")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Screening of novel HSP-inducing compounds to conserve cardiomyocyte function in experimental atrial fibrillation
Authors van Marion DMS , Hu X, Zhang D, Hoogstra-Berends F, Seerden JPG , Loen L, Heeres A, Steen H, Henning RH, Brundel BJJM
Received 12 June 2018
Accepted for publication 7 October 2018
Published 18 January 2019 Volume 2019:13 Pages 345—364
DOI https://doi.org/10.2147/DDDT.S176924
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Qiongyu Guo
Supplementary video S4: Beating heart wall of tachypaced Drosophila prepupa pretreated with GGA*-59 (same prepupa as S3, but different part of the body to view heart wall).
Views: 93
Denise MS van Marion,1,* Xu Hu,1,* Deli Zhang,1 Femke Hoogstra-Berends,2 Jean-Paul G Seerden,3 Lizette Loen,3 Andre Heeres,3,4 Herman Steen,5 Robert H Henning,2 Bianca JJM Brundel1
1Department of Physiology, Amsterdam Cardiovascular Sciences, VU University Medical Center, Amsterdam, The Netherlands; 2Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen, Groningen, The Netherlands; 3Syncom BV, Groningen, The Netherlands; 4Hanze University of Applied Sciences, Groningen, The Netherlands; 5Chaperone Pharma BV, Groningen, The Netherlands
*These authors contributed equally to this work
Background: The heat shock protein (HSP) inducer, geranylgeranylacetone (GGA), was previously found to protect against atrial fibrillation (AF) remodeling in experimental model systems. Clinical application of GGA in AF is limited, due to low systemic concentrations owing to the hydrophobic character of GGA.
Objectives: To identify novel HSP-inducing compounds, with improved physicochemical properties, that prevent contractile dysfunction in experimental model systems for AF.
Methods: Eighty-one GGA-derivatives were synthesized and explored for their HSP-inducing properties by assessment of HSP expression in HL-1 cardiomyocytes pretreated with or without a mild heat shock (HS), followed by incubation with 10 µM GGA or GGA-derivative. Subsequently, the most potent HSP-inducers were tested for preservation of calcium transient (CaT) amplitudes or heart wall contraction in pretreated tachypaced HL-1 cardiomyocytes (with or without HSPB1 siRNA) and Drosophilas, respectively. Finally, CaT recovery in tachypaced HL-1 cardiomyocytes posttreated with GGA or protective GGA-derivatives was determined.
Results: Thirty GGA-derivatives significantly induced HSPA1A expression after HS, and seven showed exceeding HSPA1A expression compared to GGA. GGA and nine GGA-derivatives protected significantly from tachypacing (TP)-induced CaT loss, which was abrogated by HSPB1 suppression. GGA and four potent GGA-derivatives protected against heart wall dysfunction after TP compared to non-paced control Drosophilas. Of these compounds, GGA and three GGA-derivatives induced a significant restoration from CaT loss after TP of HL-1 cardiomyocytes.
Conclusion: We identified novel GGA-derivatives with improved physicochemical properties compared to GGA. GGA-derivatives, particularly GGA*-59, boost HSP expression resulting in prevention and restoration from TP-induced remodeling, substantiating their role as novel therapeutics in clinical AF.
Keywords: atrial fibrillation, heat shock protein, Drosophila, proteostasis, geranylgeranylacetone
Introduction
Atrial fibrillation (AF) is the most common human cardiac arrhythmia with a prevalence of 2.7–6.1 million and 6.5–12.3 million in the USA and European Union, respectively, in 2010. This prevalence is expected to rise significantly due to the aging population.1 AF is a persistent disease, characterized by progressive electrical, structural, and contractile remodeling of cardiomyocytes, also referred to as electropathology.2,3 Current AF therapies are symptomatic and aim for rate control but do not prevent expansion of the arrhythmogenic substrate.4,5 As a consequence, most patients eventually develop longstanding persistent AF, which leads to a substantial increase in cardiac morbidity and mortality and warranty of life-long anticoagulant therapy. Despite extensive investigation of the molecular substrate of cardiac remodeling in AF, no effective therapy is available to date.
Research by us and others provide evidence that derailment of proteostasis is a key factor underlying electropathology and AF progression.6–10 Proteostasis, the homeostasis of protein production, function, and breakdown, is important for proper cell function. Accordingly, derailment of proteostasis is associated with AF but also with many age-related protein-misfolding diseases, including Alzheimer’s, Parkinson’s, and Huntington’s disease.11 Derailment of proteostasis contributes importantly to cardiomyocyte remodeling and predisposes to AF in experimental models and in AF patients.6,7,9 The most important chaperones to maintain a balanced proteostasis are the heat shock proteins (HSPs). During stress, the heat shock response (HSR) is stimulated by activation of heat shock (HS) transcription factors, of which heat shock factor 1 (HSF1) is the major regulator of HSP transcription in eukaryotes.12 HSPs consist of five HSP families, ie, HSPA (HSP70), HSPB (small HSPs), HSPC (HSP90), HSPD (HSP60), and DnaJB (HSP40), each with several family members, (specific) co-factors in various cellular localizations, with distinct and overlapping functions.13,14 The family of HSPBs is probably the most important in maintaining proteostasis in cardiomyocytes.15 Cardiomyocytes express high levels of HSPBs which localize with contractile and microtubule proteins, thereby stabilizing the cardiomyocyte structure, and conserving the contractile and electrophysiological function of the atrial cardiomyocytes.16–20 Although HSPB1 levels are induced in atrial tissue samples of patient with paroxysmal AF, HSPB1 levels get exhausted in patients with (longstanding) persistent AF. In addition, in these patients, HSPB1 levels in atrial tissue correlate inversely with the amount of structural remodeling, suggesting that the HSR becomes exhausted in time resulting in derailment of proteostasis, structural remodeling, and AF progression.10,16 In line, boosting of the endogenous HSR with drugs may constitute an emerging therapeutic strategy for clinical AF. Securing HSP levels at an adequate level may limit the expansion of the AF substrate for the induction of AF and the ensuing progression of paroxysmal to persistent AF.8 A well-known HSP-inducing compound is geranylgeranylacetone (GGA). GGA is a nontoxic acyclic isoprenoid compound with a retinoid skeleton that is originally used as an antiulcer drug in Asian countries.21,22 GGA induces HSPs in various tissues, including gastric mucosa, intestine, liver, myocardium, retina, and central nervous system.23,24 The protective effect of GGA-induced HSP expression on tachycardia-induced cardiomyocyte remodeling has been observed in atrial cardiomyocytes, and a Drosophila model for AF, suggesting that the induction of HSPs by GGA might have potential value for clinical AF.7,16,25,26 Furthermore, GGA treatment protected from cardiomyocyte remodeling and tachypacing (TP)-induced AF promotion in a dog model for (acute) atrial ischemia and in a heart failure model in rabbits.7,27,28 Notwithstanding the protective effects, the poor physicochemical properties of GGA, including its lipophilic nature (LogP value 6.54) and limited solubility, pose a serious disadvantage to its drugability. The gut mucosal distribution pattern owing to GGA’s hydrophobic character hinders its systemic bioavailability29,30 and consequently a relative high daily oral dosage of 120 mg/kg was required to treat dogs.7 To overcome these disadvantages, various GGA-derivatives with improved physicochemical properties were synthesized and tested for their ability to induce an HSR in HL-1 cardiomyocytes and confer protection against TP-induced contractile dysfunction in HL-1 cardiomyocytes and Drosophila. Furthermore, favorable GGA-derivatives were studied on their improvement of recovery from contractile dysfunction in tachypaced HL-1 cardiomyocytes.
Materials and methods
Synthesis of GGA-derivatives
The synthesis of the most potent GGA-derivatives is described in Supplementary materials.
HL-1 mouse atrial cardiomyocytes culture, TP, and CaT measurements
HL-1 atrial cardiomyocytes, derived from adult mouse atria, were obtained from Dr William Claycomb (Louisiana State University, New Orleans, USA, institutional approved MTA with laboratory of Brundel)31 and maintained as described previously.9 HL-1 cardiomyocytes, seeded on coverslips, were subjected to TP as described before7,16 and calcium transient (CaT) were measured. Detailed description can be found in Supplementary materials.
GGA and GGA-derivative treatment and HS
HL-1 cardiomyocytes, seeded into six wells plates, were treated with 10 μM GGA or GGA-derivative or control (dimethylsulfoxide, DMSO). For a more detailed description on drug and HS treatment, see Supplementary materials.
Drosophila TP and drug treatment
Wild-type W1118 Drosophila larvae were treated with 100 μM GGA or GGA-derivatives, prepupae were tachypaced, and heart wall contractions were assessed, as described in more detail in Supplementary materials.
Protein isolation and Western blot analysis
HL-1 cardiomyocytes were lysed and equal amounts of protein were analyzed by Western blot analysis as described in detail in Supplementary materials.
RNA isolation and PCR analysis
Total RNA was extracted from HL-1 cardiomyocytes and PCR analysis (for primers see Table 1) was performed according to the procedures as described in Supplementary materials.
 | Table 1 Primers for real-time reverse-transcriptase PCR |
siRNA HSPB1 knock-down
HL-1 cardiomyocytes were transfected with CD8 and siRNA-HSPB1 constructs or mock siRNA and subsequently treated with GGA or GGA-derivatives before TP and CaT measurements. Refer to Supplementary materials for a complete description of the procedures.
Statistics
Data are presented as mean ± SD or SEM. All experiments were performed at least in duplicate series. Individual group mean differences were evaluated with the Student’s t-test. Multiple-group comparisons were obtained by ANOVA with Bonferroni-corrected post hoc t-tests. A 2-tailed P<0.05 was considered statistically significant. GraphPad version 7 was used for all statistical evaluations.
Results
Synthesis of a GGA-derivative library
In order to overcome the high lipophilicity of GGA, we first truncated the east side of the molecule to geranylacetone (GA) (Figure 1). Based on this parent structure, a library of 81 compounds was prepared, considering the Lipinski rule of five32 (Table 2). Not only the east and west sides of the molecule were varied but also several bio-isosters of the central keto-moiety of GA were designed and prepared. The keto-moiety of GGA and truncated derivatives was replaced by isosteric groups, such as oxime, amide, sulfonamide, ester, hydroxyl, pyrazolone, pyrazole, and oxazole moieties. For the east and west side variations, different alkyl chains were chosen, sometimes containing additional functional groups (hydrogen bond donors and acceptors), including aromatics. The strategy used to prepare GGA-derivatives is depicted in Figure 1; see Supplementary materials for a detailed description of synthesis. Following this strategy, almost all GGA-derivatives reveal an improved LogP value, compared to the mother compound GGA (LogP 6.54), having a molecular weight below 500 (Table 2).
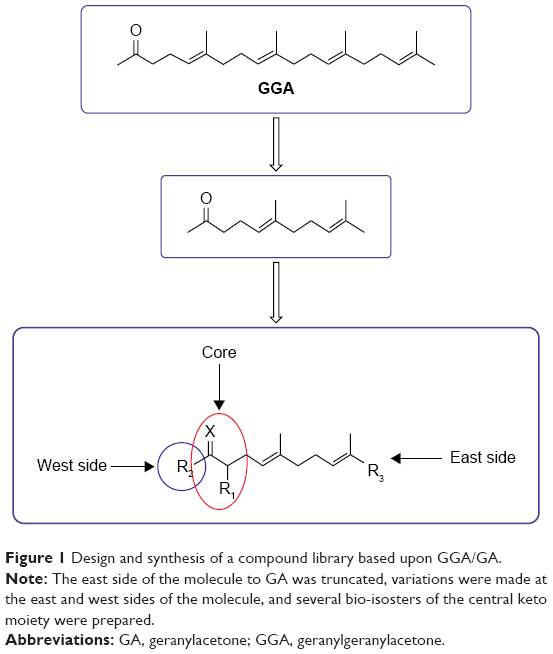 | Figure 1 Design and synthesis of a compound library based upon GGA/GA. |
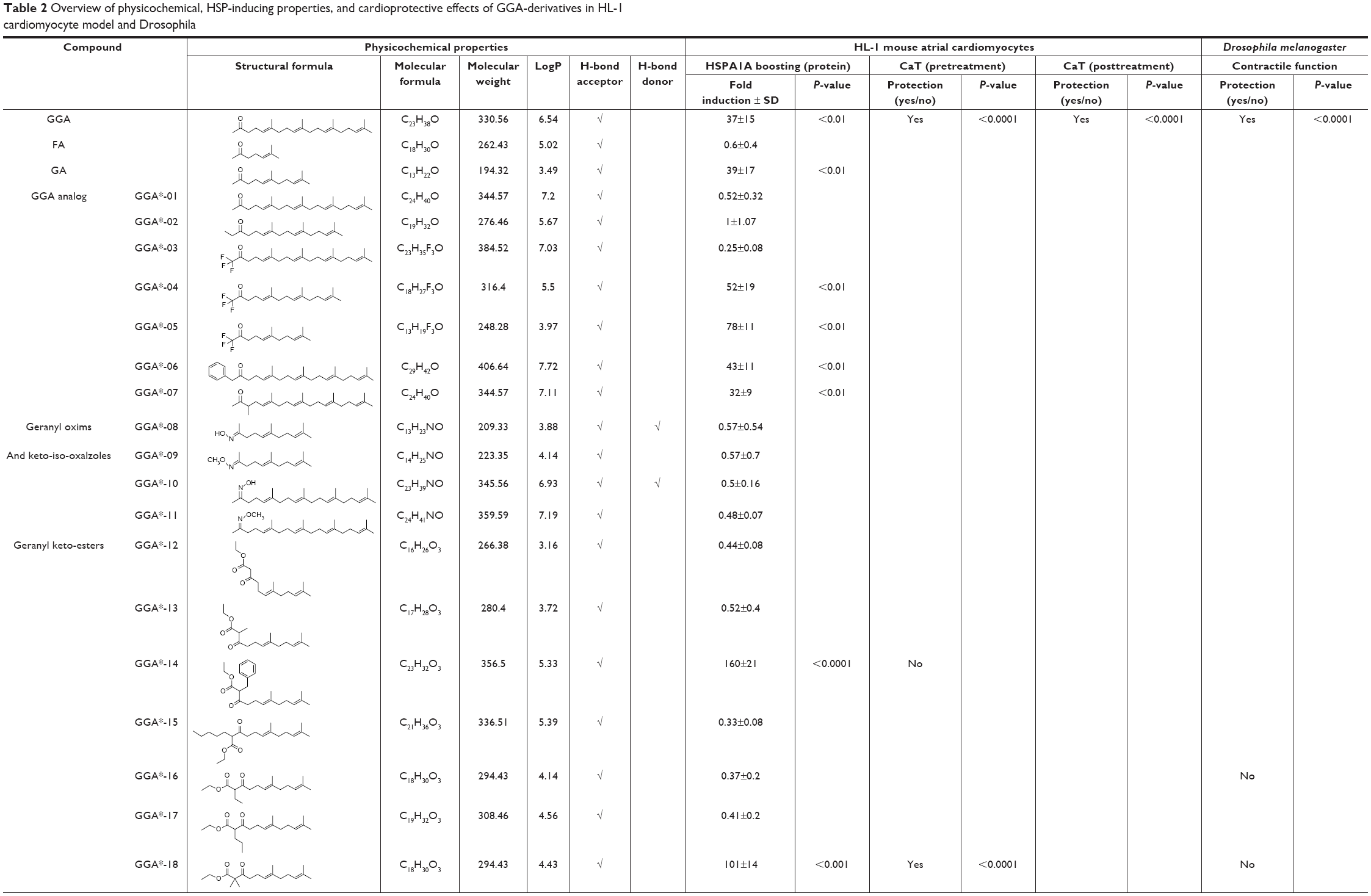 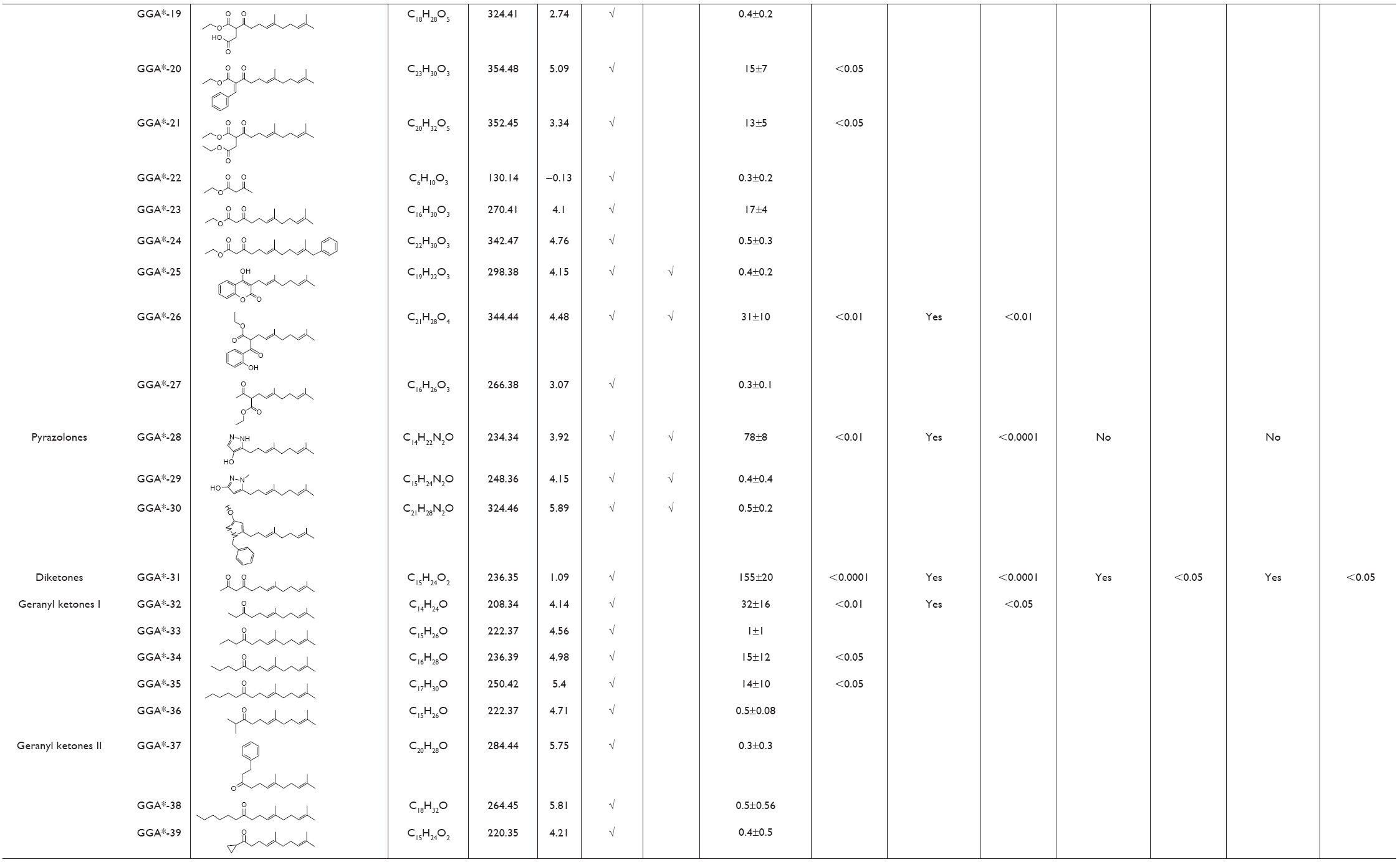 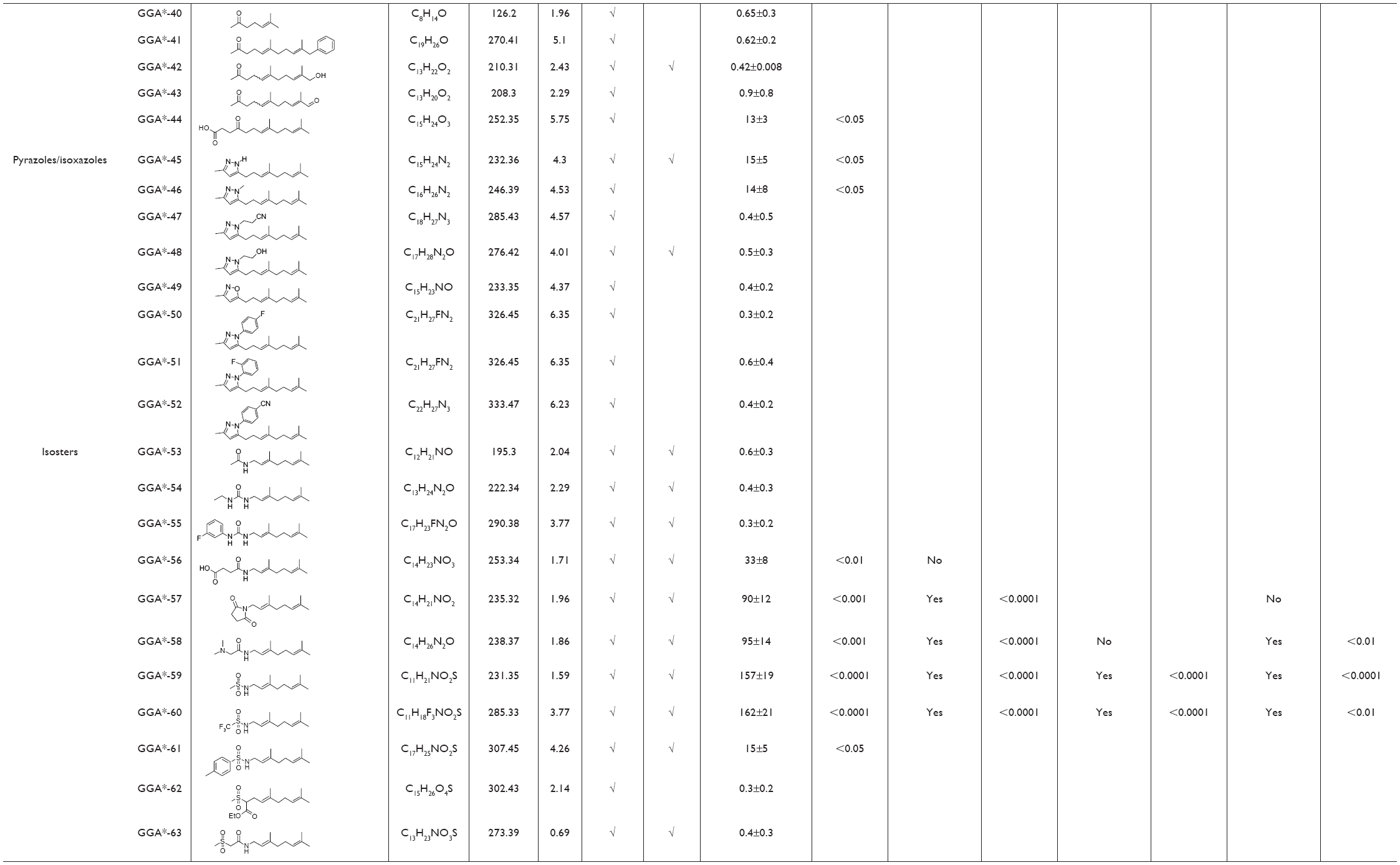 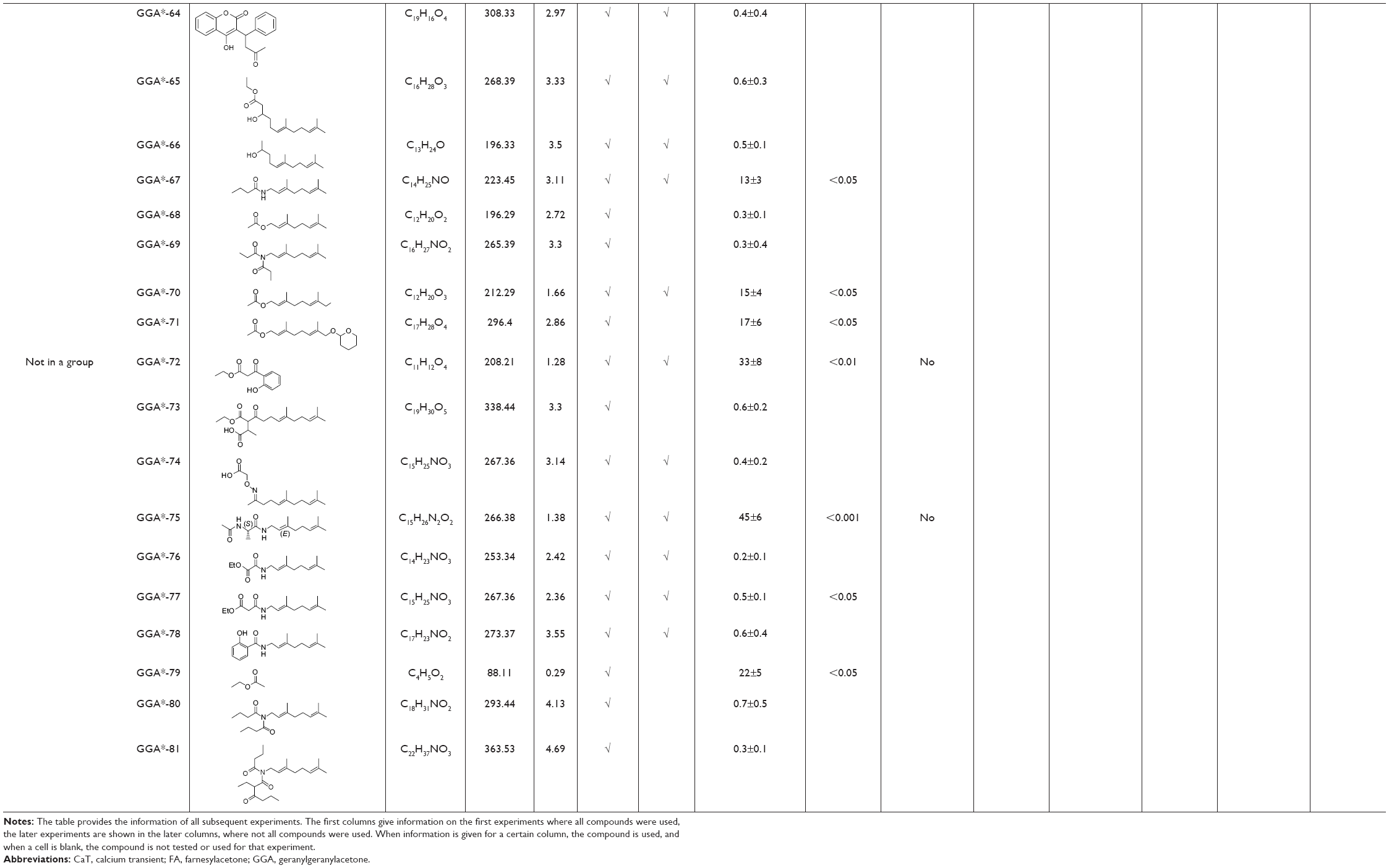 | Table 2 Overview of physicochemical, HSP-inducing properties, and cardioprotective effects of GGA-derivatives in HL-1 cardiomyocyte model and Drosophila |
GGA and GGA-derivatives boost HSF1-related HSP expression in HL-1 cardiomyocytes
HSP-inducing properties of the 81 synthesized GGA-derivatives and GGA were explored in HL-1 cardiomyocytes (Table 2). Since HSPA1A is expressed at low basal levels (compared to abundant basal levels of HSPB1) in nonstressed control HL-1 cardiomyocytes and becomes strongly upregulated upon a HS, HSPA1A was used as a read-out. When nonstressed control HL-1 cardiomyocytes were treated for 6 hours with 10 μM GGA or GGA-derivatives, a minor induction of HSPA1A expression was observed by Western blot analysis (Figure S1). Posttreatment with 10 μM GGA or eighty-one GGA-derivatives after a mild, nonlethal HS (10 minutes 44°C, 10 minutes recovery 37°C) revealed 30 derivatives that significantly elevated HSPA1A expression compared to control. The HSPA1A boosting effect of GGA-derivatives was comparable to or significantly larger compared to GGA (Figure 2A and B). The HSPA1A boosting effect could not be linked to a specific group of molecular structures of GGA-derivatives (Figure S2). Next to boosting of HSPA1A expression levels, GGA-derivatives elevated mRNA expression of HSF1-mediated HSPs, including HSPA1A, HSPB1, DNAJB1 and HSPCA, while mRNA levels of the non-HSF1-related HSPA5 were unaffected (Figure S3). Together, the data suggest that GGA and 30 GGA-derivatives boost HSP expression in HL-1 cardiomyocytes most likely via HSF1 regulation. At identical concentrations, seven GGA-derivatives boosted protein abundance of HSPA1A to levels exceeding those of GGA (Figure 2B).
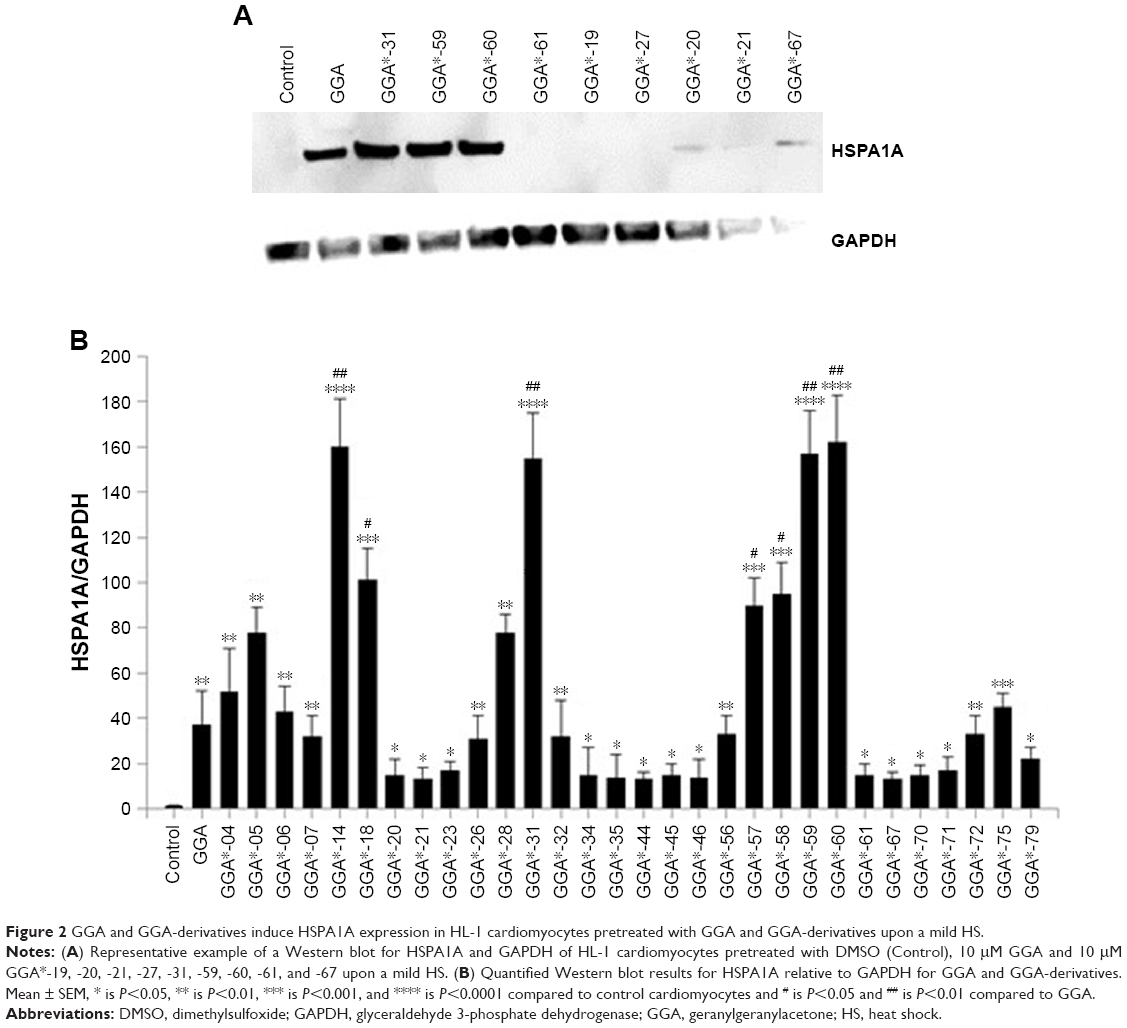 | Figure 2 GGA and GGA-derivatives induce HSPA1A expression in HL-1 cardiomyocytes pretreated with GGA and GGA-derivatives upon a mild HS. |
GGA and GGA-derivatives protect against CaT loss in tachypaced HL-1 cardiomyocytes
To examine whether GGA-derivatives, like GGA, protect against contractile dysfunction, HL-1 cardiomyocytes were pretreated with 12 GGA-derivatives with strong HSPA1A-boosting properties (Table 3) for 8 hours, followed by 8 hours TP (4.5 Hz) or normal pacing (1 Hz). Previous studies revealed that GGA induces HSPA1A expression up to 24 hours upon treatment.7,16 Contractile function was determined by measuring CaT. TP induced significant CaT loss compared to normal pacing, which was prevented by GGA and GGA*-18, -26, -28, -31, -32, -57, -58, -59, and -60 (Figure 3).
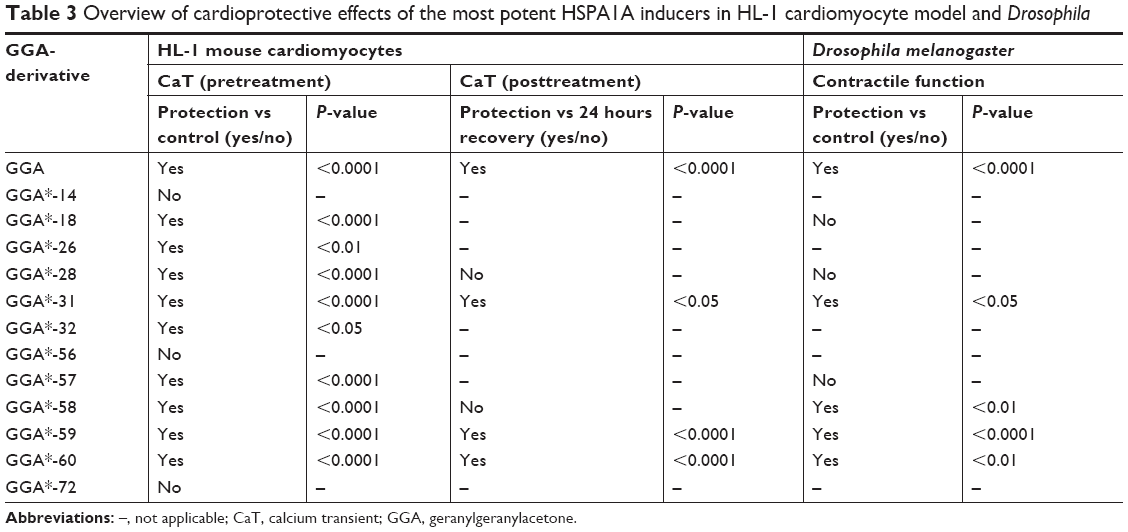 | Table 3 Overview of cardioprotective effects of the most potent HSPA1A inducers in HL-1 cardiomyocyte model and Drosophila |
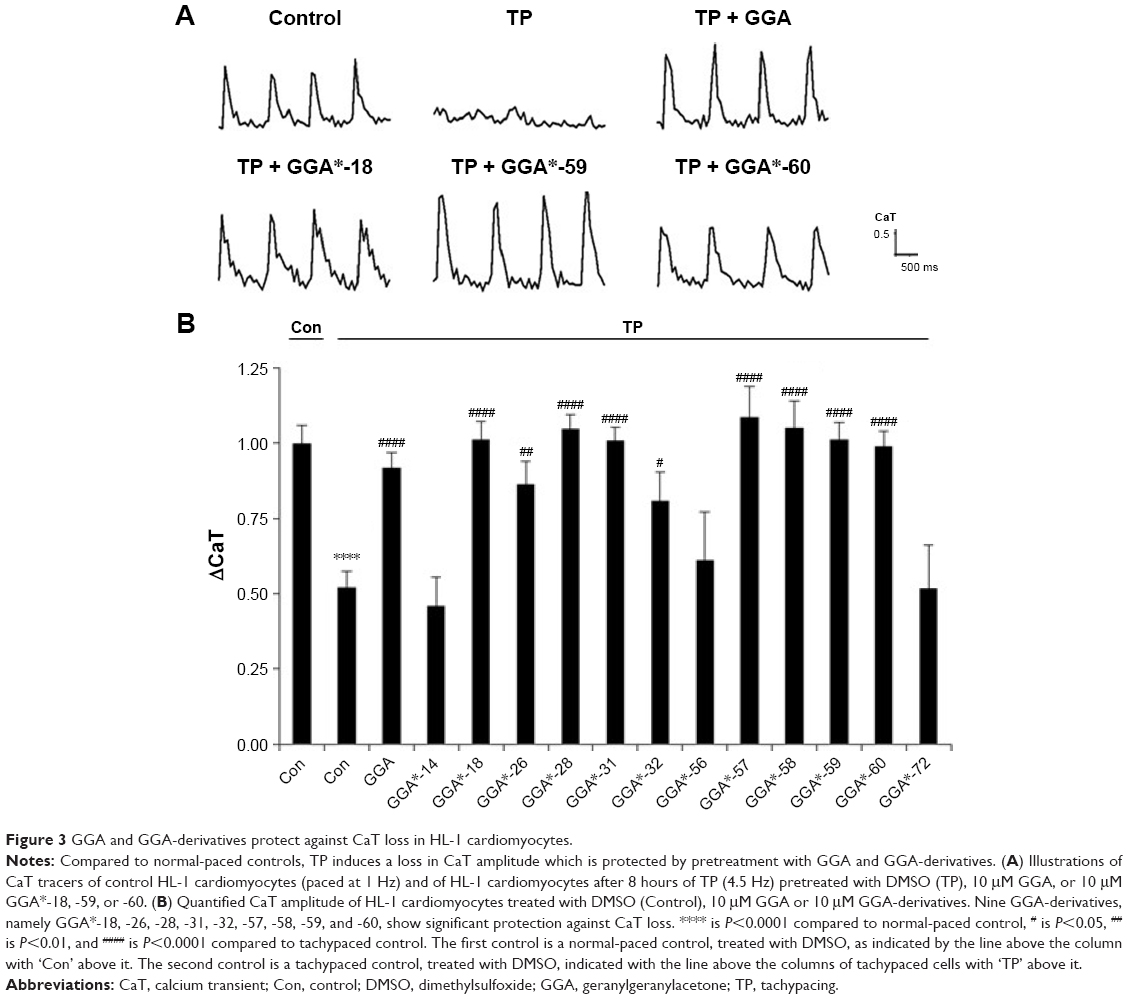 | Figure 3 GGA and GGA-derivatives protect against CaT loss in HL-1 cardiomyocytes. |
Suppression of HSPB1 abrogated the protective effect of the GGA-derivative in HL-1 cardiomyocytes
Since previous studies implicate HSPB1 as a crucial HSP in the protective effect of GGA against TP-induced CaT loss,7 we tested whether the protective effect of GGA-derivatives acts via HSPB1. Hereto, HL-1 cardiomyocytes were transfected with siRNA-HSPB1 constructs or control, followed by GGA or GGA-derivative treatment for 8 hours, and TP or nonpacing for 8 hours. siRNA treatment successfully suppressed HSPB1 24 hours after transfection (Figure 4A). siRNA-mediated suppression of HSPB1 levels completely abrogated the protective effect of GGA or GGA-derivatives (Figure 4B and C), indicating that their protective effect is dependent on HSPB1.
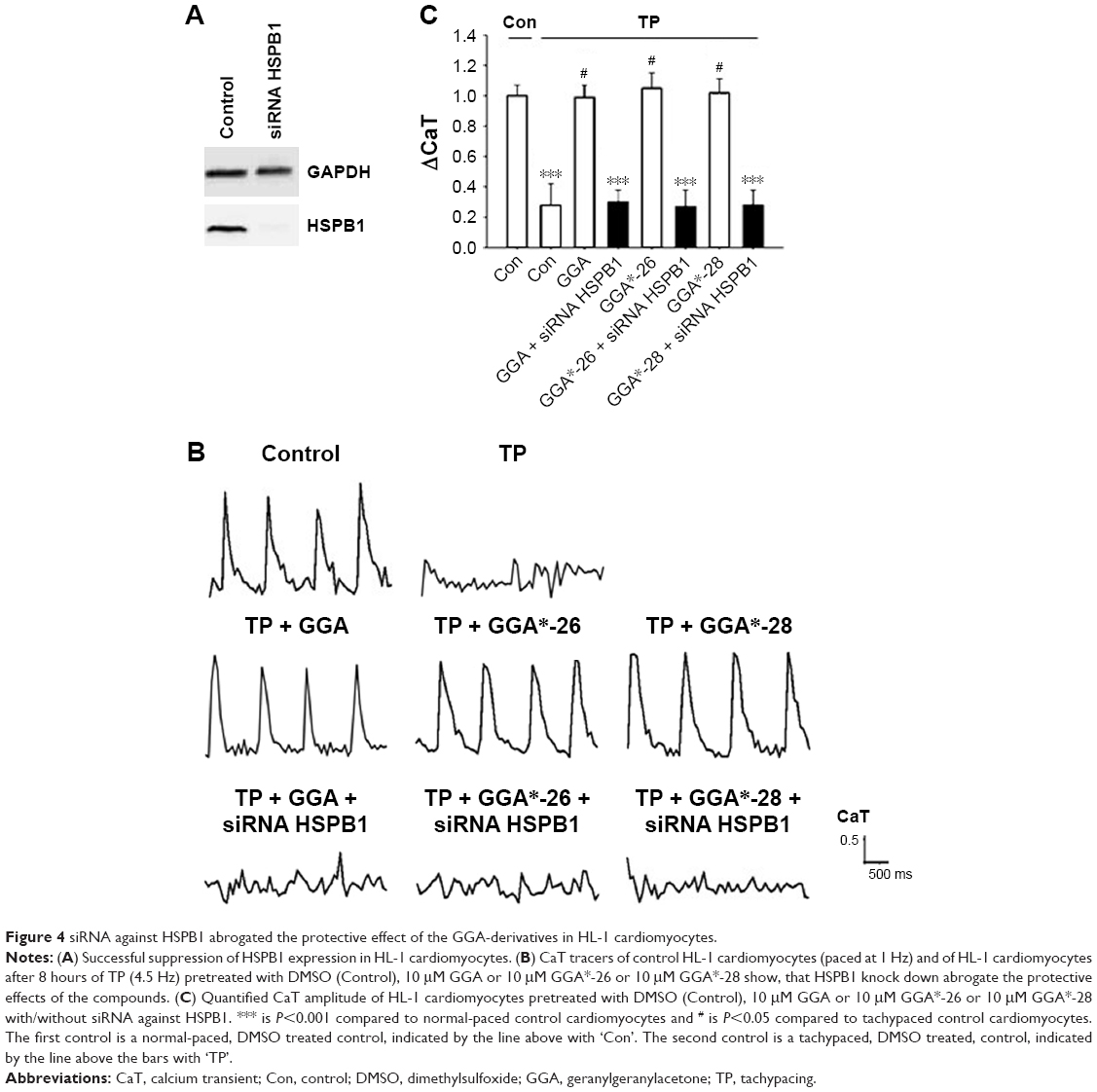 | Figure 4 siRNA against HSPB1 abrogated the protective effect of the GGA-derivatives in HL-1 cardiomyocytes. |
GGA and GGA-derivatives protect against contractile dysfunction in Drosophila
Next, we assessed whether protective GGA-derivatives, identified in HL-1 cardiomyocytes, also protected Drosophila from TP-induced contractile dysfunction. Hereto, the seven most protective GGA-derivatives (GGA*-18, -28, -31, -57, -58, -59, and -60, Table 3) and, as a negative control, one non-HSPA1A boosting GGA-derivative (GGA*-16), were examined. All compounds were without effect on basal heart function (Figure S4). TP for 20 minutes at 5 Hz induced a significant dysfunction of heart wall contractility (Figure 5 and Videos S1 and S2), which was prevented by GGA and GGA*-31, -58, -59, and -60 (Figure 5 and Videos S3 and S4). Expectedly, GGA*-16 was without effect, because of its lack of protection in HL-1 cardiomyocytes (Figure 5). These findings indicate that four GGA-derivatives protect against TP-induced contractile dysfunction in the Drosophila model for AF.
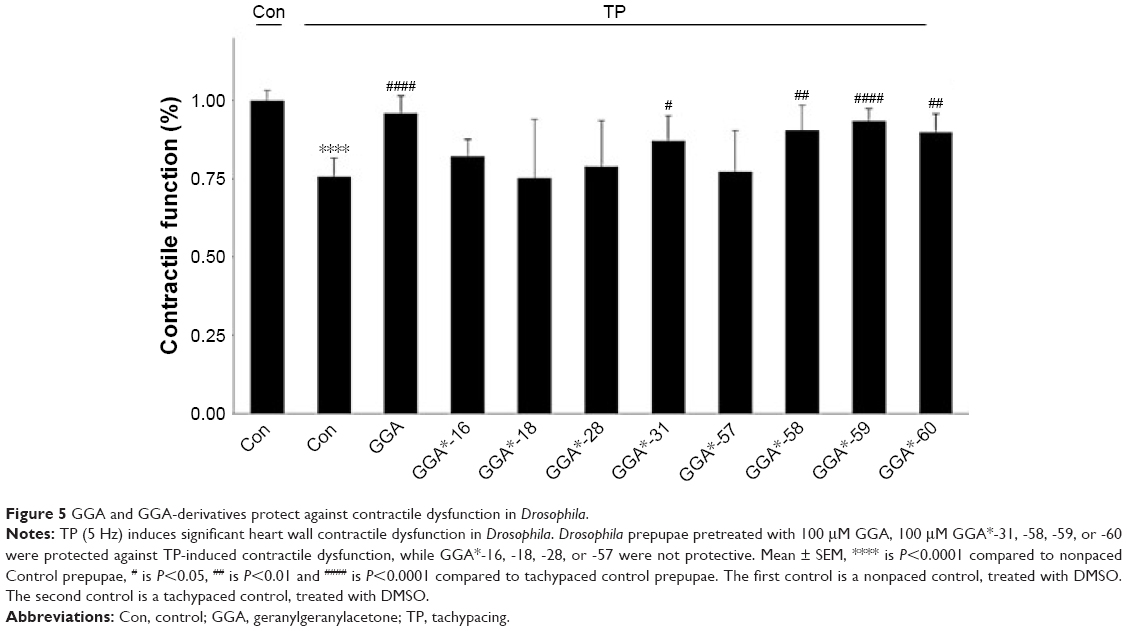 | Figure 5 GGA and GGA-derivatives protect against contractile dysfunction in Drosophila. |
GGA-derivatives restore contractile function after TP
Since most patients with AF reveal cardiomyocyte remodeling at the moment of diagnosis, compounds that restore contractile function are of high clinical relevancy. Therefore, GGA-derivatives were tested for their ability to restore CaT loss in the tachypaced HL-1 cardiomyocyte model. Hereto, HL-1 cardiomyocytes were tachypaced at 4.5 Hz for 8 hours, followed by 24 hours recovery and treatment with GGA or GGA-derivatives (GGA*-28, -31, -58, -59, and -60, Table 3). TP resulted in a significant CaT loss, which even further decreased after 24 hours recovery (Figure 6A and B). However, tachypaced HL-1 cardiomyocytes posttreated with 10 μM GGA*-31, -59, or -60, or GGA significantly restored CaT, compared to untreated HL-1 cardiomyocytes. Restoration of CaT by GGA and GGA*-59 was significantly larger than GGA*-31 and -60 at 10 μM, while GGA*-28 and -58 did not restore contractile function (Figure 6A and B). These findings indicate that GGA and GGA*-59 restore cardiomyocyte function after TP, whilst GGA*-28, -31, -58, and -60 do not or to a lesser extent.
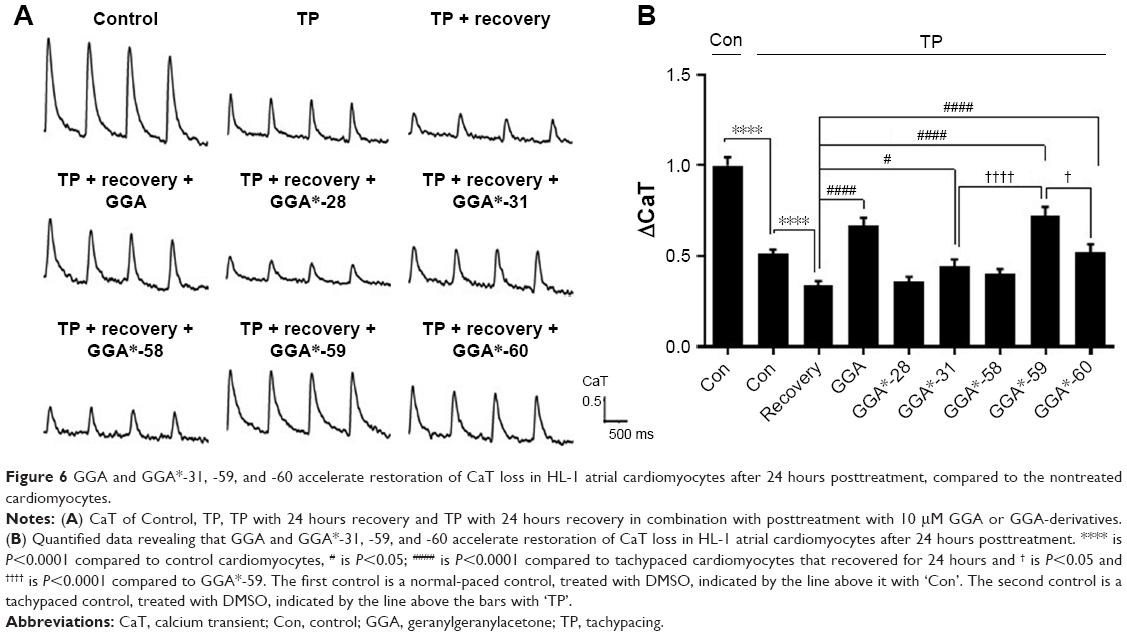 | Figure 6 GGA and GGA*-31, -59, and -60 accelerate restoration of CaT loss in HL-1 atrial cardiomyocytes after 24 hours posttreatment, compared to the nontreated cardiomyocytes. |
HSF1 activation by GGA-derivative
Finally, to address the mode of action of the top functional GGA-derivative, we assessed the effect of GGA*-59 (10 μM) on the main effector of GGA effects, ie, HSF1 phosphorylation status,33 by assessing HSF1 mobility on SDS-PAGE. HS induces activation of HSF1, as indicated by decreased mobility because of HSF1 hyperphosphorylation,34 lasting for up to 1 hour (Figure 7A and B). Treatment with GGA*-59 enhances the ratio of hyperphosphorylated HSF1/HSF1 (Figure 7A and B), thus further enhancing HSF1 activation. In accord, treatment with GGA*-59 enhances subsequent HSPA1A protein expression (Figure 7C and D). These data signify that GGA*-59 boosts HSPs via HSF1 activation.
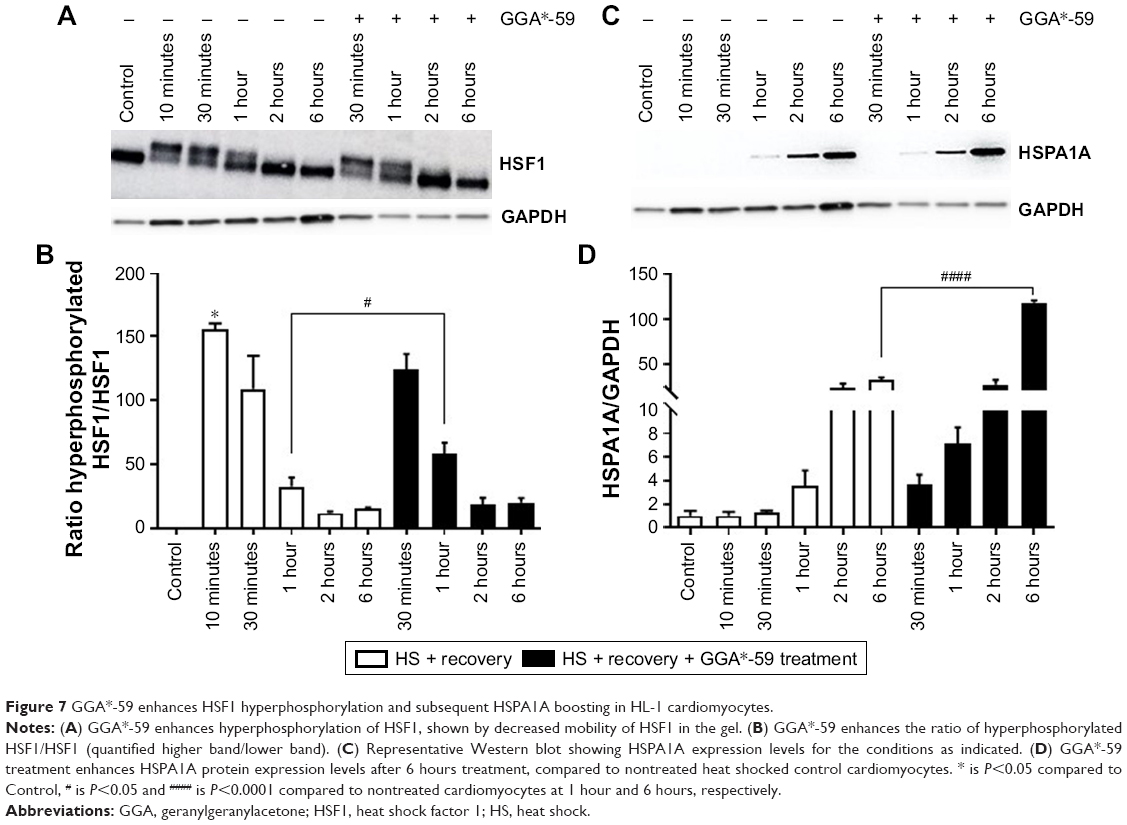 | Figure 7 GGA*-59 enhances HSF1 hyperphosphorylation and subsequent HSPA1A boosting in HL-1 cardiomyocytes. |
Discussion
Previous studies identified the HSP-booster GGA to protect against contractile dysfunction and remodeling in experimental models for AF.7,16,25,26,35 However, its lipophilic character hampers systemic bioavailability in patients.30 Since HSPs may limit AF progression and possibly reverse remodeling in clinical AF, we here report on the structural properties of novel derivatives of GGA. Eighty-one GGA-derivatives were synthesized by shortening the lipophilic backbone of GGA and modifying the east, west, and central part of the molecule. Seven out of 30 GGA-derivatives that significantly induced HSPA1A expression after a mild HS were superior to GGA, at the given dose, in terms of higher HSPA1A protein expression. Nine out of the 12 most potent HSPA1A boosters (GGA*-18, -26, -28, -31, -32, -57, -58, -59, and -60) protected against TP-induced loss of CaT in HL-1 cardiomyocytes and four GGA-derivatives (GGA*-31, -58, -59, and -60) also protected against contractile dysfunction in Drosophila prepupae, which consumed GGA-derivative supplemented food during their larval stage as a pretreatment. Protective effects of GGA-derivatives seem dependent of HSPB1, because siRNA against HSPB1 abrogates the protection from TP-induced CaT loss, as previously found for GGA.7 Intriguingly, posttreatment, ie treatment with GGA and GGA-derivatives (GGA*-31, -59, and -60 of which GGA*-59 was superior compared to GGA*-31 and -60) directly after TP for the duration of 24 hours, restored TP-induced CaT loss in HL-1 cardiomyocytes. Not all GGA-derivatives which revealed protective effects against CaT loss in HL-1 cardiomyocytes were also protective upon pretreatment in tachypaced Drosophila or could restore cardiomyocyte function upon 24 hours posttreatment (Table 3). This may be due to reduced stability of the derivative. Future studies should elucidate the pharmacokinetics of potent GGA-derivatives. Nevertheless, we identified one GGA-derivative, GGA*-59, with improved physicochemical properties, that boosts HSPs, and both protects from TP-induced contractile dysfunction, and restores this after TP. Consequently, our analysis indicates GGA*-59 as a GGA-derivative with substantial potential for clinical applications.
Improved physicochemical properties of GGA-derivatives
The clinical use of GGA to treat AF is hampered by its lipophilicity and therefore high dosages have to be used.7,36 Here we report a library of GGA-derivatives, created considering the Lipinski rule of five,32 to achieve more druggable analogs of GGA while maintaining in vitro/in vivo activity. The improved physicochemical properties of the GGA-derivatives, essentially having LogP values between 0 and 5 and molecular weight below 500, increase their solubility and are expected to improve passage of lipid bilayers, most likely resulting in increased uptake in the intestine and improved distribution to the body and cells,32 thereby improving clinical applicability.
The derivatives that boosted HSP expression and protected against and induced recovery (only GGA*-31, -59, and -60) from TP-induced contractile dysfunction were GGA*-57, -58, -59, and -60 (isosters from GGA), GGA*-14, -18, and -26 (“geranyl keto esters”) and GGA*-28 (“a geranyl ketone”). Despite the fact that the most favorable compounds are divided into groups based on the isosteric (central part) and east- and west-side modifications, we were unable to attribute this to specific structural features of the compounds. Further research/synthesis is needed to expand the hits to series of lead compounds with clinical potential.
Preserving proteostasis: possible mode of action of GGA-derivatives
Given the similarity of the actions of the efficacious GGA-derivatives to GGA effects, the most likely mode of action of our derivatives comprises their preservation of proteostasis by activation of HSF1 and subsequent boosting of HSPs expression. The observed increase in gene expression levels of various HSF1-regulated genes by GGA-derivatives, including HSPA1A, HSPB1, DNAJB1, HSPCA, and absence of increase in the HSF1-independent HSPA5, are similar to GGA. In addition, similar to GGA,33 GGA*-59 prolonged HSF1 activation, as shown by its increased hyperphosphorylation. Collectively, these data indicate that GGA-derivatives boost HSP expression by prolonging HSF1 activation, resulting in its extension of binding to the HS element (HSE) in the promotor regions of hsp genes, thus prolonging hsp gene transcription and HSP protein expression. At least, 30 GGA-derivatives significantly induced HSPA1A protein expression of which seven demonstrated improved HSP-boosting properties compared to GGA.
The precise nature of the molecular pathway by which GGA and GGA-derivatives prolong hyperphosphorylation of HSF1 is still subject of speculation. It is known that geranyl-groups act as posttranslational modifiers of proteins, and thereby regulate protein function.37 Natural occurring prenylation with C15 (farnesyl) or C20 (geranylgeranyl) isoprenoids, derived from mevalonic acid, mediates translocation of RhoA to the plasma membrane and activation of the downstream pathway.38–40 In addition, active RhoA was found to abrogate the HSF1 transcriptional activity by suppressing HSF1 binding to the HSE of hsp genes.34 Therefore, one of the possible mechanisms constitute the competition of GGA and GGA-derivatives with endogenous geranyl-groups, which could lead to inhibition of RhoA activation, resulting in enhanced binding of HSF1 to the HSE region.41,42 However, this should be further investigated by genetic ablation, competition and enhanced binding experiments.
How HSPs protect the cardiomyocytes
Irrespective of the precise molecular pathways affected by GGA and GGA-derivatives, their protective action seems to be critically dependent on boosting HSPB1, because the protective effect was abrogated by siRNA against HSPB1. In previous studies HSPB1 overexpression protected from TP-induced contractile dysfunction and structural remodeling in HL-1 and Drosophila models for AF.7,26 HSPB1 stabilizes sarcomeric proteins, including alpha-actinin and actin, prevents their disruption and enhances recovery after disruption.20,43 Furthermore, HSPB1 (co)-localizes with myosin in HL-1 cardiomyocytes and at myofilaments in human atrial myocytes and thereby potentially conserve myofibrils.16 Therefore, HSPB1 may shield the contractile proteins from AF-induced cleavage by cysteine proteases, such as calpain.26,44,45 Given that GGA-derivatives enhance HSP expression and protect from TP-induced contractile dysfunction, which is abrogated after suppression of HSPB1, we appoint HSPB1 to be one of the most important players in the cardioprotective effect of GGA-derivatives.
Therapeutic application of HSP-inducing compounds
In this study, HSP-inducing compounds were shown to prevent and reverse contractile dysfunction in experimental models for AF, which is promising for treating clinical AF, since patients diagnosed with AF already suffer from AF-related electropathology. Besides, comparable to GGA, the GGA-derivatives only boosted the HSR in cardiomyocytes pretreated with a mild, sub-lethal HS and not under nonstressed conditions, indicating that augmentation of the HSR by GGA and its derivatives is confined to stressed cells. In clinical perspective, this might indicate that side effects due to enhancing HSR are limited, if existent at all. Accordingly, it would be of interest to test suitable GGA-derivatives for future applications in the clinic. This would initially require testing of selected GGA-derivatives in a larger in vivo model for AF, such as the atrial pacemaker stimulated dog model for AF. Although the experimental models used in the current study were suitable to extract potent HSP boosters from the 81 synthesized GGA-derivatives, they do not fully reflect the complexity of electropathology in AF, making extrapolation to clinical AF beyond reach. Furthermore, pharmacokinetics need to be further explored in in vivo models to investigate bioavailability of the derivatives. Yet, GGA itself could already be further explored in clinical AF despite its less favorable physicochemical therapeutic profile, since the compound is already marketed in various Asian countries and considered a safe drug. As a proof of concept, a first study may explore whether GGA induces HSP expression in atrial tissue of AF patients and whether induced HSP expression correlates to reduced AF burden. Further research is necessary to test whether GGA or GGA-derivatives can prevent progression of clinical AF and reverse existing electropathology in clinical AF.
Conclusion
We identified multiple GGA-derivatives, especially GGA*-59, with an improved physicochemical profile and full preservation of HSP-boosting capacities, including both cardioprotective properties and acceleration of recovery from contractile dysfunction in an experimental model of AF. As such, this study substantiates our previous findings that HSP induction has high potential as a novel approach to prevent progression of AF and reverse remodeling.
Acknowledgments
This work was supported by the Dutch Heart Foundation (2013T144 and 2013T096) and the Netherlands Cardiovascular Research Initiative and Dutch Heart Foundation (CVON2014-40 DOSIS and CVON-STW2016-14728 AFFIP), the European Community, European Fund for Regional Development (Operationeel Programma Noord-Nederland 2007–2012, OP-EFRO), the Life Sciences & Health-Impulse grant (40-43100-98-008), and the Province of Groningen, Innovative Action-program Groningen (IAG3).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Disclosure
Jean-Paul G Seerden, Lizette Loen, and Andre Heeres are employees of Syncom BV. Herman Steen is the founder and CEO of Chaperone Pharma BV, a pharmaceutical company engaged in clinical development of HSP boosting drugs. The authors report no other conflicts of interest in this work.
References
Mozaffarian D, Benjamin EJ, Go AS, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–e322. | ||
de Groot NM, Houben RP, Smeets JL, et al. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: epicardial breakthrough. Circulation. 2010;122(17):1674–1682. | ||
Allessie MA, de Groot NM, Houben RP, et al. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: longitudinal dissociation. Circ Arrhythm Electrophysiol. 2010;3(6):606–615. | ||
Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11(4):275–291. | ||
Camm AJ, Lip GY, de Caterina R. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719–2747. | ||
Zhang D, Wu CT, Qi X, et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129(3):346–358. | ||
Brundel BJ, Shiroshita-Takeshita A, Qi X, et al. Induction of heat shock response protects the heart against atrial fibrillation. Circ Res. 2006;99(12):1394–1402. | ||
Lanters EA, van Marion DM, Steen H, de Groot NM, Brundel BJ. The future of atrial fibrillation therapy: intervention on heat shock proteins influencing electropathology is the next in line. Neth Heart J. 2015;23(6):327–333. | ||
Wiersma M, Meijering RAM, Qi XY, et al. Endoplasmic reticulum stress is associated with autophagy and cardiomyocyte remodeling in experimental and human atrial fibrillation. J Am Heart Assoc. 2017;6(10):e006458. | ||
Henning RH, Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. 2017;14(11):637–653. | ||
Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319(5865):916–919. | ||
Baler R, Dahl G, Voellmy R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol. 1993;13(4):2486–2496. | ||
Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12(10):842–846. | ||
Kim HJ, Hwang NR, Lee KJ. Heat shock responses for understanding diseases of protein denaturation. Mol Cells. 2007;23(2):123–131. | ||
Golenhofen N, Perng MD, Quinlan RA, Drenckhahn D. Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem Cell Biol. 2004;122(5):415–425. | ||
Brundel BJ, Henning RH, Ke L, van Gelder IC, Crijns HJ, Kampinga HH. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J Mol Cell Cardiol. 2006;41(3):555–562. | ||
Kötter S, Unger A, Hamdani N, et al. Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J Cell Biol. 2014;204(2):187–202. | ||
Bullard B, Ferguson C, Minajeva A, et al. Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem. 2004;279(9):7917–7924. | ||
Ghosh JG, Houck SA, Clark JI. Interactive domains in the molecular chaperone human alphaB crystallin modulate microtubule assembly and disassembly. PLoS One. 2007;2(6):e498. | ||
Bryantsev AL, Loktionova SA, Ilyinskaya OP, Tararak EM, Kampinga HH, Kabakov AE. Distribution, phosphorylation, and activities of Hsp25 in heat-stressed H9c2 myoblasts: a functional link to cytoprotection. Cell Stress Chaperones. 2002;7(2):146–155. | ||
Murakami M, Oketani K, Fujisaki H, Wakabayashi T, Ohgo T. Antiulcer effect of geranylgeranylacetone, a new acyclic polyisoprenoid on experimentally induced gastric and duodenal ulcers in rats. Arzneimittelforschung. 1981;31(5):799–804. | ||
Yanaka A, Zhang S, Sato D, et al. Geranylgeranylacetone protects the human gastric mucosa from diclofenac-induced injury via induction of heat shock protein 70. Digestion. 2007;75(2–3):148–155. | ||
Katsuno M, Sang C, Adachi H, et al. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102(46):16801–16806. | ||
Hirakawa T, Rokutan K, Nikawa T, Kishi K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology. 1996;111(2):345–357. | ||
Brundel BJ, Ke L, Dijkhuis AJ, et al. Heat shock proteins as molecular targets for intervention in atrial fibrillation. Cardiovasc Res. 2008;78(3):422–428. | ||
Zhang D, Ke L, Mackovicova K, et al. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for atrial fibrillation. J Mol Cell Cardiol. 2011;51(3):381–389. | ||
Sakabe M, Shiroshita-Takeshita A, Maguy A, et al. Effects of a heat shock protein inducer on the atrial fibrillation substrate caused by acute atrial ischaemia. Cardiovasc Res. 2008;78(1):63–70. | ||
Chang SL, Chen YC, Hsu CP, et al. Heat shock protein inducer modifies arrhythmogenic substrate and inhibits atrial fibrillation in the failing heart. Int J Cardiol. 2013;168(4):4019–4026. | ||
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. | ||
Nishizawa Y, Yamada K, Yamato C, Fujita T. Metabolic fate of geranylgeranylacetone. Drug Metab Pharmacokinet. 1986;1(2):171–177. | ||
Claycomb WC, Lanson NA, Stallworth BS, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95(6):2979–2984. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | ||
Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. | ||
Meijering RA, Wiersma M, van Marion DM, et al. RhoA activation sensitizes cells to proteotoxic stimuli by abrogating the HSF1-dependent heat shock response. PLoS One. 2015;10(7):e0133553. | ||
Hoogstra-Berends F, Meijering RA, Zhang D, et al. Heat shock protein-inducing compounds as therapeutics to restore proteostasis in atrial fibrillation. Trends Cardiovasc Med. 2012;22(3):62–68. | ||
Luo FC, Qi L, Lv T, et al. Geranylgeranylacetone protects mice against morphine-induced hyperlocomotion, rewarding effect, and withdrawal syndrome. Free Radic Biol Med. 2012;52(7):1218–1227. | ||
Shack S, Gorospe M, Fawcett TW, Hudgins WR, Holbrook NJ. Activation of the cholesterol pathway and Ras maturation in response to stress. Oncogene. 1999;18(44):6021–6028. | ||
Rattan S. 3-Hydroxymethyl coenzyme A reductase inhibition attenuates spontaneous smooth muscle tone via RhoA/ROCK pathway regulated by RhoA prenylation. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G962–G969. | ||
Yang J, Chen YN, Xu ZX, Mou Y, Zheng LR. Alteration of RhoA prenylation ameliorates cardiac and vascular remodeling in spontaneously hypertensive rats. Cell Physiol Biochem. 2016;39(1):229–241. | ||
Cook M, Mani P, Wentzell JS, Kretzschmar D. Increased RhoA prenylation in the loechrig (loe) mutant leads to progressive neurodegeneration. PLoS One. 2012;7(9):e44440. | ||
Sysa-Shah P, Xu Y, Guo X, et al. Geranylgeranylacetone blocks doxorubicin-induced cardiac toxicity and reduces cancer cell growth and invasion through RHO pathway inhibition. Mol Cancer Ther. 2014;13(7):1717–1728. | ||
Hashimoto K, Morishige K, Sawada K, et al. Geranylgeranylacetone inhibits lysophosphatidic acid-induced invasion of human ovarian carcinoma cells in vitro. Cancer. 2005;103(7):1529–1536. | ||
Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol. 1995;15(1):505–516. | ||
Garrido C, Bruey JM, Fromentin A, Hammann A, Arrigo AP, Solary E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J. 1999;13(14):2061–2070. | ||
Concannon CG, Orrenius S, Samali A. Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene Expr. 2001;9(4–5):195–201. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.