Back to Journals » International Journal of Nanomedicine » Volume 21
Sequential Release of mRNA Complex and T Cells by a Double-Layered Implantable Scaffold for Combination Therapy of Head and Neck Squamous Cell Carcinoma
Authors Ma P ![]() , Gao Y
, Gao Y ![]() , Wu S
, Wu S ![]() , Chen X, Fu X
, Chen X, Fu X ![]() , Cai Y, Men K
, Cai Y, Men K ![]() , Li L
, Li L ![]() , Li C
, Li C
Received 3 December 2025
Accepted for publication 5 March 2026
Published 23 March 2026 Volume 2026:21 582078
DOI https://doi.org/10.2147/IJN.S582078
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Dong Wang
Pingchuan Ma,1,* Yan Gao,2,* Shan Wu,2,* Xiayu Chen,2 Xizi Fu,2 Yuqin Cai,2 Ke Men,2 Longjiang Li,1 Chunjie Li1
1State Key Laboratory of Oral Diseases & National Center for Stomatology & National Clinical Research Center for Oral Diseases & Department of Head and Neck Oncology, West China Hospital of Stomatology, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 2Department of Biotherapy, Cancer Center and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Longjiang Li; Chunjie Li, State Key Laboratory of Oral Diseases & National Center for Stomatology & National Clinical Research Center for Oral Diseases & Department of Head and Neck Oncology, West China Hospital of Stomatology, Sichuan University, No. 14, 3rd Section of Ren Min Nan Road, Chengdu, Sichuan, 610041, People’s Republic of China, Tel +86 13708004544 ; +86 18382439003, Email [email protected]; [email protected]
Background: Head and neck squamous cell carcinoma (HNSCC) is associated with considerable morbidity and mortality, necessitating the development of novel therapeutic strategies. Adoptive cell therapy (ACT) and gene therapy are validated strategies for HNSCC treatment; however, both strategies have limitations when used alone in HNSCC and their synergistic integration requires further exploration to establish effective delivery platforms. In this study, we designed a double-layered scaffold based on photocurable hydrogel to combine mRNA gene therapy and ACT. And we aimed to validate the potential of this co-delivery scaffold in HNSCC treatment.
Methods: An implantable, double-layered, spherical scaffold was designed using a photocurable hydrogel gelatin methacryloyl (GelMA) with suitable mechanical strength and compressive properties. This scaffold incorporated primary T cells derived from mouse lymph nodes within the inner layer and a DOTAP-mPEG-PCL (DMP)/mBim mRNA-based gene therapy complex within the outer layer. Characteristics of the scaffold and delivery capacity of DMP nanoparticles were first measured. Then the active functions of both therapeutic components were tested separately. The synergistic therapeutic efficacy of the scaffold was further validated using mouse subcutaneous and mandibular invasion models. The immune activation and killing processes associated with sequential release were measured in this process.
Results: A double-layered spherical scaffold was produced and the DMP-mBim complex was characterized. The proliferation-inhibiting effect of the gene therapy complex on HNSCC cells was first demonstrated in vitro upon release, and the maintenance of T-cell bioactivity was confirmed. Results revealed the release process of two components during degradation of scaffold. The initially released DMP-mBim complex could induce immunogenic tumor cell death. Subsequently, tumor antigens generated during this process migrated into the scaffold along with the recruited dendritic cells (DCs). Activated T cells within the inner layer subsequently exerted tumor-killing effects after release. In HNSCC subcutaneous tumor and mandibular invasion models, local implantation of the double-layered scaffold effectively harnessed the synergistic effects of gene and cell therapies, inhibiting tumor growth and progression.
Conclusion: The combination of DMP-mBim gene complex and T-cell therapies represents an effective immunotherapeutic strategy, and the sequential release of mRNA gene therapy and T cells within a double-layered hydrogel prolongs antitumor efficacy. This strategy presents a potential immunotherapeutic approach for HNSCC that warrants further validation to support its future clinical translation.
Plain Language Summary: In this study, we developed an implantable spherical double-layered scaffold based on a photocurable GelMA hydrogel to combine gene and ACT. In this system, DMP-mBim gene therapy complexes are present in the outer layer, and primary T cells are present in the inner layer. Implantable scaffolds were implanted adjacent to a HNSCC mouse tumor model to perform immunogene therapy. After implantation, the outer layer was first released to exert a direct tumor-killing effect, inducing tumor cell apoptosis and antigen release. The subsequent release of activated T cells sustains an antitumor immune response. The therapeutic effects were confirmed in HNSCC subcutaneous xenograft and mandibular invasion models. This design provides a potential strategy for combined immunotherapy for the treatment of HNSCC.
Keywords: gene therapy, head and neck squamous cell carcinoma, hydrogel, implantable scaffold
Introduction
Head and neck squamous cell carcinoma (HNSCC) primarily arises from the mucosal squamous epithelium of the oral cavity, hypopharynx, larynx, and oropharynx.1 HNSCC accounts for over 800,000 new cases annually,2 and is associated with high morbidity and mortality due to its aggressive and invasive nature.3,4 Besides conventional therapies (surgery, chemotherapy, and radiotherapy), biotherapeutic approaches, particularly immunotherapy, have undergone significant innovation.5 However, both adoptive cell therapy (ACT) and immune checkpoint blockade demonstrate limited efficacy against HNSCC. The poor immune cell infiltration and insufficient target antigen expression constrained the advancement of ACT.6,7 And the heterogeneity among patients with HNSCC further impedes consistent therapeutic outcomes.8,9 Consequently, the development of combination immunotherapy strategies is essential and warrants further exploration, with gene therapy emerging as a promising approach for treating this disease.
Gene therapy involves molecular modifications at the DNA or RNA level within target cells,10 and RNA-based gene therapies have been extensively developed for cancer treatment.11 However, in HNSCC, the transient therapeutic effects that necessitate frequent complex administration and inadequate tissue penetration depth have impeded the development of gene therapy.12–14 Previous studies have indicated that gene therapy not only exerts direct antitumor effects15 but also modulates immune function,16,17 suggesting its synergistic potential with immunotherapy. This synergy from gene therapy may involve enhanced antigen release, improved T-cell bioenergetics and cytokine-mediated immunomodulation, imposing stringent requirements on the efficacy and safety of gene therapy complexes. Based on previous studies, the efficacy of the suicide gene Bim mRNA has been confirmed in other types of carcinoma, and the reported DMP cationic nanoparticles have also been demonstrated to possess adequate delivery capacity and biosafety.18 Therefore, we aim to investigate the potential of integrating DMP-Bim mRNA complexes with immune-related strategies and to develop novel delivery systems to achieve enhanced therapeutic efficacy.19
Adoptive cell therapy (ACT) is a vital component of immunotherapy that involves the administration of autologous or allogeneic antitumor immune cells.20 Potential effector cells include T cells,21 NK-cells,22 macrophages,23 and dendritic cell (DC).24 T-cell therapies have seen substantial development for HNSCC, including tumor-infiltrating lymphocyte (TIL) therapy, chimeric antigen receptor T-cell (CAR-T) therapy, and gene-engineered T-cell receptor (TCR) T-cell therapy.25,26 Nevertheless, T-cell therapies still have limitations primarily owing to the immunosuppressive microenvironment and scarcity of tumor-specific antigens.27,28 For instance, although HPV-16 E6/E7 represents a viable target for TCR T-cell therapy in preliminary studies, its applicability is restricted to HPV 16-positive patients.29 Similarly, targeting the ErbB family of CAR-T cells is complicated by their expression in healthy tissues, resulting in limited target specificity and on-target/off-tumor toxicity.30 Furthermore, the presence of immunosuppressive intratumoral regulatory T cells (Tregs) and a deficiency in effector T cells reflect the immunosuppressive landscape of HNSCC,31 impeding immune cell infiltration.32 These findings suggest that the use of T cells in combination with gene therapy may circumvent tumor microenvironment barriers33,34 and leverage tumor antigens to facilitate immune cell recruitment. To realize synergistic immunotherapy, a suitable delivery platform that can achieve co-delivery and sequentially controllable release of genetic and cellular components is crucial.
Hydrogel materials feature three-dimensional hydrophilic polymer networks with inherent physical integrity.35 Their utility in drug delivery and tissue engineering, particularly as therapeutic agents and RNA complexes, has been demonstrated during degradation.36–38 Implantable hydrogels offer advantages in local HNSCC therapy owing to their tunable mechanical strength and tissue adhesion properties.39,40 These features make them suitable for complex anatomical structures and implantation challenges within potential cavities in the head and neck regions, particularly in the co-delivery process for gene and cell therapy in double-layered system.41 The double-layered system is considered the direct and effective approach for achieving ordered release of a two-component combination, which is the advantage of it when compared to single system. Additional advantages include controlled-release kinetics, favorable biocompatibility and biodegradability profiles for localized therapy.42,43 Consequently, bilayered hydrogel scaffold systems are promising candidates for HNSCC treatment. Based on these considerations, the gelatin methacryloyl (GelMA) was suitable in our design. Compared to other kinds of hydrogels including alginate and hyaluronic acid hydrogel, GelMA hydrogels offer proper mechanical properties, controllable degradation behavior and adhesive properties, which make them more suitable to produce double-layered scaffold and be implanted in the complex anatomical spaces and tissue mobility of the head and neck region.44 Moreover, GelMA is also easy to synthesize with mature commercial options available and high reproducibility, enabling its use in our implantable experimental research.
Based on these principles, we engineered a double-layered spherical scaffold using photocurable hydrogel GelMA. This design incorporated primary T cells within the inner layer and a DOTAP-mPEG-PCL (DMP)-mBim gene therapy complex within the outer layer. The double-layered scaffold was prepared in vitro and planned to be implanted adjacent to the tumor to initiate a coordinated immune response (Figure 1). In the design, the initial degradation of the outer layer would release DMP-mBim complexes, inducing tumor cell apoptosis and antigen release while disrupting the immunosuppressive tumor microenvironment. Subsequently, the released tumor antigens could infiltrate the scaffold and recruit antigen-presenting cells (APCs), particularly dendritic cells (DCs). This triggers an immune cascade that activates encapsulated T cells. Ultimately, the released T cells would infiltrate tumor tissues, executing direct tumor killing and immunomodulatory functions to amplify the antitumor response. Through this system, we aimed to validate the potential of this co-delivery scaffold for integrating gene and cell therapies in HNSCC treatment.
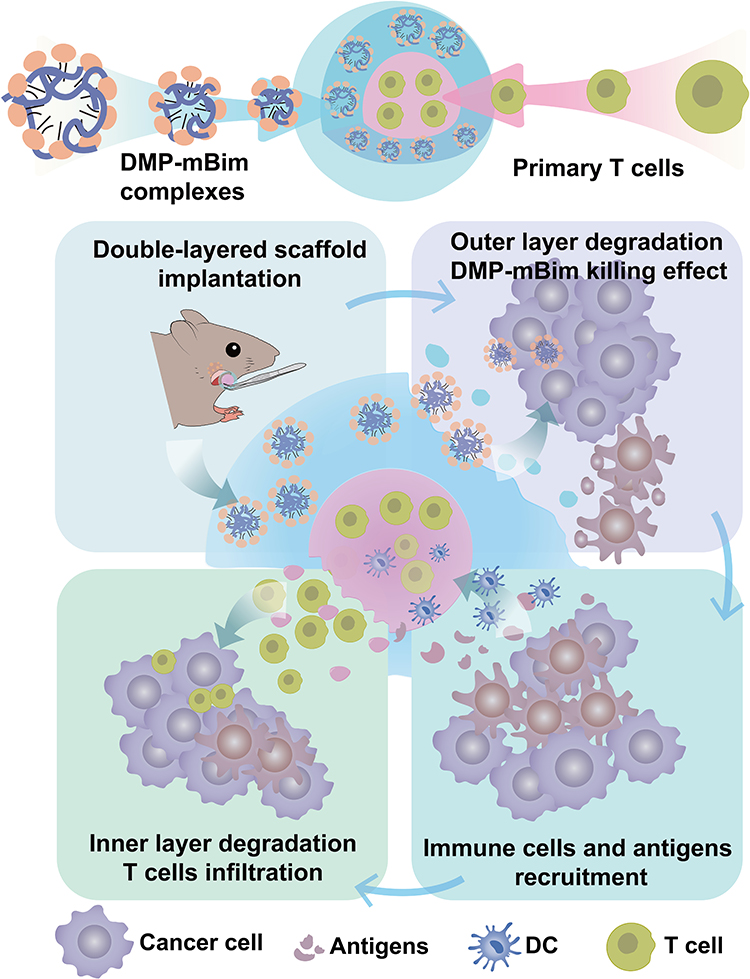 |
Figure 1 Composition of the double-layered scaffold and illustration of the immune therapy process after scaffold implantation. |
Materials and Methods
Mice and Cell Lines
The human HEK-293T cell line was obtained from the American Type Culture Collection (ATCC, Manassas, USA) and the murine head and neck squamous carcinoma cell line SCC-VII was purchased from the Cellverse Co., Ltd. (Shanghai, China). The SCC-VII cell line was cultured in RPMI 1640 medium (Gibco, Grand Island, NY, USA), and 293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM). All medium were supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, USA), penicillin (100 U/mL), and streptomycin (100 µg/mL). The cells were incubated at 37°C in a 5% CO2 atmosphere.
In this study, C3H/HeN female mice (6–8 weeks old) were obtained from SiPeiFu Biotechnology Co., Ltd. (Beijing, China). All animals were housed under specific pathogen-free (SPF) conditions in a temperature- (22 ± 1°C) and humidity-controlled (50 ± 10%) facility with a 12/12-hour light/dark cycle. Animal experiments were approved by the Institutional Review Board of West China Hospital, Sichuan University (Ethical Approval No. 20230330013) and conducted in accordance with the Guidelines and Standard Operating Procedures (SOP) for Laboratory Animals. In the animal experiments, animals were randomly assigned to different experimental groups. The investigators performing the administration and assessments were blinded to the group allocation throughout the experiment.
In this study, inhalation anesthesia with isoflurane was administered in the animal experiments, and micro-CT detection was performed using an RSA small vet anesthesia machine (RWD Life Science, Shenzhen, China). The specific anesthesia process was as follows: Mice were first placed in a clear, sealed induction chamber (V101, RWD Life Science, Shenzhen, China) connected to an RSA small vet anesthesia machine for anesthetic induction. Isoflurane (RWD Life Science, Shenzhen, China) was administered at a concentration of 3.0–3.5% (v/v) in 100% medical oxygen, with a flow rate of 1.0–1.5 L/min. Loss of the righting reflex was used as the endpoint for successful induction, which typically occurred within 60–90 seconds. The mice were quickly transferred to a nose cone system connected to an anesthesia machine. For the maintenance of surgical anesthesia, the isoflurane concentration was reduced to 1.0–1.5% (v/v), and the oxygen flow rate was 0.5–1.0 L/min. The scaffold implantation process and micro-CT detection were performed. The scaffold implantation procedure involves making an incision, inserting the scaffold, and suturing the incision site. The respiratory rate and pattern of the mice were monitored visually. Upon completion of the surgical procedure, isoflurane delivery was discontinued, and the mice were administered 100% oxygen at 1.0 L/min for at least 60s to facilitate the clearance of the anesthetic. The mice were then placed individually in clean, pre-warmed recovery cages positioned at a 45° incline to prevent airway obstructions. The bottom of the cage was lined with a heating pad set to 37°C. Recovery was defined as a full return of the righting reflex and spontaneous ambulation.
Preparation and Characterization of DMP Nanoparticles
DOTAP-mPEG-PCL (DMP) micelles were synthesized as follows. Briefly, N-[1-(2,3-dioleoyloxy) propyl]-N,N,N-trimethylammonium methylsulfate (cationic lipid DOTAP; Avanti) and mPEG-PCL polymer (Ruixibio, China) were mixed in a 1:1 weight ratio. The amount of each component used was 30 mg. The mixture was dissolved in 4 mL dichloromethane (KeLong Chemicals, China) and subsequently subjected to rotary evaporation (130rpm, N-1210, EYELA, JAPAN) for 1 h under heating (60°C) to obtain a lipid film. The film was then hydrated in 3 mL of double-distilled water at 60°C to achieve a final concentration of 20 mg/mL. The resulting nanoparticles were stored at 4°C. The mBim mRNA and DMP nanoparticle were mixed together and incubated at room temperature for 15 min to obtain DMP-mBim nanoparticle before use for transfection or other experiments. The detailed transfection process including the DMP-mBim obtain is listed behind.
The particle size distribution and zeta potential were measured using dynamic light scattering (Malvern Instruments, Worcestershire, UK). The morphological characteristics of the DMP nanoparticles were examined using transmission electron microscopy (TEM; Tecnai G2 F20 S-TWIN, FEI, USA).
In vitro Transfection Assay
The standard transfection protocol was as follows: 24 h prior to transfection, the cells were seeded in 24-well or 6-well plates (depending on the experiment) containing medium supplemented with 10% FBS. DMP nanoparticles and mRNA were combined in 100 μL of serum-free medium at specified mass ratios for 15 min. Throughout the study, a DMP: mRNA ratio of 25:1 was maintained. The complexes were then added to each well. Following a 4-h incubation at 37°C, the serum-free medium was replaced with complete medium (10% FBS), and the plates were maintained at 37°C under 5% CO2 for subsequent procedures. This transfection procedure was used in subsequent experiments.
The delivery efficiency of the DMP nanoparticles was assessed via in vitro transfection into SCC-VII murine head and neck squamous carcinoma cells. Cells were plated in 24-well plates at a density of 3×104 cells/well and incubated for 24 h at 37°C. The medium was then replaced with serum-free RPMI 1640. EGFP mRNA (TriLink Biotechnologies, USA) was used as a reporter gene for transfection after a 15-minute incubation with varying DMP quantities. The complexes were formulated at DMP: EGFP mRNA mass ratios of 5:1, 15:1, 25:1, and 35:1 using 1 μg mRNA. After 4 h, the medium was replaced with RPMI 1640 medium containing 10% FBS. Fluorescence microscopy was used to evaluate the cellular status and fluorescence intensity 24 h after transfection. The quantitative transfection efficiency was determined by flow cytometry (NovoCyte, ACEA Biosciences, USA).
Besides, the details of reagents and in vitro experiments (including gel retardation assay, cytotoxicity assay, clonogenic assay, live/dead staining, apoptosis assay and bone marrow derived dendritic cell isolation etc.) are given in Supplementary section 1.
Primary T Lymphocyte Extract and Phenotype Assessment
T lymphocytes were isolated from 6- to 10-week-old C3H/HeN mice. Prior to extraction, anti-mouse CD3 and CD28 antibodies (BioLegend, San Diego, CA, USA) were diluted in PBS to a concentration of 2 μg/mL and used to coat 6-well plates overnight at 4°C. Subsequently, the mice were euthanized, and lymph nodes from the axillary, intestinal, cervical, and inguinal regions were dissected and harvested. The lymph nodes were mechanically disrupted using plastic grinding pestles until the capsules were fully fragmented. Cell suspensions were filtered through sterile 70-μm nylon cell strainers and pelleted by centrifugation at 300 × g for 5 min. Erythrocytes were lysed using red blood cell (RBC) lysis buffer (Solarbio, China). After washing with RPMI 1640 medium, the cells were quantified for downstream applications including cultivating or manufacturing scaffolds. In the cultivating protocol, 6 × 106 cells were resuspended in 2 mL of RPMI 1640 medium in precoated plates, with interleukin-2 (BioLegend, San Diego, CA, USA) added to promote activation and proliferation. Finally, CD3 expression in the isolated T cells was analyzed using flow cytometry.
Synthesis of the Double-Layered Scaffold
The double-layered scaffold comprised an outer ordinary gelatin methacryloyl (EFL-GM-30) hydrogel and an inner porous gelatin methacryloyl hydrogel (EFL-GM-PR-001). These two kinds of hydrogels were purchased from Engineering For Life (EFL-Tech Co., Ltd., Suzhou, China). In the experiment description, the EFL-GM-30 hydrogel was referred to as GM, and the EFL-GM-PR-001 was referred to as PR. Specifically, 50 μL of PR hydrogel was added to paraffin oil to form spherical droplets. The hydrogel was then primarily crosslinked under 405 nm blue light to obtain an inner layer scaffold. Subsequently, 100 μL of GM hydrogel was added to the oil surrounding the inner scaffold. Interfacial integration enables the GM hydrogel to encapsulate the inner layer and form concentric spherical structures. Secondary crosslinking under 405 nm light (30s) was used to finalize the scaffold. The images of scaffolds were taken using scaffold with their inner layers stained with red non-diffusible hydrogel dye. The PR hydrogel contained mouse lymph node-derived T cells, whereas the outer GM layer contained the DMP-mBim complexes. To visualize the scaffold morphology, a non-diffusible dye (EFL) was loaded into the inner layer for imaging. For microstructural analysis, intact scaffolds were snap-frozen at −80°C, lyophilized for 48 h, and sectioned. The cross-sectional morphology was examined using an EVO10 scanning electron microscope (SEM; Zeiss, Oberkochen, Germany).
In vivo Evaluation of DMP-mBim Complex Induced Immunogenic Cell Death from Scaffold
We subsequently examined the immune response cascade, beginning with an assessment of immunogenic cell death (ICD) induced by DMP-mBim complex-containing scaffolds in vivo. A mandibular invasion model was established in C3H/HeN mice by injecting SCC-VII cells (3 × 105 cells in 100 μL medium) into the right masseteric space of the mouse. The implantation dates were 0. On Day 4, the mice were randomized into four treatment groups. Apart from one group treated with normal saline (NS), mice in the other three groups were implanted with different single-layer scaffolds (empty GM hydrogel (Gel), GM hydrogel containing DMP (Gel-DMP), and GM hydrogel containing DMP-mBim (70 μg) complex (Gel-DMP-mBim) under isoflurane inhalation anesthesia, as described previously. All scaffolds were 100 μL in spherical shape and were implanted to cover the tumor in the masseter region. All the subjects were euthanized on Day 8. Tumors and residual scaffolds were collected and subjected to enzymatic digestion using collagenase I (2 mg/mL) and collagenase IV (1 mg/mL) at 37°C for 2 h. The digested samples were filtered through a 70-μm nylon mesh, centrifuged, and collected. Tumor homogenates and scaffold eluates were prepared separately for analysis. Isolated cells were washed with PBS and stained with calreticulin-APC antibody (Proteintech, USA) for flow cytometric analysis. HMGB1 levels in the tumor homogenates and scaffold eluates were quantified using a murine ELISA kit (D721089-0096; Sangon Biotech, China).
Besides, we also detected the cytosolic level of ATP after DMP-mBim transfection process. SCC-VII cell were seeded in 6-well plates (1 × 105 cells/well). Following the established transfection protocol, the cells were treated with DMP or DMP-mBim complex (3 μg/well) transfection. After an additional 48-hour incubation, the cytosolic level of ATP was quantified using the ATP Assay Kit (S0026, Beyotime, China). The result was detected with the CLARIOstar Plus Full-Wavelength Microplate Reader (BMG LABTECH, Germany).
In vitro Evaluation of the SCC-VII Cell Lysate Uptake in Scaffold
To investigate whether tumor cell antigens could be recruited to the scaffolds, stained SCC-VII cell lysates were used to simulate tumor antigens in vitro. Cells were dual-stained for the plasma membrane and tubulin using DiI (Cell Plasma Membrane Staining Kit; Beyotime, China) and Tubulin-Tracker Green (Live Cell Staining Kit; Beyotime, China). After washing with Dulbecco’s phosphate-buffered saline (DPBS), the cells were resuspended at a density of 1×106 cells/mL. Oxidative lysis was performed using hypochlorous acid (37°C, 1 h), followed by 12 freeze-thaw cycles in liquid nitrogen. The suspension was sonicated (30 min) and centrifuged (1500 rpm for 3 min) to collect the stained lysates. Spherical empty GM hydrogels (100 μL) were prepared and incubated in 24-well plates with 1 mL DPBS and 1.8 mg of stained lysate. Fluorescence microscopy images were acquired at 0, 6, 24, and 48 h after incubation.
In vivo Detection of DCs Recruit Ability of Outer Layer Scaffold
Following confirmation of cell lysate uptake, DC recruitment to the scaffolds was investigated in vivo. A mandibular invasion model was established in C3H/HeN mice as described before. On Day 4 post-inoculation, three scaffold formulations were implanted to cover the tumor: empty GM hydrogel (Gel), GM hydrogel containing DMP (Gel-DMP), and GM hydrogel containing DMP-mBim complex (70 μg) (Gel-DMP-mBim). Mouse inhalation anesthesia with isoflurane was used during the surgical process, as previously described. The mice were euthanized on Day 8, and the residual scaffolds were collected. The scaffolds were enzymatically digested with collagenase (as previously described) to collect the embedded cells. The cells were stained with fluorescence-conjugated antibodies against CD11c (FITC) and CD80 (APC). DC recruitment was quantified by flow cytometry (NovoCyte, ACEA Biosciences, USA). The antibodies used in this study are listed in Supplementary Table S1.
In vivo Analysis of Immune Cells in the Double-Layered Scaffold
To evaluate the subsequent immune responses and cellular stimulation, double-layered scaffolds were implanted in C3H/HeN mice using a mandibular invasion model. On Day 4, three experimental scaffold groups were implanted under isoflurane inhalation anesthesia: empty PR + GM hydrogel (Gel/PR), PR hydrogel with T cells (3 × 106 cells) + empty GM hydrogel (Gel/PR-T cell), and PR hydrogel with T cells (3 × 106 cells) + GM hydrogel containing the DMP-mBim (70 g) complex (Gel-DMP-mBim/PR-T cell). The scaffolds comprised 50 μL of the PR inner layer and 100 μL of the GM outer layer. The mice were euthanized on Day 9, and the retrieved scaffolds were subjected to collagenase digestion. The collected cells were stained with fluorochrome-conjugated antibodies: CD3 (FITC), CD8 (APC), CD4 (PE), CD3 (FITC), CD44 (PE), CD62L (APC), CD11c (FITC), CD80 (APC), and CD86 (PE). Immune cell profiles were analyzed by flow cytometry (NovoCyte, ACEA Biosciences, USA).
Immune Activation Analysis of the Double-Layered Scaffold
Following the analysis of immune cells within the scaffold, we evaluated the immune function changes both in vitro by the co-culture of DC+T-cell and SCC-VII+DC+T-cell, and in vivo induced by the double-layered scaffolds in tumor tissues. In vitro, the method for dendritic cell (DC) extraction is detailed in the Supplementary section 1. T cells were harvested and co-cultured with either DCs or SCC-VII+DC in 6-well plates for 48 hours. The number of DC and T-cell were both 4×105 cells/well. SCC-VII cells were seeded in plates at 3×104 cells/well 48 h before the co-culture. A portion of harvested T-cells were pre-stained with CFSE Cell Proliferation Kit (C34554, Invitrogen, USA) before co-cultivation with DC or SCC-VII+DC. Other unstained T-cells were co-cultured with DC or SCC-VII+DC in other wells. After 48 hours, cells from all wells were harvested and supernatant from unstained T-cells wells were collected. The cells from CFSE-stained T cells wells were analyzed by flow cytometry to detect proliferation via the FITC signal. Cells from unstained T cells wells were stained with fluorescent-conjugated antibodies against CD11c (FITC), CD80 (APC), and CD86 (PE) to analyze the DC maturation. The level of IFN-γ and TNF-α of supernatant was detected using ELISA kit (IFN-γ: RK00019, ABclonal, China; TNF-α: ZC-39024, ZCI BIO, Chine;). The results were detected by SpectraMax absorbance reader (Molecular Devices, USA).
In vivo, a mandibular invasion model was established in C3H/HeN mice as previously described, and scaffolds were implanted on Day 4. The mice were randomly assigned to one of the four groups. Apart from the NS group, mice in other groups were implanted with different scaffolds under isoflurane inhalation anesthesia: empty PR + GM hydrogel (Gel/PR), PR hydrogel with T cells (3 × 106 cells) + empty GM hydrogel (Gel/PR-T cell), PR hydrogel with T cells (3 × 106 cells) + GM hydrogel with DMP-mBim (70 μg) complex (Gel-DMP-mBim/PR-T cell). The volume of scaffolds was same as before. On Day 9, the mice were euthanized, and the tumors were harvested. The tumors were digested with collagenase I (2 mg/mL) and collagenase IV (1 mg/mL). The resulting cell suspensions were stained with fluorescent-conjugated antibodies against CD3 (FITC), CD4 (PE), CD8 (APC), CD25 (APC), CD69 (PE), CD11c (FITC), CD80 (APC), and CD86 (PE). Flow cytometry was used to analyze the immune cell populations and their activation markers (NovoCyte, ACEA Biosciences, USA).
In vivo Evaluation of the Treatment Effect of the Double-Layered Scaffold in SCC-VII Subcutaneous Tumor Model
The treatment efficacy of the complete double-layered scaffold system was evaluated using a subcutaneous SCC-VII tumor model in C3H/HeN mice. SCC-VII cells (2 × 106 cells in 100 μL) were subcutaneously injected into 6–8-week-old female C3H/HeN mice. The mice were randomly divided into five groups: NS, Gel/PR, Gel-DMP-mBim/PR, Gel/PR-T cell, and Gel-DMP-mBim/PR-T cell. Except for the NS group, mice in the other groups were implanted with different double-layered scaffolds on Days 4 and 9 under isoflurane anesthesia. The compositions of the scaffold systems were as follows: Gel/PR indicates an empty PR hydrogel (inner layer) + empty GM hydrogel (outer layer). Gel-DMP-mBim/PR represents an empty PR hydrogel for the inner layer + GM hydrogel containing the DMP-mBim (70 μg) complex for the outer layer. Gel/PR-T cell were PR hydrogels with T cells (3 × 106 cells), and empty GM hydrogels were used for the outer layer. Gel-DMP-mBim/PR-T cell was PR hydrogel with T cells (3 × 106 cells) + GM hydrogel containing DMP-mBim (70 μg) complex. All hydrogels were implanted adjacent to subcutaneous tumors. Tumor volume was measured throughout the experiment and calculated using the following formula: volume = 0.5 × length × width2. The body weights of each mouse were recorded. On Day 11, all mice were euthanized, and the tumor tissues were collected, weighed, and photographed. The major organs were harvested for hematoxylin-eosin staining. Tumors from a subset of mice in each group were fixed in 4% paraformaldehyde for histological analyses. The remaining tumors were digested with collagenase I (2 mg/mL) and collagenase IV (1 mg/mL). The resulting cell suspensions were stained with fluorescent-conjugated antibodies against CD3 (FITC), CD8 (APC), CD4 (PE), CD11c (FITC), CD80 (APC), and CD86 (PE). The samples were analyzed by flow cytometry.
In vivo Evaluation of the Treatment Effect of the Double-Layered Scaffold in SCC-VII Mandible-Invasion Model
We further evaluated the therapeutic efficacy of the double-layered scaffold system in an SCC-VII mandibular invasion model using C3H/HeN mice. The model was established as above. The group assignments and hydrogel designs were consistent with those of the subcutaneous tumor models. On Day 4, the scaffolds were implanted into the tumor site under isoflurane inhalation anesthesia. Tumor volume (calculated as volume = 0.5 × length × width2) and body weights were monitored. On Day 10, the mice were euthanized, and whole-body images were captured.
For flow cytometry analysis, tumor tissues from a subset of mice were collected and digested with collagenase I (2 mg/mL) and collagenase IV (1 mg/mL). The cells were stained with fluorescence-conjugated antibodies against CD3 (FITC), CD8 (APC), CD4 (PE), CD11c (FITC), CD80 (APC), and CD86 (PE). In another subset, the intact heads were excised and fixed in 4% paraformaldehyde for histological analyses. Major organs were harvested and fixed for safety assessments.
In addition, micro-computed tomography (micro-CT) imaging (SkyScan 1276, Bruker-microCT, Kontich, Belgium) was performed during the study and post-sample collection (70 kV, 200 µA, pixel size 15 μm, 2048×2048 resolution, 1 mm filter, 0.4 degrees rotation step) under isoflurane inhalation anesthesia. The bone volume fraction (BV/TV) was quantified to assess bone resorption, and three-dimensional (3D) reconstruction of mouse heads was performed. Zygomatic bone destruction was scored as follows: 0, normal; 1, asymmetric; 2, hairline fracture; 3, complete separation; and 4, destruction of >1/3 of the zygoma. The scores were evaluated by two independent researchers.
Multiplex Immunohistochemistry Analysis
Multiplex immunohistochemistry (mIHC) and hematoxylin-eosin (H&E) staining was performed on formalin-fixed paraffin-embedded tissue sections collected from the heads of the mice. The tissues were first decalcified in 0.5 M EDTA, followed by standard dehydration and clearing procedures. Suitable tissue sites were selected for paraffin embedding and 3–4-μm-thick coronal sections were prepared for staining. Parts of the sections were stained with hematoxylin-eosin (H&E). Others were analyzed by mIHC. mIHC was performed using a TSA 4-color kit (AFIHC024; AiFang Biological, Hunan, China) according to the manufacturer’s instructions. This analysis primarily focused on the distribution of T cells and dendritic cells (DCs). Deparaffinized slides were incubated overnight at 4°C with primary antibodies. The horseradish peroxidase (HRP)-conjugated anti-rabbit/mouse secondary antibody provided in the kit was applied for 20 min, followed by the development of the corresponding tyramide (TYR) fluorescent dye. After each staining cycle, the sections were treated with an antibody elution buffer at 37°C for 40 min before the next antibody was applied. The primary antibodies and corresponding TSA dyes used were CD3 (ab16669, Abcam, UK)/TYR520, CD8 (ab217344, Abcam, UK)/TYR570, CD11c (ab219799, Abcam, UK)/TYR520, and CD80 (ab215166, Abcam, UK)/TYR570. After all antibody staining steps, DAPI (S36964, Invitrogen) was used for nuclear counterstaining. Whole-section fluorescence images were acquired using a SLIDEVIEW VS200 system (Olympus, Japan) for subsequent analysis.
Statistical Analysis
In this study, data are presented as mean ± standard deviation (SD), and P < 0.05 was defined as statistically significant. Experimental data were analyzed using GraphPad Prism 8.0 (GraphPad Software, USA) by one-way ANOVA or Student’s t-test. Figures were prepared using Adobe Photoshop and Illustrator software.
Results
Preparation and Characterization of DMP
DMP cationic micelles were prepared via the self-assembly of the cationic lipid DOTAP and amphiphilic block copolymer mPEG-PCL. The resulting nanoparticles exhibited an average particle size of 25.76 ± 0.85 nm and a zeta potential of 59.9 ± 4.6 mV, as measured by dynamic light scattering (Figure 2A), confirming their nanoscale dimensions and positive surface charge. TEM revealed a spherical morphology with an approximate diameter of 30 nm, which is consistent with the size distribution measurements (Figure 2B). The cytotoxicity of DMP nanoparticles was assessed in 293T cells (Figure 2C). Compared to the gold standard transfection reagent PEI25K, which had an IC50 of less than 75 μg/mL, DMP displayed significantly lower cytotoxicity with an IC50 of 1000 μg/mL, demonstrating its acceptable biosafety profile. Besides, the hemolysis test also revealed that the percentage hemolysis of DMP was significantly lower than that of PEI25K (Figure 2D), with less hemolysis of red blood cells in DMP. The critical micelle concentration (CMC) of DMP was calculated as 0.0210 mg/mL (Figure 2E).
 |
Figure 2 Characteristics of DMP nanoparticles and the proliferation-inhibiting effect of the DMP-mBim complex in vitro. (A) Size distribution and zeta potential of DMP nanoparticles. (B) Transmission electron microscopy (TEM) photomicrographs of DMP stained with phosphotungstic acid solution (scale bar: 50 nm). (C) Cell viability MTT assay of DMP and PEI25K in 293T cells. (D) Hemolysis test of DMP nanoparticles, Triton X-100 was used as positive control, and normal saline was used as negative control (***P<0.001, ****P<0.0001). (E) Critical micelle concentration evaluation of DMP using pyrene as the fluorescent dye. (F) Murine Bim mRNA (mBim) obtained by the in vitro transcription method was detected by electrophoresis. (G) Gel-retardation assay of mRNA and DMP at different weight ratios. (H) RNase protection electrophoresis assay of the DMP-mRNA complex. (I) Transfection efficiency of DMP-EGFP in SCC-VII cells (scale bar: 100 μm). (J) Bim levels in SCC-VII cells after DMP-mBim complex transfection (***P<0.001). (K) MTT assay of SCC-VII cells after DMP-mBim transfection and representative images. The cell viability and inhibition rate were also calculated (scale bar: 100 μm, ****P<0.0001). (L) Proliferation-inhibition ability of the DMP-mBim complex on SCC-VII cells evaluated by clonogenic assay (****P<0.0001). (M) Apoptosis-inducing ability of DMP-mBim complex in SCC-VII cells using Annexin V/PI staining by flow cytometry (****P<0.0001). (N) Live/dead staining evaluation of SCC-VII cells treated with the DMP-mBim complex (scale bar: 100 μm, ****P<0.0001). |
Gel electrophoresis was performed to confirm mBim mRNA transcription (Figure 2F). The binding capacity and optimal transfection ratio of DMP and mRNA were evaluated using a gel-retardation assay (Figure 2G). The mRNA band was completely retarded when the DMP: mRNA weight ratio was 25:1. Under this condition, the RNase protection assay revealed that whereas naked mRNA was rapidly degraded by RNase, the DMP-mRNA complex remained intact (Figure 2H), confirming the protective capacity of DMP against enzymatic degradation. Besides, we quantified the transfection efficiency of DMP-mRNA complexes in SCC-VII cells across a range of weight ratios (Figures 2I and S1). We found that even if the 5:1 ratio could neutralize the negatively charged mRNA, the transfection efficiency in SCC-VII cells was not satisfactory (34.13% ± 1.26%). Similar results were obtained for the 15:1 ratio. In contrast, a 25:1 ratio yielded an optimal transfection efficiency of 64.2% ± 0.92% with low cytotoxicity, which is consistent with our previous experience. Therefore, the ratio of 25:1 (DMP: mRNA) was selected for the subsequent experiments. Collectively, these results demonstrate that DMP exhibits qualified binding and RNase protection abilities toward mRNA with acceptable biosafety and transfection of SCC-VII cells was also effective.
DMP-mBim Complex Inhibits SCC-VII Cell Proliferation in vitro
Murine Bim mRNA (mBim) was synthesized via in vitro transcription, and its expression in transfected SCC-VII cells was quantified using quantitative real-time PCR (qRT-PCR). The results revealed an average of 4288.6-fold increase in Bim mRNA expression compared to that in the untreated group as calculated in relative mRNA level (Figure 2J), confirming the high transfection efficiency of the DMP system and its ability to enhance gene expression.
Subsequently, the antiproliferative effects of the DMP-mBim complex were assessed. As shown in Figure 2K, cell viability in the DMP-mBim-treated group was significantly reduced to 55.13% ± 2.30%, compared to 85.02% ± 5.83% in the DMP group (P < 0.0001). This inhibitory effect was further corroborated by the clonogenic assay (Figure 2L), where the number of clones in the DMP-mBim group (50.25 ± 13.60) was markedly lower than that in the DMP (134.5 ± 13.40, P < 0.0001) and untreated groups (141.5 ± 11.09, P < 0.0001).
To further investigate the mechanism of growth inhibition, we assessed apoptosis induction using Annexin V/PI and live/dead staining. As shown in Figure 2M, the apoptosis rate in the DMP-mBim group reached 11.29% ± 1.45%, which was significantly higher than the 2.95 ± 0.40% observed in the DMP group (P < 0.0001). Consistent results were obtained via live/dead staining (Figure 2N), where the percentage of red fluorescent area (indicating dead cells), was significantly higher in the DMP-mBim group (4.3% ± 0.22%) than in the DMP-only group (0.35% ± 0.16%, P < 0.0001). These findings demonstrate that the DMP-mBim complex effectively delivered mRNA, induced apoptosis, and inhibited cell proliferation in SCC-VII cells.
Preparation and Characteristics of the Double-Layered Spherical Scaffold
The design and preparation of the implantable double-layered spherical scaffold are illustrated in Figure 3A, which demonstrates a simple and tunable fabrication procedure. Two types of gelatin methacryloyl (GelMA) hydrogels were used to construct the inner and outer layers. Initially, 50 μL of porous GelMA (PR) solution was added to mineral oil and photocrosslinked to form the inner layer. This layer was designed to encapsulate the T cells within a spherical structure. Subsequently, 100 μL of ordinary GelMA (GM) solution was added around the inner layer to encase it, followed by a second photocuring step to form an outer layer. The resulting double-layered scaffold was transferred to PBS and washed three times to remove residual oil.
 |
Figure 3 Preparation of the double-layered scaffolds. (A) Illustration of the preparation of a double-layered scaffold. (B) Image of the whole intact double-layered scaffold and the cross-section of the scaffold. The yellow line and the black line indicated in the cross-section picture refer to the diameter of inner layer and the difference in outer and inner layer diameters. (C) Images of intact double-layered scaffolds of different sizes. (D) Images of the double-layered scaffold under pressure and after pressure relief. The black arrows on the pictures indicates the direction of pressure and relief. (E) Double-layered scaffold held using surgical forceps. (F) Scanning electron microscopy (SEM) images of the inner and outer layers of the scaffold (scale bar: 100 μm). (G) Image of double-layered scaffold degraded in vitro at different collagenase concentrations. (H) Volume and images of the scaffold on different days after subcutaneous implantation. |
As shown in Figure 3B, the intact structure and cross-section of the scaffold were visualized. The inner layer measured approximately 5 mm in diameter, and the total scaffold size was approximately 8 mm. Moreover, this synthesis method exhibited good reproducibility, with no significant differences in diameter or shape observed between scaffolds from different batches (Figure S2). Figure 3C illustrates the fabrication of scaffolds with various volumes, indicating that the method allows for size customization to accommodate different application requirements. The scaffold exhibited adhesive and compressive properties (Figure 3D) and maintained its spherical shape even after compression. These features ensure stability at the implantation site and resistance to deformation caused by movement. In addition, the scaffold demonstrated excellent elasticity and resilience (Figure 3E), allowing easy manipulation using surgical forceps. The microstructure and morphology of both scaffold layers were examined using SEM after freeze-drying (Figure 3F). According to the results, the pore size of the inner layer was approximately 100 μm, which was larger than that of the outer layer (approximately 30 μm). The interface between the two layers is clearly demarcated by a visible transition zone (white dotted line), highlighting successful integration while preserving functional differences between the layers.
The degradation profiles of the double-layered scaffolds were assessed in vitro and in vivo. As shown in Figure 3G, the scaffolds incubated in collagenase IV solutions (various concentrations) demonstrated sequential degradation, with the outer layer degrading first, followed by the inner layer. In vivo, subcutaneous implantation of color-stained hydrogels (inner layer: red; outer layer: blue) in mice allowed for the monitoring of degradation over time (Figure 3H). By Day 4, partial degradation of the outer layer was observed. By Day 7, most of the outer layer had been resorbed, and degradation of the inner layer had commenced. The entire degradation process lasted for 14 days, and after 10 days, the residual scaffold was minimal. This degradation tendency revealed the degradation profile of the double-layered scaffold and this degradation process aligns with our design for sequential components release. In summary, a double-layered spherical scaffold with mechanical stability, tunable porosity, and biodegradability was successfully fabricated, making it suitable for implantation and controlled drug/cell delivery in therapeutic applications.
Controlled Release and Activity of the DMP-mBim Complex from the Scaffold
In our design, the outer layer of scaffold contained the gene therapy complex DMP-mBim, and we expected to confirm that the complex could still exert therapeutic effects after release from the scaffold. The performance of the released DMP-mBim was evaluated under both short-term (1 d) and long-term (7 d) release conditions. As shown in Figure 4A and B, the DMP-EGFP complex released after 1 d maintained an average transfection efficiency of 38.88%, indicating preserved bioactivity. In the long-term release study, the DMP-EGFP (10 μg) complex continuously released active cargo from Day 2 to Day 6, with transfection efficiency ranging from 8.17% (Day 1) to 16.50% (Day 5) (Figure 4C and D). Notably, detectable transfection efficiency was observed even on the final 2 days of release, supporting the activity of this complex. Furthermore, mRNA quantification revealed that up to 73.08% of the originally loaded DMP-mBim complex was detected after 7 days (Figure 4E). To assess the functional integrity of the released mRNA, qRT-PCR analysis was performed following short-term release, which showed a significant increase in Bim mRNA levels in the treated SCC-VII cells compared to the untreated group (Figure 4F). These findings indicate that the DMP-mBim complex in the outer layer of the scaffold could protect mRNA from degradation for approximately 7 days and release most of the mRNA in a controlled manner with bioactivity.
 |
Figure 4 Transfection efficiency and treatment effect of DMP-mBim complex after release from scaffold. (A and B) Representative fluorescence images of SCC-VII cells transfected with supernatants of the outer layer of the scaffold containing the DMP-EGFP complex in short-term release (scale bar: 50 μm) and detection of efficiency by flow cytometry. (C and D) Representative fluorescence images of SCC-VII cells transfected with supernatants of the outer layer of the scaffold containing the DMP-EGFP complex in long-term release (scale bar: 100 μm) and the calculated transfection efficiency. (E) Cumulative RNA-release rate in the outer layer scaffold. (F) Bim levels in SCC-VII cells after treatment with supernatants of the outer layer of the scaffold containing DMP-mBim complex (***P<0.001). (G and H) MTT assay of SCC-VII cells after treatment with the supernatants of the outer layer of the scaffold containing DMP-mBim complex (scale bar: 100 μm, ****P<0.0001, **P<0.01). (I) Live/dead staining evaluation of SCC-VII cells treated with the supernatants of the outer layer of the scaffold (scale bar: 100 μm, ****P<0.0001). |
Based on these results, the antitumor efficacy of the released DMP-mBim was evaluated in vitro. Hydrogel-encapsulated DMP-mBim (3 μg) was enzymatically degraded using collagenase, and the collected supernatants were applied to SCC-VII cells in 96-well plates. As shown in Figure 4G and H, the MTT assay demonstrated that the cell viability of the treatment group decreased to 42.26% ± 12.44%, which was significantly lower than that of the Gel-DMP control group (P < 0.01). This inhibitory effect was further corroborated by live/dead staining (Figure 4I), where the Gel-DMP-mBim group exhibited a markedly higher percentage area of red fluorescence (6.53% ± 0.66%) than the untreated (0.09% ± 0.02%, P < 0.0001) and Gel-DMP groups (0.59% ± 0.08%, P < 0.0001). These results demonstrate that after biodegradation, the DMP-mBim complex could be released from the outer layer of the scaffold and still perform transfection, despite some loss during degradation. The released complex also induced increased transcript levels of Bim in cells, inhibiting in vitro tumor cell growth.
Characteristics and Release of T Cells in the Inner Layer Scaffold
For the inner scaffold component, porous GelMA (PR) was selected because of its structural suitability for cellular encapsulation and cultivation. Lymphocytes were isolated from C3H/HeN mouse lymph nodes, and flow cytometry revealed a high proportion of CD3+ cells (88.75%; Figure 5A), indicating successful primary T-cell isolation from the lymph nodes. Primary T cells were then stimulated for 3 days in anti-mouse CD3/CD28-precoated 6-well plates with IL-2 supplementation, followed by a 5-day expansion period. Cultured cells maintained 93.38% CD3+ expression, confirming successful in vitro culturing.
 |
Figure 5 Characteristics of T cells in the inner layer scaffold. (A) Successful collection and cultivation of mouse primary T cells. (B) The number of released T cells in the supernatant of the inner layer of the scaffolds in collagenase. (C) Representative flow cytometry images of CFSE-stained T cells before and after culturing in the medium or in different scaffolds for 5 days (ns, not significant). (D) Representative flow cytometry statistics showing the characteristics of T cells cultured in medium or different scaffolds (CD4, CD8, and CD25 markers on T cells and T-cell subsets (Tcm, CD44+ CD62L+; Tem, CD44+ CD62L−, ns, not significant, **P<0.01). |
T-cell release kinetics from inner layer scaffolds were evaluated using double-layered constructs (inner layer: 3×106 cells in PR-GelMA; outer layer: empty ordinary GelMA). The scaffolds were immersed in 1 mL of medium (10% FBS) containing 1 μg/mL collagenase IV. Daily supernatant collection with medium replenishment revealed minimal cell release during the initial degradation of the outer layers. From Day 4, accelerated inner layer degradation yielded increased cell counts, peaking between Days 4–7 with complete degradation by Day 8 (Figure 5B). This sequential degradation profile enables controlled T-cell release.
The proliferation capacity under various conditions was assessed using CFSE staining. The stimulated T cells were stained with CFSE and cultured in ordinary 1640 medium or in PR hydrogel with different outer layers (Gel/PR-T cell, Gel-DMP-mBim/PR-T cell). All samples exhibited a proliferation rate of > 99% after 5 days (Figure 5C). These results indicate that the inner layer scaffold could support T-cell proliferation without the influence of outer layer components. Next, T-cell functionality was compared between scaffold and conventional cultures. First, we examined the CD4+/CD8+ ratio (Figures 5D and S3). There was no significant difference between the three groups, as the ratio was approximately 0.8 (0.8209 vs. 0.8148 vs. 0.8598, P > 0.1). This indicates that the hydrogel culture did not alter the T-cell phenotype. Furthermore, T-cell activation was assessed using CD3 and CD25. The percentage of CD25+ T cells was 73.70% in the medium culture group. The percentages in the other two groups were 72.87% and 72.46% (P>0.1), respectively. In addition, similar central memory T-cell (Tcm; CD44⁺CD62L⁺) proportions were also confirmed by the results. Effector memory T cells (Tem, CD44+ CD62L−) were higher in the medium T-cell group than in the hydrogel culture group (P < 0.01). These results revealed that inner layer scaffold cultivation preserved the T-cell phenotype, activation state, and Tcm proportions, while modestly reducing the Tem fractions, confirming its feasibility for therapeutic T-cell storage and delivery.
The Antigenicity Death Induction of Tumor and Antigens Recruiting by Outer Layer Scaffold
After evaluating the individual components of our double-layered scaffold system, we investigated the overall immune response elicited by the scaffold both in vivo and in vitro. We first assessed the ICD-inducing capability of the DMP-mBim complex. Different scaffolds were implanted near tumors in a murine mandibular invasion model as described in materials and methods. On Day 4 post-implantation, tumor cells were harvested, and the proportion of calreticulin-positive (CRT) cells was significantly higher in the Gel-DMP-mBim group (22.82%) than in the NS group (0.33%, P < 0.0001) (Figures 6A, 6B, and S4A). Similarly, HMGB1 secretion was elevated in the Gel-DMP-mBim group (1.856 ng/mL) compared to that in the NS group (0.458 ng/mL, P < 0.01) (Figure 6C). The in vitro detection of ATP also exhibits a higher cytosolic level of ATP after transfection (Figure 6D). These results indicate that DMP-mBim released from the scaffold effectively induces ICD in tumor tissues, potentially enhancing antigen-presenting cell (APC) activation and promoting antigen presentation.
 |
Figure 6 Immune response of DMP-mBim in outer layer scaffold. (A) Representative flow cytometry images of CRT+ cells in tumors after Gel-DMP-mBim implantation in mice. (B) Quantification of CRT+ cells in tumors of different groups (****P<0.0001). (C) Expression levels of HMGB1 in tumors of different groups (**P<0.01, *P<0.05). (D) Levels of ATP in SCC-VII cells with or without DMP-mBim transfection (***P<0.001, **P<0.01). (E) Representative fluorescence images showing the entry of tumor lysates into the outer layer of the scaffold (scale bar: 100 μm; white lines represent the edge of the scaffold). (F) Levels of HMGB1 detected in scaffolds of different groups (*P<0.05) and the quantification of CRT+ cells in the scaffolds (**P<0.01, *P<0.05). (G) Representative flow cytometry images and quantification of CD80+ DCs in different groups in vivo (***P<0.001, **P<0.01). |
To further examine whether tumor-derived antigenic substances could be captured by the surrounding scaffold, we simulated this process in vitro using lysates from SCC-VII cells pre-labeled with tubulin (a cytoskeleton marker) and DiI (a cell membrane marker). As shown in Figure 6E, no fluorescence signals were observed in the scaffolds at 0 h. However, red fluorescence appeared at the scaffold boundaries after 6 h. At 24 h, both membrane fragments (red) and cytoplasmic contents (green) were detected within the scaffold, and the signal intensity increased further at 48 h. This pattern suggests that the outer layer of the scaffold can receive both membrane and cytoplasmic components released by the tumor cells. Subsequently, we validated the antigen-capture process in vivo. Four days after scaffold implantation, residual scaffolds were collected, and the levels of CRT⁺ cells and HMGB1 were assessed using flow cytometry and ELISA, respectively (Figures 6F, and S4B). The Gel-DMP-mBim group exhibited a significantly higher proportion of CRT + cells (57.63%) than the Gel-DMP group (39.49%, P < 0.05) and elevated HMGB1 levels (1.482 ng/mL, P < 0.05). These findings support the conclusion that tumor-derived antigens can be retained in the outer layer of the scaffold, providing a reservoir for subsequent immune activation studies. Finally, we hypothesized that antigen accumulation in the scaffold facilitates the recruitment and maturation of APCs, particularly dendritic cells (DCs). Flow cytometry detection of CD11c and CD80 revealed increased proportions of CD80⁺CD11c⁺ DCs in the Gel-DMP-mBim group (58.82%) than in the Gel-DMP (50.92%, P < 0.01) and Gel groups (43.58%, P < 0.001) (Figures 6G and S4C). These results further confirmed that the outer layer scaffold not only captured tumor antigens but also promoted DC recruitment and maturation, facilitating downstream immune responses.
Activated T Cells Released from the Inner Layer of the Scaffold
Following dendritic cell (DC) recruitment to the scaffolds, we hypothesized that DCs would activate T cells and subsequently mediate tumoricidal effects. To evaluate whether immune responses within the outer scaffold could facilitate the function of inner-layered T cells, different double-layered scaffolds were implanted adjacent to tumors in the mandibular invasion model. Residual scaffolds were harvested after 5 days for flow cytometric analysis. As shown in Figures 7A and S5, the Gel-DMP-mBim/PR-T cell group exhibited significantly higher percentages of CD80⁺CD11c⁺ (P < 0.05) and CD86⁺CD11c⁺ cells (P < 0.01) than the Gel/PR-T cell group. These results confirmed post-implantation DC recruitment, with the outer layer of DMP-mBim complexes playing a pivotal role in this process. Analysis of T-cell subsets (Figure 7B) revealed a decreased CD4 +/CD8 + ratio (0.29 vs. 1.52; P < 0.05) in the Gel-DMP-mBim/PR-T cell group, indicating enhanced CD8 + cytotoxic T lymphocyte (CTL) infiltration. Furthermore, this group demonstrated a higher proportion of central memory T cells (Tcm: 47.86% vs. 37.96%; P < 0.05; Figure 7C), suggesting preferential differentiation toward memory phenotypes for sustained immune response. Meanwhile, the mature DC and activated T-cell were also confirmed in vitro. It exhibited significantly higher percentages of CD86⁺CD11c⁺ (P < 0.001) and CD80⁺CD11c⁺ mature DCs (P < 0.0001) in SCC-VII+DC+T cell group than that in DC+T cell group (Figure S6A). And compared with co-cultured with only DC, the T-cell that was co-cultured with DC and SCC-VII exhibited higher percentages of proliferated cells (25.86% vs. 17.48%) (Figure S6B), which implied a higher degree of functional activation of T cells. Besides, the elevated cytokine, including IFN-γ (1213 vs 1118 pg/mL, P<0.001) and TNF-α (403.4 vs 349.5 pg/mL, P<0.05) were also confirmed the activation of T-cell and the DC maturation (Figure S6C). Thus, the scaffold design facilitated sequential immune activation involving antigens, DCs, and T cells, ultimately modifying the tumor’s immune microenvironment.
 |
Figure 7 Activation and release of T cells in inner layer scaffolds. (A) Representative flow cytometry images and quantification of mature DCs in scaffolds in different groups (****P<0.0001, **P<0.01, *P<0.05). (B) Representative flow cytometry images and quantification of CD4+ and CD8+ T cells in scaffolds in different groups (***P<0.001, *P<0.05). (C) Representative flow cytometry images and quantification of Tcm in scaffolds in different groups (**P<0.01, *P<0.05). (D–F) Representative flow cytometry images and quantification of T cells and DCs in the tumor after treatment with the double-layered scaffold (****P<0.0001, ***P<0.001, **P<0.01, *P<0.05). |
The Tumor Infiltration of Activated T-Cells Released from Scaffolds
As the final step in the immune response, we evaluated whether the immune cells in the scaffold infiltrated tumor tissues after being released from the scaffold. Different double-layered scaffolds were implanted in a mouse mandible-invasion model. As shown in Figures 7D and S7, the CD4+/CD8+ T-cell ratio in the tumor decreased to 1.03 in the Gel-DMP-mBim/PR-T cell group, and that ratio was 2.39 in the Gel/PR-T cell group or 3.205 in Gel/PR group, respectively (P < 0.05). The proportion of CD3+CD69+ cells was 28.65% in the Gel-DMP-mBim/PR-T cell group, which exceeded that in the Gel/PR group (24.69%, P < 0.05) (Figure 7E). The presence of T cells is essential for infiltration, and DMP-mBim primarily augments this process. Moreover, as shown in Figure 7F, an elevated proportion of CD86+CD11c+ (27.06%) and CD80+CD11c+ (28.1%) cells was detected in the Gel-DMP-mBim/PR-T cell group, indicating scaffold-mediated maturation and recruitment of DCs into the tumor. These changes support the hypothesis that sequential scaffold degradation enables T-cell tumor infiltration, ultimately reprogramming the tumor microenvironment (TME) to establish durable, multistage antitumor immunity.
The Inhibition Effect of the Double-Layered Scaffold System in SCC-VII Subcutaneous Tumor Models
Following the verification of immune responses, we evaluated the therapeutic potential of the double-layered scaffolds in C3H/HeN mice bearing SCC-VII subcutaneous tumors. Mice were randomized into five groups (Figure 8A), and apart from the NS group, mice in the other four groups were implanted with different double-layered scaffolds: Gel/PR, Gel-DMP-mBim/PR, Gel/PR-T cell, and Gel-DMP-mBim/PR-T cell near the tumors. Tumor growth curves revealed significant inhibition in the Gel-DMP-mBim/PR-T cell group compared to the other groups (P < 0.05; Figure 8B). Early phase attenuation occurred in both DMP-mBim-containing groups, although only the T-cell-incorporated scaffold maintained sustained suppression throughout the late phase (Figure 8C), demonstrating essential inner layer contributions. Implantation of an empty hydrogel did not show any obvious therapeutic effects in this experiment. Terminal tumor weights corroborated the volumetric data: Gel-DMP-mBim/PR-T cell tumors (0.39 g) were significantly lighter than Gel-DMP-mBim/PR (1.20 g; P < 0.05) and Gel/PR (1.77 g; P < 0.0001) groups (Figure 8D). No significant intergroup differences among the treatment groups in body weight were observed (Figure 8E). These results establish that whereas individual components confer partial efficacy, the integrated Gel-DMP-mBim/PR-T cell scaffold induces synergistic and sustained antitumor activity in a subcutaneous tumor model.
 |
Figure 8 Treatment effect of double-layered scaffold in C3H/HeN subcutaneous tumor models. (A) Illustration of the experimental process schedule. (B) Tumor growth curve measured in each group with different treatments (****P<0.0001, *P<0.05). (C) Isolated tumors from all groups at the end of the experiment. (D) Average tumor weight at the treatment endpoint (****P<0.0001, *P<0.05). (E) Weight of mice during the experiment (**P<0.01). (F) Quantification of T-cells in the tumor after treatment with different double-layered scaffolds (*P<0.05). (G) Representative flow cytometry images and quantification of DCs in the tumor after treatment with different double-layered scaffolds (****P<0.0001). (H) Immunohistochemical evaluation of tumor tissue in each group using different antibodies or TUNEL staining (scale bar: 100μm). |
Subsequent flow cytometric analysis of tumor-infiltrating immune cells revealed tumor microenvironment alterations in the subcutaneous model. Although the CD8 + T-cell proportions showed no significant intergroup differences, the CD4 +/CD8 + ratio provided critical evidence (Figures 8F, S8 and S9). The Gel-DMP-mBim/PR-T cell group exhibited a significantly lower ratio (1.142) than the Gel-DMP-mBim/PR (5.348; P < 0.05) and Gel/PR (8.019; P < 0.001) groups, indicating scaffold-mediated modulation of T-cell distribution. Mature DC analysis (Figure 8G) demonstrated elevated CD80⁺CD11c⁺ populations in the Gel-DMP-mBim/PR-T cell group (21.16%) compared to the Gel/PR-T cell (14.27%; P < 0.0001) and Gel-DMP-mBim/PR (8.93%; P < 0.0001) groups, with parallel trends for CD86⁺CD11c⁺ cells. These findings confirmed that the bilayer scaffold significantly restructured the TME cellular composition, with both the outer layer DMP-mBim complexes and inner layer T cells contributing to sustained immunomodulation and antitumor activity. Complementary IHC analysis (Figure 8H) revealed enhanced Bim expression in the Gel-DMP-mBim/PR-T cell and Gel-DMP-mBim/PR groups, which was corroborated by the elevated number of TUNEL + apoptotic cells. These results validate the efficiency and pro-apoptotic function of DMP-mBim. Notably, the reduction in CD31⁺ microvessel density confirmed the antiangiogenic effects of the Gel-DMP-mBim/PR-T cell group. CD8 + T-cell infiltration was minimal in groups lacking T-cell-loaded scaffolds (NS, Gel/PR, and Gel-DMP-mBim/PR) but was substantially increased in T-cell-containing scaffolds. The tendency of CD4+ signals was not significantly different among the five groups; however, it was consistent with the flow cytometry results, showing that the inner layer scaffold did not increase the proportion of CD4+ T cells. Analysis of inflammatory markers (Figure S10) showed that the important inflammatory cytokines TNF-α and IFN-γ were significantly upregulated in the Gel-DMP-mBim/PR-T cell group. Collectively, these data demonstrate that the double-layered scaffold induced potent antitumor responses through coordinated immune remodeling and targeted gene delivery, with no significant toxicity observed in the major organs (Figure S11).
The Inhibition Effect of the Double-Layered Scaffold System in SCC-VII Mandible Invasion Models
Given the clinical significance of mandibular invasion in head and neck squamous cell carcinoma, we evaluated the therapeutic potential of double-layered scaffolds in this aggressive animal model. Following the subcutaneous tumor results, scaffolds were implanted in mandibular invasion models using the surgical procedure illustrated in Figure 9A. Group allocation was the same as that in the previous experiment and the weights of the mice are shown in Figure 9B. Tumor volumetry revealed significantly suppressed growth in the Gel-DMP-mBim/PR-T cell group compared to the controls (P < 0.01; Figure 9C). The final tumor volumes were 220.44 mm3 (Gel-DMP-mBim/PR-T cell), 292.44 mm3 (Gel-DMP-mBim/PR; P<0.01), and 472.32 mm3 (Gel/PR; P < 0.0001), with growth curves shown in Figure 9D. Tumor weight analysis and macroscopic examination (Figure 9E and F) corroborated the volumetric data, demonstrating maximal efficacy in the Gel-DMP-mBim/PR-T cell group. Intermediate effects were observed in both the Gel-DMP-mBim/PR and Gel/PR-T cell groups. These findings established that whereas individual scaffold components confer partial efficacy, the integrated system produces synergistic tumor suppression in mandible-invasion models.
 |
Figure 9 Treatment effect of double-layered scaffold in C3H/HeN mandible-invasion models. (A) Illustration of the experimental process schedule and implantation operation of the scaffold in the mandible-invasion model. (B) Weight of mice during the experiments (ns, not significant). (C) Tumor growth curve measured in each group with different treatments (****P<0.0001, **P<0.01). (D) Tumor growth tendency in different groups. (E) Representative images of the masseter region of mice from each group. (F) Weight of tumors in each group at the end of the experiment (**P<0.01, *P<0.05). (G) Three-dimensional reconstructed micro-CT images of the skulls of mice in each group, including side and top views. (H) The calculated bone volume fraction (BV/TV) at the end of the experiment in each group and zygomatic bone destruction scores in each group (***P<0.001, **P<0.01, *P<0.05). (I) Representative flow cytometry images and quantification of T cells and DCs in the tumor after treatment with the double-layered scaffold (****P<0.0001, ***P<0.001, **P<0.01, *P<0.05). |
Mandibular bone integrity was assessed using micro-CT to quantify tumor-induced destruction at the end of the experiment period. Representative reconstruction images of each group are shown in Figure 9G, which also reflects tumor growth. The bone volume fraction (BV/TV) and zygomatic arch destruction scores revealed significant mandibular integrity in the Gel-DMP-mBim/PR-T cell group (Figure 9H). The NS and Gel/PR groups exhibited extensive mandibular resorption, whereas intermediate destruction was observed in the Gel-DMP-mBim/PR and Gel/PR-T cell groups. BV/TV measurements confirmed this trend (55.55% vs. 49.69% in NS; P < 0.001). We also noticed that the bone invasion model of SCC-VII had specific destruction of the zygomatic arch; therefore, we included zygoma destruction scores (Figure 9H). The scores were judged 6 and 10 days after cell injection according to the micro-CT results. Six days after cell injection, the mice in the NS and Gel/PR groups had high scores, indicating obvious destruction of the zygomatic arch, whereas the scores of the Gel-DMP-mBim/PR-T cell group were low. A similar tendency was observed according to the condition at the 10 days timepoint. Meanwhile, the same tendency and results were also verified by H&E staining of head sections (Figure S12). The tumor volume and bone resorption were much higher in the NS group, accompanied by thinning and destruction of the mandibular bone surface. And in the Gel-DMP-mBim/PR-T cell group, the tumor volume was smaller and the mandible-tumor surface was complete. These results indicate that Gel-DMP-mBim/PR-T cell scaffold implantation suppressed tumor growth and inhibited tumor progression in the mandible-invasion model. The existence of only the outer and inner layer scaffolds could still provide a part of the therapeutic effect after implantation.
Furthermore, immune conditions and different immune cells were assessed using flow cytometry (Figures 9I and S13). The results revealed a substantially reduced CD4 +/CD8 + ratio in the Gel-DMP-mBim/PR-T cell group (3.22) compared to that in the Gel-DMP-mBim/PR (10.91, P < 0.01) and Gel/PR (37.49, P < 0.0001) groups, indicating enhanced CTL infiltration into the tumor. The proportion of CD80+CD11c+ cells increased to 18.88% in the Gel-DMP-mBim/PR-T cell group and 16.44% in the Gel/PR-T cell group. However, the proportion of the Gel/PR group was only 12.1% (P < 0.0001). A similar difference was observed in the number of CD86+CD11c+ cells between the two groups. The proportion of CD86+CD11c+ cells in the Gel-DMP-mBim/PR-T cell group was 20.05%, which was higher than that in the Gel/PR group (12.72%; P < 0.0001). These results indicate that implantation of the double-layered scaffold could supply more CTLs to remodel the immune cell composition in the tumor environment and promote the cancer-immunity cycle to exert an antitumor effect in vivo.
Apoptosis and the detailed distribution of immune cells were explored using multiplex immunohistochemical staining (Figure 10). TUNEL staining of whole head sections in different groups revealed that treatment with the Gel-DMP-mBim/PR-T cell scaffold induced apoptosis and suppressed tumor growth in vivo. The apoptotic cells were distributed both locally and regionally. CD3+ CD8+ T cells and CD80+ CD11c+ DCs were also observed in these regions. Positive T cells and DCs were most abundant in the Gel-DMP-mBim/PR-T cell group. Fewer positive regions were observed in the Gel/PR-T cell group. The number of positive regions was lowest in the NS and Gel/PR groups, respectively. We propose that this spatial correlation indicates localized immune activation that drives tumor apoptosis. This finding is consistent with the basic characteristics of immune processes. No obvious pathological abnormalities were observed in any of the major organs (Figure S14). Based on this, we demonstrated that the Gel-DMP-mBim/PR-T cell scaffold synergistically combined gene therapy and T-cell immunotherapy, remodeling the TME to suppress tumor progression while preserving mandibular integrity in HNSCC mandible-invasion models.
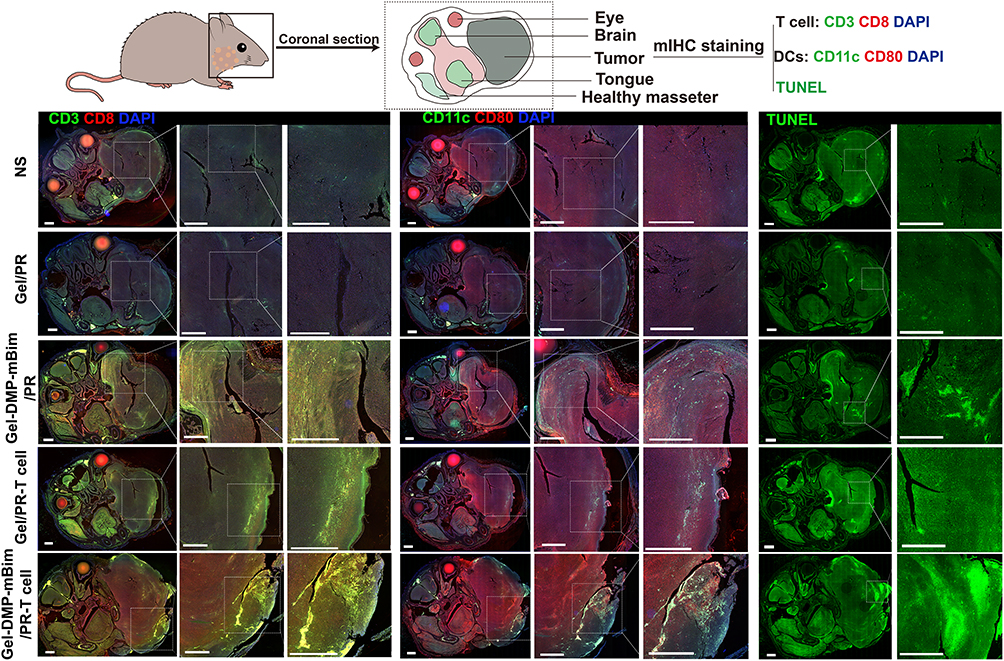 |
Figure 10 Representative multiplex immunohistochemistry staining images in the coronal slice of the mouse head in different treatments showing the distribution of T cells (CD3+CD8+), DCs (CD11c+CD80+), and TUNEL signals (scale bar: 1 mm). |
Discussion
In this study, we engineered a double-layered Gel-DMP-mBim/PR-T cell scaffold comprising an outer layer loaded with DMP-mBim gene therapy complexes and an inner layer encapsulating primary lymph node-derived T cells. Following implantation, sequential scaffold degradation first releases DMP-mBim complexes to induce tumor apoptosis and ICD, which subsequently enables T-cell-DC interactions that promote CTL tumor infiltration. This combinatorial approach inhibited tumor cell growth in vitro and HNSCC tumor growth in both SCC-VII subcutaneous and mandibular invasion models.
Animal model selection is critical in HNSCC research, as it requires accurate disease progression recapitulation and therapeutic applicability.45 Subcutaneous xenograft models remain prevalent owing to their technical accessibility and immunological relevance,46 with SCC-VII cells extensively validated in such models.47 However, this model fails to reproduce local invasion patterns and exhibits anatomical discordance with primary tumor sites. Therefore, intraoral orthotopic transplantation models have been used in some studies.48 Cells were injected into the tongue, cheek, or floor of the mouth of mice to provide a platform for researchers to focus on the pathogenesis or treatment of HNSCC rather than tumorigenesis and cancer prevention.49 However, surgical complexity and implantation difficulties limit the utility of intraoral orthotopic models. To overcome these drawbacks, an orthotopic transplantation model outside the mouth has been selected in several studies to evaluate the mandibular invasion characteristics of HNSCC.50,51 Tumors develop in the masseter region and enable quantification of mandibular destruction during tumor progression.52,53 Given these considerations, we selected both subcutaneous and mandibular invasion models. And compared to the previous mandible invasion model, we have attempted to apply this model to implantable scaffold therapy and achieved therapeutic results. This dual approach provides sufficient implantation space for double-layered scaffolds, enabling the detection of stepwise immune responses to therapy. They also provide conditions for the direct assessment of tumor progression and bone invasion.
Hydrogels have been adopted as therapeutic platforms in numerous studies because of their three-dimensional structural scaffolds and capacity to confer diverse antitumor functions through the incorporation of specific components.54,55 Numerous studies have aimed to design drug delivery systems that use distinct agents to achieve synergistic therapeutic outcomes.56 For example different types of chemical drugs/nanocomplexes and hydrogel formulations of multiple chemical drugs/nanocomplexes are used for combination therapy of tumor growth.57,58 Similar approaches include the use of gambogic acid within thermosensitive hydrogels for localized delivery to enhance antitumor immunity.59 These studies primarily use hydrogels for uniform component drug release, typically focusing on immunostimulants or chemotherapeutic agents. In this study, we integrated bioactive gene therapy complexes and immune cells (T cells) into a double-layered scaffold system. Ordinary GelMA hydrogel served as the delivery matrix for the DMP-mRNA complexes and was selected for its appropriate in vivo biodegradation rate. These complexes demonstrated effective cell transfection capabilities after hydrogel degradation. A porous GelMA (PR) hydrogel was selected because of its porous architecture to encapsulate T cells within its inner layer, with cell activation preceding release. The suitable mechanical strength and adhesive properties of GelMA enabled us to perform implantable procedures in the highly mobile head and neck region, whereas other hydrogel materials are better suited for injection applications.60 And our results established sequential release kinetics, with DMP-mBim complexes being released initially, followed by the activation of T cells. Flow cytometry confirmed the immune-activating functionality of the double-layered scaffolds. Meanwhile the use of DMP to construct the gene therapy complex was based on our previous study that confirmed the delivery efficiency and mature synthetic process compared to other nanoparticles including liposome system.61,62 We have also found that in the 25:1 transfection ratio, the DMP/mRNA complex possesses the potential to escape lysosomes and a lower transfection ratio does not facilitate this capability according to the results. The DMP-mRNA complexes were also proved to be taken up by tumor cells via lipid raft-mediated endocytosis in the cellular mechanism evaluation.63
The use of a double-layered scaffold containing distinct elements to perform different functions is not a new concept in biotherapy.64 This strategy has mostly been applied in wound-healing studies, such as a self-adaptive double-layer hydrogel (the inner layer for irregular wounds and the outer layer for providing mechanical protection) and a diabetic double-layer hydrogel (the inner layer for generating hydroxyl radicals and the outer layer for clearing reactive oxygen species).65,66 However, hydrogel systems with distinct compositions at different treatment stages remain uncommon in cancer immunotherapy. One proposed double-layered system featured an outer layer containing sorafenib-adsorbed graphene oxide nanoparticles to reprogram tumor-associated macrophages through the initial sorafenib release,67 whereas an anti-CD47 antibody within the inner layer sustained an antitumor effect in breast cancer. In this study, we engineered distinct functions for each scaffold layer. The DMP-mBim complex exerts a tumor-killing effect, and the outer layer of the scaffold provides conditions for dendritic cell (DC) recruitment and antigen accumulation. Our analyses revealed an increased recruitment of mature DCs within the scaffold, leading to in situ T-cell activation. T cells subsequently released from the inner layer modulate the tumor microenvironment (TME). The continuous supply of tumor antigens and DCs establishes the scaffold as a localized reservoir of T cells adjacent to the tumor site during treatment. Through this sequential release, this double-layered scaffold could combine the advantages of gene therapy and ACT to remodel the immune environment around the immunosuppressive HNSCC atmosphere and establish an immune cycle. Through cyclical repetition, this double-layered scaffold system introduces a novel paradigm for integrating gene and immune cell therapies into a HNSCC treatment platform. Meanwhile, this mechanism also suggests potential challenges in translating this therapeutic strategy to clinical practice, as the tumor-suppressive microenvironment in humans is more complex. How to effectively activate the aforementioned immune cycle within the human immunosuppressive microenvironment remains a critical question for future validation.
In recent years, immunotherapy, particularly ACT, has shown significant progress in the treatment of hematological malignancies.68 However, current T-cell receptor-engineered T-cell therapy and chimeric antigen receptor T-cell therapy have limitations in HNSCC field.69 Although some novel target antigens (eg, MUC1 and the ErbB family) have been proposed, the scarcity of tumor-specific antigens and the immunosuppressive tumor microenvironment impede their advancement.70,71 Meanwhile, the implementation of mRNA-based gene therapy for HNSCC also remains a challenge. Although gene therapy can directly induce tumor cell death or modulate the immune environment,72 sustaining durable antitumor effects is difficult when tumor-suppressor genes are delivered via conventional administration.61 To address these constraints, we propose the integration of immune cell therapy with mRNA gene therapy as a viable strategy against tumor invasion. In this study, the pro-apoptotic DMP-mBim complex, released initially from the outer scaffold layer, induced ICD in tumors, as evidenced by the increased number of calreticulin-positive cells and elevated HMGB1 levels. Subsequently, the system activates the tumor microenvironment, facilitating the infiltration of T cells released from the inner scaffold layer. This combined approach generated sustained antitumor effects in murine subcutaneous xenograft and mandibular invasion models. Treatment efficacy was demonstrated by significant tumor growth inhibition and mandibular integrity in the Gel-DMP-mBim/PR-T cell group, with outcomes notably absent in the hydrogel groups receiving single therapy. These results establish that the sequential release of mRNA gene therapy and T cells within a double-layered scaffold prolongs antitumor efficacy, providing a novel combinatorial strategy against HNSCC.
There are also some limitations present in our research. Firstly, the use of limited female-mouse models may not fully reflect the heterogeneity and complexity of HNSCC, particularly with regard to the tumor microenvironment, anatomical structures and disease subtypes defined by specific molecular expression patterns. The present findings should be considered preliminary, and further safety evaluations in larger animal models or well-designed clinical trials are necessary before definitive therapeutic claims can be made. Secondly, considering the inherent differences between murine primary T-cells and human immune cells, we are not yet able to propose a double-layered scaffold therapy strategy that is fully in alignment with the current standard of care for HNSCC. Future studies will be essential to bridge this translational gap and better assess the clinical feasibility of this approach. Despite these limitations, this study introduces a novel strategy that synergistically combines gene therapy and ACT within a sequentially releasing scaffold. This innovative platform demonstrates broad applicability and offers promising potential for translatable clinical practice and combination immunotherapy in solid tumors.
Conclusions
In conclusion, we developed an implantable, double-layered GelMA scaffold for HNSCC therapy, with an outer layer encapsulating the DMP-mBim gene complex and an inner layer housing primary T cells. The therapeutic effect of the system was achieved through the sequential release of these components. The antitumor efficacy of the gene complex and T-cell viability were confirmed in vitro experiments. In vivo, the scaffold biodegraded in a controlled manner, eliciting a localized immune response. The initial release of DMP-mBim complexes could induce immunogenic cell death in tumor cells, releasing antigens that then infiltrated the scaffold to recruit DCs. These mature DCs would subsequently activate the T cells in the inner layer. The activated T cells were then released to infiltrate and eliminate tumors, thereby remodeling the tumor microenvironment. In both murine subcutaneous and mandibular invasion models, this scaffold significantly inhibited tumor growth. This co-delivery strategy highlights the potential of integrating gene and cell therapies for advancing future HNSCC clinical practice, although further safety evaluations and validations are necessary.
Data Sharing Statement
All data needed to evaluate the conclusions in this paper are presented in the main text and/or the Supplementary Information. The original data are available from the corresponding author (Chunjie Li) upon request.
Ethical Statement
Animal experiments were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Institutional Review Board of West China Hospital, Sichuan University (Ethical Approval No. 20230330013) and conducted in accordance with the Guidelines and Standard Operating Procedures (SOP) for Laboratory Animals.
Acknowledgments
The authors thank Yaojia Zhou ([email protected], Animal Laboratory Center, West China Hospital, Sichuan University) for the help with micro-CT scanning and analysis.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work is supported by the Regional Innovation and Development Joint Fund Key Project of the National Natural Science Foundation of China (U24A20735), the National Natural Science Foundation of China (82473289, 82372735), the Key Research Program of Science and Technology Department of Sichuan (23NSFJQ0104) and the Science and Technology Project of Sichuan Provincial Health Commission (24LCYJZD10).
Disclosure
The authors report no potential conflicts of interest in this work.
References
1. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Author correction: Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2023;9(1):4. doi:10.1038/s41572-023-00418-5
2. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–27. doi:10.3322/caac.21834
3. Zhang J, Ni Z, Zhang Y, et al. DAZAP1 phase separation regulates mitochondrial metabolism to facilitate invasion and metastasis of oral squamous cell carcinoma. Cancer Res. 2024;84(22):3818–3833. doi:10.1158/0008-5472.can-24-0067
4. Kraaijenga SA, Oskam IM, van Son RJ, et al. Assessment of voice, speech, and related quality of life in advanced head and neck cancer patients 10-years+ after chemoradiotherapy. Oral Oncol. 2016;55:24–30. doi:10.1016/j.oraloncology.2016.02.001
5. Sahu M, Suryawanshi H. Immunotherapy: the future of cancer treatment. J Oral Maxillofac Pathol. 2021;25(2):371. doi:10.4103/0973-029x.325257
6. Chow LQM, Haddad R, Gupta S, et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the Phase Ib keynote-012 expansion cohort. J Clin Oncol. 2016;34(32):3838–3845. doi:10.1200/jco.2016.68.1478
7. Kitamura N, Sento S, Yoshizawa Y, Sasabe E, Kudo Y, Yamamoto T. Current trends and future prospects of molecular targeted therapy in head and neck squamous cell carcinoma. Int J Mol Sci. 2020;22(1):240. doi:10.3390/ijms22010240
8. Iivanainen S, Ahvonen J, Knuuttila A, Tiainen S, Koivunen JP. Elevated CRP levels indicate poor progression-free and overall survival on cancer patients treated with PD-1 inhibitors. ESMO Open. 2019;4(4):e000531. doi:10.1136/esmoopen-2019-000531
9. Qiao XW, Jiang J, Pang X, et al. The evolving landscape of PD-1/PD-L1 pathway in head and neck cancer. Front Immunol. 2020;11:1721. doi:10.3389/fimmu.2020.01721
10. Gregg JR, Thompson TC. Considering the potential for gene-based therapy in prostate cancer. Nat Rev Urol. 2021;18(3):170–184. doi:10.1038/s41585-021-00431-x
11. Lei S, Zhang X, Li J, et al. Current progress in messenger RNA-based gene therapy. J Biomed Nanotechnol. 2020;16(7):1018–1044. doi:10.1166/jbn.2020.2961
12. Wang L, Vuletic I, Deng D, et al. Bifidobacterium breve as a delivery vector of IL-24 gene therapy for head and neck squamous cell carcinoma in vivo. Gene Ther. 2017;24(11):699–705. doi:10.1038/gt.2017.74
13. Wang K, Gao Y, Wu S, et al. Dual-mRNA delivery using tumor cell lysate-based multifunctional nanoparticles as an efficient colon cancer immunogene therapy. Int J Nanomed. 2024;19:4779–4801. doi:10.2147/ijn.s452548
14. Hanna GJ, Liu H, Jones RE, et al. Defining an inflamed tumor immunophenotype in recurrent, metastatic squamous cell carcinoma of the head and neck. Oral Oncol. 2017;67:61–69. doi:10.1016/j.oraloncology.2017.02.005
15. Kajiyama T, Serada S, Fujimoto M, et al. SOCS1 gene therapy for head and neck cancers: an experimental study. Anticancer Res. 2022;42(7):3361–3372. doi:10.21873/anticanres.15823
16. Bray D, Yu SZ, Koprowski H, et al. Combination nonviral interleukin 2 gene therapy and external-beam radiation therapy for head and neck cancer. Arch Otolaryngol Head Neck Surg. 2003;129(6):618–622. doi:10.1001/archotol.129.6.618
17. Adappa ND, Sung CK, Choi B, Huang TG, Genden EM, Shin EJ. The administration of IL-12/GM-CSF and Ig-4-1BB ligand markedly decreases murine floor of mouth squamous cell cancer. Otolaryngol Head Neck Surg. 2008;139(3):442–448. doi:10.1016/j.otohns.2008.05.001
18. Huang J, Wang K, Wu S, et al. Tumor cell lysate-based multifunctional nanoparticles facilitate enhanced mRNA delivery and immune stimulation for melanoma gene therapy. Mol Pharm. 2024;21(1):267–282. doi:10.1021/acs.molpharmaceut.3c00826
19. Zou W, Huo B, Tu Y, et al. Metabolic reprogramming by chemo-gene co-delivery nanoparticles for chemo-immunotherapy in head and neck squamous cell carcinoma. Acta Biomater. 2025;199:361–373. doi:10.1016/j.actbio.2025.04.031
20. Moreno V, Hernandez T, de Miguel M, Doger B, Calvo E. Adoptive cell therapy for solid tumors: chimeric antigen receptor T cells and beyond. Curr Opin Pharmacol. 2021;59:70–84. doi:10.1016/j.coph.2021.05.004
21. Papa S, Adami A, Metoudi M, et al. Intratumoral pan-ErbB targeted CAR-T for head and neck squamous cell carcinoma: interim analysis of the T4 immunotherapy study. J Immunother Cancer. 2023;11(6):e007162. doi:10.1136/jitc-2023-007162
22. Charap AJ, Enokida T, Brody R, et al. Landscape of natural killer cell activity in head and neck squamous cell carcinoma. J Immunother Cancer. 2020;8(2):e001523. doi:10.1136/jitc-2020-001523
23. Klichinsky M, Ruella M, Shestova O, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–953. doi:10.1038/s41587-020-0462-y
24. Mitchell DA, Sayour EJ, Reap E, et al. Severe adverse immunologic reaction in a patient with glioblastoma receiving autologous dendritic cell vaccines combined with GM-CSF and dose-intensified temozolomide. Cancer Immunol Res. 2015;3(4):320–325. doi:10.1158/2326-6066.cir-14-0100
25. Stevanović S, Helman SR, Wunderlich JR, et al. A Phase II study of tumor-infiltrating lymphocyte therapy for human papillomavirus-associated epithelial cancers. Clin Cancer Res. 2019;25(5):1486–1493. doi:10.1158/1078-0432.ccr-18-2722
26. Park YP, Jin L, Bennett KB, et al. CD70 as a target for chimeric antigen receptor T cells in head and neck squamous cell carcinoma. Oral Oncol. 2018;78:145–150. doi:10.1016/j.oraloncology.2018.01.024
27. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. doi:10.1038/s41408-021-00459-7
28. Rosewell Shaw A, Porter CE, Watanabe N, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther. 2017;25(11):2440–2451. doi:10.1016/j.ymthe.2017.09.010
29. Doran SL, Stevanović S, Adhikary S, et al. T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: a first-in-human, phase I/II study. J Clin Oncol. 2019;37(30):2759–2768. doi:10.1200/jco.18.02424
30. Papa S, van Schalkwyk M, Maher J. Clinical evaluation of ErbB-targeted CAR T-cells, following intracavity delivery in patients with ErbB-expressing solid tumors. Methods Mol Biol. 2015;1317:365–382. doi:10.1007/978-1-4939-2727-2_21
31. Jie HB, Gildener-Leapman N, Li J, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. 2013;109(10):2629–2635. doi:10.1038/bjc.2013.645
32. Solomon B, Young RJ, Rischin D. Head and neck squamous cell carcinoma: genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin Cancer Biol. 2018;52(Pt 2):228–240. doi:10.1016/j.semcancer.2018.01.008
33. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185–199. doi:10.1038/s41573-019-0051-2
34. Gao Y, Men K, Pan C, et al. Functionalized DMP-039 hybrid nanoparticle as a novel mRNA vector for efficient cancer suicide gene therapy. Int J Nanomed. 2021;16:5211–5232. doi:10.2147/ijn.s319092
35. Cao H, Duan L, Zhang Y, Cao J, Zhang K. Current hydrogel advances in physicochemical and biological response-driven biomedical application diversity. Signal Transduct Target Ther. 2021;6(1):426. doi:10.1038/s41392-021-00830-x
36. Zhong R, Talebian S, Mendes BB, et al. Hydrogels for RNA delivery. Nat Mater. 2023;22(7):818–831. doi:10.1038/s41563-023-01472-w
37. Chung TW, Tyan YC, Lin SW, Yang MH, Liu YH, Wang RP. Developing photothermal-responsive and anti-oxidative silk/dopamine nanoparticles decorated with drugs which were incorporated into silk films as a depot-based drug delivery. Int J Biol Macromol. 2021;185:122–133. doi:10.1016/j.ijbiomac.2021.06.084
38. Yin Y, Li X, Ma H, et al. In situ transforming RNA nanovaccines from polyethylenimine functionalized graphene oxide hydrogel for durable cancer immunotherapy. Nano Lett. 2021;21(5):2224–2231. doi:10.1021/acs.nanolett.0c05039
39. Wang Z, Liu Z, Wang S, et al. Implantation of hydrogel-liposome nanoplatform inhibits glioblastoma relapse by inducing ferroptosis. Asian J Pharm Sci. 2023;18(3):100800. doi:10.1016/j.ajps.2023.100800
40. Hasani-Sadrabadi MM, Sarrion P, Pouraghaei S, et al. An engineered cell-laden adhesive hydrogel promotes craniofacial bone tissue regeneration in rats. Sci Transl Med. 2020;12(534):eaay6853. doi:10.1126/scitranslmed.aay6853
41. Li Y, Thambi T, Lee DS. Co-delivery of drugs and genes using polymeric nanoparticles for synergistic cancer therapeutic effects. Adv Healthc Mater. 2018;7(1):1700886. doi:10.1002/adhm.201700886
42. Fu Z, Li H, Xue P, et al. Implantable bioresponsive hydrogel prevents local recurrence of breast cancer by enhancing radiosensitivity. Front Bioeng Biotechnol. 2022;10:881544. doi:10.3389/fbioe.2022.881544
43. Abasalizadeh F, Moghaddam SV, Alizadeh E, et al. Correction to: Alginate-based hydrogels as drug delivery vehicles in cancer treatment and their applications in wound dressing and 3D bioprinting. J Biol Eng. 2020;14:17. doi:10.1186/s13036-020-00239-0
44. Kurian AG, Singh RK, Patel KD, Lee JH, Kim HW. Multifunctional GelMA platforms with nanomaterials for advanced tissue therapeutics. Bioact. Mater. 2022;8:267–295. doi:10.1016/j.bioactmat.2021.06.027
45. Supsavhad W, Dirksen WP, Martin CK, Rosol TJ. Animal models of head and neck squamous cell carcinoma. Vet J. 2016;210:7–16. doi:10.1016/j.tvjl.2015.11.006
46. Li S, Wang D, Cheng J, et al. A photodynamically sensitized dendritic cell vaccine that promotes the anti-tumor effects of anti-PD-L1 monoclonal antibody in a murine model of head and neck squamous cell carcinoma. J Transl Med. 2022;20(1):505. doi:10.1186/s12967-022-03707-x
47. Jin S, Li M, Chang H, et al. The m6A demethylase ALKBH5 promotes tumor progression by inhibiting RIG-I expression and interferon alpha production through the IKKε/TBK1/IRF3 pathway in head and neck squamous cell carcinoma. Mol Cancer. 2022;21(1):97. doi:10.1186/s12943-022-01572-2
48. Barasch A, Li H, Rajasekhar VK, et al. Photobiomodulation effects on head and neck squamous cell carcinoma (HNSCC) in an orthotopic animal model. Support Care Cancer. 2020;28(6):2721–2727. doi:10.1007/s00520-019-05060-0
49. Chen F, Lang L, Yang J, et al. SMAC-armed oncolytic virotherapy enhances the anticancer activity of PD1 blockade by modulating PANoptosis. Biomark Res. 2025;13(1):8. doi:10.1186/s40364-025-00726-w
50. Furuta H, Osawa K, Shin M, et al. Selective inhibition of NF-κB suppresses bone invasion by oral squamous cell carcinoma in vivo. Int, J, Cancer. 2012;131(5):E625–E635. doi:10.1002/ijc.27435
51. Martin CK, Werbeck JL, Thudi NK, et al. Zoledronic acid reduces bone loss and tumor growth in an orthotopic xenograft model of osteolytic oral squamous cell carcinoma. Cancer Res. 2010;70(21):8607–8616. doi:10.1158/0008-5472.can-10-0850
52. Ma P, He M, Lian H, et al. Systemic and local administration of a dual-siRNA complex efficiently inhibits tumor growth and bone invasion in oral squamous cell carcinoma. Mol Pharm. 2024;21(2):661–676. doi:10.1021/acs.molpharmaceut.3c00802
53. Tada Y, Kokabu S, Sugiyama G, et al. The novel IκB kinase β inhibitor IMD-0560 prevents bone invasion by oral squamous cell carcinoma. Oncotarget. 2014;5(23):12317–12330. doi:10.18632/oncotarget.2640
54. Zhang J, Chen C, Li A, et al. Immunostimulant hydrogel for the inhibition of malignant glioma relapse post-resection. Nat Nanotechnol. 2021;16(5):538–548. doi:10.1038/s41565-020-00843-7
55. Correa S, Grosskopf AK, Lopez Hernandez H, et al. Translational applications of hydrogels. Chem Rev. 2021;121(18):11385–11457. doi:10.1021/acs.chemrev.0c01177
56. Wang H, Chen Y, Wei R, et al. Synergistic chemoimmunotherapy augmentation via sequential nanocomposite hydrogel-mediated reprogramming of cancer-associated fibroblasts in osteosarcoma. Adv Mater. 2024;36(15):e2309591. doi:10.1002/adma.202309591
57. Wu Y, Chang X, Yang G, et al. A physiologically responsive nanocomposite hydrogel for treatment of head and neck squamous cell carcinoma via proteolysis-targeting chimeras enhanced immunotherapy. Adv Mater. 2023;35(12):e2210787. doi:10.1002/adma.202210787
58. Haider M, Jagal J, Ali Alghamdi M, et al. Erlotinib and curcumin-loaded nanoparticles embedded in thermosensitive chitosan hydrogels for enhanced treatment of head and neck cancer. Int J Pharm. 2024;666:124825. doi:10.1016/j.ijpharm.2024.124825
59. Chen X, Chen DR, Liu H, et al. Local delivery of gambogic acid to improve anti-tumor immunity against oral squamous cell carcinoma. J Control Release. 2022;351:381–393. doi:10.1016/j.jconrel.2022.09.010
60. Tsai LH, Young TH, Yen CH, Yao WC, Chang CH. Intratumoral thermo-chemotherapeutic alginate hydrogel containing doxorubicin loaded PLGA nanoparticle and heating agent. Int J Biol Macromol. 2023;251:126221. doi:10.1016/j.ijbiomac.2023.126221
61. Li J, Men K, Gao Y, et al. Single micelle vectors based on lipid/block copolymer compositions as mRNA formulations for efficient cancer immunogene therapy. Mol Pharm. 2021;18(11):4029–4045. doi:10.1021/acs.molpharmaceut.1c00461
62. Lei S, Zhang X, Men K, et al. Efficient colorectal cancer gene therapy with IL-15 mRNA nanoformulation. Mol Pharm. 2020;17(9):3378–3391. doi:10.1021/acs.molpharmaceut.0c00451
63. Lei S, Gao Y, Wang K, et al. An implantable double-layered spherical scaffold depositing gene and cell agents to facilitate collaborative cancer immunotherapy. ACS nano. 2025;19(18):17653–17673. doi:10.1021/acsnano.5c01366
64. Hu Y, Hu S, Zhang S, et al. A double-layer hydrogel based on alginate-carboxymethyl cellulose and synthetic polymer as sustained drug delivery system. Sci Rep. 2021;11(1):9142. doi:10.1038/s41598-021-88503-1
65. Xie L, Liu R, Li J, et al. A multifunctional and self-adaptive double-layer hydrogel dressing based on chitosan for deep wound repair. Int J Biol Macromol. 2023;253(Pt 6):127033. doi:10.1016/j.ijbiomac.2023.127033
66. Tan M, Zeng J, Zhang FZ, et al. Double-layer hydrogel with glucose-activated two-stage ROS regulating properties for programmed diabetic wound healing. ACS Appl Mater Interfaces. 2023;15(44):50809–50820. doi:10.1021/acsami.3c10607
67. Huang L, Zhang Y, Li Y, et al. Time-programmed delivery of sorafenib and anti-CD47 antibody via a double-layer-gel matrix for postsurgical treatment of breast cancer. Nanomicro Lett. 2021;13(1):141. doi:10.1007/s40820-021-00647-x
68. Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi:10.1056/NEJMoa1914347
69. Pan K, Farrukh H, Chittepu V, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41(1):119. doi:10.1186/s13046-022-02327-z
70. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol. 2020;17(1):1–12. doi:10.1038/s41423-019-0306-1
71. Nguyen A, Johanning G, Shi Y. Emerging novel combined CAR-T cell therapies. Cancers. 2022;14(6):1403. doi:10.3390/cancers14061403
72. Zeng C, Zhang C, Walker PG, Dong Y. Formulation and delivery technologies for mRNA vaccines. Curr Top Microbiol Immunol. 2022;440:71–110. doi:10.1007/82_2020_217
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.