")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Semaglutide May Alleviate Hepatic Steatosis in T2DM Combined with NFALD Mice via miR-5120/ABHD6
Authors Li R , Ye Z, She D, Fang P, Zong G , Hu K, Kong D, Xu W, Li L, Zhou Y, Zhang K, Xue Y
Received 3 August 2022
Accepted for publication 6 October 2022
Published 12 October 2022 Volume 2022:16 Pages 3557—3572
DOI https://doi.org/10.2147/DDDT.S384884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Ran Li,1 Zhengqin Ye,1 Dunmin She,2,3 Ping Fang,1 Guannan Zong,1 Kerong Hu,1 Dehong Kong,1 Wei Xu,1 Ling Li,1 Yun Zhou,1 Keqin Zhang,1 Ying Xue1
1Department of Endocrinology and Metabolism, Tongji Hospital, School of Medicine, Tongji University, Shanghai, People’s Republic of China; 2Clinical Medical College, Yangzhou University, Yangzhou, People’s Republic of China; 3Department of Endocrinology, Northern Jiangsu People’s Hospital Affiliated to Yangzhou University, Yangzhou, People’s Republic of China
Correspondence: Ying Xue; Keqin Zhang, Department of Endocrinology and Metabolism, Tongji Hospital, School of Medicine, Tongji University, No. 389, Xincun Road, Shanghai, 200065, People’s Republic of China, Tel +86-021-66111061, Email [email protected]; [email protected]
Objective: Although the pathogenesis of non-alcoholic fatty liver disease (NAFLD) has been extensively studied, the role of its underlying pathogenesis remains unclear, and there is currently no approved therapeutic strategy for NAFLD. The purpose of this study was to observe the beneficial effects of Semaglutide on NAFLD in vivo and in vitro, as well as its potential molecular mechanisms.
Methods: Semaglutide was used to treat type 2 diabetes mellitus (T2DM) combined with NAFLD mice for 12 weeks. Hepatic function and structure were evaluated by liver function, blood lipids, liver lipids, H&E staining, oil red staining and Sirius staining. The expression of α/β hydrolase domain-6 (ABHD6) was measured by qPCR and Western blotting in vivo and in vitro. Then, dual-luciferase reporter assay was performed to verify the regulation of the upstream miR-5120 on ABHD6.
Results: Our data revealed that Semaglutide administration significantly improved liver function and hepatic steatosis in T2DM combined with NAFLD mice. Furthermore, compared with controls, up-regulation of ABHD6 and down-regulation of miR-5120 were found in the liver of T2DM+NAFLD mice and HG+FFA-stimulated Hepa 1– 6 hepatocytes. Interestingly, after Semaglutide intervention, ABHD6 expression was significantly decreased in the liver of T2DM+NAFLD mice and in HG+FFA-stimulated Hepa 1– 6 hepatocytes, while miR-5120 expression was increased. We also found that miR-5120 could regulate the expression of ABHD6 in hepatocytes, while Semaglutide could modulate the expression of ABHD6 through miR-5120. In addition, GLP-1R was widely expressed in mouse liver tissues and Hepa 1– 6 cells. Semaglutide could regulate miR-5120/ABHD6 expression through GLP-1R.
Conclusion: Our data revealed the underlying mechanism by which Semaglutide improves hepatic steatosis in T2DM+NAFLD, and might shed new light on the pathological role of miR-5120/ABHD6 in the pathogenesis of T2DM+NAFLD.
Keywords: Semaglutide, non-alcoholic fatty liver disease, type 2 diabetes mellitus, α/β hydrolase domain-6, microRNA-5120
Introduction
Non-alcoholic fatty liver disease (NAFLD) is recognized as a common chronic disease worldwide, with a global prevalence of 25%.1 Clinically, there is a close bidirectional correlation between NAFLD and metabolic syndrome,1 especially type 2 diabetes mellitus (T2DM).2 Considering the serious adverse effects of NAFLD on patients,3 a wide variety of different therapies are being studied for the management of NAFLD, and several novel drugs have emerged as potential candidates for the treatment of NAFLD/NASH.4 However, to date, there are no drugs approved for the treatment of NAFLD other than weight reduction and lifestyle interventions.5 Therefore, therapeutic agents and drug targets for patients with NAFLD need to be further explored.
In recent years, glucagon-like peptide-1 receptor agonists (GLP-1RAs) have been reported to alleviate hepatic lipid accumulation in both clinical and basic studies.6–16 Exenatide, a GLP-1 mimetic for the treatment of T2DM, has shown beneficial effects on hepatic lipid accumulation and fibrosis in T2DM patients.6–8 Basic studies have suggested that Exenatide may ameliorate liver lipid content by improving de novo lipogenesis,9 stimulating hepatic lipid oxidation10 and inhibiting inflammation.11 Liraglutide, a modified analogue of human GLP-1,12 has been reported to protect against NAFLD and non-alcoholic steatohepatitis (NASH) in human and experimental studies 13–16 The underlying mechanisms of these benefits may be through multiple modalities, such as reducing hepatic lipid synthesis,16 limiting hepatic lipotoxicity,16 alleviating hepatic inflammation,16 activating mitophagy,15 etc. Semaglutide, a new weekly treatment for T2DM, has a more pronounced metabolic effect than Liraglutide.17–19 It is a modification of Liraglutide that highly enhances anti-enzyme degradation and reduces the rate of renal clearance by replacing alanine at position 8 with α-aminoisobutyric acid and adding lysine at position 26.18 A 72-week, double-blind Phase 2 trial (NCT02970942), including 320 NASH patients who received once-daily subcutaneous Semaglutide, showed that compared with placebo, Semaglutide treatment significantly increased the percentage of patients with NASH resolution, whereas it did not increase the percentage of patients with an improvement in fibrosis stage.20 Moreover, Semaglutide has been illustrated to markedly reduce levels of alanine aminotransferase (ALT) and high-sensitivity C-reactive protein in clinical trials in obesity and/or T2DM subjects.21 In addition, Semaglutide robustly reduced hepatomegaly and liver biochemical markers in Gubra-Amylin NASH (GAN) diet-induced obese (DIO) -NASH mice after 8 weeks of Semaglutide treatment.22 Although the above-mentioned studies have suggested that Semaglutide has ameliorating effects on NAFLD, the underlying mechanisms of these benefits have not been confirmed.
α/β hydrolase domain-6 (ABHD6) is a newly identified 32kDa integral membrane protein capable of hydrolyzing monoacylglycerol (MAG).23,24 Previous studies have reported ABHD6 is primarily involved in regulating endocannabinoid signaling in the brain.23,25,26 However, ABHD6 has recently been shown to play a potential role in the pathogenesis of various metabolic diseases, such as metabolic syndrome,27 insulin secretion disorder,28 obesity and T2DM.29 Interestingly, the expression of ABHD6 has been shown to be ubiquitous (eg, liver, white adipose tissue, small intestine and kidney).24,27 Thomas et al27 have demonstrated that antisense oligonucleotide (ASO) knockdown of ABHD6 expression in peripheral tissues can reduce body weight, improve liver fat accumulation and other metabolic disorders in mice fed a high-fat diet (HFD). Furthermore, the enhanced adipose browning and augmented brown adipose function in ABHD6-knockout mice are at least partially due to the accumulation of 1-monoacylglycerol (1-MAG), which activates peroxisome proliferator-activated receptor alpha (PPARα) and peroxisome proliferator activated receptor gamma (PPARγ).29 Taken together, the inhibition of ABHD6 may have potential therapeutic values in HFD-induced metabolic syndrome, obesity and NAFLD. Therefore, we evaluated the therapeutic effects of Semaglutide on NAFLD combined with T2DM mice, and explored the potential effects of Semaglutide on ABHD6 expression in the liver, thus providing an effective target for the treatment of NAFLD.
Materials and Methods
Animals
The animal study was performed in accordance with Tongji Hospital requirements and approved by the Animal Ethics Committee of Tongji Hospital, Tongji University School of Medicine (2021-DW-(003)). Male C57BL/6J mice (8 weeks old) were purchased from the Model Animal Research Center of Nanjing University. All mice were housed in specific pathogen-free environment in the Experimental Animal Center of Tongji Hospital, Shanghai. After 1 week of adaptive feeding, thirty mice were randomly divided into three groups (n = 10 in each group): Control group, T2DM+NAFLD group and T2DM+NAFLD+Semaglutide group (Semaglutide group). The control mice were fed with a regular diet; the other two groups were fed with a high fat diet (HFD) (42% Kcal fat, Trophic Animal Feed High-Tech Co., Ltd, Haian, JS, China). After 4 weeks, mice of T2DM+NAFLD group and Semaglutide group were intraperitoneally injected with streptozotocin (STZ; 100mg/kg; Sigma-Aldrich, St. Louis, MO, USA). Mice with fasting glucose >11.1 mmol/L were considered as a successful T2DM model.
Four weeks after STZ injection, mice in Semaglutide group were subcutaneously injected with Semaglutide once a week (novo nordisk, CPH, Denmark; 0.42mg/kg/w) for 12 weeks; while mice of the control group and T2DM+NAFLD group received an equal amount of sterile water. Body weight and fasting blood-glucose of all mice were measured weekly.
Intraperitoneal insulin tolerance tests (IPITTs) and oral glucose tolerance tests (OGTTs) were performed after 6 and 16 hours of fasting, respectively. Mice were given 0.75 IU/kg insulin (novo nordisk, CPH, Denmark) by intraperitoneal injection, or gavaged with 2g/kg glucose (SINE, Shanghai, China). Blood glucose of all mice was measured at 0, 15, 30, 60, 90 and 120 minutes, respectively.
Cell Culture
Hepa 1–6 mouse hepatoma cell line was obtained from National Collection of Authenticated Cell Cultures (China) and cultured in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Gibco, USA) and 1% penicillin/streptomycin (KeyGEN, Nanjing, JS, China). Hepa 1–6 cells were grown at 37°C in a 5% CO2 humidified atmosphere and starved for 12 hours. The cells subsequently exposed to 5.5mM glucose as control group, 30 mM high glucose+1mM free fatty acid (0.33mM palmitic acid, 0.67mM oleic acid) as HG+FFA group, and HG+FFA with 4μg/mL Semaglutide Injection as HG+FFA+Sema group for 48h.30
For cell transfection, miR-5120 mimics, miR-5120 inhibitors, GLP-1R siRNA, and their negative controls (Sangon Biotech, Shanghai, China) were transfected into Hepa 1–6 cells for 48h using Lipofectamine™ 3000 reagent (Invitrogen, Carlsbad, CA, USA). In order to investigate whether Semaglutide could regulate the expression of ABHD6 through miR-5120, and the role of GLP-1R in this pathway, Hepa 1–6 cells were divided into the following groups: 1) Control group (miR-5120 inhibitors control+ GLP-1R siRNA control); 2) HG+FFA group (miR-5120 inhibitors control+ GLP-1R siRNA control); 3) HG+FFA+Sema group (miR-5120 inhibitors control+GLP-1R siRNA control); 4) HG+FFA+Sema+GLP-1R SiRNA group (miR-5120 inhibitors control+GLP-1R siRNA); 5) HG+FFA+Sema+miR-5120 inhibitors group (miR-5120 inhibitors +GLP-1R siRNA control). Hepa 1–6 cells were transfected with miR-5120 inhibitors, GLP-1R SiRNA and its negative control for 8h. After that, the solution was changed with or without the mixture of HG+FFA or HG+FFA and Sema for an additional 48h. The sequences of miR-5120 mimic, miR-5120 inhibitor, GLP-1R siRNA and their controls are shown in Supplementary Table 1.
Biochemical Measurements
The levels of liver function, blood lipids and liver lipids were tested using an ELISA kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, JS, China), including alanine aminotransferase (ALT), aspartate aminotransferase (AST), triacylglycerol (TG), total cholesterol (TC), FFA, Low-density lipoprotein cholesterol (LDL-C) and High-density lipoprotein cholesterol (HDL-C).
Morphological Studies
Liver tissues of above mice were sectioned for staining. Hematoxylin and Eosin (H&E) and Sirius red staining were used to observe the morphological changes and detect collagen deposition, respectively. Collagen area was quantitatively analyzed by Image J. Oil red O staining was used to observe lipid droplets in frozen sections of liver tissue and Hepa 1–6 cells. The method for immunofluorescence staining of cells was as follows. Hepa 1–6 cells were fixed with paraformaldehyde and then incubated overnight with GLP-1R antibody (#NBP1-97308, 1:100, Novus Biologicals, Littleton, CO, USA). After cleaning, the cells were further incubated with Alexa-488 goat anti-rabbit (#111-545-003, 1:200, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) in darkness for 1 hour and finally stained with 4’,6-diamidino-2-phenylindole (DAPI, Bosterbio, CA, USA). The representative images were repeated in at least three independent experiments.
RNA Isolation and Quantitative Real-Time PCR (qPCR)
Total RNA was extracted from liver tissues and Hepa 1–6 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). MiRNA was extracted from serum or cell media using miRcute Serum/plasma miRNA isolation kit (TIANGEN, Beijing, China). 0.5μg RNA was transcribed into cDNA using a reverse transcription (RT) kit (Takara RR037A, Tokyo, Japan), and the volume of the transcription reaction was 10 μL. Gene-specific primers for RT are shown in Supplementary Table 2. PCR reaction systems were carried out on a QuantStudio7Flex System (ABI, Waltham, MA, USA) using SYBR® Premix Ex TaqTM (TAKARA RR820A, Tokyo, Japan). For mRNA analysis, 36b4 was used as an internal control. For miRNA analysis, U6 and miR-16 were used as internal controls for tissues or cells, and serum or cell media, respectively. The results were analyzed using 2−ΔΔCt method.
Western Blotting
Proteins of cells and tissues were extracted with RIPA Lysis Buffer (EpiZyme, Shanghai, China), and quantified with bicinchoninic acid (BCA, Abcam, Cambridge, MA, USA). Equal amounts of protein (25μg/lane) were separated by electrophoresis, and then transferred into membranes with a pore size of 0.2μm (Merck Millipore, Billerica, MA, USA). The membranes were blocked, and then incubated with the following antibodies: ABHD6 rabbit antibody (#97573, 1:1000, Cell Signaling TECHNOLOGY, Danvers, MA, USA), CD36 goat antibody (#Q3UAI3, 1:2000, R&D Systems, Minneapolis, MA, USA), PPARγ mouse antibody (#sc-7273, 1:500, Santa Cruz, CA, USA), GLP-1R rabbit antibody (#26196-1-AP, 1:500, Proteintech, Wuhan, HB, China) or β-actin mouse antibody (#AF0003, 1:5000, Beyotime, Shanghai, China). Secondary antibodies were as follows: horseradish peroxidase (HRP) labeled goat anti-rabbit IgG (#A0208, 1:1000, Beyotime, Shanghai, China), goat anti-mouse IgG (#A0216, 1:1000, Beyotime, Shanghai, China) or rabbit anti-goat IgG (#SA00001-4, 1:2000, Proteintech, Wuhan, HB, China). Bands were detected using an enhanced chemiluminescence assay (ECL, NCM Biotech, Suzhou, JS, China) according to the manufacturer’s instructions. The representative blot bands were repeated at least three times.
Dual Luciferase Report Assay
NM_001331064.1 was selected as the transcript of ABHD6. The interaction between the 3’- Untranslated Region (UTR) of Abhd6 and miR-5120 was predicted through targetscan (http://www.targetscan.org/), and two binding sites (TCCCCAAA and GCCCCAAA) were found. Moreover, mutation sequences of the binding sites (AGGGGTTT and CGGGGTTT) were designed.
Human Embryonic Kidney (HEK)-293T cells were obtained from National Collection of Authenticated Cell Cultures (China) and plated onto the 96-well plates. The cells were transfected with 0.2 μg Abhd6 wild type (WT) plasmid or Abhd6 mutant (MUT) plasmid (OBiO Technology, Shanghai, China) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) at 37°C in the presence of miR-5120 mimics or the mimics control (NC) separately. After 48 hours of transfection, the Firefly (FL) and Renilla luciferase (RL) activities, and FL/RL ratios were measured using the Dual-Glo Luciferase Assay System (Promega Corporation, Madison, WI, USA) in a Titertek-Berthold Detection System (Berthold Technologies, Wildbad, Germany).
Bioinformatics Analysis
The gene expression datasets of GSE199121, GSE199105 and GSE165855 were downloaded from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo). We used the GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) analysis to identify the differentially expressed genes (DEGs). The criteria of DEGs were considered to be upregulated fold change (FC) >1 and P-value of <0.1. Heatmap was drawn using the Complex heatmap R package (R Version 3.4). Venn diagrams were generated by an online tool (http://www.bioinformatics.com.cn/).
Statistical Analysis
Statistical analysis was performed using SPSS 22.0 software. All data were expressed as mean ± SD. Variables that did not meet the normal distribution were log-transformed to satisfy the normal distribution. The statistical differences in mean values between two groups were assessed by unpaired two-tailed Student’s t-test. One-way analysis of variance (ANOVA) followed by LSD post hoc test or Dunnett’s T3 post hoc multiple comparison test was performed to compare significant differences among three or more groups. P-values <0.05 was set as statistical significance.
Results
Semaglutide Reduces Body Weight, Improves Glucose Intolerance and Serum Lipid Mice with T2DM Accompanied by NAFLD
To investigate the effects of Semaglutide on glycolipid metabolism in T2DM combined with NAFLD mice, we first established a T2DM + NAFLD mouse model and treated with Semaglutide (0.42mg/kg/w) subcutaneously every week for 12 weeks. Both body weight and blood glucose in T2DM+NAFLD group were higher than those in control group, while both blood glucose and body weight in Semaglutide group were lower than those in T2DM+NAFLD group (Figure 1A and B).
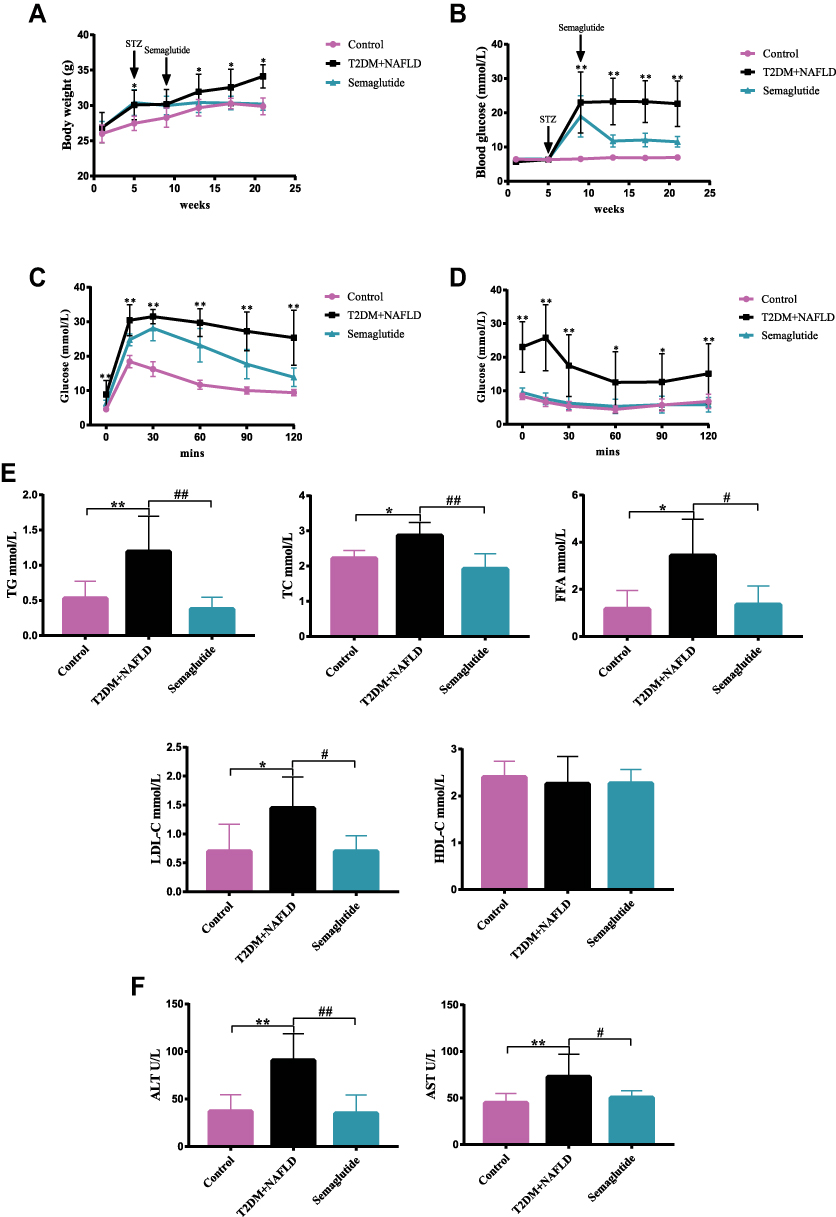 |
Figure 1 Semaglutide improves glucolipid metabolism in T2DM combined with NAFLD mice. (A and B) Body weight (A) and blood glucose (B) of mice at different weeks (n = 10). (C and D) OGTT (C) and IPITT (D) results (n = 10) were measured. (E) Serum TG, TC, FFA, LDL-C and HDL-C levels of mice were measured (n = 10). (F) Serum ALT and AST levels of mice were measured (n = 10). Data are expressed as mean ± SD, *P < 0.05, **P < 0.01 vs control group; #P < 0.05, ##P < 0.01 vs T2DM+NAFLD group. |
OGTTs and IPITTs were performed at the end of 12th week of Semaglutide treatment to evaluate glucose metabolism and insulin resistance (IR) status. OGTT results are shown in Figure 1C. Glucose tolerance was impaired in T2DM+NAFLD mice, which were significantly improved after Semaglutide administration. IPITT results indicated that although the lowest plasma glucose levels were reached at 60 min in all three groups of mice, blood glucose level of T2DM+ NAFLD mice was consistently significantly higher than that of the control group (Figure 1D). These results confirmed that Semaglutide could significantly improve glucose tolerance in T2DM+NAFLD mice.
In addition, liver functions and serum lipid profiles among the three groups were measured. Compared with the control group, serum TG, TC, FFA, LDL-C, AST and ALT were significantly increased in the T2DM+NAFLD group, while they were dramatically decreased after Semaglutide treatment (Figure 1E and F and Supplementary Table 3). There was no statistical significance in HDL-C among the three groups.
Semaglutide Mitigated Hepatic Lipid Accumulation in vivo and in vitro
To determine whether Semaglutide alleviated hepatic steatosis in vivo, the liver weight and hepatic lipids were evaluated in the control group, T2DM+ NAFLD group and Semaglutide group (Figure 2A and B). Liver weight, body weight and liver weight/body weight ratio were all significantly increased in T2DM+NAFLD group compared with the control group, whereas they were all remarkably reduced after Semaglutide treatment (Figure 2A and Supplementary Table 3). Moreover, hepatic TG and FFA in the T2DM+NAFLD group were significantly higher than those in control group, while hepatic TG and FFA were significantly decreased after Semaglutide treatment (Figure 2B and Supplementary Table 3). Although there was no significant difference in hepatic TC between T2DM+NAFLD group and Semaglutide group, a decreasing trend still occurred in Semaglutide group (Figure 2B and Supplementary Table 3). The H&E staining and Oil Red O staining showed hepatocyte swelling, noticeable lipid droplets and inflammatory cells in T2DM+NAFLD mice compared to the control group. Semaglutide treatment obviously alleviated the above pathological changes in the T2DM+NAFLD group (Figure 2C). To further explore the mechanism by which Semaglutide alleviated hepatic steatosis (TG accumulation) in NAFLD+T2DM mice, the expressions of mRNA and protein related to lipid metabolism were measured. As shown in Figure 2D, mRNA levels of fatty acid uptake (eg, cluster of differentiation 36 [Cd36], fatty acid binding protein 4 [Fabp4] and Pparg) were all significantly increased in the liver tissues of the T2DM+NAFLD group compared to the control group, whereas the above gene expressions were all dramatically decreased after Semaglutide treatment. Conversely, the mRNA level of fatty acid oxidation (such as Pparα) in the liver tissues of T2DM + NAFLD mice was significantly lower than that of the control mice, while Semaglutide treatment dramatically increased the expression of Pparα in T2DM + NAFLD mice. Moreover, the changes in protein expression of CD36 and PPARγ among the three groups were consistent with the changes in mRNA expression in liver tissues (Figure 2E).
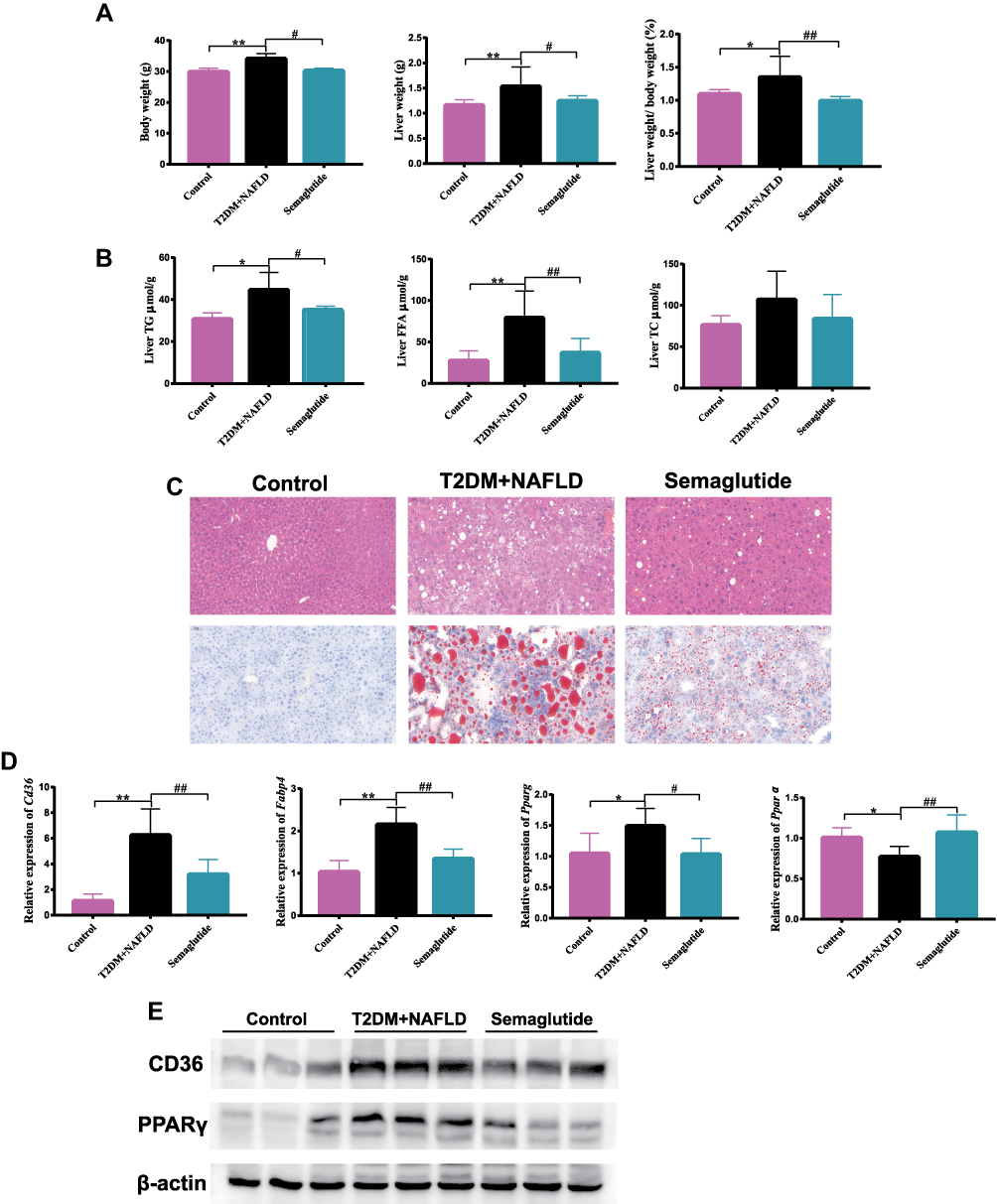 |
Figure 2 Semaglutide alleviates hepatic lipid accumulation by decreasing fatty acid uptake and increasing fatty acid oxidation in the liver of T2DM combined with NAFLD mice and Hepa 1–6 cells stimulated by HG+FFA. (A) Body weight, liver weight (g) and liver weight/body weight (%) of mice (n = 10). (B) liver TG, TC and FFA of mice (n = 10) were measured. (C) H&E and Oil red O staining of liver tissues (200×). (D) The mRNA expression of Cd36, Fabp4, Pparg and Pparα in liver tissues (n = 6). (E) The protein expression of CD36 and PPARγ in liver tissues. Data are expressed as mean ± SD, *P < 0.05, **P < 0.01 vs control group; #P < 0.05, ##P < 0.01 vs T2DM+NAFLD group. |
We also examined the effect of Semaglutide on lipid accumulation in Hepa 1–6 cells of the control group, HG+FFA group and Semaglutide group. Oil red O staining observed that lipid droplets in Hepa 1–6 cells were significantly increased in HG+FFA group compared with the control group, while Semaglutide treatment remarkably reduced lipid accumulation (Figure 3A). In addition, the changes in Cd36, Fabp4, Pparg and Pparα mRNA expression, as well as CD36 and PPARγ protein levels in the three groups of Hepa 1–6 cells were consistent with the changes in the expressions of above genes and proteins in the liver tissues of mice among the three groups (Figure 3B and C). These results indicated that Semaglutide could decrease hepatic lipid accumulation in hepatocytes in vitro after HG+FFA stimulation. Taken together, the above results suggested that Semaglutide could significantly ameliorate hepatic steatosis both in vivo and in vitro.
 |
Figure 3 Semaglutide alleviates hepatic lipid accumulation by decreasing fatty acid uptake and increasing fatty acid oxidation in Hepa 1–6 cells stimulated by HG+FFA. (A) Oil red O staining of Hepa 1–6 cells (200×). (B) The mRNA expression of Cd36, Fabp4, Pparg and Pparα in hepatocytes (n = 6). (C) The protein expression of CD36 and PPARγ in hepatocytes. (D) Sirius Red staining of liver tissues (400×). A semi-quantification analysis of the staining intensity of Sirius Red in liver tissues of mice was performed (n = 6). (E) The mRNA expression of Col1a1 and Col3a1 in liver tissues (n = 6). (F) The mRNA expression of Col1a1 and Col3a1 in hepatocytes (n = 6). Data are expressed as mean ± SD, *P < 0.05, **P < 0.01 vs control group; #P < 0.05, ##P < 0.01 vs T2DM+NAFLD group. |
Furthermore, analysis of Sirius red staining results demonstrated that liver fibrosis significantly increased in the T2DM+NAFLD group compared with the normal mice. However, Semaglutide intervention improved the fibrosis in T2DM+NAFLD mice (Figure 3D). Furthermore, in comparison with the control group, a significant increase of the mRNA expressions of collagen type I alpha 1 chain (Col1a1) and collagen type III alpha 1 chain (Col3a1) was observed in T2DM+NAFLD group. However, it obviously dropped after using Semaglutide (Figure 3E). The changes in the mRNA levels of Col1a1 and Col3a1 in the three groups of Hepa 1–6 cells (Figure 3F) were consistent with the changes in above gene expressions in the liver tissues of mice among the three groups, indicating that Semaglutide treatment significantly attenuated hepatic collagen deposition in vivo and in vitro.
Semaglutide Reduces ABHD6 Expression in the Liver of T2DM + NAFLD Mice
To explore the potential molecular mechanisms for the diagnosis and treatment of NAFLD, the DEGs in liver tissues of mice between the normal group and NAFLD group were identified in GSE199121, GSE199105 and GSE165855 datasets based on bioinformatics analysis. FCs with up-regulation >1 and P value <0.05 were applied to define DEGs between the normal and NAFLD groups. Nine upregulated DEGs overlapped across all three datasets (Figure 4A). A heatmap was presented to show that in the GSE199121 dataset, 9 overlapping genes were significantly up-regulated in the liver tissues of NAFLD group compared with the normal group (Figure 4B). We selected Abhd6 among these 9 up-regulated DEGs for further study. Previous studies have revealed that ABHD6, a MAG hydrolase, may be involved in the pathogenesis of metabolic syndrome, insulin secretion and obesity.27–29 Inhibition of ABHD6 leads to accumulation of 1-MAG that regulates insulin secretion, insulin sensitivity, and lipid browning through activation of PPARα and PPARγ.28,29 Considering that Semaglutide could dramatically affect both PPARγ and Pparα expression in the liver of T2DM + NAFLD mice and HG+FFA stimulated Hepa 1–6 cells (Figures 2D, E and 3B, C), we speculate that Semaglutide may also be involved in the regulation of ABHD6. We found that the mRNA and protein expressions of ABHD6 were significantly up-regulated in the T2DM+NAFLD group compared with the controls, whereas those were notably down-regulated after Semaglutide treatment (Figure 5A and B). Moreover, the changes in ABHD6 mRNA and protein expression in the three groups of Hepa 1–6 cells were consistent with the above changes in liver tissues among the three groups of mice (Figure 5C and D).
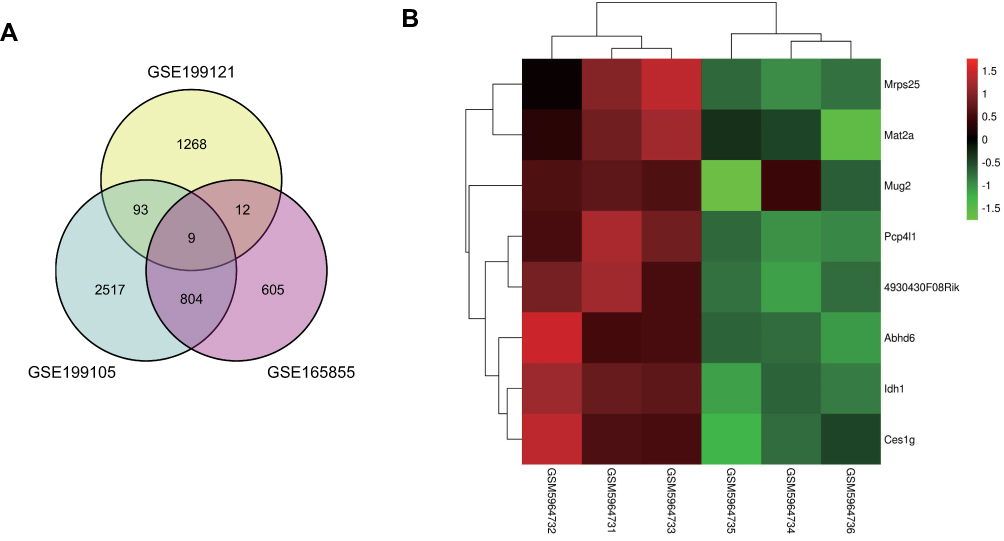 |
Figure 4 Identification of DEGs in NAFLD mouse model based on bioinformatics analysis. (A) Venn diagram for overlapping DEGs of the liver tissues among control and NAFLD group from the three cohort profile sets (GSE199121, GSE199105, GSE165855). (B) A Heatmap of DEGs between the normal and NAFLD group from the GSE199121 dataset, where DEGs overlapped with the other two datasets. Normal group was GSM5964731, GSM5964732 and GSM5964733; NAFLD group was GSM5964734, GSM5964735 and GSM5964736. Statistically significant DEGs were defined based on gene upregulation FC > 1 and P < 0.1 as the cut-off criteria. |
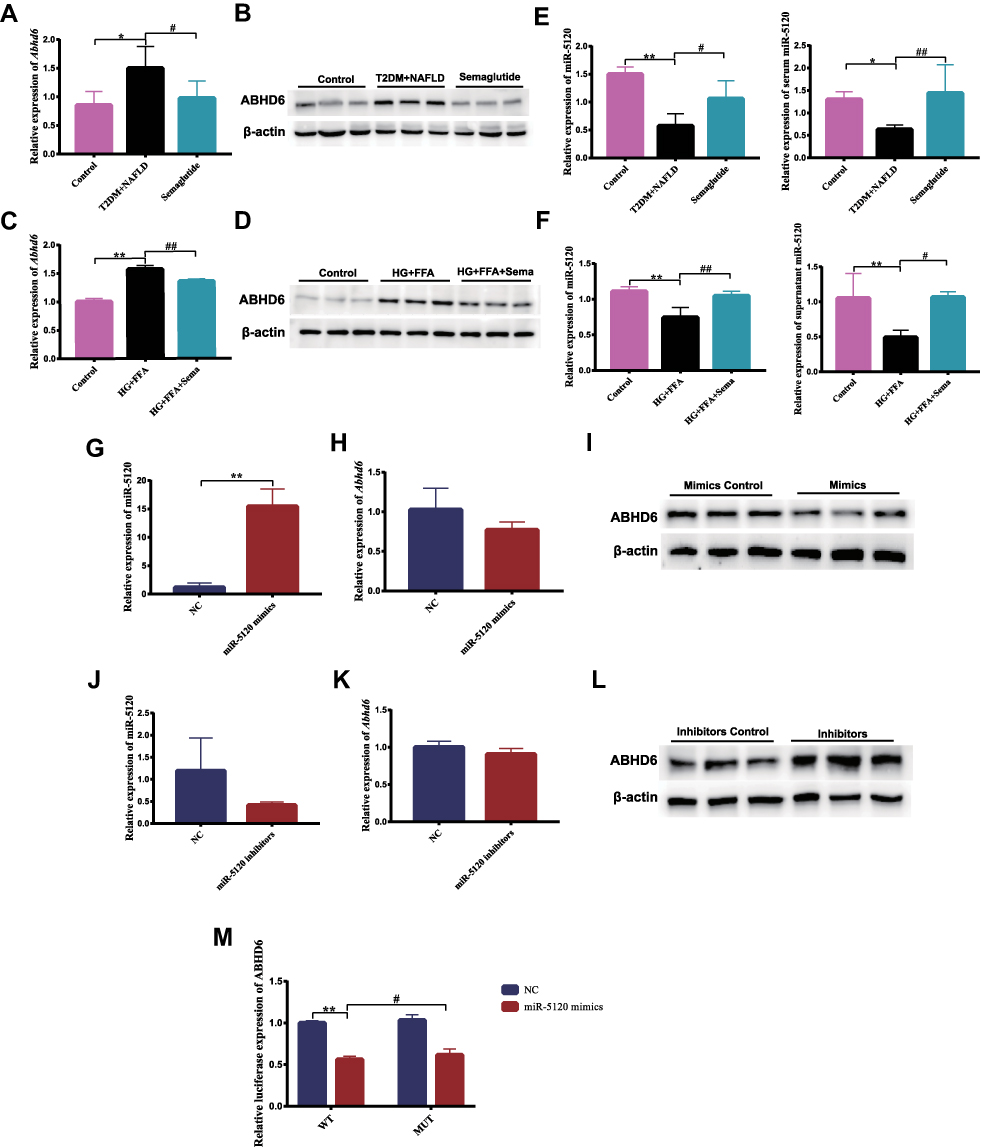 |
Figure 5 Semaglutide decreases ABHD6 and increases miR-5120 expression in the liver of T2DM combined with NAFLD mice and in hepatocytes stimulated by HG+FFA. (A) The mRNA expression of Abhd6 in liver tissues (n = 6). (B) The protein expression of ABHD6 in liver tissues. (C) The mRNA expression of Abhd6 in hepatocytes (n = 6). (D) The protein expression of ABHD6 in hepatocytes. (E) The expression of hepatic and serum miR-5120 was detected (n = 6). (F) miR-5120 mRNA levels of Hepa 1–6 cells and supernatant were measured (n = 6). (G) The expression of miR-5120 in Hepa 1–6 cells activated by miR-5120 mimics was measured by qPCR (n = 6). (H) The mRNA expression of Abhd6 in Hepa 1–6 cells stimulated by miR-5120 mimics was detected (n = 6). (I) ABHD6 protein level in Hepa 1–6 cells stimulated by miR-5120 mimics was determined. (J) The expression of miR-5120 in Hepa 1–6 cells suppressed by miR-1a inhibitors was measured (n = 6). (K) The mRNA expression of Abhd6 in Hepa 1–6 cells stimulated by miR-5120 inhibitors was detected (n = 6). (L) ABHD6 protein level in Hepa 1–6 cells stimulated by miR-1a inhibitors was determined. (M) Dual-Luciferase reporter assay verified that ABHD6 was a target gene of miR-5120 (n = 3). Data are expressed as mean ± SD, *P < 0.05, **P < 0.01 vs control group; #P < 0.05, ##P < 0.01 vs T2DM+NAFLD group. |
MiR-5120 Regulates the Expression of ABHD6
Recent studies have revealed that microRNAs (miRNAs) play critical roles in lipid metabolism by degrading or suppressing the translation of its target genes.31–33 In this study, miR-5120 was predicted to match the 3’ UTR region of Abhd6 and regulate the expression of ABHD6 by a network bioinformatics tool (http://www.targetscan.org/). We further determined that the expression of hepatic and serum miR-5120 was significantly decreased in the T2DM+NAFLD group compared with the control mice, whereas Semaglutide treatment dramatically up-regulated the expression of miR-5120 (Figure 5E). Moreover, we detected the changes in miR-5120 expression in Hepa 1–6 cells and supernatants (including control group, HG+FFA stimulation group and Semaglutide-treated group). We found that the changes in miR-5120 expression in cells and supernatants were completely consistent with the aforementioned changes in miR-5120 expression in liver tissues and serum (Figure 5F).
MiR-5120 mimics and miR-5120 inhibitors were used to investigate the effect of miR-5120 on ABHD6 expression in Hepa 1–6 cells. Compared with the control group, miR-5120 mimics could significantly increase miR-5120 expression in Hepa 1–6 cells (Figure 5G), while the mRNA and protein expressions of ABHD6 were significantly reduced in Hepa 1–6 cells treated with miR-5120 mimics (Figure 5H and I). MiR-5120 expression showed a downward trend after miR-5120 inhibitors treatment in Hepa 1–6 cells (Figure 5J). Although there was no significant change in mRNA expression of Abhd6 in Hepa 1–6 cells between the miR-5120 inhibitors group and the control group (Figure 5K), the expression of ABHD6 protein was remarkably increased in Hepa 1–6 cells treated with miR-5120 inhibitors compared with the control group (Figure 5L).
The direct interaction of miR-5120 with WT and mutant Abhd6 3’UTR was studied using a dual-luciferase reporter assay. Luciferase activity of Abhd6 3’UTR was significantly reduced upon the treatment with miR-5120 mimics, and the inhibitory effect of miR-5120 on Abhd6 3’UTR was attenuated by the mutation of the binding site (Figure 5M). These results indicated that ABHD6 was a target of miR-5120.
Semaglutide Affects the Expression of ABHD6 Through miR-5120 in vitro
In order to explore whether Semaglutide can regulate the expression of ABHD6 through miR-5120, we measured the expression of ABHD6 in Hepa 1–6 cells stimulated by HG+FFA+Sema plus miR-5120 inhibitors. Our data showed that compared with the control group, the expression of miR-5120 in Hepa 1–6 cells of HG+FFA group was significantly decreased (Figure 6A), whereas the gene and protein expression of ABHD6 in Hepa 1–6 cells of HG+FFA group was remarkably increased (Figure 6B and C). Conversely, after Semaglutide treatment, the expression of miR-5120 was significantly increased in Hepa 1–6 cells compared with the HG+FFA group (Figure 6A), while the gene and protein expression of ABHD6 was remarkably decreased in Hepa 1–6 cells (Figure 6B and C). The effect of Semaglutide on ABHD6 gene and protein expression in HG+FFA stimulated Hepa 1–6 cells was completely eliminated after treatment with miR-5120 inhibitors (Figure 6B and C). The above results indicated that Semaglutide could regulate ABHD6 expression in hepatocytes through miR-5120.
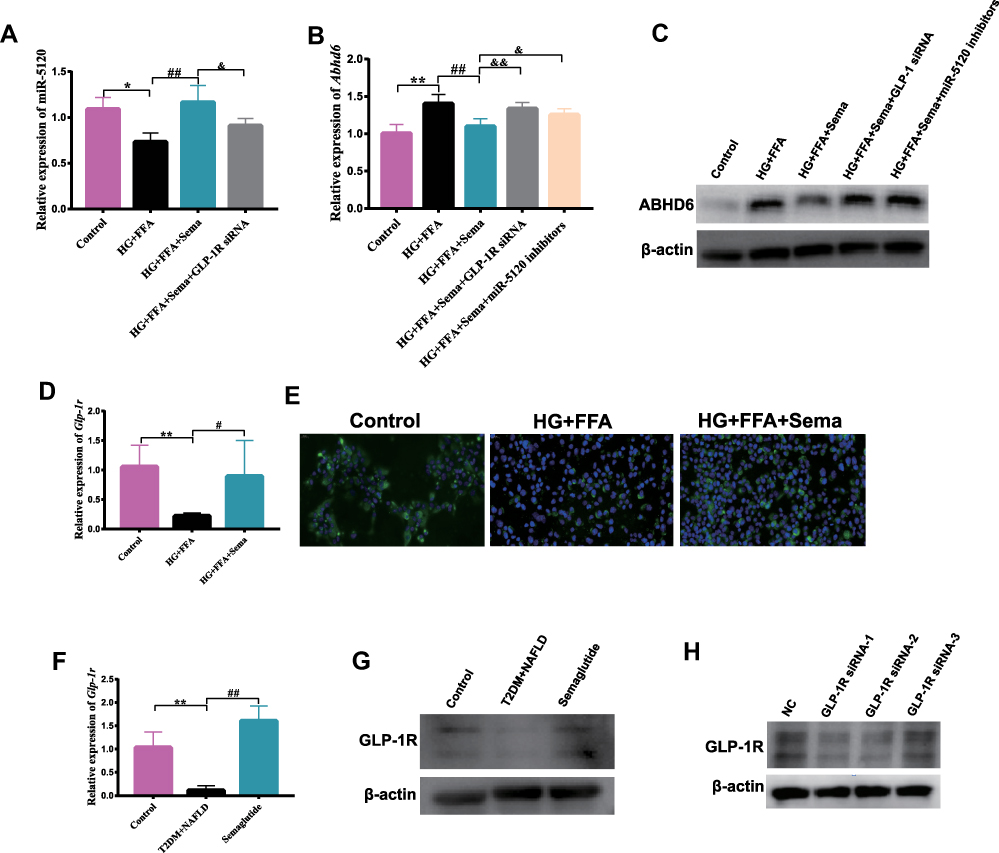 |
Figure 6 Semaglutide directly acts on hepatic GLP-1R to regulate miR-5120/ABHD6 expression, and affects ABHD6 expression through miR-5120 in vitro. (A) The expression of miR-5120 in Hepa 1–6 cells stimulated by HG+FFA+Sema plus GLP-1R siRNA was detected (n = 6). (B) The mRNA expression of Abhd6 in Hepa 1–6 cells stimulated by HG+FFA+Sema plus GLP-1R siRNA or miR-5120 inhibitors was detected (n = 6). (C) ABHD6 protein expression in Hepa 1–6 cells stimulated by HG+FFA+Sema plus GLP-1R siRNA or miR-5120 inhibitors was measured by Western blotting. (D) The mRNA expression of Glp-1r in hepatocytes was measured (n = 6). (E) The protein expression of GLP-1R in hepatocytes was measured by immunofluorescence staining (400×). (F) The mRNA expression of Glp-1r in liver tissues (n = 6). (G) The protein expression of GLP-1R in liver tissues was measured. (H) The protein expression of GLP-1R in Hepa 1–6 cells after transfection with three GLP-1R SiRNAs. Data are expressed as mean ± SD, *P < 0.05, **P < 0.01 vs Control group; #P < 0.05, ##P < 0.01 vs T2DM+NAFLD group or HG+FFA group; &P < 0.05, &&P < 0.01 vs HG+FFA+Sema group. |
Semaglutide Regulates miR5120/ABHD6 Expression Through GLP-1R in vitro
To explore whether Semaglutide can directly act on hepatocytes through GLP-1R, we determined the expression of GLP-1R in the liver of mice and Hepa 1–6 hepatocytes by qPCR, Western blotting, and immunofluorescence staining. GLP-1R expression was significantly decreased in HG+FFA stimulated hepatocytes and the liver of T2DM + NAFLD mice, whereas Semaglutide intervention alleviated the decline of GLP-1R expression (Figure 6D–G). To determine whether Semaglutide directly regulated miR-5120/ABHD6 in hepatocytes through GLP-1R, GLP-1R expression in Hepa 1–6 cells was inhibited by GLP-1R siRNA. Three GLP-1R siRNAs were synthesized, and Western blotting analysis showed that GLP-1R siRNA-1 and 2 had the most obvious blocking effect on GLP-1R expression in Hepa 1–6 cells (Figure 6H). Our study also indicated that the effects of Semaglutide on increasing miR-5120 expression and decreasing ABHD6 expression were abolished after the inhibition of GLP-1R in Hepa 1–6 hepatocytes by GLP-1R siRNA-1 (Figure 6A–C). We speculated that Semaglutide might regulate the expression of miR-5120/ABHD6 in hepatocytes by binding to GLP-1R.
Discussion
In this study, we proposed that Semaglutide could significantly improve glycolipid metabolism in a mouse model of T2DM+NAFLD induced by HFD combined with STZ, and further elucidated the molecular mechanism of Semaglutide on hepatic lipid metabolism. Here, we demonstrated for the first time that Semaglutide, upon binding to GLP-1R on hepatocytes, might regulate hepatocyte steatosis in vivo and in vitro through the miR-5120/ABHD6 pathway.
Previous studies have suggested that GLP-1RAs, represented by Exenatide and Liraglutide, can effectively reduce hepatic lipid accumulation in vivo and vitro,6–16 which is a new potential treatment for NAFLD, especially in NAFLD combined with T2DM.1 Numerous studies have explored the regulatory mechanism of Exenatide and Liraglutide in hepatic lipid deposition.9–11,15,16 For example, Exenatide ameliorates sirtuin 1 (SIRT1)-mediated lipogenesis by activating the 5’ AMP-activated protein kinase (AMPK) pathway and simultaneously inhibiting SREBP-1c. Moreover, Exenatide also requires SIRT1 to improve liver inflammation.9 In addition, Exenatide has been reported to improve hepatic IR and fatty acid β-oxidation in NASH rats by increasing PPARγ and protein kinase A (PKA) activity, and activating PKA-dependent PPARα activity.10 Yu et al15 illustrated that Liraglutide reduced lipid accumulation, alleviated mitochondrial dysfunction, and enhanced the elimination of abnormal mitochondria, thereby inhibiting NLRP3 inflammasome and pyroptosis in palmitate/lipopolysaccharide-treated hepatocytes.
Semaglutide, a novel long-acting GLP-1RA, has a more pronounced metabolic effect than Liraglutide.17–19 In a phase 3b open-label trial for 577 patients with T2DM (HbA1c 7.0–11.0%), Semaglutide was found to be superior to liraglutide in reducing body weight and HbA1c after 30 weeks of treatment.17 Recent clinical and experimental studies have shown that Semaglutide can dramatically improve liver function and lipid accumulation in NAFLD or NASH.20–22 However, so far, the regulatory mechanism of Semaglutide for NAFLD remains to be further studied. Our study evaluated the therapeutic effect of Semaglutide on hepatic fat accumulation in NAFLD combined with T2DM mice. Our research found that Semaglutide treatment could significantly reduce body weight, improve glucose intolerance, and decrease serum lipid in T2DM+NAFLD mice. Notably, Semaglutide also significantly reduced liver weight, improved liver function, hepatic lipid accumulation and hepatic fibrosis in T2DM + NAFLD mice. The therapeutic dose of Semaglutide in mice is equivalent to the maximum tolerated dose in humans, according to the instructions for Semaglutide, primarily because rodents tend to require higher doses of the drug to be effective. In addition, there was no statistical significance in HDL-C among the three groups in our study. Impaired HDL-C is one of the main drivers of atherosclerosis, the key-prognostic cardiovascular (CV) factor of both NAFLD and T2DM.34 The results of HDL-C in our animal model of T2DM combined with NALFD do not match the results of previous clinical studies, probably because animal model cannot completely mirror human illness. Although many diverse animal models are currently used in the field of NAFLD, the ideal preclinical model, suitable for all ideal characteristics of all possible study types, may not yet exist.35
Our study further explored the potential regulatory mechanism of Semaglutide on hepatic steatosis. Previous studies have suggested that GLP-1RAs may exert effects on hepatic lipid metabolism through direct and indirect mechanisms.10,36–38 Tanaka et al36 reported that Exenatide ameliorated hepatic steatosis by reducing appetite and enhancing lipid utilization in adipose tissue of NAFLD rats. However, Zhou et al37 demonstrated that Liraglutide reduced hepatic lipid accumulation and liver injury in T2DM rats by activating the expression of PPARα through a GLP-1R dependent AMPK pathway. To date, the expression of GLP-1R in the liver remains controversial.36–38 Our study validated the mRNA and protein expression of GLP-1R in liver tissue and in hepatocytes. Furthermore, we found that the inhibition of GLP-1R blocked the regulatory effect of Semaglutide on miR-5120/ABHD6 expression in FFA+HG-stimulated Hepa 1–6 cells. Therefore, we speculated that Semaglutide might alleviate hepatic steatosis by directly acting on GLP-1R in hepatocytes of T2DM and NAFLD mice, thereby activating its downstream pathway.
One previous study has reported that the suppression of ABHD6 can prevent diet-induced obesity and increase 1-MAG levels, enhancing adipose browning and brown adipose function through PPARa/γ activation.29 ABHD6 was significantly upregulated in the liver of HFD-fed mice, whereas ABHD6 knockdown significantly reduced the accumulation of hepatic TG.27 Our study indicated that the expression of ABHD6 in the T2DM+NAFLD group was significantly higher than that in the control group, and Semaglutide treatment could significantly reduce the expression of ABHD6 in the liver of T2DM+NAFLD mice. We speculate that Semaglutide may alleviate hepatic steatosis by reducing the expression of ABHD6 in the liver of NAFLD mice with T2DM.
A large number of abnormally expressed miRNAs have been found to be the underlying pathogenic factors of NAFLD, T2DM, and hyperlipidemia, and become novel targets for the treatment of metabolic diseases.31–33 Ng et al31 proposed that the inhibition of miRNA-24 expression in the liver of HFD-fed male C57Bl/6 mice resulted in the up-regulation of insulin-induced gene 1(Insig1), thereby decreasing hepatic lipid accumulation and hyperlipidemia. In addition, overexpression of miRNA-206 in the liver of obese mice promoted insulin signaling and inhibited hepatic lipogenesis by inducing degradation of protein tyrosine phosphatase, non-receptor type 1 (PTPN1).32
Using network bioinformatics tools, we predicted that miR-5120 could regulate the expression of ABHD6. Our data revealed for the first time that miR-5120 expression was significantly decreased in both the liver of T2DM+NAFLD mice and HG+FFA stimulated Hepa 1–6 cells compared with each control group, while Semaglutide significantly up-regulated miR-5120 expression in vivo and in vitro. Meanwhile, we found that the changes in miR-5120 expression in serum and cell supernatant were fully consistent with those in liver tissues and hepatocytes. We further demonstrated that miR-5120 mimics significantly inhibited the expression of ABHD6 in Hepa 1–6 cells, while miR-5120 inhibitors significantly increased the expression of ABHD6 in hepatocytes. Dual luciferase reporter genes indicated that Abhd6 3’UTR was directly regulated by miR-5120. Moreover, miR-5120 inhibitors blocked the effect of Semaglutide in reducing ABHD6 expression in HG+FFA-stimulated hepatocytes. Therefore, we speculate that Semaglutide may ameliorate hepatic lipogenesis through the regulation of ABHD6 expression by miR-5120.
Conclusions
In summary, Semaglutide could effectively alleviate hepatic steatosis, and might be expected to be a candidate for the intervention of T2DM accompanied by NAFLD. Moreover, we demonstrated for the first time that miR-5120 could regulate ABHD6 expression in hepatocytes, and Semaglutide could regulate ABHD6 expression in HG+FFA-stimulated hepatocytes through miR-5120. In addition, we also determined GLP-1R expression in hepatocytes, and found that Semaglutide directly regulated miR-5120/ABHD6 expression in hepatocytes by binding to GLP-1R. Therefore, we speculate that Semaglutide might ameliorate hepatic steatosis through the miR-5120/ABHD6 pathway. miR-5120/ABHD6 may become a potential therapeutic target for T2DM with NAFLD, and serum miR-5120 may also become a novel diagnostic marker for T2DM accompanied by NAFLD.
Abbreviations
NAFLD, non-alcoholic fatty liver disease; T2DM, type 2 diabetes mellitus; HG, high glucose; FFA, free fatty acid; ABHD6, α/β hydrolase domain-6; GLP-1RAs, glucagon-like peptide-1 receptor agonists; NASH, non-alcoholic steatohepatitis; GAN, Gubra-Amylin; DIO, diet-induced obese; MAG, monoacylglycerol; ASO, antisense oligonucleotide; HFD, high-fat diet; 1-MAG, 1-monoacylglycerol; PPARα, peroxisome proliferator-activated receptor alpha; PPARγ, peroxisome proliferator activated receptor gamma; STZ, streptozotocin; IPITTs, intraperitoneal insulin tolerance tests; OGTTs, oral glucose tolerance tests; ALT, alanine aminotransferase; AST, aspartate aminotransferase; TG, triacylglycerol; TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; DAPI, 4’,6-diamidino-2-phenylindole; RT, reverse transcription; H&E, hematoxylin and eosin; HRP, horseradish peroxidase; ECL, enhanced chemiluminescence assay; BCA, bicinchoninic acid; HEK, human embryonic kidney; WT, wild type; MUT, mutant; GEO, Gene Expression Omnibus; DEGs, the differentially expressed genes; SD, standard deviation; ANOVA, one-way analysis of variance; IR, insulin resistance; Col1a1, collagen type I alpha 1 chain; Col3a1, collagen type III alpha 1 chain; AMPK, 5’ AMP-activated protein kinase; PKA, protein kinase A; PTPN1, protein tyrosine phosphatase, non-receptor type 1; SIRT1, sirtuin.
Data Availability statement
All data and material have been presented in the manuscript. No outliers were omitted from the main statistical analysis. Related information is available under request to the corresponding author.
Ethics Approval
All animal experiments were approved by the Animal Ethics Committee of Tongji Hospital, Tongji University School of Medicine (2021-DW-(003)). This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication, 8th Edition, 2011), and Animal Research: Reporting of In Vivo Experiments guidelines. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Funding
This study was sponsored by the National Natural Science Foundation of China (Grant No. 82270877, 81974105), Cultivation project for National Natural Science Foundation of Shanghai Tongji Hospital.
Disclosure
The authors declare that they have no conflict of interest.
References
1. Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. Lancet. 2021;397(10290):2212–2224. doi:10.1016/S0140-6736(20)32511-3
2. Kwok R, Choi KC, Wong GL, et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: a prospective cohort study. Gut. 2016;65(8):1359–1368. doi:10.1136/gutjnl-2015-309265
3. Younossi ZM, Golabi P, de Avila L, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J Hepatol. 2019;71(4):793–801. doi:10.1016/j.jhep.2019.06.021
4. Negi CK, Babica P, Bajard L, Bienertova-Vasku J, Tarantino G. Insights into the molecular targets and emerging pharmacotherapeutic interventions for nonalcoholic fatty liver disease. Metabolism. 2022;126:154925. doi:10.1016/j.metabol.2021.154925
5. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol. 2018;53(3):362–376. doi:10.1007/s00535-017-1415-1
6. Gastaldelli A, Repetto E, Guja C, et al. Exenatide and dapagliflozin combination improves markers of liver steatosis and fibrosis in patients with type 2 diabetes. Diabetes Obes Metab. 2020;22(3):393–403. doi:10.1111/dom.13907
7. Unsal İO, Calapkulu M, Sencar ME, Cakal B, Ozbek M. Evaluation of NAFLD fibrosis, FIB-4 and APRI score in diabetic patients receiving exenatide treatment for non-alcoholic fatty liver disease. Sci Rep. 2022;12(1):283. doi:10.1038/s41598-021-04361-x
8. Shao N, Kuang HY, Hao M, Gao XY, Lin WJ, Zou W. Benefits of exenatide on obesity and non-alcoholic fatty liver disease with elevated liver enzymes in patients with type 2 diabetes. Diabetes Metab Res Rev. 2014;30(6):521–529. doi:10.1002/dmrr.2561
9. Xu F, Li Z, Zheng X, et al. SIRT1 mediates the effect of GLP-1 receptor agonist exenatide on ameliorating hepatic steatosis. Diabetes. 2014;63(11):3637–3646. doi:10.2337/db14-0263
10. Svegliati-Baroni G, Saccomanno S, Rychlicki C, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31(9):1285–1297. doi:10.1111/j.1478-3231.2011.02462.x
11. Saad ZA, Khodeer DM, Zaitone SA, Ahmed AAM, Moustafa YM. Exenatide ameliorates experimental non-alcoholic fatty liver in rats via suppression of toll-like receptor 4/NFκB signaling: comparison to metformin. Life Sci. 2020;253:117725. doi:10.1016/j.lfs.2020.117725
12. Cho YM, Fujita Y, Kieffer TJ. Glucagon-like peptide-1: glucose homeostasis and beyond. Annu Rev Physiol. 2014;76:535–559. doi:10.1146/annurev-physiol-021113-170315
13. Rahman K, Liu Y, Kumar P, et al. C/EBP homologous protein modulates liraglutide-mediated attenuation of non-alcoholic steatohepatitis. Lab Invest. 2016;96(8):895–908. doi:10.1038/labinvest.2016.61
14. Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387(10019):679–690. doi:10.1016/S0140-6736(15)00803-X
15. Yu X, Hao M, Liu Y, et al. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur J Pharmacol. 2019;864:172715. doi:10.1016/j.ejphar.2019.172715
16. Zhou JY, Poudel A, Welchko R, et al. Liraglutide improves insulin sensitivity in high fat diet induced diabetic mice through multiple pathways. Eur J Pharmacol. 2019;861:172594. doi:10.1016/j.ejphar.2019.172594
17. Capehorn MS, Catarig AM, Furberg JK, et al. Efficacy and safety of once-weekly semaglutide 1.0mg vs once-daily liraglutide 1.2mg as add-on to 1-3 oral antidiabetic drugs in subjects with type 2 diabetes (SUSTAIN 10). Diabetes Metab. 2020;46(2):100–109. doi:10.1016/j.diabet.2019.101117
18. Hedrington MS, Tsiskarishvili A, Davis SN. Subcutaneous semaglutide (NN9535) for the treatment of type 2 diabetes. Expert Opin Biol Ther. 2018;18(3):343–351. doi:10.1080/14712598.2018.1439014
19. O’Neil PM, Birkenfeld AL, McGowan B, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392(10148):637–649. doi:10.1016/S0140-6736(18)31773-2
20. Newsome PN, Buchholtz K, Cusi K, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384(12):1113–1124. doi:10.1056/NEJMoa2028395
21. Newsome P, Francque S, Harrison S, et al. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment Pharmacol Ther. 2019;50(2):193–203. doi:10.1111/apt.15316
22. Møllerhøj MB, Veidal SS, Thrane KT, et al. Hepatoprotective effects of semaglutide, lanifibranor and dietary intervention in the GAN diet-induced obese and biopsy-confirmed mouse model of NASH. Clin Transl Sci. 2022;15:1167–1186. doi:10.1111/cts.13235
23. Cao JK, Kaplan J, Stella N. ABHD6: its place in endocannabinoid signaling and beyond. Trends Pharmacol Sci. 2019;40(4):267–277. doi:10.1016/j.tips.2019.02.002
24. Poursharifi P, Madiraju SRM, Prentki M. Monoacylglycerol signalling and ABHD6 in health and disease. Diabetes Obes Metab. 2017;19(Suppl 1):76–89. doi:10.1111/dom.13008
25. Marrs WR, Blankman JL, Horne EA, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13(8):951–957. doi:10.1038/nn.2601
26. Marrs WR, Horne EA, Ortega-Gutierrez S, et al. Dual inhibition of alpha/beta-hydrolase domain 6 and fatty acid amide hydrolase increases endocannabinoid levels in neurons. J Biol Chem. 2011;286(33):28723–28728. doi:10.1074/jbc.M110.202853
27. Thomas G, Betters JL, Lord CC, et al. The serine hydrolase ABHD6 Is a critical regulator of the metabolic syndrome. Cell Rep. 2013;5(2):508–520. doi:10.1016/j.celrep.2013.08.047
28. Zhao S, Mugabo Y, Iglesias J, et al. α/β-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metab. 2014;19(6):993–1007. doi:10.1016/j.cmet.2014.04.003
29. Zhao S, Mugabo Y, Ballentine G, et al. α/β-hydrolase domain 6 deletion induces adipose browning and prevents obesity and type 2 diabetes. Cell Rep. 2016;14(12):2872–2888. doi:10.1016/j.celrep.2016.02.076
30. Yang X, Wu S, Feng Z, Yi G, Zheng Y, Xia Z. Combination therapy with semaglutide and rosiglitazone as a synergistic treatment for diabetic retinopathy in rodent animals. Life Sci. 2021;269:119013. doi:10.1016/j.lfs.2020.119013
31. Ng R, Wu H, Xiao H, et al. Inhibition of microRNA-24 expression in liver prevents hepatic lipid accumulation and hyperlipidemia. Hepatology. 2014;60(2):554–564. doi:10.1002/hep.27153
32. Wu H, Zhang T, Pan F, et al. MicroRNA-206 prevents hepatosteatosis and hyperglycemia by facilitating insulin signaling and impairing lipogenesis. J Hepatol. 2017;66(4):816–824. doi:10.1016/j.jhep.2016.12.016
33. Dang Y, Xu J, Zhu M, Zhou W, Zhang L, Ji G. Gan-Jiang-Ling-Zhu decoction alleviates hepatic steatosis in rats by the miR-138-5p/CPT1B axis. Biomed Pharmacother. 2020;127:110127. doi:10.1016/j.biopha.2020.110127
34. Bonizzi A, Piuri G, Corsi F, Cazzola R, Mazzucchelli S. HDL dysfunctionality: clinical relevance of quality rather than quantity. Biomedicines. 2021;9:7. doi:10.3390/biomedicines9070729
35. Jahn D, Kircher S, Hermanns HM, Geier A. Animal models of NAFLD from a hepatologist’s point of view. Biochim Biophys Acta Mol Basis Dis. 2019;1865(5):943–953. doi:10.1016/j.bbadis.2018.06.023
36. Tanaka K, Masaki Y, Tanaka M, et al. Exenatide improves hepatic steatosis by enhancing lipid use in adipose tissue in nondiabetic rats. World J Gastroenterol. 2014;20(10):2653–2663. doi:10.3748/wjg.v20.i10.2653
37. Zhou R, Lin C, Cheng Y, et al. Liraglutide Alleviates Hepatic Steatosis and Liver Injury in T2MD Rats via a GLP-1R Dependent AMPK Pathway. Front Pharmacol. 2020;11:600175. doi:10.3389/fphar.2020.600175
38. Panjwani N, Mulvihill EE, Longuet C, et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(-/-) mice. Endocrinology. 2013;154(1):127–139. doi:10.1210/en.2012-1937
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.