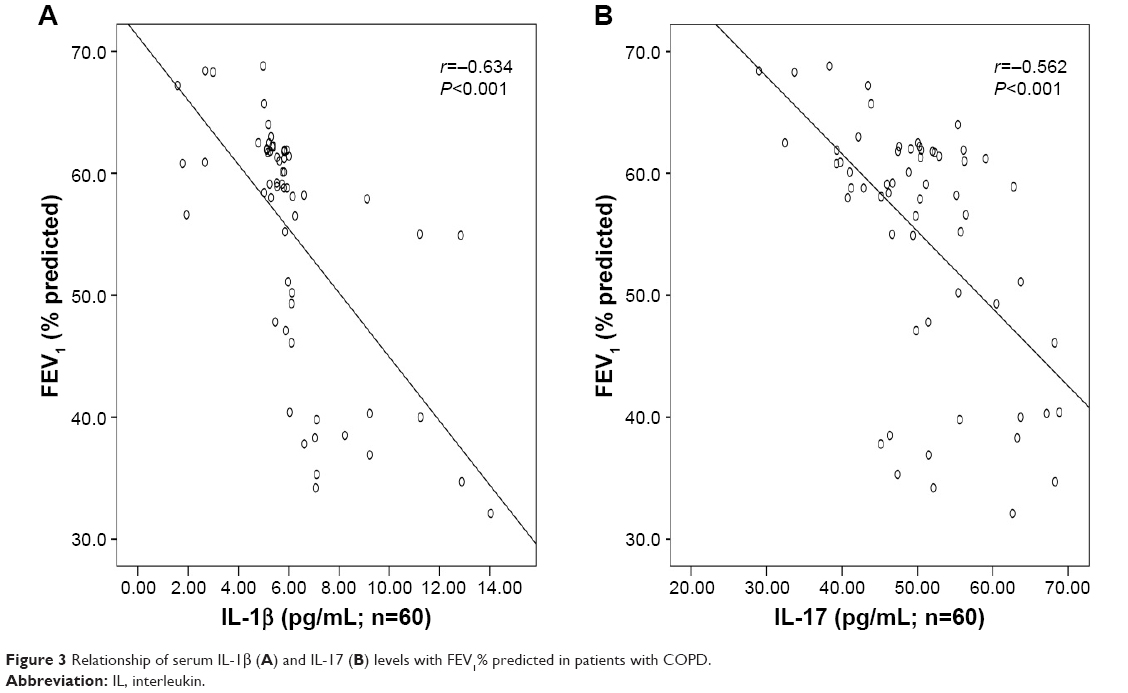 | Figure 3 Relationship of serum IL-1β (A) and IL-17 (B) levels with FEV1% predicted in patients with COPD. |
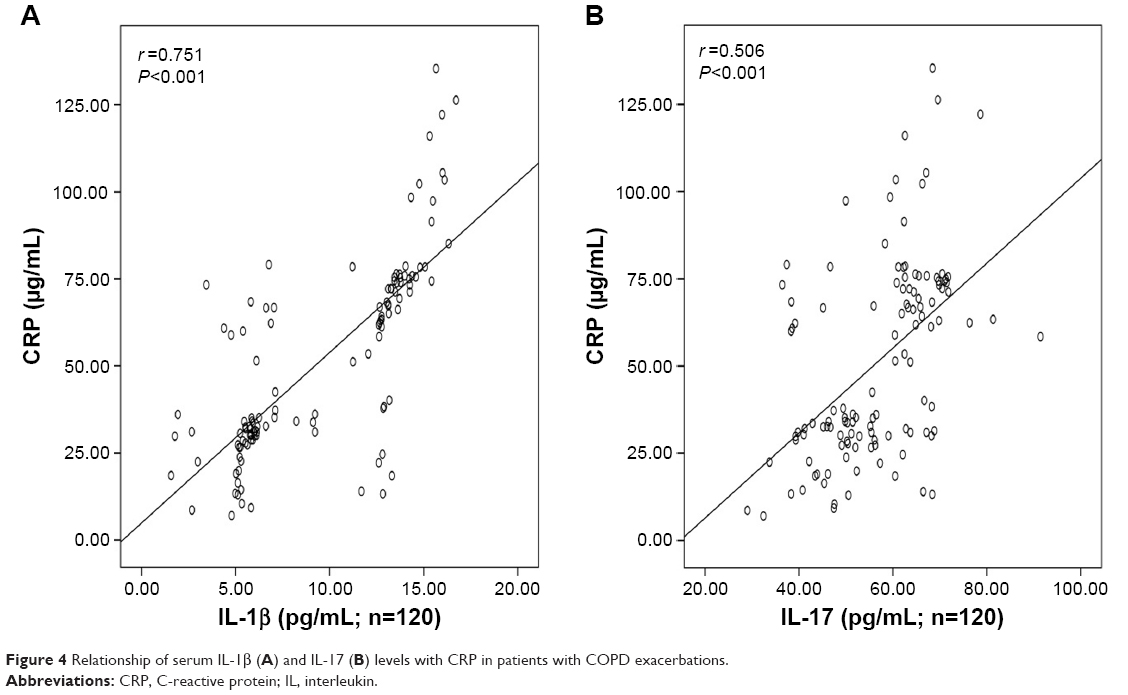 | Figure 4 Relationship of serum IL-1β (A) and IL-17 (B) levels with CRP in patients with COPD exacerbations. |
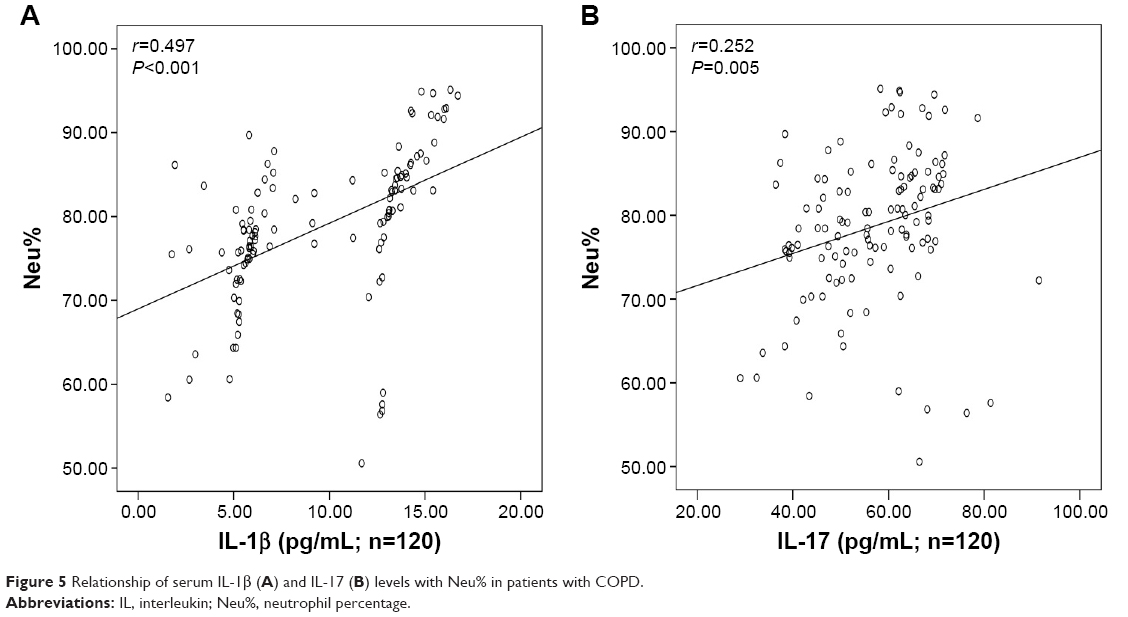 | Figure 5 Relationship of serum IL-1β (A) and IL-17 (B) levels with Neu% in patients with COPD. |
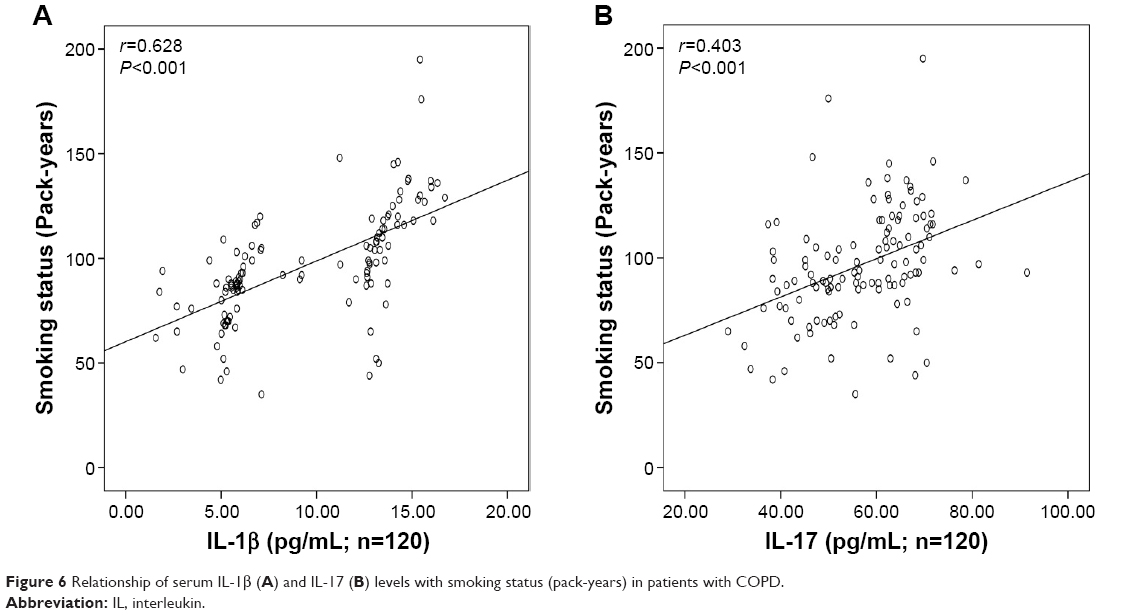 | Figure 6 Relationship of serum IL-1β (A) and IL-17 (B) levels with smoking status (pack-years) in patients with COPD. |
Discussion
There are several important findings of this study, which are 1) serum IL-1β and IL-17 levels were significantly higher in patients with COPD than those in the healthy control group; 2) levels of these two inflammatory mediators in the serum were associated with the important clinical parameters in COPD, including degree of airflow limitation, smoking status, CRP and Neu% in serum; and 3) serum IL-1β levels were correlated positively with IL-17, and more importantly, IL-1β might be an important factor contributing to elevation of IL-17 in patients with COPD. Therefore, serum IL-1β or IL-17 may be an important biomarker for distinguishing patients with COPD from healthy subjects, which helps in evaluating the severity of COPD and predicting the clinical outcomes.
COPD is characterized by anomalous and persistent inflammation, and neutrophils are the most prominent inflammatory cells infiltrating into the airway.6,30 Moreover, in a CS-induced pulmonary inflammation model in mice, neutrophils infiltrated first and increased with time in the BLF and lung tissue.18 Accumulated neutrophils can produce reactive oxygen species and release a variety of proinflammatory mediators and enzymes including NE and myeloperoxidase, promoting the occurrence and development of chronic bronchitis and emphysema.7–9
In addition, neutrophils can produce and release neutrophil extracellular traps (NETs) in patients with COPD.31–33 NETs are web-like structures containing DNA, histones, NE and myeloperoxidase,34 and they can prime macrophages to produce a precursor form of the inflammatory cytokine IL-1β (pro-IL-1β).35 NETs collaborate with another activation signal, such as cholesterol crystals or heat shock protein, promoting the release of IL-1β together.35,36 This theory supports our results that serum IL-1β was increased and associated with Neu% in patients with COPD. Previous studies also confirmed that IL-1β secretion was increased in S-COPD and exacerbating COPD.12,13 In return, IL-1β initiated and maintained neutrophil airway inflammation in COPD, mainly involving the following two mechanisms: first, IL-1β stimulated normal human bronchial epithelial cells to produce many inflammatory cytokines, such as IL-6 and IL-8, two putative cytokines promoting neutrophil recruitment and activation;17 second, IL-1β could promote the expression of IL-17 in the lung by increasing the number of IL-17–producing T lymphocytes (αβ T cells and γδ T cells),18,35 and IL-17 was recognized as a key regulator of neutrophils. Therefore, neutrophils mediate the formation of IL-1β that facilitates the neutrophil recruitment into airways, creating a vicious circle of neutrophil airway inflammation and contributing to the progressive development of COPD.
IL-17 expression and the number of IL-17+cells were increased in the bronchial mucosa and sputum of COPD patients in previous studies.20–23 Furthermore, Zhang et al recently reported that serum IL-17 was higher in COPD compared to that in healthy subjects and positively correlated with CRP and negatively correlated with FEV1% predicted,37 which is in accordance with our results. Another study showed that the serum concentrations of IL-17 in the patients with S-COPD were lower than those in the healthy controls, but serum IL-17 was higher in AE-COPD than that in S-COPD or healthy nonsmokers.38 These differences between studies may be attributed to different medications for COPD patients and COPD stages and severity. Moreover, IL-17 is mainly secreted by IL-17–producing T-lymphocytes including αβ T cells and γδ T cells, and these two kinds of cells can be induced by IL-1β in the lung tissue and BLF in patients with COPD.18,35 Consequently, it is proposed that IL-1β may be an important factor leading to increased expression of IL-17 in patients with COPD, and this was consistent with our result of the regression analysis.
This study has limitations. First, its relatively small size affected the power to detect associations between systemic markers and clinical parameters. Second, this study was carried out as a cross-sectional evaluation, and thus, alterations in serum inflammatory cytokines are correlative, but not predictive. Above all, the medications for patients with COPD were not investigated, and the influence of inhaled corticosteroid and bronchodilators on the systemic levels of these two cytokines could not be evaluated.
Conclusion
Our data indicate that concomitant measurements of IL-1β and IL-17 in serum may provide a novel biomarker of COPD. Serum IL-1β and IL-17 levels may be valuable as a marker of neutrophilic inflammation, chronic airflow obstruction and an acute exacerbation. Blockade of IL-1β or IL-17 could be a valid strategy for the prevention and control of COPD.
Acknowledgments
We express our gratitude to Jie Liu for specimen collection and Xiao Kuang for date processing and statistical analysis.
This work was supported by the National Natural Science Foundation of China (Grant No 81572284) and the Important Research and Development Plan of Hunan Provincial Science and Technology Department (Grant No 2015SK20662).
Disclosure
The authors declare no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Cosio MG, Majo J, Cosio MG. Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest. 2002;121(Suppl 5):160S–165S. | ||
Snider GL. Understanding inflammation in chronic obstructive pulmonary disease: the process begins. Am J Respir Crit Care Med. 2003;167(8):1045–1046. | ||
Terashima T, Wiggs B, English D, Hogg JC, van Eeden SF. Phagocytosis of small carbon particles (PM10) by alveolar macrophages stimulates the release of polymorphonuclear leukocytes from bone marrow. Am J Respir Crit Care Med. 1997;155(4):1441–1447. | ||
Stampfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol. 2009;9(5):377–384. | ||
O’Donnell R, Breen D, Wilson S, Djukanovic R. Inflammatory cells in the airways in COPD. Thorax. 2006;61(5):448–454. | ||
Profita M, Sala A, Bonanno A, et al. Chronic obstructive pulmonary disease and neutrophil infiltration: role of cigarette smoke and cyclooxygenase products. Am J Physiol Lung Cell Mol Physiol. 2010;298(2):L261–L269. | ||
Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;48(5):531–539. | ||
Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):71–86. | ||
Slack J, McMahan CJ, Waugh S, et al. Independent binding of interleukin-1 alpha and interleukin-1 beta to type I and type II interleukin-1 receptors. J Biol Chem. 1993;268(4):2513–2524. | ||
Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol. 2011;127(1):153–160, 160 e151–159. | ||
Pauwels NS, Bracke KR, Dupont LL, et al. Role of IL-1alpha and the Nlrp3/caspase-1/IL-1beta axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. 2011;38(5):1019–1028. | ||
Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184(6):662–671. | ||
Churg A, Zhou S, Wang X, Wang R, Wright JL. The role of interleukin-1beta in murine cigarette smoke-induced emphysema and small airway remodeling. Am J Respir Cell Mol Biol. 2009;40(4):482–490. | ||
Couillin I, Vasseur V, Charron S, et al. IL-1R1/MyD88 signaling is critical for elastase-induced lung inflammation and emphysema. J Immunol. 2009;183(12):8195–8202. | ||
Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32(4):311–318. | ||
Khan YM, Kirkham P, Barnes PJ, Adcock IM. Brd4 is essential for IL-1beta-induced inflammation in human airway epithelial cells. PLoS One. 2014;9(4):e95051. | ||
D’Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. EurRespir J. 2005;26(2):204–213. | ||
Sichelstiel A, Yadava K, Trompette A, et al. Targeting IL-1beta and IL-17A driven inflammation during influenza-induced exacerbations of chronic lung inflammation. PLoS One. 2014;9(2):e98440. | ||
Chang Y, Nadigel J, Boulais N, et al. CD8 positive T cells express IL-17 in patients with chronic obstructive pulmonary disease. Respir Res. 2011;12:43. | ||
Di Stefano A, Caramori G, Gnemmi I, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157(2):316–324. | ||
Doe C, Bafadhel M, Siddiqui S, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138(5):1140–1147. | ||
Eustace A, Smyth LJ, Mitchell L, Williamson K, Plumb J, Singh D. Identification of cells expressing IL-17A and IL-17F in the lungs of patients with COPD. Chest. 2011;139(5):1089–1100. | ||
Laan M, Cui ZH, Hoshino H, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999; 162(4):2347–2352. | ||
Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21(4):467–476. | ||
Zuniga LA, Jain R, Haines C, Cua DJ. Th17 cell development: from the cradle to the grave. Immunol Rev. 2013;252(1):78–88. | ||
Divo M, Cote C, de Torres JP, et al. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(2):155–161. | ||
Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59(7):574–580. | ||
Mackay AJ, Donaldson GC, Patel AR, Jones PW, Hurst JR, Wedzicha JA. Usefulness of the Chronic Obstructive Pulmonary Disease Assessment Test to evaluate severity of COPD exacerbations. Am J Respir Crit Care Med. 2012;185(11):1218–1224. | ||
Pilette C, Colinet B, Kiss R, et al. Increased galectin-3 expression and intra-epithelial neutrophils in small airways in severe COPD. Eur Respir J. 2007;29(5):914–922. | ||
Grabcanovic-Musija F, Obermayer A, Stoiber W, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res. 2015;16:59. | ||
Pedersen F, Marwitz S, Holz O, et al. Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD patients. Respir Med. 2015;109(10):1360–1362. | ||
Wright TK, Gibson PG, Simpson JL, McDonald VM, Wood LG, Baines KJ. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology. 2016;21(3):467–475. | ||
Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. | ||
Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349(6245):316–320. | ||
Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis-induced neutrophil extracellular traps activate human macrophages. J Innate Immun. 2013;5(6):591–602. | ||
Zhang L, Cheng Z, Liu W, Wu K. Expression of interleukin (IL)-10, IL-17A and IL-22 in serum and sputum of stable chronic obstructive pulmonary disease patients. COPD. 2013;10(4):459–465. | ||
Jin Y, Wan Y, Chen G, et al. Treg/IL-17 ratio and Treg differentiation in patients with COPD. PLoS One. 2014;9(10):e111044. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.