Back to Journals » Drug Design, Development and Therapy » Volume 10
Selective CDK7 inhibition with BS-181 suppresses cell proliferation and induces cell cycle arrest and apoptosis in gastric cancer
Authors Wang B, Liu Q, Cao J, Chen J, Liu Z
Received 9 April 2015
Accepted for publication 15 August 2015
Published 16 March 2016 Volume 2016:10 Pages 1181—1189
DOI https://doi.org/10.2147/DDDT.S86317
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Prof. Dr. Wei Duan
Bo-Yong Wang, Quan-Yan Liu, Jun Cao, Ji-Wei Chen, Zhi-Su Liu
Department of General Surgery, Zhongnan Hospital of Wuhan University, Wuhan, Hubei, People’s Republic of China
Abstract: Cyclin-dependent kinase (CDK) family members have been considered as attractive therapeutic targets for cancer. In this study, we aim to investigate the anticancer effects of a selective CDK7 inhibitor, BS-181, in gastric cancer (GC) cell line. Human GC cells (BGC823) were cultured with or without BS-181 at different concentrations for 24–72 hours. BS-181 significantly reduced the activity of CDK7 with downregulation of cyclin D1 and XIAP in GC cells. Treatment with BS-181 induced cell cycle arrest and apoptosis. The expression of Bax and caspase-3 was significantly increased, while Bcl-2 expression was decreased in cells treated with BS-181. In addition, the inhibition of CDK7 with BS-181 resulted in reduced rates of proliferation, migration, and invasion of gastric cells. Those results demonstrated the anticancer activities of selective CDK7 inhibitor BS-181 in BGC823 cells, suggesting that CDK7 may serve as a novel therapeutic target or the treatment of GC.
Keywords: selective CDK7 inhibitor, gastric cancer, BS-181, anticancer activities
Introduction
Gastric cancer (GC) is one of the most common types of malignant cancer in the world.1 Despite a decrease in the past several decades, it still remains the second leading cause of cancer death.2 Current choices of treatment for GC include chemotherapy, surgery, and radiation therapy. However, unfortunately, the overall prognosis of the disease is disappointing, especially for those with advanced GC. Chemotherapy resistance and metastasis largely contribute to the failure of curative treatment. Thus, it is of great necessity and importance to develop new drugs.
Cyclin-dependent kinases (CDKs) are a large group of serine/threonine protein kinases that have central roles in controlling the cell cycle and transcription. Activation of specific CDKs is required for the appropriate progression through the cell cycle and into the next stage of the cell cycle. CDK–cyclin complexes were also identified as conserved components of the RNA polymerase II (Pol II) transcriptional machinery.3 Two nonoverlapping sets of CDKs have been defined in metazoans: CDK1, CDK2, CDK4, and CDK6, which regulate the cell division, and CDK8 and CDK9, which are dedicated to transcription control. CDK7 is the only one that cannot be easily classified. Phosphorylation of the first group of CDKs is mediated by CDK-activating kinase that has three subunits including CDK7, cyclin H, and MAT1.4
Additionally, as an essential component of the transcription factor TFIIH, CDK7 is also involved in transcription initiation by phosphorylating the COOH-terminal domain of the largest subunit of RNA pol II.5 Thus, CDK7 is becoming an attractive target for anticancer treatment. Inhibition of CDK7 activity is expected to inhibit both transcription and cell progression, therefore, leading to an antitumor effect. The first generation of CDK inhibitors has entered late-stage clinical trials but so far has only shown modest activity because of unfavorable pharmacology.6 Most of the CDK inhibitors target the highly conserved kinase domain and could inhibit multiple CDK family members, which in part explain the undesirability of those trials. Hence, the identification of more selective and potent CDK inhibitors is imperative. BS-181, a pyrazolo [1,5-α] pyrimidine-derived compound, is a novel selective inhibitor of CDK7 by computer-aided drug design. In the previous study, it was demonstrated that BS-181 inhibited the phosphorylation of the PolII C-terminal domain (CTD) at Ser,5 which is known as a CDK7 substrate, thus promoting cell cycle arrest and apoptosis to inhibit the growth of cancer cells in a breast cancer cell line.7 Furthermore, compared to roscovitine, a broad-spectrum CDK inhibitor, BS-181 exhibits a substantially higher preference for CDK7. The results mentioned earlier strongly indicated the potential of BS-181 in anticancer treatment. However, no literature has shown the antitumor effect of BS-181 in GC.
In the present study, we investigated whether BS-181 inhibited the tumor growth and metastatic behavior of GC in vitro as well as in vivo and the underlying mechanisms.
Materials and methods
The Ethics Committee of Zhongnan Hospital of Wuhan University approved this study (2014-02-ZN012).
Cell culture
The biological effects of BS-181 on GC have been investigated using BGC823. BGC823 cells were purchased from The Medical Experiment Center of Zhongnan Hospital of Wuhan University (Wuhan, Hubei, People’s Republic of China). BGC823 cells were cultured in F-12 Ham medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (MP Bio-medicals, Solon, OH, USA). Cultures were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Reagents
BS-181 was purchased from Biofavor Biotech Service Co., Ltd. (Wuhan, Hubei, People’s Republic of China). Cyclin D1 (sc-753) antibody was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA); PolII (N-20) and PolII phosphoserine-5 (ab5131) antibodies were purchased from Santa Cruz Biotechnology Inc.; Bcl-2 (#2870p) antibody was purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA); Bax (BS-2538) antibody was purchased from Bioworld Technology, Inc. (MN, USA). Caspase-3 antibody was purchased from Sanying (Wuhan, People’s Republic of China); XIAP (ab86227) antibody was purchased from Abcam (Shanghai, People’s Republic of China). Anti-β-actin and horseradish peroxidase-labeled secondary antibodies were purchased from Boster (Wuhan, Hubei, People’s Republic of China); the concentrations of all antibodies were used according to the manufacturers’ instruction.
Kinase assays in vitro
As previously described,7 kinase assays were performed to evaluate the inhibitory effects of BS-181 on CDK activities in vitro. Kinase assays were carried out using substrate peptides purchased from ProQinase GmbH (Freiburg, Germany). A luciferase assay (PKLight assay; Cambrex, UK) was used to determine ATP remaining at the end of the kinase reaction according to the manufacturer’s protocols.
Western blot analysis
Whole-cell lysates were prepared from BGC823 cells, and standard Western blot analysis was performed. Protein concentrations were detected using Bradford dye (Bio-Rad Laboratories Inc., Hercules, CA, USA). Equal amounts of protein extracts (10 μg) were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The membrane was probed with specific primary antibodies including XIAP (1:300), cyclin D1 (1:300), BCl-2 (1:800), Bax (1:600), and caspase-3 (1:600). The protein bands were normalized with β-actin, and all the blots were quantified with Software Quantity One (Bio-Rad Laboratories Inc.).
Cell cycle and apoptosis analysis
Cells were seeded on plates in Dulbecco’s Modified Eagle’s Medium containing 10% fetal calf serum. After incubation with BS-181 for 12 hours, 24 hours, 48 hours, and 72 hours at 0 μM, 1 μM, 10 μM, and 20 μM, cells were harvested, rinsed with cold PBS, and fixed with 70% ice-cold ethanol for 30 minutes and then incubated with propidium iodide (PI) for 30 minutes prior to flow cytometry (BD Biosciences, San Jose, CA, USA). Cell apoptosis was determined by flow cytometry using Annexin V-FITC/PI (BD Biosciences) double staining assay. Annexin V-positive and PI-negative cells were identified as apoptotic cells. The apoptotic rate was determined using CellQuest software (FCM, BD Biosciences).
Cell viability assay
Cell viability was detected using Cell Counting Kit (CCK-8 kit, Dojindo Laboratory, Japan) according to supplier’s introductions. Briefly, BGC823 cells were seeded at 104 cells per well for 48 hours with or without BS-181. Then, the absorbance was detected at 450 nm (reference at 650 nm) in each well.
Cell migration and invasion assay
To evaluate cell migration, BGC823 cells (1×105/mL, in minimum essential medium supplemented with 1% fetel bovine serum) were placed into top chamber of transwell migration chambers (8 μm pores Transwell inserts, BD BioCoat™). The lower chamber was filled with 0.1% bovine serum albumin–DMEM without serum. After 24 hours, unmigrated cells were removed from the upper surface of transwell membrane, and migrated cells in the lower chamber were fixed, stained, photographed, and counted. To assess invasion in vitro, invasion assays were done under the same conditions as the transwell migration assays, but in matrigel-coated transwells.
Animal preparation and human tumor xenografts
Sixty-seven-week-old male BALB/c-nu mice were purchased from the Hubei Center for Disease Control and Prevention (Wuhan, Hubei, People’s Republic of China). All animal experiments were performed in the Animal Research Center of Wuhan University (Wuhan, Hubei, People’s Republic of China) and were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of Wuhan University. The Ethics Committee of Zhongnan Hospital of Wuhan University approved the animal study (20140120ZN01). Animals were raised under controlled conditions with a temperature of 24°C±2°C and a relative humidity of 50%±15%.
Human tumor xenografts were established as previously described.7 In total, 5×106 BGC823 cells (0.1 mL) were injected subcutaneously into the flank of the mice. Tumor measurements were performed two times per week, and volumes were calculated using the formula: tumor size = (length [mm] × width2 [mm])/2. Finally, 30 mice (tumor volume 100–200 mm3) were selected and randomly assigned into three groups. As previously described,7 BS-181 was prepared in 10% dimethyl sulfoxide/50 mM HCl/5% Tween 20/85% saline. Mice received BS-181 injection (ip) twice daily at indicated doses (BS-181 [10 mg/kg/d or 20 mg/kg/d] or roscovitine [20 mg/kg/d]) for a total of 14 days. Control mice were injected with vehicles. Animal weights and tumor volume were measured each day throughout the 14-day treatment. In addition, all rats were kept for another 30 days for survival observation. Mice were injected intraperitoneally twice daily with BS-181 at 5 mg/kg or 10 mg/kg.
Statistical analysis
Comparisons among all groups were performed with the one-way analysis of variance test or unpaired Student’s t-test. The survival analysis was estimated with the Kaplan–Meier method. The significance of comparison was calculated with the log-rank test. A two-tailed P<0.5 was considered significant. All data were analyzed using SPSS software version 21.0 (StataCorp LP, College Station, TX, USA).
Results
BS-181-inhibited gastric cancer cell proliferation, migration, and invasion
To evaluate the antiproliferative ability of BS-181 in GC, several different cell lines including MKN28, SGC-7901, AGS, and BGC823 were treated with increasing concentrations of BS-181 for 48 hours. CCK-8 assay showed that GC cell growth was inhibited by BS-181, with inhibitory concentration (IC50) ranging from 17 to 22 μM. For normal gastric epithelial RGM-1 cell line, IC50 was 6.5 μM (Table 1). In addition, we investigated the effects of BS-181 on cell migration and invasion ability. As expected, BS-181 significantly inhibited cell migration and invasion ability in a dose-dependent manner (P<0.05, respectively; Figure 1).
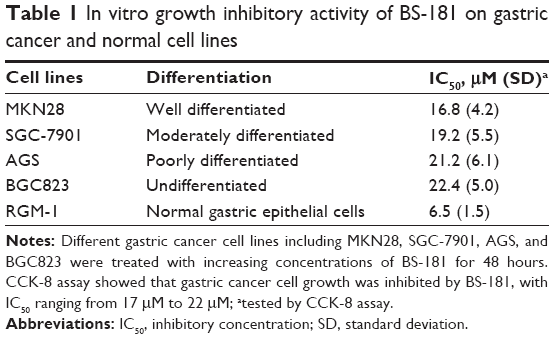 | Table 1 In vitro growth inhibitory activity of BS-181 on gastric cancer and normal cell lines |
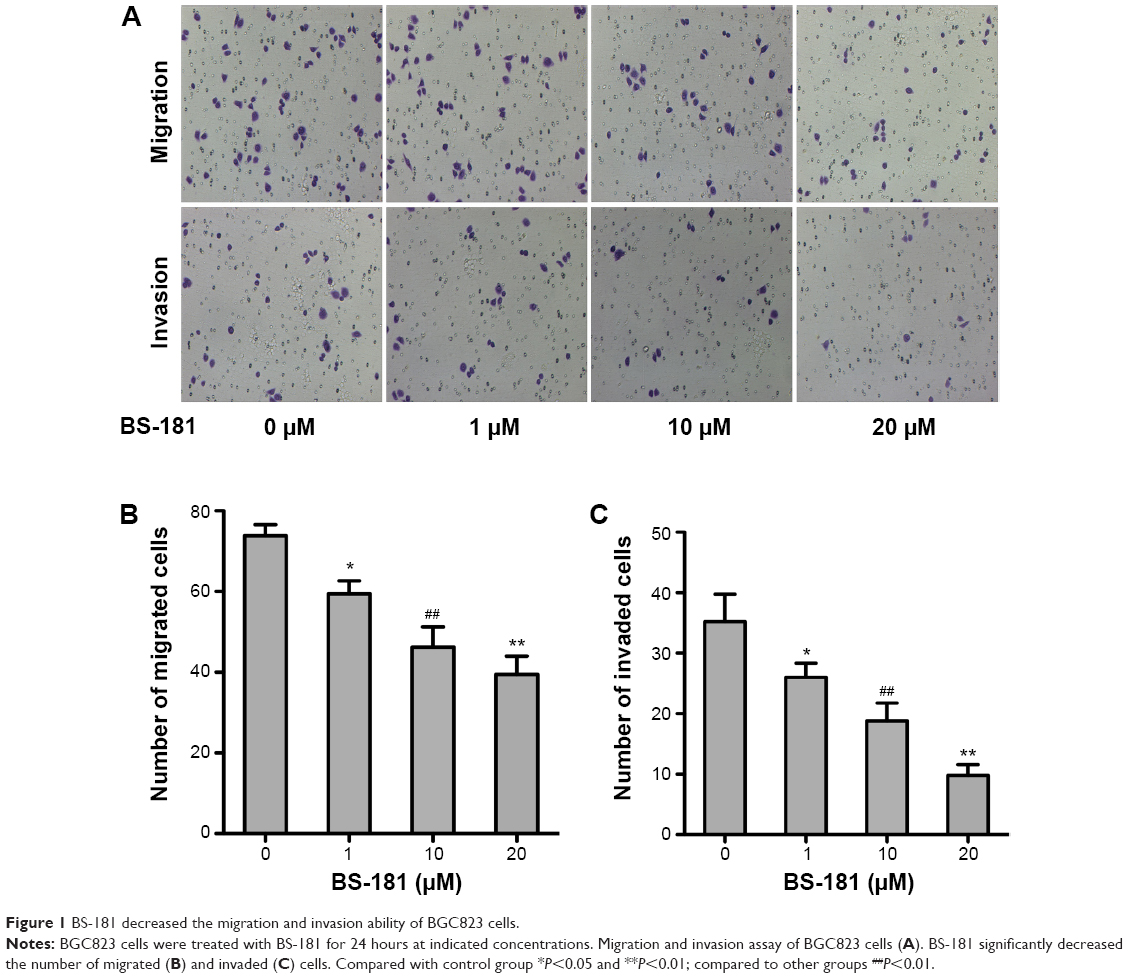 | Figure 1 BS-181 decreased the migration and invasion ability of BGC823 cells. |
BS-181-induced cell apoptosis and cell cycle arrest in gastric cancer cells
Cell apoptosis was determined using flow cytometry. Significant increases in apoptotic cells were observed in BS-181-treated BGC823 cells compared to control (P<0.05, respectively; Figure 2A). Our results also showed that BS-181-induced cell apoptosis in a dose- and time-dependent manner. Additionally, caspase-3 and Bax expressions have been significantly increased, while Bcl-2 level has been reduced in cells treated with BS-181 compared to control (P<0.05, respectively) (Figure 2B). These results indicated that BS-181 could induce apoptosis in GC cells. Furthermore, the inhibition of CDK7 activity led to a significant reduction of key antiapoptotic protein XIAP and cell cycle regulator cyclin D1 (P<0.05) (Figure 2C). Thus, BS-181 may regulate cell apoptosis and cell cycle progression via downregulating XIAP and cyclin expression in BGC823 cells. In the present study, cell cycle distribution was analyzed by flow cytometry (Figure 3). Treatment of BS-181 showed an increase in cells in G0/G1, accompanied by a reduction of cell population in S and G2/M phases. These results indicated that BS-181-induced cell cycle arrest in the G0/G1 phase and delayed the progression of the cell cycle.
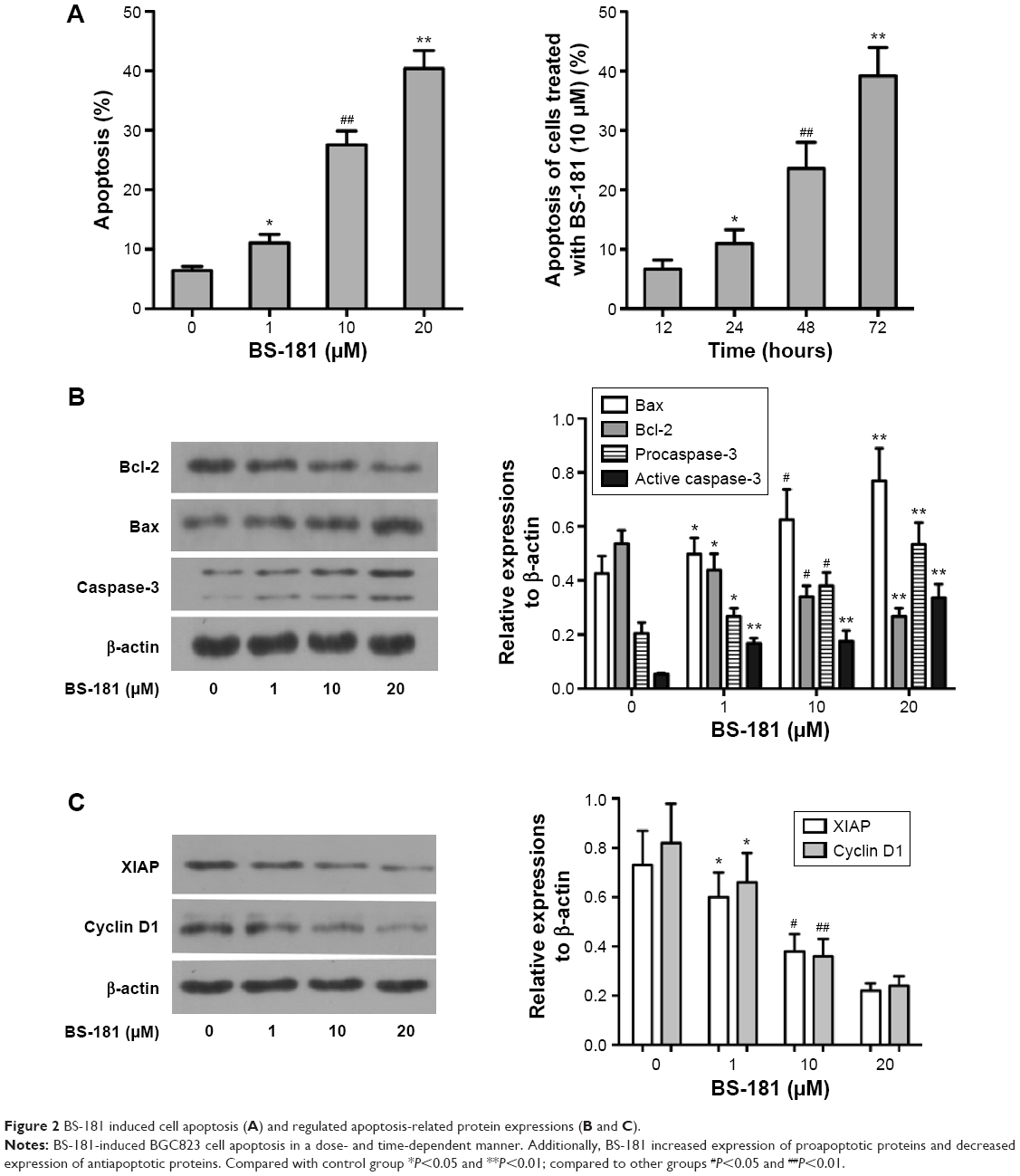 | Figure 2 BS-181 induced cell apoptosis (A) and regulated apoptosis-related protein expressions (B and C). |
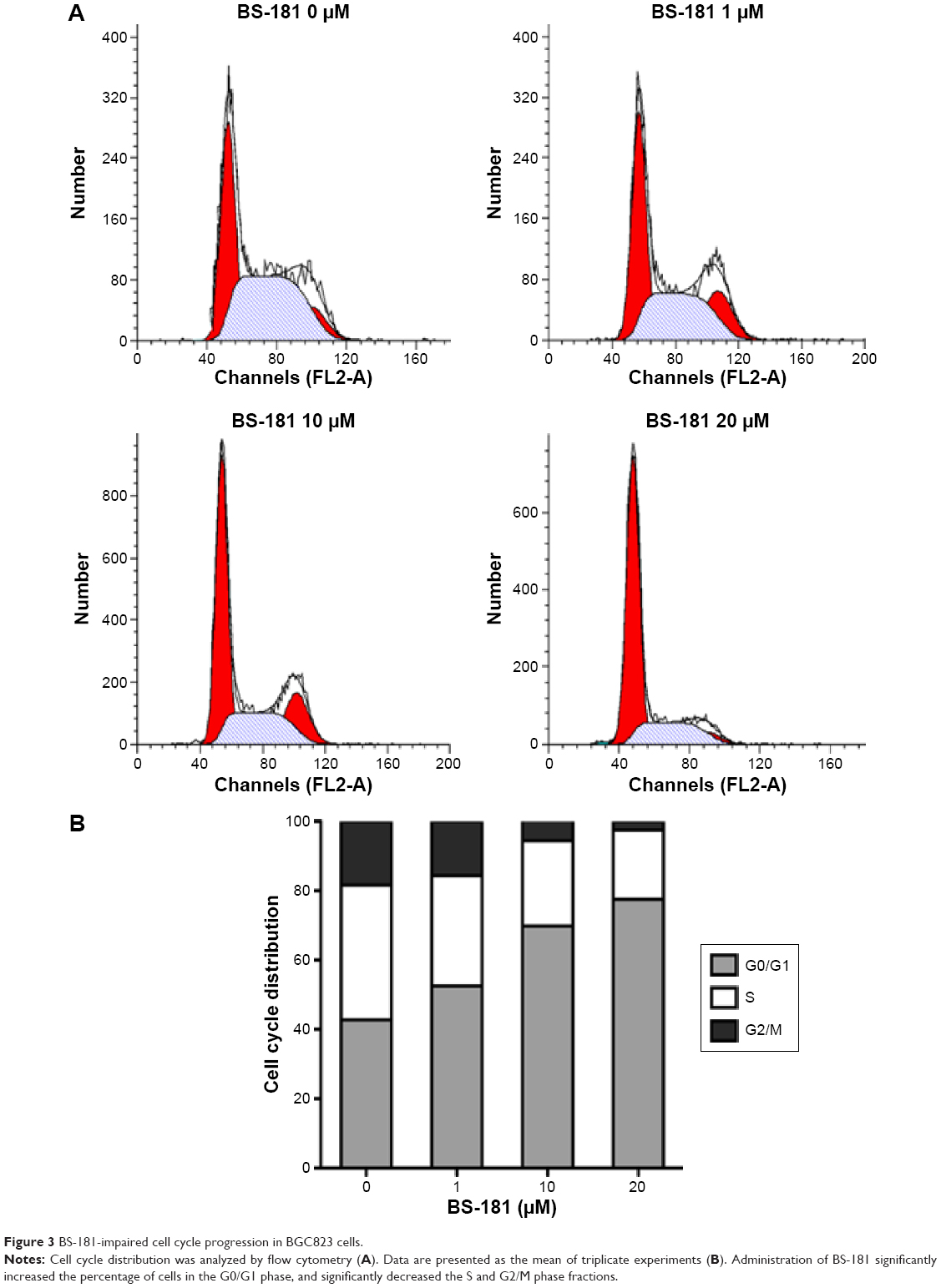 | Figure 3 BS-181-impaired cell cycle progression in BGC823 cells. |
BS-181-inhibited CDK7 activity in gastric cancer cells
BS-181 was synthesized as a specific CDK7 inhibitor. In this study, we also confirmed that BS-181 is a specific inhibitor of CDK7. As shown in Table 2, BS-181-inhibited CDK7 activity with an IC50 =0.019 μM, while the IC50 achieved with roscovitine was 0.48 μM. In addition, the IC50 values for inhibition of other CDKs by BS-181 were >1 μM, which is much more higher than that of CDK7. In addition, immunoblotting showed that BS-181 inhibited the phosphorylation of the RNA polymerase II CTD at the well-established CDK7 phosphorylation site serine 5 (P-Ser5) (Figure 4).
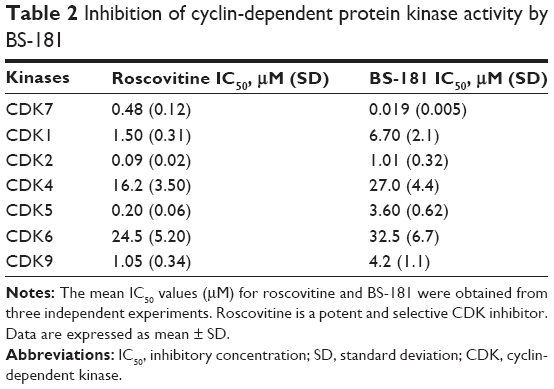 | Table 2 Inhibition of cyclin-dependent protein kinase activity by BS-181 |
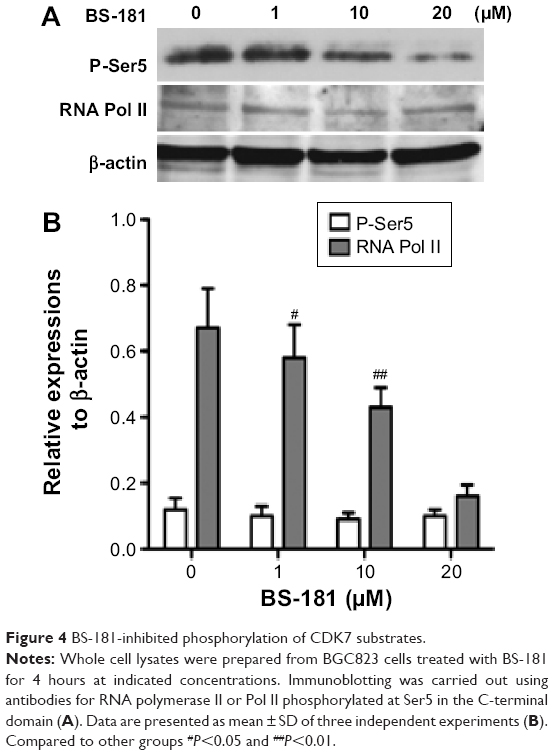 | Figure 4 BS-181-inhibited phosphorylation of CDK7 substrates. |
BS-181-inhibited tumor growth in vivo and increased survival rate
As previously described,7 the maximum tolerated single dose for BS-181 given intraperitoneally was 30 mg/kg/d, and a dose of 10 mg/kg/d or 20 mg/kg/d was well-tolerated in BALB/c-nu mice. In this study, mice received intraperitoneal injection of BS-181 twice daily with 5 mg/kg/d or 10 mg/kg/d to give daily doses of 10 mg/kg or 20 mg/kg, over a period of 2 weeks. In addition, another group of 15 rats received roscovitine (20 mg/kg/d) injection for a total of 14 days. We observed that tumor growth was significantly inhibited by BS-181 in a dose-dependent manner compared to the control group (P<0.05, respectively) (Figure 5A). However, there was no significant difference in mice body weights between groups during a 14-day observation (Figure 5B). This indicated that there was no apparent toxicity at a daily dose of 10 mg/kg or 20 mg/kg. In addition, all animals were kept for another 30 days for survival observation. As seen in Figure 5C, eight of ten mice (80%) died in the control group, five of ten (50%) died in the roscovitine group, while six of ten (60%, 10 mg/kg/d) and three of ten (30%, 20 mg/kg/d) mice died in BS-181-treated groups. The overall difference in survival rate between rats treated with or without BS-181 was significant (P<0.05, respectively).
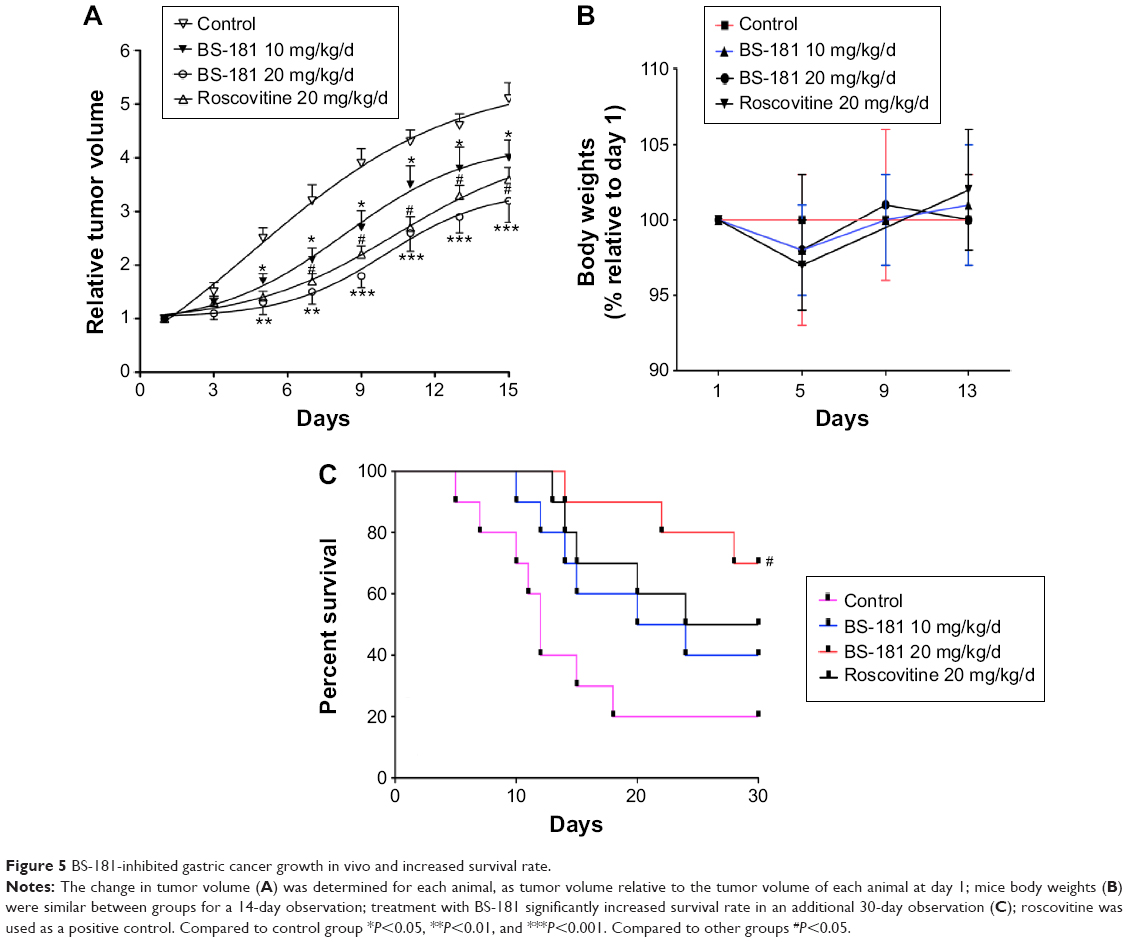 | Figure 5 BS-181-inhibited gastric cancer growth in vivo and increased survival rate. |
Discussion
GC represents an important burden on human health, with hundreds of thousands of newly diagnosed cases and related deaths in the world per year. Thus, the needs for improved understanding of the disease at the basic molecular level and the search for new effective drugs are imperative. CDK7, which plays critical roles in the regulation of transcription and cell cycle, is now an attractive target for the development of anticancer drugs.8 As a member of the transcriptional CDKs, CDK7 facilitates efficient transcriptional initiation, pause release, and elongation by phosphorylating the CTD of RNA Pol II. Moreover, CDK7 is also known to regulate the cell cycle of metazoans by controlling the activation of CDKs through T-loop phosphorylation.9 Inhibition of CDK7 perturbs transcriptional elongation, resulting in the downregulation of short-lived antiapoptotic proteins, such as Mcl-1 and XIAP, and cell cycle regulators such as cyclin D1.10 Previous study has shown that BS-181 selectively inhibited CDK7 by blockage of phosphorylation of the CDK7 substrate, Ser5, in the Pol II COOH-terminal domain, thus leading to anticancer effects in breast cancer cells.7 In the present study, we examined the antitumor effect of BS-181 in GC both in vitro and in vivo. We demonstrated that BS-181 strongly inhibited tumor cell growth in different GC cell lines of which the differentiation levels range from well-differentiated to undifferentiated. Considering that the undifferentiated GC has generally a worse prognosis, BGC823 cells were selected for the rest of our studies at cellular and molecular levels. Moreover, the results of in vivo studies demonstrate that BS-181 reduced the tumor volume in a time- and dose-dependent manner. To further explore the underlying mechanisms of cell growth inhibition, we conducted a cell cycle analysis by flow cytometry, which reveals that GC cells treated with BS-181 accumulated in the G0/G1 phase of the cell cycle, whereas the number of cells in S and G2/M phases significantly decreased. This coincided with the downregulation of cyclin D1 demonstrated by Western blotting. Cyclin D1 is a key regulator of G1 to S phase progression of the cell cycle. Accumulation of cyclin D1 in early to mid-G1 phase leads to activation of the kinases that phosphorylate and inactivate the tumor suppressor Rb, which is necessary for cell cycle progression through the G1 to S phases.11 Previous studies show that blockage of cyclin D1 prevented from cell cycle from proceeding into S phase,12 which is in concordance with the result of our studies. Cyclin D1 overexpression was found in a series of tumor types, including GC,13 breast cancer14 and colon cancer,15 and is a prognostic indicator associated with poor survival of cancer.16 Our results suggest that the downregulation of cyclin D1 can be part of the mechanisms in which BS-181 suppressed cell proliferation and tumor growth of GC.
Apart from the interference in the cell cycle, inhibition of CDK7 by BS-181 also induced cell apoptosis in GC cell lines, as shown by cytometry in our present study. Additionally, immunoblotting shows downregulation of XIAP, and expression of Bcl-2 was also reduced by BS-181. Bcl-2 is the prototype of a class of genes that contribute to tumor progression by inhibiting cell apoptosis. The family of Bcl-2-related proteins constitutes one of the apoptosis-regulatory gene products acting at the effector stage of apoptosis.17 X chromosome-linked inhibitor of apoptosis (XIAP) is a key member of the newly discovered family of intrinsic inhibitors of apoptosis (IAP) proteins. Unlike Bcl-2 that elicits the antiapoptotic effects by regulating the intrinsic pathway, IAP proteins act mainly by blocking cell death directly binding and inhibiting initiator as well as caspases. XIAP is known as a potent inhibitor of caspases-3, -7, and -9.18 As our results show, both Bax, a member of Bcl-2 family, and caspase-3 were reduced by BS-181. Thus, induction of apoptosis via Bcl-2 and XIAP pathways might contribute to the antitumor effects of BS-181 in GC.
Metastasis is responsible for most of the cancer mortalities. The capability of cell migration and invasion is considered to be one of the important determinants in the process of metastasis. In the current study, we found that BS-181 could decrease the GC cell migration and invasion in vitro. It was demonstrated that in breast cancer cells, cyclin D1 was required for TGF β-mediated migration,19 and higher Bcl-2 levels were also shown to be correlated with better outcomes in metastatic breast cancer.20 Based on observations of our present study, we postulated that the antiinvasion and antimigration effect of BS-181 might be partly due to the modulation of cell cycle and antiapoptotic mechanism. However, further evidences are required.
Conclusion
In conclusion, our study demonstrates that BS-181, the selective inhibitor of CDK7, prevents cell growth both in vitro and in vivo, induces the G1 arrest and apoptosis, and inhibits cell migration and invasion of GC. Therefore, BS-181 provides potent and selective CDK7 inhibitor with the potential as an antigastric cancer agent.
Acknowledgment
The authors would like to thank the Center for Medical Experiment (Zhongnan Hospital of Wuhan University, Wuhan, People’s Republic of China) for technical assistance.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Bertuccio P, Chatenoud L, Levi F, et al. Recent patterns in gastric cancer: a global overview. Int J Cancer. 2009;125(3):666–673. | ||
Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. | ||
Bregman DB, Pestell RG, Kidd VJ. Cell cycle regulation and RNA polymerase II. Front Biosci. 2000;5:D244–D257. | ||
Harper JW, Elledge SJ. The role of Cdk7 in CAK function, a retro-retrospective. Genes Dev. 1998;12(3):285–289. | ||
Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118(pt 22):5171–5180. | ||
Kaliszczak M, Patel H, Kroll SH, et al. Development of a cyclin-dependent kinase inhibitor devoid of ABC transporter-dependent drug resistance. Br J Cancer. 2013;109(9):2356–2367. | ||
Ali S, Heathcote DA, Kroll SH, et al. The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 2009;69(15):6208–6215. | ||
Larochelle S, Merrick KA, Terret ME, et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25(6):839–850. | ||
Chipumuro E, Marco E, Christensen CL, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159(5):1126–1139. | ||
Wesierska-Gadek J, Borza A, Komina O, Maurer M. Impact of roscovitine, a selective CDK inhibitor, on cancer cells: bi-functionality increases its therapeutic potential. Acta Biochim Pol. 2009;56(3):495–501. | ||
Zhao J, Pestell R, Guan JL. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol Biol Cell. 2001;12(12):4066–4077. | ||
Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7(5):812–821. | ||
Feakins RM, Nickols CD, Bidd H, Walton SJ. Abnormal expression of pRb, p16, and cyclin D1 in gastric adenocarcinoma and its lymph node metastases: relationship with pathological features and survival. Hum Pathol. 2003;34(12):1276–1282. | ||
Sutherland RL, Musgrove EA. Cyclins and breast cancer. J Mammary Gland Biol Neoplasia. 2004;9(1):95–104. | ||
Bondi J, Bukholm G, Nesland JM, Bukholm IR. Expression of non-membranous beta-catenin and gamma-catenin, c-Myc and cyclin D1 in relation to patient outcome in human colon adenocarcinomas. APMIS. 2004;112(1):49–56. | ||
Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220(2):292–296. | ||
Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3(6):614–620. | ||
Holcik M, Gibson H, Korneluk RG. XIAP: apoptotic brake and promising therapeutic target. Apoptosis. 2001;6(4):253–261. | ||
Dai M, Al-Odaini AA, Fils-Aime N, et al. Cyclin D1 cooperates with p21 to regulate TGFbeta-mediated breast cancer cell migration and tumor local invasion. Breast Cancer Res. 2013;15(3):R49. | ||
Smerage JB, Budd GT, Doyle GV, et al. Monitoring apoptosis and Bcl-2 on circulating tumor cells in patients with metastatic breast cancer. Mol Oncol. 2013;7(3):680–692. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.