")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Self-nanomicellizing solid dispersion of edaravone: part I – oral bioavailability improvement
Authors Parikh A, Kathawala K, Tan CC , Garg S , Zhou XF
Received 8 January 2018
Accepted for publication 24 April 2018
Published 5 July 2018 Volume 2018:12 Pages 2051—2069
DOI https://doi.org/10.2147/DDDT.S161940
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Georgios Panos
Ankit Parikh, Krishna Kathawala, Chun Chuan Tan, Sanjay Garg, Xin-Fu Zhou
School of Pharmacy and Medical Sciences, University of South Australia, Adelaide, SA, Australia
Background: Edaravone (EDR) is known for its free radical scavenging, antiapoptotic, antinecrotic, and anticytokine effects in neurological and non-neurological diseases. It is currently available clinically as Radicava® and Radicut®, intravenous medications, recently approved for the treatment of amyotrophic lateral sclerosis and cerebral infarction. However, the oral use of EDR is still restricted by its poor oral bioavailability (BA) due to poor aqueous solubility, stability, rapid metabolism, and low permeability. The present study reports the development of novel EDR formulation (NEF) using self-nanomicellizing solid dispersion (SNMSD) strategy with the aim to enable its oral use.
Materials and methods: The selection of a suitable carrier for the development of NEF was performed based on the miscibility study. The optimization of EDR-to-carrier ratio was conducted via kinetic solubility study after preparing SNMSDs using solvent evaporation technique. The drug–polymer carrier interaction and self-nanomicellizing properties of NEF were investigated with advanced characterization studies. In vitro permeation, metabolism, and dissolution study was carried out to examine the effect of the presence of a carrier on physicochemical properties of EDR. Additionally, the dose-dependent pharmacokinetic study of NEF was conducted and compared with the EDR suspension.
Results: Soluplus® (SOL) as a carrier was selected based on the potential for improving aqueous solubility. The NEF containing EDR and SOL (1:5) resulted in the highest enhancement in aqueous solubility (17.53-fold) due to amorphization, hydrogen bonding interaction, and micellization. Moreover, the NEF demonstrated significant improvement in metabolism, permeability, and dissolution profile of EDR. Furthermore, the oral BA of NEF showed 10.2-, 16.1-, and 14.8-fold enhancement compared to EDR suspension at 46, 138, and 414 µmol/kg doses.
Conclusion: The results demonstrated that SNMSD strategy could serve as a promising way to enhance EDR oral BA and NEF could be a potential candidate for the treatment of diseases in which oxidative stress plays a key role in their pathogenesis.
Keywords: edaravone, Soluplus®, nanotechnology, oral bioavailability, metabolism, permeability
Introduction
Edaravone (EDR) is a well-established potent antioxidant and strong free radical scavenging drug, particularly in Japan since 2001 after receiving approval for acute ischemic stroke.1,2 Recently, it garnered global attention for gaining approval in the United States and Japan for amyotrophic lateral sclerosis (ALS) in years 2017 and 2015, respectively.3 Chemically, it is 3-methyl-1-phenyl-2-pyrazolin-5-one and belongs to the therapeutic class of central nervous system agents.4 The therapeutic activity of EDR is due to the removal of hydroxyl radical, modulation of inflammatory processes as well as neurodegenerative processes, apoptotic and necrotic cell death, nitric oxide production, and matrix metalloproteinase levels.1,2 The unique feature of EDR is access to the brain through blood–brain barrier due to its high lipophilicity and low molecular weight (174.203 g/mol); thus, its activity is not limited to vascular compartment.5 Its therapeutic potential against both neurological and non-neurological diseases was already proven from several in vitro and in vivo experiments including stroke,5–7 Alzheimer’s disease,8,9 epilepsy,10 pneumococcal meningitis,11 motor neuron disease,7,12,13 spinal cord injury,14 myocardial injury,15 liver injury,16 renal injury,17,18 lung injury,19 cardiac fibrosis and dysfunction,20 chronic nephropathy,18 and retinal diseases.21
The oral route of administration is the most preferred by patients considering their compliance and comfort. Accurate and flexible dosing, easy production, economic, and higher stability are the key reasons for most frequent use of oral route for drug administration.22 EDR was proved to be orally active against the Alzheimer’s disease8 and cerebral aneurysms.23 Moreover, Treeway, a biotech company, developed an unrevealed oral formulation of EDR to conduct clinical trials for ALS, which received orphan designation from regulatory agencies including European Medicines Agency (2014)24 and US Food and Drug Administration (USFDA) (2015).25 However, the current approved and available forms of EDR in the market are Radicava® and Radicut® (Mitsubishi Tanabe Pharmaceuticals, Osaka, Japan), the intravenous infusion solutions for parenteral administration. Rong et al26 reported the poor absolute oral bioavailability (BA) (5.23%) of EDR. Besides, it was designated as class IV drug as per Biopharmaceutics Classification System (BCS) due to its poor aqueous solubility (1.85±0.15 mg/mL) and permeability (Peff =3.18±0.0706×10−7 cm/s).26,27 EDR is recognized as a substrate of P-glycoprotein (Pgp). Thus, despite being highly lipophilic in nature, poor intestinal permeability was reported with EDR. Pgp plays a major role in limiting drug absorption by enhancing Pgp-mediated efflux and consequently reducing the oral BA of EDR.26 The poor oral BA was the key reason for the failure of a clinical trial of potential candidates against alzheimer disease such as curcumin.28,29 Therefore, there is a need to conduct the clinical trials for its safety and efficacy study at large scale for the development of EDR as a therapeutic for the treatment of diseases in which oxidative stress plays a key role in their pathogenesis. To improve the oral BA of EDR, the complexation of EDR with hydroxypropyl-sulfobutyl-β-cyclodextrin showed promising results by displaying a 10.3-fold improvement by improving its solubility, dissolution, and permeability.26 However, the impact on aqueous stability as well as rapid metabolism was not studied. Furthermore, its commercial translation was not realized possibly due to the high molecular weight of cyclodextrin, the slow process of complexation, and the unstable condition of processing media.30 Besides, the novel oral delivery system of EDR based on co-solvency and pH modification technology showed limited success by improving oral BA by 5.71-fold.27 Based on this, the liquid and solid lipid-based nanosystem (LNS) of EDR revealed 10.79- and 9.29-fold improvement in oral BA of EDR compared to EDR suspension, respectively, due to significant improvement in solubility, modulation of Pgp efflux pump, and inhibition of uridine 5-diphospho-glucuronosyl-transferase (UGT) enzymes.31 However, solid dispersion-based formulation showed improvement in solubility, stability, and BA compared to lipid-based formulations in case of clopidogrel napadisilate.32 Also, the issues associated specifically with self-microemulsifying drug delivery system could restrict its chronic use including high surfactant concentration, which causes gastrointestinal irritation and chemical instabilities of drugs.33 Hence, a novel oral formulation of EDR is necessary to offer a superior oral BA compared to currently available alternatives, suitability for chronic use, and have commercialization potential. Self-nanomicellizing solid dispersion (SNMSD) is the combination of widely accepted solid dispersions and nanomicelles’ strategies to improve the oral BA of challenging drugs. The dramatic improvement in oral BA of drugs including curcumin34,35 and ritonavir,36 belonging to BCS class IV using SNMSD strategy by improving solubility, stability, and intestinal permeability.27,31 Use of SNMSD strategy to enhance the oral BA of EDR is yet to be studied as per our knowledge. Thus, it was explored to develop novel EDR formulation (NEF). The work reported here aimed to develop NEF using the SNMSD strategy and assessed for its solubility, stability, dissolution, metabolism, permeability, and BA.
Materials and methods
Materials
EDR was purchased from Aladdin Industrial Corporation (Shanghai, China). Soluplus® (SOL) and CAVASOL® W7 HP PHARMA (hydroxypropyl-β-cyclodextrin [HP-β-CD]) were gifted by BASF Australia Ltd (Melbourne, VIC, Australia) and Chemiplas Australia Pty Ltd (Melbourne, VIC, Australia), respectively. Potassium pyrophosphate, Tyrode’s solution, ascorbic acid, sodium carboxymethylcellulose, polyvinylpyrrolidone K 30 (PVP K30), polyethylene glycol (PEG) 4000, poloxamer 188, poloxamer 407, concentrated hydrochloric acid, perchloric acid, formic acid, disodium hydrogen phosphate, citric acid, uridine diphosphate glucuronic acid (UDPGA) trisodium salt, alamethicin, D-glucaric acid 1,4-lactone, and DMSO-d3 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Acetic acid, phosphoric acid, sucrose, magnesium chloride, and sodium hydroxide pellets were obtained from Chem-Supply (Gillman, SA, Australia), boric acid was obtained from Optigen Scientific (Port Adelaide, SA, Australia), saline was obtained from Pfizer, Inc. (New York, NY, USA), and Pierce BCA Protein Assay Kit was obtained from Thermo Fisher Scientific (Waltham, MA, USA). High-performance liquid chromatography (HPLC) grade ethanol and methanol were purchased from EMD Millipore (Billerica, MA, USA). High-purity water using a Milli-Q water purification system (Millipore Ultrapure Water System; EMD Millipore) was used throughout the study. All other reagents were of analytical grade.
Formulation development and optimization of NEF
Selection of a suitable carrier for the development of NEF
The carrier selection was performed based on the miscibility study as described previously.37 The solutions of polymeric carriers including SOL, HP-β-CD, PVP K30, PEG 4000, poloxamer 407, and poloxamer 188 were prepared at different concentrations including 2.5, 5, 7.5, and 10% (w/v) using water. An excess amount of EDR was added to each polymeric solution in glass vials and allowed to shake in a mechanical shaker (Axyos Technologies, Brisbane, QLD, Australia) for 24 h with 150 rpm at ambient temperature.27 Subsequently, each sample was filtered through 0.45 μm polyvinyl difluoride (PVDF) syringe filter and diluted with appropriate dilution by mobile phase. For the assay of EDR, the analytical method from the Japanese Pharmacopeia was used as per the previous study with minor modification.75 The analysis of samples was conducted using the ultraviolet (UV)–visible spectrophotometer detector – HPLC method with a mobile phase consisting of water, methanol, and acetic acid (100:100:1) with 1 mL/min flow rate and 20 μL injection volume at 240 nm.27
Preparation of SNMSD using solvent evaporation (SE) technique
SE method was used to prepare the SNMSDs of EDR and SOL, a selected polymer from miscibility study, as described by Wang et al.38 EDR and SOL at the ratio of 1:1, 1:2.5, 1:5, 1:7.5, 1:10, 1:12.5, and 1:15 were dissolved in ethanol. Subsequently, the mixture was allowed to evaporate under the vacuum (500–600 mbar) using a rotatory evaporator at 55°C–60°C. SNMSDs were then scraped using a spatula, pulverized in the mortar-pestle and passed through 250 μm sieve. All prepared SNMSDs were kept overnight in the desiccator for drying and stored in a refrigerator at 4°C until use for further studies.
Kinetic solubility study
The selection of SNMSD as a NEF was based on the kinetic solubility study. SNMSDs or EDR was added in an excess amount to 2 mL of simulated intestinal fluid (SIF), pH 6.8, in glass vials.39 The solubility study was conducted as described previously, and samples were collected at predetermined time intervals.27
Characterization of NEF
Comparison of solubility of NEF with unformulated EDR and physical mixture (PM)
To prepare PM, EDR and SOL in the ratio of 1:5 were mixed using pestle mortar until a homogeneous mixture was obtained and passed through 250 μm sieve.40 The improvement in aqueous solubility of EDR with PM and NEF was investigated in simulated body fluids (SBFs) such as simulated gastric fluid (SGF) (pH 1.2), SIF (pH 6.8 and pH 7.4), and water as described previously and compared with the EDR.27
Determination of active content in solid dispersion
The NEF equivalent to 1 mg of EDR was dissolved in 1 mL of methanol. The solution was further analyzed by using previously developed HPLC method. The loading ability and loading efficiency were determined based on the weight ratio of EDR to SOL in NEF and the amount of EDR loaded in the NEF from the initial value used for the preparation, respectively.41
Investigating drug–polymer interaction with advanced characterization
Differential scanning calorimetry (DSC) study
DSC analysis was carried out by using Discovery DSC (Model 2920; TA Instruments, New Castle, DE, USA). DSC was performed for the samples including EDR, SOL, PM, and NEF. The samples (2–4 mg) were taken in the aluminum open crucible, and an empty crucible was used as blank. Thermograms of each sample were recorded at the heating rate of 10°C/min, in the temperature range of 40°C–250°C under the flow (50 mL/min) of nitrogen gas.31,39
X-ray diffraction (XRD) study
XRD patterns were recorded for the samples including EDR, SOL, PM, and NEF to study the solid characteristics. The XRD instrument (PANalytical, Empyrean X-ray diffractometer) was used, and diffractograms were taken by using CuKα radiation (λ=1.5418 Å), 40 kV voltage, and 40 mA current. The scanning rate was 2° per minute in the 2θ of 2°–50°.31,39
Scanning electron microscopy (SEM) study
Surface morphology was studied by using an instrument, Zeiss Microscopy Merlin with GEMINI II column. SEM study was carried out for the samples including EDR, SOL, and NEF. Each sample was mounted on the double-sided adhesive tape. The photomicrographs were obtained at the voltage of 0.7 kV and examined at the magnification of 2,000×.31,39
Fourier transform infrared (FTIR) spectroscopy study
FTIR spectroscopy was carried out using a conventional potassium bromide (KBr) pellet method and a PerkinElmer Spectrum 400 spectrometer. The study was performed for the samples including native EDR, SOL, PM, and NEF. Briefly, 2–4 mg of samples were mixed with 100–150 mg of KBr and made pellet using the pressure of 8 tonnes for 10–15 s. The KBr pellet was placed in the sample holder. FTIR spectra were obtained between 400 and 4,000 cm−1 at the scanning rate of 2 cm−1.38
1H nuclear magnetic resonance (NMR) study
Bruker AVANCE III 500 MHz was used to record the 1H NMR spectra at the frequency of 500 MHz. The samples including EDR and NEF were prepared by dissolving in DMSO-d3 followed by transferring to thin 5 mm diameter tubes prior to measurement. The data analysis was performed using the Topspin 3.2 software.42
Assessment of self-nanomicellizing properties of NEF
Preparation of EDR-loaded micelles solution of NEF
An accurate quantity of NEF (equivalent to 15 mg EDR) was dispersed in 10 mL of water and kept on the magnetic stirrer for stirring at 500 rpm up to 3 h. The resultant mixture was then filtered through 0.45 μm PVDF filter to get the homogeneous micellar solution and to remove the unloaded EDR.43
Characterization of EDR-loaded micelles
The Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) was used to measure the particle size, polydispersity index (PDI), and zeta potential of the above prepared micellar solution based on the principle of photon correlation spectroscopy. In addition, a Philips CM 100 Transmission Electron Microscope fitted with an SIS MegaView II CCD camera was used to determine morphological characteristics using transmission electron microscopy (TEM) analysis. Briefly, a tiny drop of micellar solution was kept on the copper grid followed by drying overnight and analyzed by the Image Analysis Software.
To determine drug loading and entrapment efficiency of micelles, EDR-loaded micellar solution was freeze-dried. Before freeze drying, the micellar solution was kept in a deep freezer maintained at −80°C for 24 h (MDF-U74V-PE; Panasonic Healthcare Co., Ltd., Tokyo, Japan). The sample was then freeze-dried using lyophilizer (Labconco Corporation, Kansas City, MO, USA) at −44°C and 8×10−3 M bar for 36 h. The freeze-dried powder was further dissolved in methanol and analyzed by using previously developed HPLC method. The percentage of drug loading and percentage of entrapment efficiency were determined based on the weight ratio of EDR in micelles to total mass of micelles and the amount of EDR loaded in micelles from the initial mass of EDR used for the preparation of micelles, respectively.43
Stability study
Stability study of EDR-loaded micelles was performed at various stability conditions including hydrolytic, thermal, and photolytic, up to 24 h as described previously.27 For hydrolytic condition, 2 mL of each solution (pH 2–10) was filled in the glass bottle and covered with aluminum foil to protect from light and kept at 25°C. The solution was kept under various conditions, which included 4°C, 25°C/60% relative humidity (RH), and 40°C/75% RH for determining the stability against temperature. The EDR micellar solution was kept in the KBF ICH 720 (E2) – a constant climate chamber as per the International Council for Harmonization (ICH) guideline to determine the stability when exposed to light. The samples were taken out at predetermined time points and analyzed with HPLC.27
Physical stability study of NEF as per ICH guideline
The stability study of NEF was carried out by evaluating the percentage of drug content and aqueous solubility up to 8 weeks as per ICH guideline.27 The stability conditions include 4°C, 25°C/60% RH, and 40°C/75% RH. The NEF was filled in the glass vials, sealed, and stored in the stability chamber after covering with aluminum foil. The aliquots were collected at predetermined time intervals and analyzed by HPLC method.
In vitro permeation and metabolism study of NEF
In vitro permeation and metabolism study were carried out as per our previous study.27,31 Male Wister rats (180–200 g) were used to prepare everted sacs of gut for the assay. Each sac was filled with 2 mL of Tyrode’s solution having the EDR concentration of 287.02 μmol with or without borneol (1.30 mmol) and NEF (equivalent to EDR concentration 287.02 μmol). The amount of EDR and EDR glucuronide (EDR-G) metabolites and the rate of permeation were determined and compared.
In vitro dissolution study
A dissolution study was performed for EDR and NEF by using United States Pharmacopeia (USP) type II (paddle) apparatus at 37°C and 50 rpm.34 EDR and NEF were placed in the dissolution flask containing 900 mL of dissolution media including SGF (pH 1.2) and SIF (pH 6.8). The samples were collected and replaced with an equal volume of the dissolution media at different time points 5, 15, 30, 45, 60, 90, and 120 min. Each sample was further filtered through 0.45 μm PVDF syringe filters followed by dilution if required with mobile phase. The amount of the drug released at each time point was determined by previously developed HPLC method. SNMSD could be categorized under controlled release formulation; the use of various mathematical models in describing drug release from solid dispersion including zero-order, first-order, Higuchi, Hixson–Crowell, and Korsmeyer–Peppas models was well recognized and reported.44–47 Specifically, a solid dispersion using SOL could improve dissolution due to solid-state transformation as well as micellization. The use of zero-order, first-order, Higuchi, Hixson–Crowell, and Korsmeyer–Peppas models has been successfully described for drug release from SOL-based solid dispersion.47,48 Thus, the results obtained from the HPLC analysis were fitted into the different release kinetic models including first order, zero order, Hixson–Crowell, Higuchi matrix, and Korsmeyer–Peppas to determine the release kinetic. In addition, the release profile of native EDR and NEF was compared by calculating the dissimilarity (f1) and similarity (f2) factors to understand the impact of pH on release. The formulas to determine the best suitable release kinetic model and dissimilarity and similarity factors are mentioned in Table S1. Dissolution profiles are considered to be similar if the f1 and f2 values are <15 and 50–100, respectively.
In vivo pharmacokinetic study
The animal study was carried out as per the previous report using the male Sprague Dawley rats (300±25 g) and was approved by the Animal Ethics Committee of the University of South Australia under South Australian Animal Welfare Act 1985.27 The dose-dependent pharmacokinetic study of NEF was conducted and compared with the EDR suspension prepared using 0.5% sodium carboxymethylcellulose. Based on our previous publications, the standard deviation (SD) was determined within the 20% of mean. Thus, the sample size n=6 was calculated for the detection of difference of 25% between groups at the β level of 80% and the α level of 5%.27,31 Four groups of rats (n=6) were orally administered with the EDR suspension of 172 μM/kg and NEF equivalent to 46, 138, and 414 μM/kg of EDR. The plasma samples have been collected at the predetermined time interval of 0, 5, 15, 30, 45, 60, 90, 12, 180, 240, 300, 360, 420, and 480 min. EDR was extracted from the plasma by using the mixture of Mcllvaine buffer (pH 5.4) and dichloromethane-n-pentane (3:7, v/v) as mentioned in our study.27 EDR concentration in each sample was measured using the liquid chromatography-tandem mass spectrometry method, and Phoenix WinNonlin software was used to calculate the pharmacokinetic parameters.27 The study has been conducted for 8 h as the level of EDR was below detection limit after 8 h.
Statistical analysis
Mean and SD were used to present all values. The statistical analysis of data was conducted using GraphPad Prism 6. Data were evaluated for normal distribution first using Shapiro–Wilk test. Based on the results, further analysis was performed using Student’s t-test for two groups, or one-way or two-way analysis of variance (ANOVA) for multiple groups. P-values <0.05 were considered statistically significant.
Results and discussion
Development of formulation and optimization of NEF
Poor solubility, dissolution, metabolism, and permeability are determined as challenges that resulted in poor oral BA of EDR.27 The SNMED strategy is the combination of solid dispersion and nanomicelles strategies, which are the part of the most exciting area to curb the issues that restrict drug’s absorption and displayed a potential for overall improvement in the BA. It is the system where drug molecule disperses in the polymeric carrier similar to solid dispersion-based formulation.49,50 The role of polymeric carrier is critical for drug solubilization, stabilization, improving dissolution, permeability, and absorption.34,51 Therefore, miscibility study was used to finalize the most suitable polymer for the development of NEF. Furthermore, the ratio of drug to carrier was chosen based on the kinetic solubility study. Also, the solubility of optimized NEF was compared with a PM of EDR and carrier with the similar amount used in NEF to evaluate the impact of the presence of polymeric carrier on the solubility profile of EDR.
Selection of suitable carrier for the development of NEF
A number of carriers were shortlisted from the literature based on their potential to overcome the hurdles in the development of a novel oral delivery system such as PVP K30, SOL, PEG 4000, poloxamer 188, and poloxamer 407.38,39 In addition to abovementioned polymers, HP-β-CD was considered as a control based on its proven ability to improve the solubility and dissolution of EDR.52 The reported aqueous solubility of EDR was 1.85±0.15 mg/mL.27 The increment in the aqueous solubility of EDR was observed with increasing the percentage of polymer content (w/v). The regression coefficient (R2) values for PVP K30, SOL, PEG 4000, HP-β-CD, poloxamer 188, and poloxamer 407 are 0.997, 0.994, 0.951, 0.971, 0.917, and 0.989, respectively. The polymers such as PVP K30 and SOL suggested the best linear relationship in the enhancement of solubility with the polymer concentration compared to other polymers. Also, 10% SOL showed 4.67-fold improvement in the aqueous solubility of EDR, whereas with the same concentration of HP-β-CD, PVP K30, PEG 4000, poloxamer 188, and poloxamer 407 presented 3.49-, 2.22-, 1.73-, 1.74-, and 2.14-fold enhancement, respectively (Figure 1A). The solubility enhancement of EDR with SOL is statistically significantly greater than HP-β-CD, which justified the selection of SOL for the development of NEF.
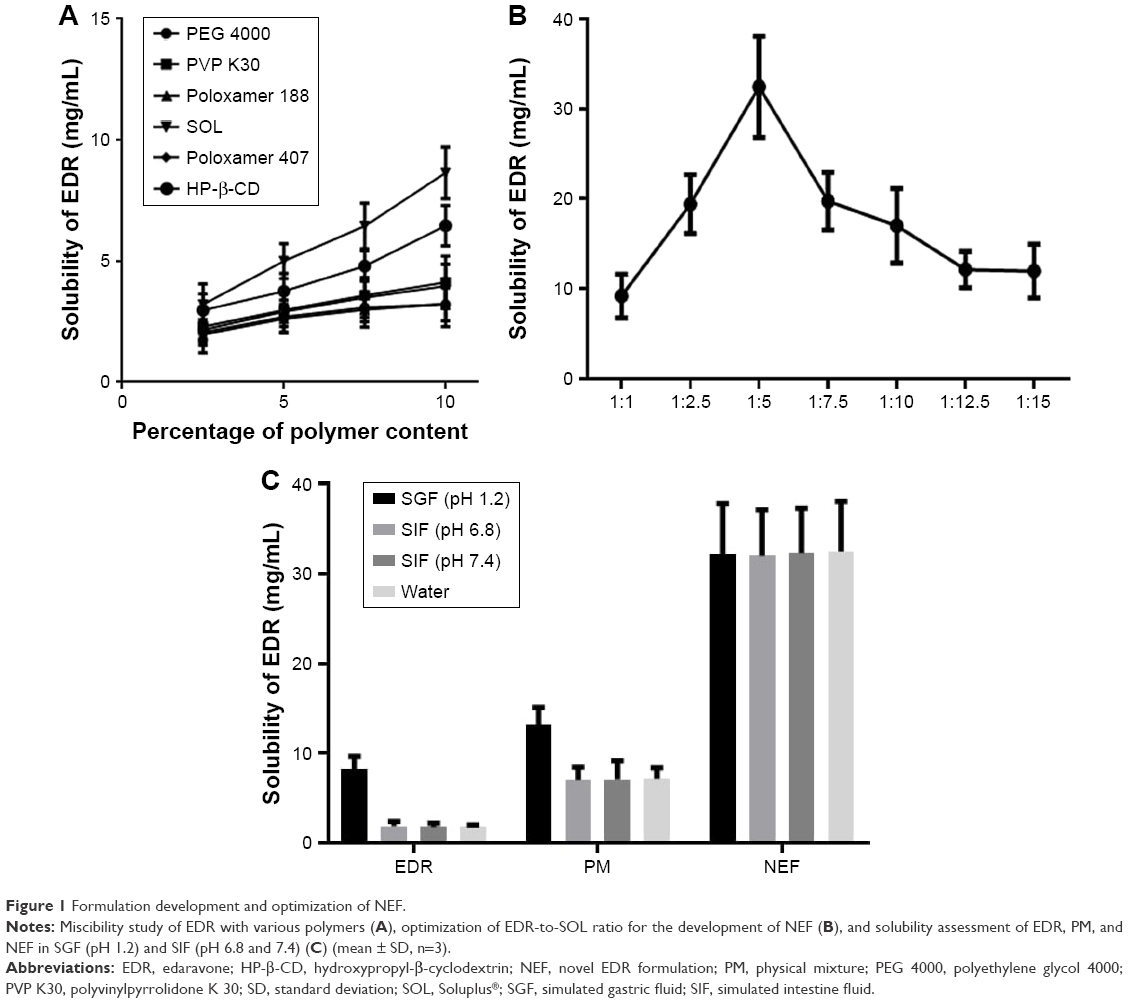 | Figure 1 Formulation development and optimization of NEF. |
There are a number of reasons based on the literature and experiments which might explain the superior performance of SOL over another carrier including hydrogen bonding interactions with EDR as it has two hydrogen donor and two acceptor groups while EDR has two hydrogen donor groups and ability to form solid solution with EDR, which makes EDR available in a dissolved state,53 can act as a precipitation inhibitor, keeps the supersaturated condition for at least 24 h based on the results of kinetic solubility study,39 and can favor micellar solubilization.39
Kinetic solubility study
The ratio of EDR to SOL was optimized based on the kinetic solubility study. The maximum and equilibrium solubility and time for precipitation were determined for each ratio. The SD showing highest solubility without precipitation was considered for the development of NEF.39 The crystalline EDR displayed equilibrium aqueous solubility and maximum solubility (1.85±0.15 mg/mL27 and 2.29±0.32 mg/mL, respectively). Additionally, the precipitation was seen after 30 min, which could be a reason for the difference in the values of maximum solubility and equilibrium solubility. With a good agreement to miscibility study, the solubility of EDR was significantly enhanced with the increasing concentration of SOL in up to 1:5 EDR-to-SOL ratio. Approximately 5- and 11-fold improvements in aqueous solubility were observed with SDs having 1:1 and 1:2.5 ratios, while the SD with 1:5 ratio showed the highest solubility enhancement (17.5-fold) compared to all ratios used for the optimization study (Figure 1B). Interestingly, lower solubility was detected with 1:7.5, 1:10, 1:12.5, and 1:15 ratios compared to 1:5 ratio but significantly higher in comparison with crystalline EDR with ~11-, 9-, 6.6-, and 6.5-fold, respectively. The decreasing solubility after certain drug-to-polymer ratio could be due to reaching beyond supersaturation stage, which was also observed in the previous study with SOL, PEG 4000, and PVP K30 polymers.38 No precipitation was noticed with all ratios, suggesting the critical role of SOL as a precipitation inhibitor by maintaining supersaturation condition for the desired period. The binary solid dispersion system of EDR and SOL with 1:5 ratio was considered as an NEF.
Characterization of NEF
Comparison of solubility of NEF with unformulated EDR and PM
There was a significant enhancement in aqueous solubility of EDR observed with PM (3.88-fold) and NEF (17.53-fold) (Figure 1C). The NEF displayed 4.51-fold greater solubility improvement compared to PM. Partial amorphization could be a potential reason for the low solubility of EDR with PM compared to NEF.54 The solubility of EDR in SGF (pH 1.2) was significantly greater (4.46-fold), while SIF (pH 6.8 and 7.4) showed an insignificant difference compared to solubility in water. The PM and NEF showed significant enhancement in the solubility of EDR in all SBFs. The exceptional performance of NEF was further investigated with advanced characterization study of NEF.
Determination of active content in solid dispersion
NEF is an amorphous solid dispersion, which can be defined as a molecular mixture of poorly water-soluble drugs and hydrophilic carriers.55 The manufacturing process of solid dispersion should ideally allow entire amount of EDR to be mixed with SOL to form NEF. The drug loading and efficiency as applied to other formulations such as nanoparticles will be not applied with solid dispersion-based formulations. To determine the proportion of EDR in NEF to understand the loss of EDR in manufacturing, we have used methanol technique. The proportion of EDR in NEF was 16.52%±0.16% (w/w), which is 99.03%±0.42% of the initial amount. No significant loss of EDR was observed during the manufacturing by SE technique.
Investigating drug–polymer interaction with advanced characterization
Change in solid-state characteristics from crystalline to amorphous, hydrogen bonding interaction, and micellization are perhaps potential reasons for the dramatic improvement in EDR aqueous solubility with NEF.31 To evaluate our hypothesis, DSC, XRD, and SEM studies were conducted to determine if any change happened in solid-state characteristics of EDR and SOL when compared to optimized NEF.31,39 Also, the possibility of hydrogen bonding between EDR and SOL was assessed from FTIR and 1H NMR analyses.41 The micellization property of NEF was studied by evaluating its aqueous solution for the parameters such as particle size, zeta potential, and PDI by zetasizer and TEM analyses.56
DSC study
The thermal behavior of EDR, SOL, PM, and NEF is shown in Figure 2A. The characteristic melting endothermic peak at 128.39°C was recorded with pure EDR, which confirmed the crystalline nature of EDR used in the preparation of developing NEF. SOL did not display any thermal event due to amorphous nature of the polymer reported in the literature.57 Less intense endothermic peak was seen in the case of PM, which might be due to the partial amorphization because of the force used in mixing during preparation with mortar and pestle or dilution effect of SOL. The disappearance of the sharp melting endothermal peak of EDR in the thermogram of NEF revealed the complete alteration of the solid state of EDR from crystalline to amorphous.
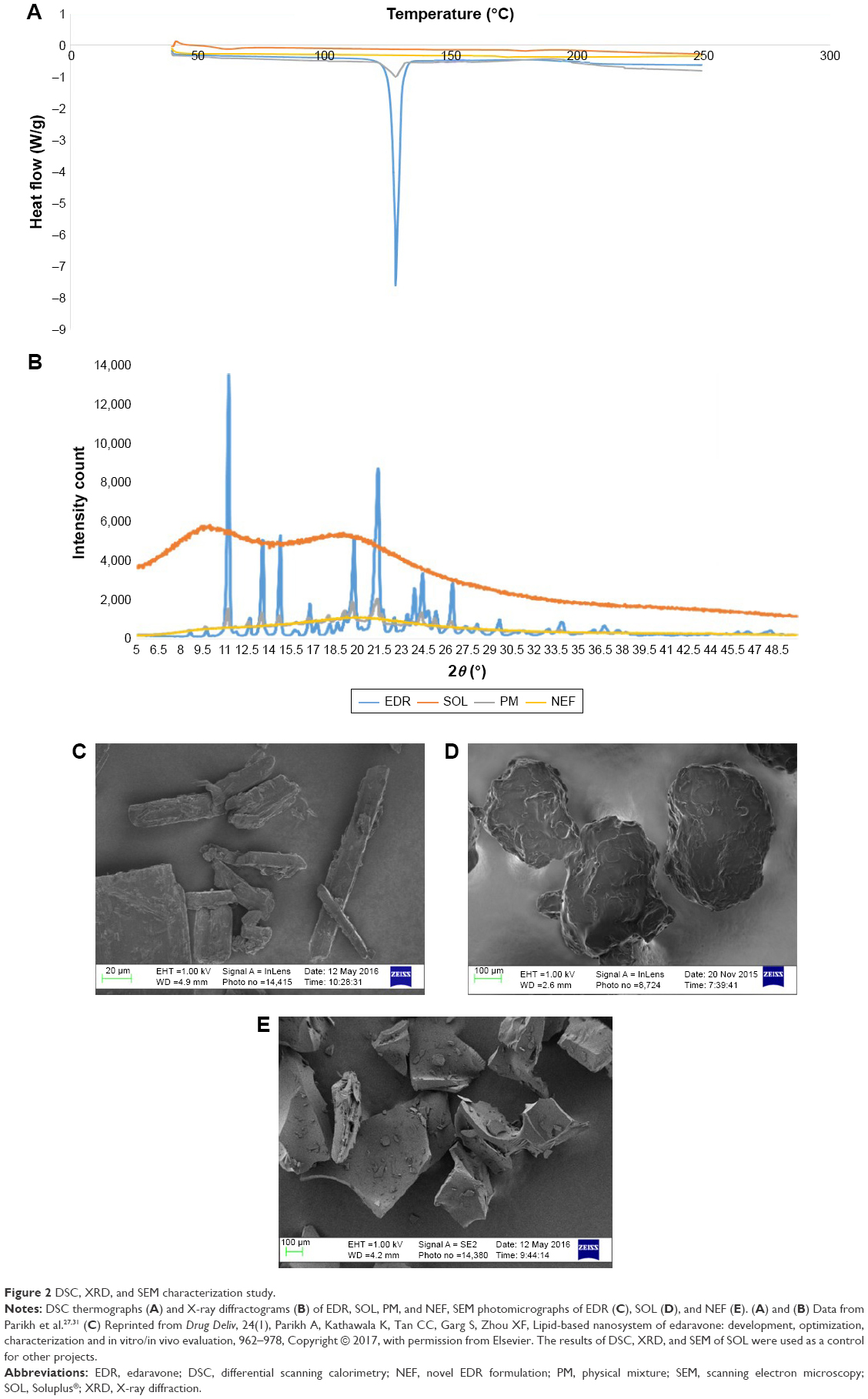 | Figure 2 DSC, XRD, and SEM characterization study. |
XRD study
The solid-state characteristic of NEF was further confirmed with XRD study by comparing diffractograms of pure EDR, SOL, PM, and NEF. Pure EDR showed sharp characteristics peaks at 11.2, 13.7, 14.7, 20.1, and 21.4 2θ assuring its crystalline nature, whereas amorphous nature of SOL was confirmed from its diffractogram without any characteristic peaks (Figure 2B). The decrease in the intensity of all characteristic peaks of pure EDR in the diffractogram of PM verified the results of partial conversion to amorphous or dilution effect of SOL from DSC study (Figure 2B). However, diffractogram of NEF exhibited apparently absent of all peaks of pure EDR, which indicated complete amorphization of EDR in NEF (Figure 2B). SOL has been previously shown to have the unique capability of interacting with the drug molecules via H-bonds as it has two each of hydrogen donor and acceptor groups in each chain.58 SOL could interact with the EDR crystal surfaces, especially with the 2θ values of 11.2, 13.7, 14.7, 20.1, and 21.4° and disruption of the crystal lattice and order.59 The results of XRD analysis show a good agreement with the DSC study.
SEM study
The SEM analysis was performed to determine the surface micrographs of pure EDR, SOL, and NEF. The large agglomerate particles with ordered size and shape were observed in the micrograph of pure EDR, which indicated its crystalline nature (Figure 2C). Moreover, irregular-shaped particles were seen in the micrograph of SOL revealing its amorphous nature (Figure 2D). Furthermore, the micrograph of NEF demonstrated particles with relatively rough surface and disordered shape, which specified the amorphous nature of EDR in NEF without any traces of crystalline EDR (Figure 2E). The surface characteristics of NEF facilitate the quick and efficient dissolution of NEF upon contact with aqueous fluids.60 The confirmation of complete amorphization of EDR in NEF from SEM results is consistent with the results of DSC and XRD analyses.
FTIR and 1H NMR study
The hydrogen bond interaction could be possible between EDR and SOL due to the presence of hydrogen donor and acceptor chemical groups in their chemical structure.27,43 Therefore, the interaction between EDR and SOL was investigated using FTIR and 1H NMR. FTIR spectra of pure EDR, SOL, PM, and NEF are displayed in Figure 3A–D, respectively. Pure EDR (Figure S1) showed peaks at 3,200–3,600 cm−1 corresponding to N−H and O−H stretching, 1,580–1,627 cm−1 in regard to C=N stretching, and 2,850–3,100 for aliphatic and aromatic C−H stretching. SOL displayed peaks representing O−H stretching and aliphatic C−H stretching, which were similar to EDR. The stretching corresponding to carbonyl group was additionally observed at 1,670–1,820 cm−1 in SOL spectra. The shifting or broadening of peaks between 3,200 and 3,600 cm−1 was witnessed in FTIR spectra of NEF, which indicated the involvement of chemical groups present in EDR and SOL such as N−H and O−H in hydrogen bonding. Additionally, a similar observation was seen between 1,670 and 1,820 cm−1 that confirm the participation of carbonyl group of SOL in hydrogen bonding. Shifting of chemical shifts was also observed in the results of 1H NMR of NEF compared to pure EDR, which could be considered as a proof of hydrogen bonding interaction between the drugs and polymer as per the previous publication (Figure 3E and F and Table S2).41 SOL has two each of hydrogen donor and acceptor groups while EDR has two hydrogen donor groups. As the optimized ratio of EDR to SOL for the development of NEF is 1:5, five times more SOL molecules are present in the NEF, which could facilitate six hydrogen bonding interactions. From the results of FTIR and NMR studies, the strong hydrogen bonding interaction between EDR and SOL was confirmed, which could interact with the EDR crystal surfaces, especially with the 2θ values of 11.2, 13.7, 14.7, 20.1, and 21.4 and disruption of the crystal lattice and order. The formation of hydrogen bond between drug and polymer in preparation could restrict the solid-state change from amorphous to crystalline during the storage and provide physical stability to the formulation.61 Similar results have been reported previously in solid dispersion-based preparations.41,60
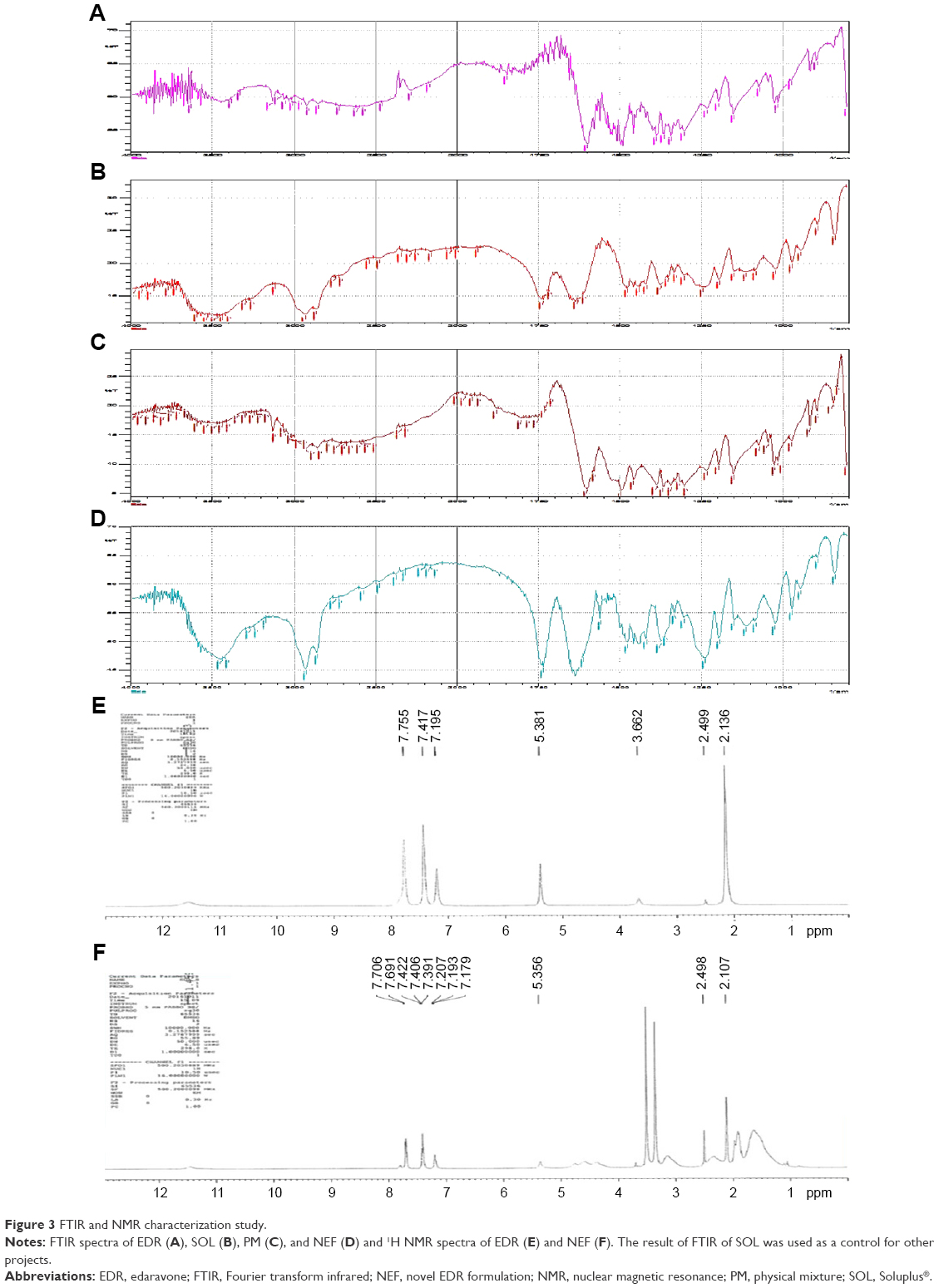 | Figure 3 FTIR and NMR characterization study. |
Assessment of self-nanomicellizing properties of NEF
SOL, a graft copolymer of poly(vinyl caprolactam)–poly(vinyl acetate)–poly(ethylene glycol), could form micelles over the concentration of 7.6 mg/L.56 The micellar system of SOL showed dramatic improvement in oral BA of drugs such as quercetin56 and cyclosporine A43 and the therapeutic efficacy of drugs such as doxorubicin,62 α-lipoic acid,63 and carvedilol.64 Thus, the self-nanomicellizing properties of NEF after dissolving NEF in aqueous media were investigated. The particle size is the most critical parameter as <100 nm could enhance cellular uptake of drugs and also facilitate the transport across the intestinal membrane by paracellular or transcellular routes.56 The average particle diameter of 73.46±3.15 nm, the PDI of 0.12±0.04, and the zeta potential of −4.98±1.56 mV were observed using zetasizer. A TEM photograph of micelles of NEF revealed the spherical droplets with the good agreement of particle size measured by zetasizer (Figure 4A). Additionally, micellar formulation remained stable with and without 20-fold dilution with dissolution media such as SGF (pH 1.2) and SIF (pH 6.8) after 24 h. The narrow size distribution, which prevents particle growth due to Ostwald ripening, could also justify the good stability of micelles in dissolution media.65 The percentage of drug loading and percentage of entrapment efficiency of micelles were 0.143 and 95.46, respectively.
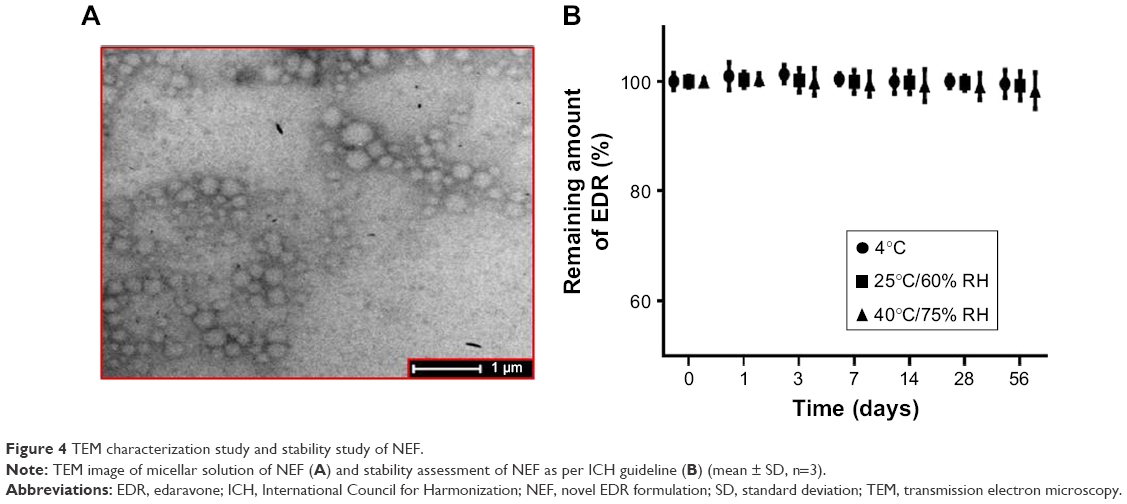 | Figure 4 TEM characterization study and stability study of NEF. |
Moreover, the uniform dispersion of NEF in various aqueous buffer systems from pH 2 to 8, SBF, and water confirmed nearly complete encapsulation of EDR in micelles. The complete encapsulation could provide protection to EDR against the degradation or metabolism in the presence of gastrointestinal contents.66 Thus, the stability of EDR in micelles at different pH and temperatures, in the presence of light, and in SBF such as SGF (pH 1.2) and SIF (pH 6.8) was examined for 24 h. There was no significant degradation (<3% degradation) of EDR that was detected in all conditions, while crude EDR showed dramatic degradation at alkaline conditions (pH 8–10).27 These results confirmed the complete amorphization, strong hydrogen bonding interaction, and micellization, which made NEF an excellent candidate for further development.
Physical stability study of NEF as per ICH guideline
No significant difference in the percentage of drug content and the aqueous solubility of EDR was witnessed (Figure 4B). Therefore, it might be stored at ≤40°C for at least 8 weeks. The strong hydrogen bond interaction between EDR and SOL could favor the physical stability of EDR in NEF and prevent the conversion of solid state from amorphous to crystalline.61
In vitro metabolism and permeation study
EDR was previously recognized as a substrate of UGT enzymes.26 Thus, it rapidly undergoes extensive metabolism, which results in poor oral BA.27,31 The inhibition of glucuronidation is the critical requirement in improving the oral BA of EDR. Borneol is known for its inhibitory effect on glucuronidation and considered as a positive control in investigating the inhibitory effect of excipients on glucuronidation.67 Thus, similar to our previous studies, borneol was considered as a positive control in the current study.27,31
The novel oral delivery system and LNS of EDR showed 5.71- and 10.79-fold improvements in the oral BA of EDR, respectively, due to the inhibitory effect of surfactants on glucuronidation and the Pgp efflux of EDR during metabolism and permeability.27,31 Based on such information, the impact of SOL on the glucuronidation of EDR was studied in rat liver microsomes. The significant inhibitory effect of SOL (45.64%) compared to borneol (37.56%) was observed (Figure 5A). The potential of SOL-based NEF was further tested on the permeability and metabolism of EDR during permeation across the small intestine using the everted intestine sac technique. The inhibitory effect of SOL was also witnessed as NEF displayed 2.73-fold greater transfer amount of EDR compared to crude EDR and statistically significant compared to borneol (Figure 5B). Additionally, NEF presented 41.67 and 15% less amount of EDR-G on the serosal side compared to crude EDR and borneol, respectively (Figure 5C). Moreover, it presented 59.9% and 44.2% fewer molar ratio of EDR-G to EDR compared to crude EDR and borneol, respectively, which indicated a statistically significant effect of SOL present on the NEF glucuronidation of EDR (Figure 5D). There were 2.96-fold higher transfer rate of EDR from NEF and 2.65-fold greater with borneol compared to crude EDR (Figure 5E). The inhibitory effect of SOL on Pgp efflux and <100 nm of micelles generated from NEF could also enhance the permeability of EDR across the small intestine.56 The results confirmed the significant improvement in the metabolism and permeability profile of EDR with NEF due to the inhibitory effect of SOL on glucuronidation.
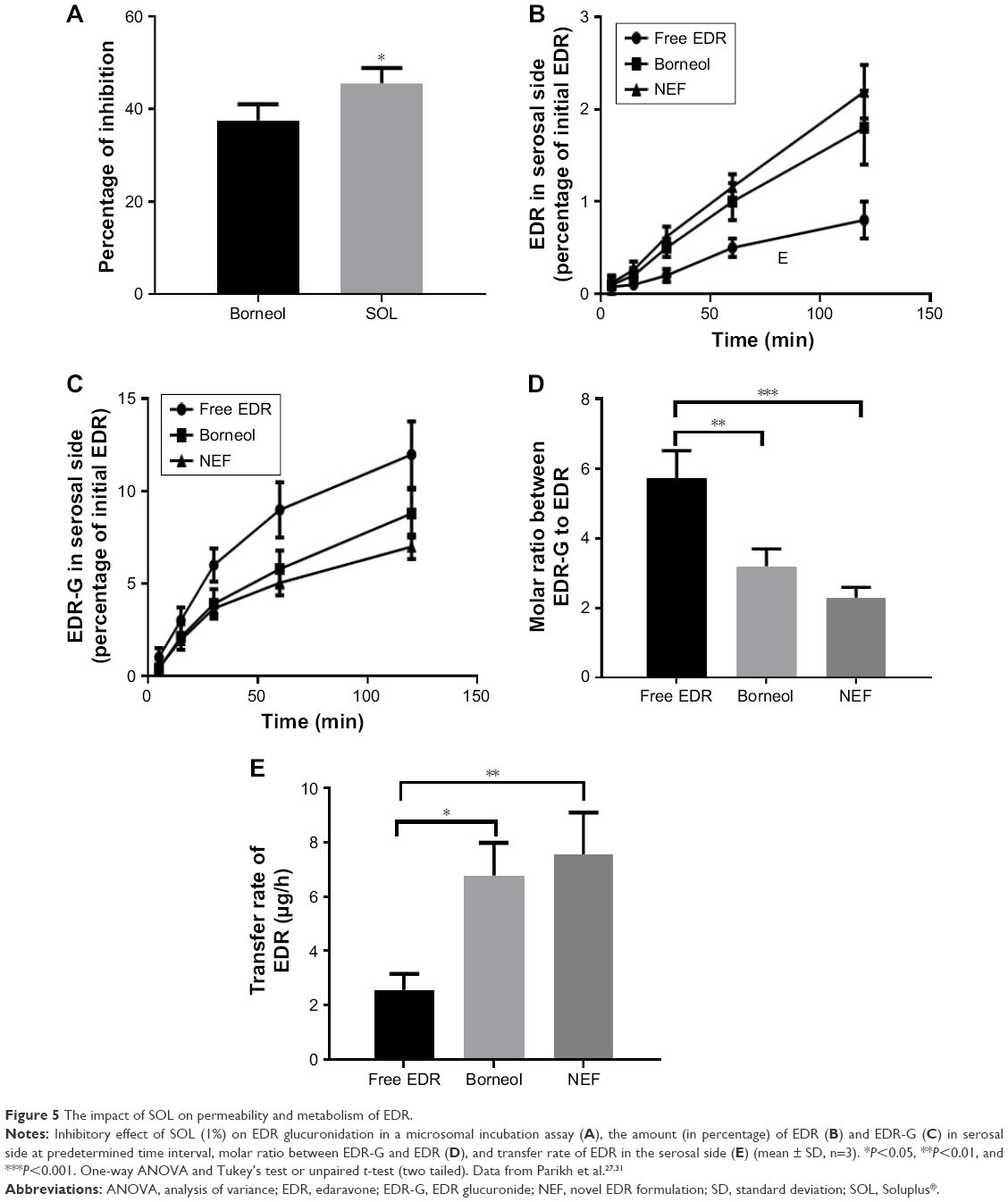 | Figure 5 The impact of SOL on permeability and metabolism of EDR. |
In vitro drug dissolution
The result of dissolution study of EDR and NEF is shown in Figure 6A. EDR showed 100% release within 15 min in SGF compared to 85% release in 120 min in SIF, which indicated pH-dependent release. It was further examined by comparing the dissolution profiles using f1 and f2 factors. The determined f2 value (similarity factor) and f1 value (dissimilarity factor) are 14 and 26, respectively, which confirmed the impact of pH of the media on EDR release. As per our previous report, EDR exhibited significantly higher pH-dependent solubility in SGF (8.26±1.42 mg/mL) than in SIF (1.89±0.51 mg/mL).31 The difference in dissolution results of crude EDR in SGF and SIF could be based on solubility data. Moreover, crude EDR followed Hixson–Crowell and Korsmeyer–Peppas release kinetic models in SGF and SIF, respectively (Table 1). Additionally, in SIF, it followed an anomalous transport (non-Fickian) mechanism, which could be controlled by factors other than diffusion. The sedimentation and wetting of the EDR at interface could control the release of EDR in SIF.
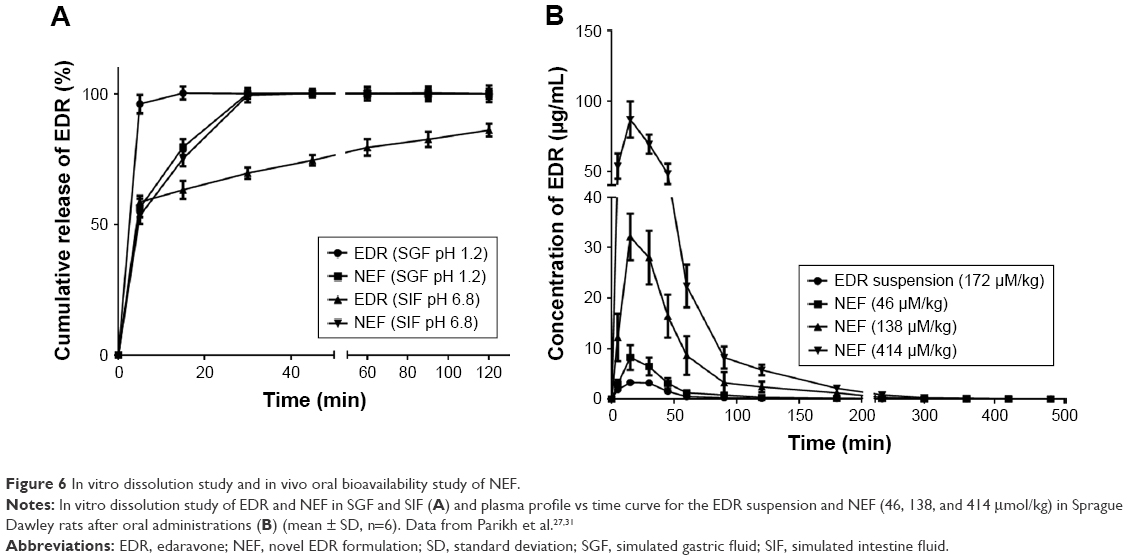 | Figure 6 In vitro dissolution study and in vivo oral bioavailability study of NEF. |
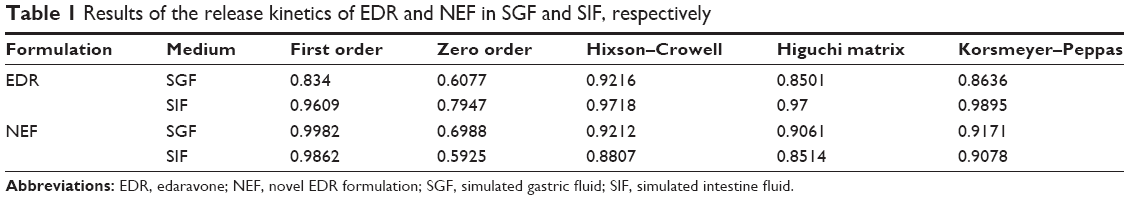 | Table 1 Results of the release kinetics of EDR and NEF in SGF and SIF, respectively |
The dissolution of NEF in SGF and SIF was 100% within 15 and 30 min, respectively. There was a significant improvement in the dissolution of NEF detected in SIF compared to crude EDR, whereas no dramatic change was witnessed between the dissolution profile of crude EDR and NEF in SGF except at 5 min. The amorphization, hydrogen bonding interaction, and micellization could play a vital role in the improvement of dissolution behavior of EDR from NEF compared to crude EDR. Additionally, the f2 (65) and f1 (3) values confirmed the similar dissolution profiles of EDR in SGF and SIF, which suggested the minimum impact of pH on EDR release from NES. Moreover, the first-order release kinetic model was best fitted with the dissolution profiles of NEF. Thus, the rate of EDR release depends on the amount of EDR retained in NEF.68
EDR exhibited pH-dependent solubility in SGF (8.26±1.42 mg/mL) and SIF (1.89±0.51 mg/mL), respectively.31 We have used 900 mL of dissolution media including SGF (pH 1.2) and SIF (pH 6.8) in the dissolution study. The fast release of EDR and EDR from NEF could be due to sufficient solubility and the large amount of dissolution media. Moreover, NEF is the amorphous solid dispersion, which can be defined as molecular mixture of EDR and SOL. Based on DSC and XRD results, there was no significant amount of free EDR present in NEF. Moreover, during the process of micelles’ formation after dispersing NEF into dissolution media, EDR can be either encapsulated in micelles or remained in unloaded form. Based on the results of drug loading and entrapment efficiency of micelles, ~96% of EDR was found encapsulated in micelles. Thus, there should be a nonsignificant contribution from unloaded EDR in dissolution results. Additionally, a number of studies of solid dispersion-based formulation containing SOL showed the quick release of drugs (within 5 min).60,69,70 It could be due to the ability of SOL of forming solid solutions, which makes the drug available in a dissolved state in quick time, resulting in an improved BA, once in the body. Moreover, SOL is hydrophilic in nature resulting in a better wettability of the drug. Furthermore, micellization is another mechanism that could contribute to the quick release of drug.39
In vivo pharmacokinetic study
The developed formulation in part I of the study would undergo the assessment of dose-dependent therapeutic effect against the Alzheimer’s disease in part II of the study. Thus, three different doses of NEF were considered for the pharmacokinetic study. The plasma concentration against time, pharmacokinetic parameters, and statistical analysis of pharmacokinetic parameters are displayed in Figure 6B and Tables 2 and S3, respectively. A significant increase in Cmax was observed at all doses of NEF compared to EDR suspension, while NEF did not show any difference in Tmax. These results are consistent with the result of dissolution study as NEF displayed quick release in both media similar to EDR suspension release in SGF. Also, NEF showed significantly longer t1/2 compared to EDR suspension, which could be due to the inhibitory effect of SOL on EDR glucuronidation. No significant difference in t1/2 value between the NEF groups with different doses was observed. Moreover, NEF exhibited dramatic increment in AUC0–t and 10.24- (46 μM/kg), 16.08- (138 μM/kg), and 14.78- (414 μM/kg) fold enhancement in relative BA compared to EDR suspension. There was a significant difference in BA noticed between the groups of NEF with doses 46–138 and 414 μM/kg. EDR displays high protein binding (91.0%–91.9%), and it is possible that at the higher doses, protein-binding sites are saturated so that EDR remains free and available for metabolism and excretion.71,72 The similar dose-dependent absorption via intraperitoneal route was also reported.73
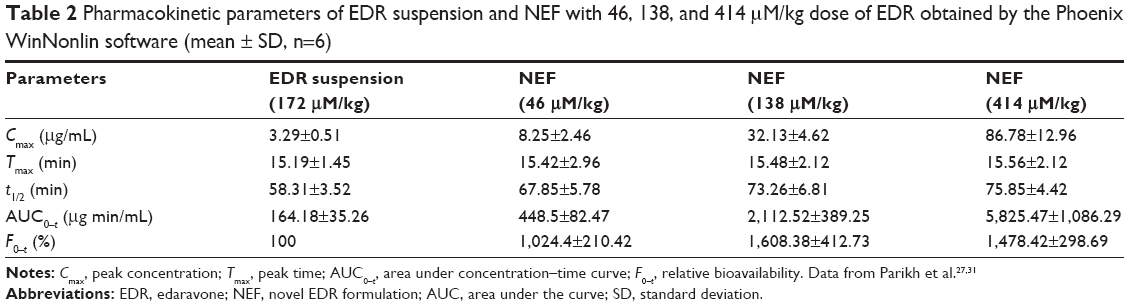 | Table 2 Pharmacokinetic parameters of EDR suspension and NEF with 46, 138, and 414 μM/kg dose of EDR obtained by the Phoenix WinNonlin software (mean ± SD, n=6) |
The solubilizing amount of EDR in the vehicle used for pharmacokinetic study might significantly affect the outcome. Thus, determination of solubilized EDR in a total of 1.5 mL of vehicle used for oral administration in rats was performed. The dissolved part of EDR in the supplied vehicle was 31 and 100% with EDR suspension and NEF, respectively, for all doses. The presence of solubilized EDR in greater amount could be one of the reasons for significant improvement in oral BA.27 Moreover, with regard to dissolution results, both NEF and EDR showed fast dissolution (100%) within 15 min in the 900 mL of dissolution media. The reported fluid content in the gastrointestinal tract of the fasted rat is 3.2±1.8 mL.74 Therefore, EDR could precipitate in the rat stomach, even though it exhibits an exceptional dissolution profile in SGF. Similar results were observed in the kinetic solubility study where precipitation of EDR was noticed after 30 min in the absence of SOL. SOL is well recognized with its characteristic as a precipitation inhibitor by maintaining supersaturation. Thus, NEF could perform better in terms of in vivo dissolution. Moreover, we have shown that NEF displayed a 2.4-fold decrease in the glucuronidation of EDR and a 2.73-fold greater transfer amount of EDR across the small intestine compared to nonformulated EDR. The inhibitory effect of SOL on glucuronidation, Pgp efflux, and <100 nm of micelles generated from NEF could also enhance the metabolism and permeability of EDR across the small intestine. Also, improvement in dissolution, metabolism, and permeability profile could have played a vital role in the exceptional performance of NEF in the pharmacokinetic study compared to EDR suspension as per our previous reports.27,31
Conclusion
The poor oral BA of EDR due to poor aqueous solubility, dissolution, and permeability could hamper its optimum therapeutic use. To improve its BA, EDR-loaded NEF was developed using the SNMSD-based strategy. The impact on physicochemical characteristics of EDR, such as aqueous solubility, stability, and dissolution, as well as metabolism and permeability, was assessed. SOL as a polymer was selected based on its potential for improving solubility and stability. The optimized formulation, NEF, showed 17.5-fold improvement in SGFs. The mechanism of improvement of solubility and stability was extensively studied, and amorphization, hydrogen bonding interaction, and micellization were determined as significant contributors. Moreover, NEF showed better performance in in vitro metabolism and intestinal permeability study compared to crude EDR. Furthermore, oral pharmacokinetic studies in rats showed that NEF was ~16-fold bioavailable compared to unformulated EDR. The exceptional enhancement of oral BA of NEF compared to EDR suspension could be due to an improvement in solubility, dissolution, and permeability. Our studies proved that the developed NEF is a promising candidate for further study in oxidative stress-associated diseases.
Acknowledgments
The authors would specially like to acknowledge Fujian Kangshimei Co, China, for the financial support for present research, the University President’s Scholarships (UPS) from University of South Australia for AP for his doctorate study, Vacation Research Scholarships from the University of South Australia for CCT, and NHMRC fellowship for X-FZ. The authors would like to acknowledge the technical support provided by Lyn Waterhouse from Adelaide Microscopy, The University of Adelaide, for TEM analysis; Dr Andrew C Robinson, Dr Nobuyuki Kawashima, and Dr Steve McInnes from Future Industries Institute, University of South Australia, for XRD, SEM, and DSC studies, respectively; H Md Morshed Alam (BASF Australia Ltd) for providing samples of SOL; and Stephen Philip, a PhD candidate, from School of Pharmacy and Medical Sciences, the University of South Australia, for NMR study. The staff members of Reid animal house facility including Alex Whittaker, Ruth Brogan, Jayne Skinner, Alysha Servin, Jess Parken, and Becky Nitschke from the University of South Australia are acknowledged for their generous support in animal work.
Disclosure
AP, X-FZ, and SG are the named inventors of Chinese patent 200610149832.9. Fujian Kangshimei Co, China, owns the intellectual property. The authors report no other conflicts of interest in this work.
References
Tanaka M, Sugimura N, Fujisawa A, Yamamoto Y. Stabilizers of edaravone aqueous solution and their action mechanisms. 1. Sodium bisulfite. J Clin Biochem Nutr. 2017;61(3):159–163. | ||
Shao F, Hu XL, Liu X, Shan MT. A novel LC-MS-MS method with an effective antioxidant for the determination of edaravone, a free-radical scavenger in dog plasma and its application to a pharmacokinetic study. J Chromatogr Sci. 2017;55(6):595–602. | ||
Tanaka M, Motomiya S, Fujisawa A, Yamamoto Y. Stabilizers of edaravone aqueous solution and their action mechanisms. 2. glutathione. J Clin Biochem Nutr. 2017;61(3):164–168. | ||
Ali ZK, Baker DE. Formulary drug review: edaravone. Hosp Pharm. 2017;52(11):732–736. | ||
Kikuchi K, Uchikado H, Miyagi N, et al. Beyond neurological disease: new targets for edaravone (review). Int J Mol Med. 2011;28(6):899–906. | ||
Kikuchi K, Takeshige N, Miura N, et al. Beyond free radical scavenging: beneficial effects of edaravone (Radicut) in various diseases (review). Exp Ther Med. 2012;3(1):3–8. | ||
Watanabe K, Tanaka M, Yuki S, Hirai M, Yamamoto Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J Clin Biochem Nutr. 2018;62(1):20–38. | ||
Jiao SS, Yao XQ, Liu YH, et al. Edaravone alleviates Alzheimer’s disease-type pathologies and cognitive deficits. Proc Natl Acad Sci U S A. 2015;112(16):5225–5230. | ||
Reeta KH, Singh D, Gupta YK. Edaravone attenuates intracerebroventricular streptozotocin-induced cognitive impairment in rats. Eur J Neurosci. 2017;45(7):987–997. | ||
Liu Z, Yang C, Meng X, Li Z, Lv C, Cao P. Neuroprotection of edaravone on the hippocampus of kainate-induced epilepsy rats through Nrf2/HO-1 pathway. Neurochem Int. 2018;112:159–165. | ||
Li Z, Ma QQ, Yan Y, et al. Edaravone attenuates hippocampal damage in an infant mouse model of pneumococcal meningitis by reducing HMGB1 and iNOS expression via the Nrf2/HO-1 pathway. Acta Pharmacol Sin. 2016;37(10):1298–1306. | ||
Ikeda K, Iwasaki Y. Edaravone, a free radical scavenger, delayed symptomatic and pathological progression of motor neuron disease in the wobbler mouse. PLoS One. 2015;10(10):e0140316. | ||
Takei K, Watanabe K, Yuki S, Akimoto M, Sakata T, Palumbo J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(Supp11):5–10. | ||
Ishii H, Petrenko AB, Sasaki M, et al. Free radical scavenger edaravone produces robust neuroprotection in a rat model of spinal cord injury. Brain Res. 2018;1682:24–35. | ||
Zhang Y, Liao H, Zhong S, et al. Effect of N-n-butyl haloperidol iodide on ROS/JNK/Egr-1 signaling in H9c2 cells after hypoxia/reoxygenation. Sci Rep. 2015;5:11809. | ||
Uchiyama M, Tojo K, Yazawa T, Ota S, Goto T, Kurahashi K. Edaravone prevents lung injury induced by hepatic ischemia-reperfusion. J Surg Res. 2015;194(2):551–557. | ||
Zhang S, Chen ZX, Jiang YY, et al. [Intervention of edaravone against renal injury induced by acute paraquat poisoning in rats]. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2017;35(6):408–413. Chinese. | ||
Sattarinezhad E, Panjehshahin MR, Torabinezhad S, et al. Protective effect of edaravone against cyclosporine-induced chronic nephropathy through antioxidant and nitric oxide modulating pathways in rats. Iran J Med Sci. 2017;42(2):170–178. | ||
Zhang Z, Luo Z, Bi A, et al. Compound edaravone alleviates lipopolysaccharide (LPS)-induced acute lung injury in mice. Eur J Pharmacol. 2017;811:1–11. | ||
Zhang WW, Bai F, Wang J, et al. Edaravone inhibits pressure overload-induced cardiac fibrosis and dysfunction by reducing expression of angiotensin II AT1 receptor. Drug Des Devel Ther. 2017;11:3019–3033. | ||
Masuda T, Shimazawa M, Hara H. Retinal diseases associated with oxidative stress and the effects of a free radical scavenger (edaravone). Oxid Med Cell Longev. 2017;2017:9208489. | ||
Aleksovski A, Dreu R, Gasperlin M, Planinsek O. Mini-tablets: a contemporary system for oral drug delivery in targeted patient groups. Expert Opin Drug Deliv. 2015;12(1):65–84. | ||
Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Reactive oxygen species modulate growth of cerebral aneurysms: a study using the free radical scavenger edaravone and p47phox(−/−) mice. Lab Invest. 2009;89(7):730–741. | ||
Anonymous [Webpage on the Internet]. Public Summary of Opinion on Orphan Designation: Edaravone for the Treatment of Amyotrophic Lateral Sclerosis. 2015. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/orphans/2015/02/human_orphan_001489.jsp&mid=WC0b01ac058001d12b. Accessed May 01, 2016. | ||
Anonymous [Webpage on the Internet]. Orphan Drug Designations and Approvals. 2015; Available from: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=465514. Accessed May 01, 2016. | ||
Rong WT, Lu YP, Tao Q, et al. Hydroxypropyl-sulfobutyl-beta-cyclodextrin improves the oral bioavailability of edaravone by modulating drug efflux pump of enterocytes. J Pharm Sci. 2014;103(2):730–742. | ||
Parikh A, Kathawala K, Tan CC, Garg S, Zhou XF. Development of a novel oral delivery system of edaravone for enhancing bioavailability. Int J Pharm. 2016;515(1–2):490–500. | ||
Ringman JM, Frautschy SA, Teng E, et al. Oral curcumin for Alzheimer’s disease: tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res Ther. 2012;4(5):43. | ||
Mecocci P, Polidori MC. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim Biophys Acta. 2012;1822(5):631–638. | ||
Paradkar A, Ambike AA, Jadhav BK, Mahadik KR. Characterization of curcumin-PVP solid dispersion obtained by spray drying. Int J Pharm. 2004;271(1–2):281–286. | ||
Parikh A, Kathawala K, Tan CC, Garg S, Zhou XF. Lipid-based nanosystem of edaravone: development, optimization, characterization and in vitro/in vivo evaluation. Drug Deliv. 2017;24(1):962–978. | ||
Kim DW, Kwon MS, Yousaf AM, et al. Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate. Carbohydr Polym. 2014;114:365–374. | ||
Akula S, Gurram AK, Devireddy SR. Self-microemulsifying drug delivery systems: an attractive strategy for enhanced therapeutic profile. Int Sch Res Notices. 2014;2014:964051. | ||
Seo SW, Han HK, Chun MK, Choi HK. Preparation and pharmacokinetic evaluation of curcumin solid dispersion using Solutol(R) HS15 as a carrier. Int J Pharm. 2012;424(1–2):18–25. | ||
Song IS, Cha JS, Choi MK. Characterization, in vivo and in vitro evaluation of solid dispersion of curcumin containing d-alpha-tocopheryl polyethylene glycol 1000 succinate and mannitol. Molecules. 2016;21(10):E1386. | ||
Sinha S, Ali M, Baboota S, Ahuja A, Kumar A, Ali J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech. 2010;11(2):518–527. | ||
Kyaw Oo M, Mandal UK, Chatterjee B. Polymeric behavior evaluation of PVP K30-poloxamer binary carrier for solid dispersed nisoldipine by experimental design. Pharm Dev Technol. 2017;22(1):2–12. | ||
Wang W, Song Y, Petrovski K, et al. Development of intramammary delivery systems containing lasalocid for the treatment of bovine mastitis: impact of solubility improvement on safety, efficacy, and milk distribution in dairy cattle. Drug Des Devel Ther. 2015;9:631–642. | ||
Lim SM, Pang ZW, Tan HY, Shaikh M, Adinarayana G, Garg S. Enhancement of docetaxel solubility using binary and ternary solid dispersion systems. Drug Dev Ind Pharm. 2015;41(11):1847–1855. | ||
Leonardi D, Salomon CJ. Unexpected performance of physical mixtures over solid dispersions on the dissolution behavior of benznidazole from tablets. J Pharm Sci. 2013;102(3):1016–1023. | ||
Li J, Lee IW, Shin GH, Chen X, Park HJ. Curcumin-Eudragit(R) E PO solid dispersion: a simple and potent method to solve the problems of curcumin. Eur J Pharm Biopharm. 2015;94:322–332. | ||
Bala V, Rao S, Li P, Wang S, Prestidge CA. Lipophilic prodrugs of SN38: synthesis and in vitro characterization toward oral chemotherapy. Mol Pharm. 2016;13(1):287–294. | ||
Yu H, Xia D, Zhu Q, Zhu C, Chen D, Gan Y. Supersaturated polymeric micelles for oral cyclosporine A delivery. Eur J Pharm Biopharm. 2013;85(3 pt B):1325–1336. | ||
Lee YS, Song JG, Lee SH, Han HK. Sustained-release solid dispersion of pelubiprofen using the blended mixture of aminoclay and pH independent polymers: preparation and in vitro/in vivo characterization. Drug Deliv. 2017;24(1):1731–1739. | ||
Obaidat RM, Tashtoush BM, Awad AA, Al Bustami RT. Using supercritical fluid technology (SFT) in preparation of tacrolimus solid dispersions. AAPS PharmSciTech. 2017;18(2):481–493. | ||
Verma S, Rudraraju VS. Disintegration mediated controlled release supersaturating solid dispersion formulation of an insoluble drug: design, development, optimization, and in vitro evaluation. AAPS PharmSciTech. 2015;16(1):85–97. | ||
Altamimi MA, Neau SH. Investigation of the in vitro performance difference of drug-Soluplus(R) and drug-PEG 6000 dispersions when prepared using spray drying or lyophilization. Saudi Pharm J. 2017;25(3):419–439. | ||
Fule R, Amin P. Development and evaluation of lafutidine solid dispersion via hot melt extrusion: investigating drug-polymer miscibility with advanced characterization. Asian J Pharm Sci. 2014;9(2):92–106. | ||
Mendonca LM, Machado Cda S, Teixeira CC, Freitas LA, Bianchi ML, Antunes LM. Comparative study of curcumin and curcumin formulated in a solid dispersion: evaluation of their antigenotoxic effects. Genet Mol Biol. 2015;38(4):490–498. | ||
Vo CL, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85(3 pt B):799–813. | ||
Lian X, Dong J, Zhang J, et al. Soluplus((R)) based 9-nitrocamptothecin solid dispersion for peroral administration: preparation, characterization, in vitro and in vivo evaluation. Int J Pharm. 2014;477(1–2):399–407. | ||
Jian Zenga YR, Chengliang Z, Shuqin Y, Wen-Hua C. Preparation and physicochemical characteristics of the complex of edaravone with hydroxypropyl-cyclodextrin. Carbohydr Polym. 2010;83:5. | ||
Paaver U, Tamm I, Laidmae I, et al. Soluplus graft copolymer: potential novel carrier polymer in electrospinning of nanofibrous drug delivery systems for wound therapy. Biomed Res Int. 2014;2014:789765. | ||
Bahl D, Bogner RH. Amorphization of indomethacin by co-grinding with neusilin US2: amorphization kinetics, physical stability and mechanism. Pharm Res. 2006;23(10):2317–2325. | ||
Vasconcelos T, Marques S, das Neves J, Sarmento B. Amorphous solid dispersions: rational selection of a manufacturing process. Adv Drug Deliv Rev. 2016;100:85–101. | ||
Dian L, Yu E, Chen X, et al. Enhancing oral bioavailability of quercetin using novel Soluplus polymeric micelles. Nanoscale Res Lett. 2014;9(1):2406. | ||
Shi NQ, Lai HW, Zhang Y, et al. On the inherent properties of Soluplus and its application in ibuprofen solid dispersions generated by microwave-quench cooling technology. Pharm Dev Technol. Epub 2016 Nov 16. | ||
Lu J, Cuellar K, Hammer NI, et al. Solid-state characterization of Felodipine-Soluplus amorphous solid dispersions. Drug Dev Ind Pharm. 2016;42(3):485–496. | ||
Mirza S, Miroshnyk I, Heinamaki J, et al. Crystal morphology engineering of pharmaceutical solids: tabletting performance enhancement. AAPS PharmSciTech. 2009;10(1):113–119. | ||
Fule RA, Meer TS, Sav AR, Amin PD. Artemether-Soluplus hot-melt extrudate solid dispersion systems for solubility and dissolution rate enhancement with amorphous state characteristics. J Pharm (Cairo). 2013;2013:151432. | ||
Yuan X, Xiang TX, Anderson BD, Munson EJ. Hydrogen bonding interactions in amorphous indomethacin and its amorphous solid dispersions with poly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-vinyl acetate) studied using (13)c solid-state NMR. Mol Pharm. 2015;12(12):4518–4528. | ||
Jin X, Zhou B, Xue L, San W. Soluplus((R)) micelles as a potential drug delivery system for reversal of resistant tumor. Biomed Pharmacother. 2015;69:388–395. | ||
Alvarez-Rivera F, Fernandez-Villanueva D, Concheiro A, Alvarez-Lorenzo C. Alpha-Lipoic acid in Soluplus((R)) polymeric nanomicelles for ocular treatment of diabetes-associated corneal diseases. J Pharm Sci. 2016;105(9):2855–2863. | ||
Wegmann M, Parola L, Bertera FM, et al. Novel carvedilol paediatric nanomicelle formulation: in-vitro characterization and in-vivo evaluation. J Pharm Pharmacol. 2016;69(5):544–553. | ||
Jacobs C, Kayser O, Muller RH. Nanosuspensions as a new approach for the formulation for the poorly soluble drug tarazepide. Int J Pharm. 2000;196(2):161–164. | ||
Lu Y, Park K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int J Pharm. 2013;453(1):198–214. | ||
Zhou J, Zhou M, Yang FF, et al. Involvement of the inhibition of intestinal glucuronidation in enhancing the oral bioavailability of resveratrol by labrasol containing nanoemulsions. Mol Pharm. 2015;12(4):1084–1095. | ||
Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. | ||
Homayouni A, Sadeghi F, Varshosaz J, Afrasiabi Garekani H, Nokhodchi A. Promising dissolution enhancement effect of Soluplus on crystallized celecoxib obtained through antisolvent precipitation and high pressure homogenization techniques. Colloids Surf B Biointerfaces. 2014;122:591–600. | ||
Lavra ZM, Pereira de Santana D, Re MI. Solubility and dissolution performances of spray-dried solid dispersion of efavirenz in Soluplus. Drug Dev Ind Pharm. 2017;43(1):42–54. | ||
Li H, Xu K, Wang Y, et al. Phase I clinical study of edaravone in healthy Chinese volunteers: safety and pharmacokinetics of single or multiple intravenous infusions. Drugs R D. 2012;12(2):65–70. | ||
Hanlon N, Coldham N, Gielbert A, et al. Absolute bioavailability and dose-dependent pharmacokinetic behaviour of dietary doses of the chemopreventive isothiocyanate sulforaphane in rat. Br J Nutr. 2008;99(3):559–564. | ||
Ito H, Wate R, Zhang J, et al. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp Neurol. 2008;213(2):448–455. | ||
McConnell EL, Basit AW, Murdan S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J Pharm Pharmacol. 2008;60(1):63–70. | ||
Komiyama Y. The Ministry of Health, Labour and Welfare Ministerial notification no. 519. The Minister of Health, Labour and Welfare, Japan; 2012. Available from: http://jpdb.nihs.go.jp/kyokuhou/JP16e-1.pdf. Accessed June 18, 2018. |
Supplementary materials
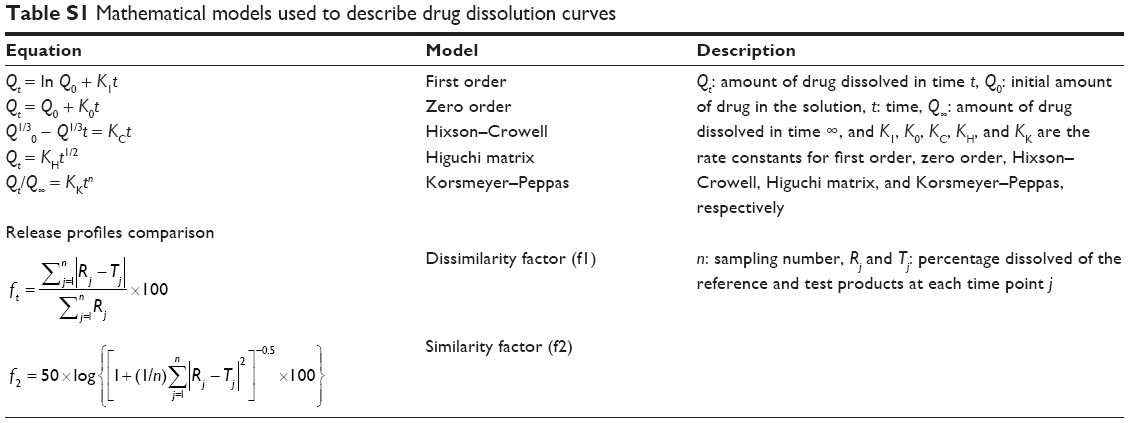 | Table S1 Mathematical models used to describe drug dissolution curves |
 | Figure S1 Structure of EDR (3-methyl-1-phenyl-2-pyrazolin-5-one). |
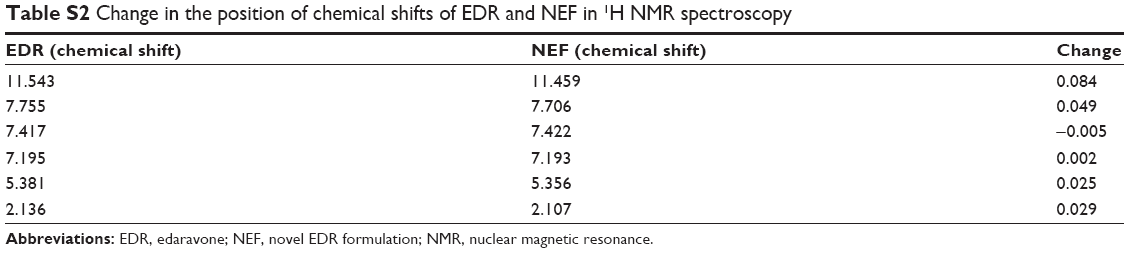 | Table S2 Change in the position of chemical shifts of EDR and NEF in 1H NMR spectroscopy |
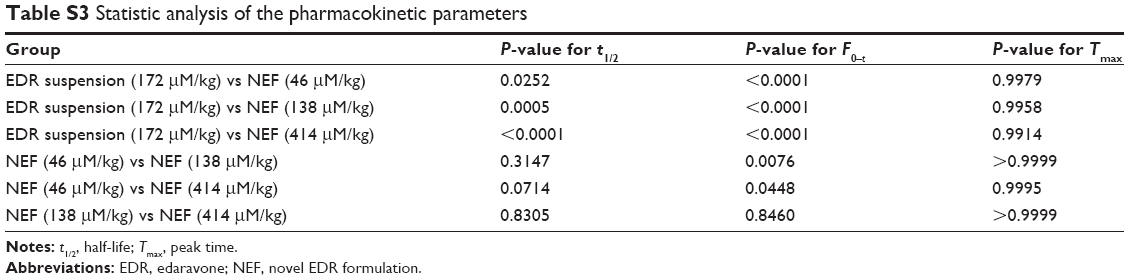 | Table S3 Statistic analysis of the pharmacokinetic parameters |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.