")
Back to Journals » International Journal of Women's Health » Volume 7
Sclerostin inhibition: a novel therapeutic approach in the treatment of osteoporosis
Authors Shah A, Shoback D, Lewiecki EM
Received 6 January 2015
Accepted for publication 9 February 2015
Published 2 June 2015 Volume 2015:7 Pages 565—580
DOI https://doi.org/10.2147/IJWH.S73244
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Elie Al-Chaer
Arti D Shah,1 Dolores Shoback,1,2 E Michael Lewiecki3,4
1Division of Endocrinology and Metabolism, Department of Medicine, University of California, San Francisco, CA, USA; 2Endocrine Research Unit, Department of Medicine, Veterans Affairs Medical Center, San Francisco, CA, USA; 3University of New Mexico School of Medicine, Albuquerque, NM, USA; 4New Mexico Clinical Research & Osteoporosis Center, Albuquerque, NM, USA
Abstract: Osteoporosis and osteoporosis-related fractures are growing problems with the aging population and are associated with significant morbidity and mortality. At this time, other than parathyroid hormone analogs, all therapies for osteoporosis are antiresorptive. Therefore, researchers have focused efforts on development of more anabolic therapies. Understanding of the Wnt signaling pathway, which is critical for skeletal development, and the role of sclerostin in inhibition of Wnt signaling has led to the discovery of a novel therapeutic approach in the treatment of osteoporosis – sclerostin inhibition. In this review, we discuss the biology of Wnt signaling and sclerostin inhibition. We then discuss human disorders of decreased sclerostin function and animal models of sclerostin inhibition. Both have served to elucidate the effects of decreased sclerostin levels and function – increased bone mass and strength and fewer fractures. In addition, we review data from Phase I and II studies of the two humanized sclerostin monoclonal antibodies, romosozumab and blosozumab, both of which have had positive effects on bone mineral density. We conclude with a discussion of the ongoing Phase III studies of romosozumab. The available data support the potential for neutralizing sclerostin monoclonal antibodies to serve as anabolic agents in the treatment of osteoporosis.
Keywords: osteoporosis, sclerostin, Wnt signaling, anabolic therapies, romosozumab
Introduction
Osteoporosis is a disease characterized by decreased bone mineral density (BMD) and poor bone quality, with deterioration of skeletal microarchitecture, predisposing to fracture.1,2 With the aging of the population, osteoporosis has become a growing medical and socioeconomic problem that is associated with significant morbidity and mortality, mainly as a result of hip fractures. By 2020, there are expected to be 14 million patients with osteoporosis in the United States (US) and over 61 million patients with increased risk for fracture due to low bone mass.3 Furthermore, the costs associated with osteoporosis are significant. Based on prior fracture and US Census data (from 1998 to 2004), there were predicted to be over two million fractures in 2005, as predicted by a Markov state-transition model, costing in excess of $17 billion with the majority of costs incurred by inpatient and long-term care.3,4 By 2025, the annual rate of fractures is projected to increase by 50% from 2005, and annual costs are expected to reach $25 billion.3,4 Interestingly, compared to 2005, there is also expected to be an ~175% increase in the costs related to osteoporosis and fractures in Hispanics living in the US by 2025 with similar increases in other ethnic groups.4 Therefore, prevention, detection, and treatment of osteoporosis are essential in reducing the personal and socioeconomic burden of osteoporotic fractures.
The most common cause of osteoporosis is estrogen deficiency in postmenopausal women, who typically have a high rate of bone remodeling with an imbalance of bone resorption in excess of bone formation. Worldwide, osteoporosis affects about one-tenth of women by age 60 years and two-fifths of women by age 80 years.5 Based on 2010 census and prevalence data, the estimated prevalence of osteoporosis in 2014 in women in nine industrialized countries was 9–38%.6 And in the US, it is estimated that 20% of white women age 50 and older are osteoporotic.7 In the year 2000, there were an estimated nine million fractures worldwide, the greatest number of which occurred in Europe.8 Furthermore, at the age of 50, a white woman has a 17% lifetime chance of a hip fracture and 16% lifetime chance of a vertebral fracture.3
The two approaches to treatment of osteoporosis are decreasing bone resorption (“antiresorptive” therapy) and increasing bone formation (“anabolic” therapy). Currently, most therapies are antiresorptive. These include the bisphosphonates, selective estrogen receptor modulators, estrogen preparations, and denosumab.9–12 However, there are only two available treatments that increase bone formation – teriparatide, which is the active 34-residue amino-terminal fragment of parathyroid hormone (PTH), and intact PTH (1–84), formerly available for osteoporosis treatment outside the US. While teriparatide and/or recombinant human (rh) PTH (1–84) have been shown to increase bone formation,13 there are some disadvantages associated with their use. First, PTH and its fragments must be administered as a daily self-injection and must be kept refrigerated. Second, the use of teriparatide in the US is limited to 2 years, and the duration of use of rhPTH (1–84) in the European Union was recommended to be 18–24 months.13 Third, the teriparatide label contains a black box warning about the potential risk of osteosarcoma due to an observed increase in the incidence of osteosarcoma in rats that was dependent on dose and treatment duration.13 Fourth, while these agents increase bone formation, they also eventually lead to an increase in bone resorption, as reflected by a rise in bone resorption markers.13 Therefore, researchers have investigated other anabolic agents for the treatment of osteoporosis that might offer greater patient convenience and a more favorable balance of benefits and risks.
With the discovery of the central role of the Wnt/low density lipoprotein receptor-related protein 5 (LRP5)/β-catenin pathway in the maintenance of skeletal mass and in the pathogenesis of diseases of low and high bone mass, increasing attention has focused on determining how activation of this pathway might be modulated to advantage in the treatment of osteoporosis. Sclerostin, an endogenous inhibitor of Wnt signaling, is an important regulator of bone formation.10,14 Sclerostin production results in decreased osteoblastic bone formation.10,14 Inhibition of sclerostin, which can be expected to increase osteoblastic bone formation, has emerged as a potential strategy in the management of osteoporosis. Models of human disease support the development of anti-sclerostin therapy, with loss-of-function mutations in the SOST gene resulting in low or absent sclerostin levels and high bone mass.10 Understanding of expression and activity of sclerostin has led to the development of humanized monoclonal antibodies (MAbs) against sclerostin, including romosozumab (AMG 785, Amgen Inc., Thousand Oaks, CA, USA) and blosozumab (LY2541546, Eli Lilly and Company, Indianapolis, IN, USA). This review summarizes the current state of information available on both antibodies.
Biology of sclerostin and the Wnt signaling pathway
Sclerostin is a 190-amino acid secreted glycoprotein made predominantly by osteocytes, but also by cementocytes and mineralized hypertrophic chondrocytes.2,15 Structurally, the protein has a cysteine-knot like domain and a semi-flexible loop, which is involved in the inhibition of Wnt signaling.15
Wnt signaling is crucial to both bone development and the regulation of bone mass.16,17 Wnt proteins bind to a receptor complex that includes a member of the frizzled family of seven-transmembrane receptors and either LRP5 or LRP6 (Figure 1A).16 This results in phosphorylation of the cytoplasmic tail of LRP5 or LRP6, which allows the protein, axin, to bind there.16 Binding of axin inhibits the activity of glycogen synthase kinase (GSK)-3β, which normally phosphorylates and targets β-catenin for degradation in the proteasome.16 Increased cytoplasmic levels of β-catenin lead to its nuclear translocation, binding to DNA binding proteins, and activation of target gene promoters.16 Wnt signaling in bone leads to osteoblast differentiation, proliferation, function and survival2,18 and hence, increased bone mass.17,19
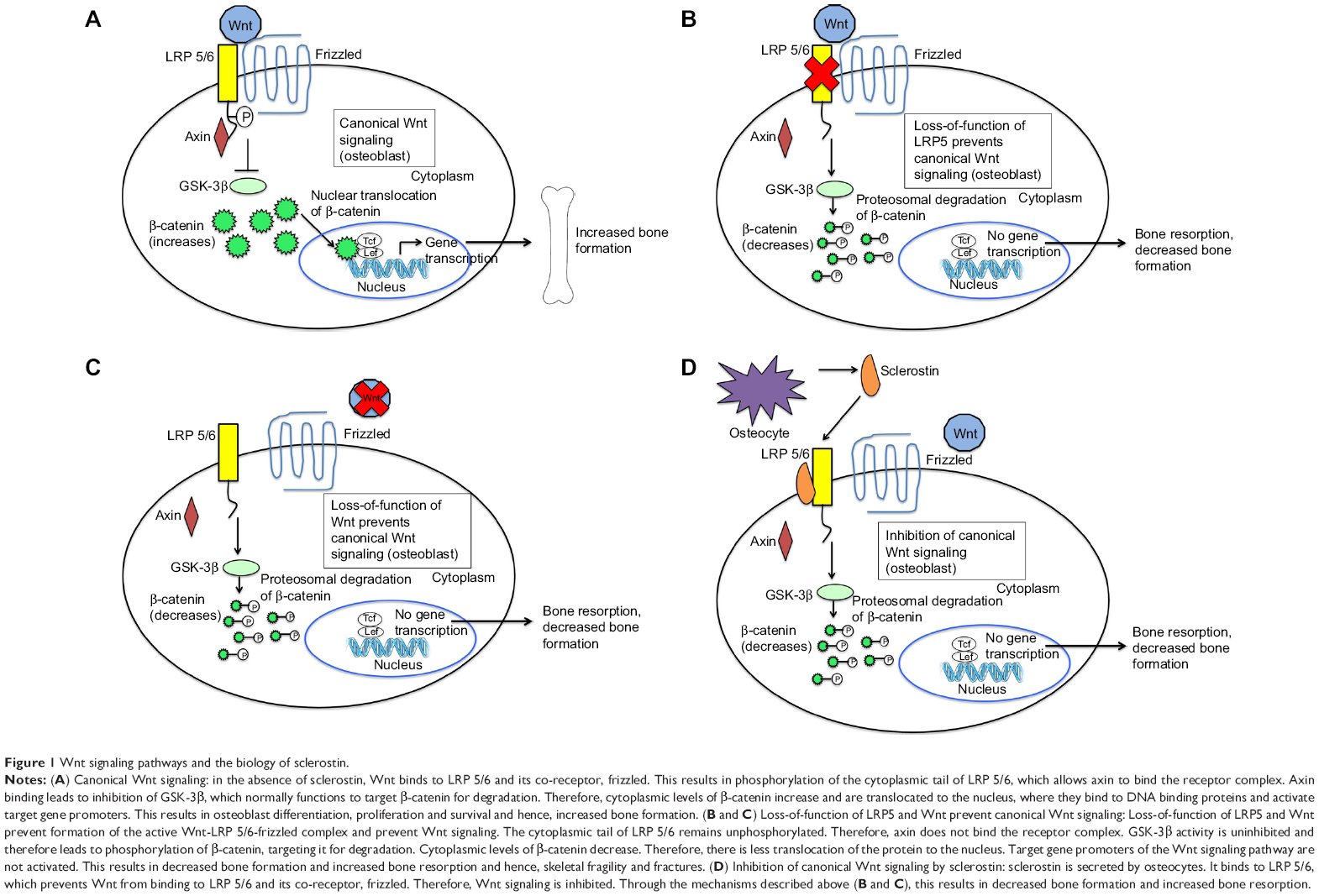 | Figure 1 Wnt signaling pathways and the biology of sclerostin. |
Loss-of-function mutations in the Wnt signaling pathway result in skeletal fragility and decreased bone mass (Figure 1B and C). Osteoporosis-pseudoglioma syndrome, an autosomal recessive disorder characterized by visual loss and skeletal fragility, is caused by loss-of-function mutations in the LRP5 gene, which expresses the protein LRP5. This impairs Wnt signaling and results in reduced BMD, skeletal fragility, and fractures.20 This finding has been replicated in LRP5 knockout (KO) mice, which exhibit abnormally low bone mass.21 Furthermore, homozygous loss-of-function mutations in the wingless-type mouse mammary tumor virus integration site family, member 1 (WNT1) gene, which expresses the protein Wnt1, have been found in autosomal recessive osteogenesis imperfecta, while autosomal dominant mutations in the WNT1 gene have been found in familial cases of early-onset osteoporosis.22 Therefore, various WNT1 gene mutations cause different forms of bone fragility. These studies highlight the importance of LRP5 and Wnt1 in Wnt signaling and skeletal development.
Mechanisms that modulate the levels of Wnt signaling are critically important in determining bone mass. Sclerostin is one key Wnt pathway regulator. Sclerostin inhibits bone morphogenetic protein (BMP)-stimulated bone formation by antagonizing Wnt signaling (Figure 1D).23 Sclerostin acts by binding to LRP5 and/or LRP6, thereby impairing further signaling through β-catenin stabilization, as described above. This decreases osteoblastic activities, including new bone formation and mineralization.16,23,24 Sclerostin is primarily secreted by osteocytes embedded in the bone matrix.25 In addition, sclerostin also blocks maturation of osteocytes and regulates the expression of genes involved in bone matrix mineralization.25 In late osteoblasts and preosteocytes, sclerostin leads to downregulation of phosphate regulating endopeptidase homolog, X-linked (PHEX).26 Through its endopeptidase activity, PHEX degrades peptides that bind to nascent bone mineral and inhibit mineral deposition. Therefore, downregulation of PHEX results in decreased bone mineralization.26 Given the inhibitory effect of sclerostin on osteoblast function and bone formation, blocking the activity of sclerostin seems to be a promising approach in the treatment of osteoporosis.
Nature’s own experiment
Human disorders of sclerostin expression and activity underscore the importance of the protein in bone. One such disorder is sclerosteosis, a rare autosomal recessive condition of generalized skeletal overgrowth characterized by facial distortion, tall stature, hand malformations including syndactyly of the digits, radial deviation of terminal phalanges, and dysplastic or absent nails (Figure 2A).27 These patients may develop increased intracranial pressure, which can lead to sudden death, the primary cause of early mortality in patients with sclerosteosis.27 Cases have primarily been reported in the Afrikaner population in South Africa.27 Van Buchem disease, another rare autosomal recessive disorder, has radiological features similar to those of sclerosteosis with thickening of the skull, mandible, clavicles, ribs, and diaphyses of long bones (Figure 2B).27,28 It is primarily found in residents of an isolated village in the Netherlands.27 In both disorders, skull and facial bone thickening can lead to entrapment of cranial nerves, which can manifest as hearing loss, facial palsy, optic nerve atrophy, and anosmia.2,27 The course of both diseases tends to stabilize in adulthood.29
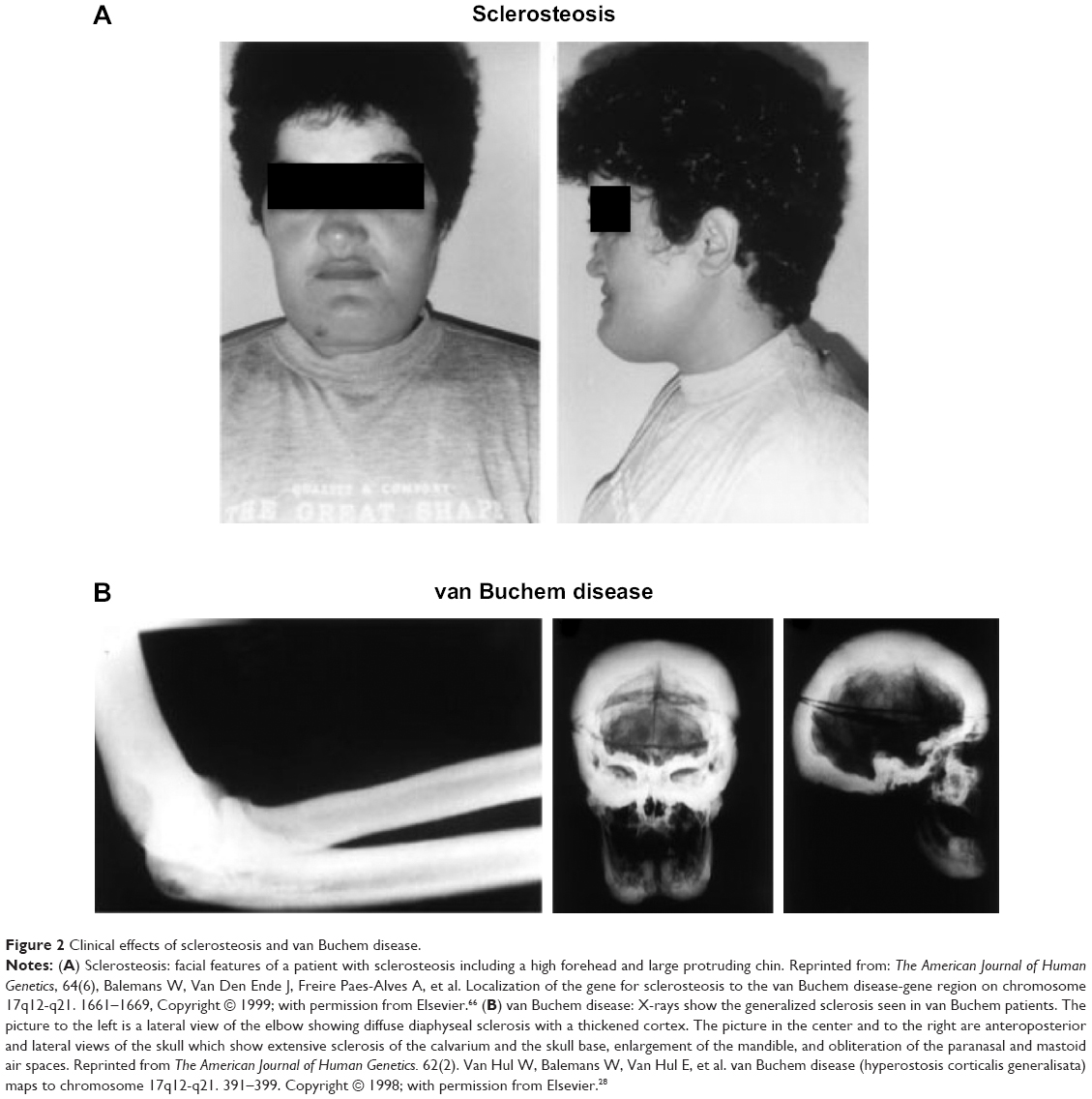 | Figure 2 Clinical effects of sclerosteosis and van Buchem disease. |
In both conditions, mutations have been mapped to the chromosome 17q12-q21 region.27 This is the region of the SOST gene whose product is sclerostin. Studies in patients with sclerosteosis and van Buchem disease have reported deletions, nonsense mutations, and splice site mutations in SOST, which then lead to absence of or decreased levels of sclerostin and hence, increased bone mass due to lack of modulation of Wnt signaling in the skeleton.27,30–32
Patients with van Buchem disease have higher BMD and higher levels of bone formation markers, procollagen type 1 amino-terminal propeptide (P1NP), compared with carriers of one mutant allele and controls, and sclerostin levels are inversely correlated with serum P1NP and BMD.29,33 Patients with sclerosteosis also have higher levels of P1NP compared with their heterozygous relatives and controls, although interestingly these levels decline with age.34 Heterozygous carriers of one mutant SOST allele have significantly higher levels of P1NP compared to controls, implying a gene-dose effect.34 Furthermore, heterozygous individuals have high BMD, but they rarely experience fractures and do not suffer from symptoms and complications of the disease.34–36 These observations in patients with reduced sclerostin suggest that interventions that modulate sclerostin levels might be effective in the treatment of osteoporosis. Interestingly, clinical manifestations are more severe in sclerosteosis than in van Buchem disease, likely due to the total absence of detectable circulating sclerostin in the former condition and low but detectable levels in patients with van Buchem disease.2,29,34
Replicating human disease in an animal model
Shortly after reports of human disorders of sclerostin production and their clinical phenotypes, SOST KO mice were developed to better understand the effects of sclerostin deficiency on bone. Li et al demonstrated that both male and female SOST KO mice have high bone mass with increases in BMD, bone volume, bone formation, and bone strength.37 These mice have increased bone mass, both in the lumbar vertebrae and long bones.37 Micro-computed tomography (micro-CT) of the femur showed that there was increased trabecular and cortical bone volume.37 By histomorphometric analysis, there was a significant increase in osteoblast surface with no change in osteoclast surface, and the bone formation rates were increased in trabecular, periosteal, and endocortical bone envelopes.37 SOST KO mice had significantly higher levels of osteocalcin, a bone formation marker.37 Another study also demonstrated that SOST KO mice had increased bone mass in the appendicular skeleton and cranium and tended to add bone periosteally.38 Furthermore, SOST KO mice are also resistant to mechanical unloading induced-bone loss.39 On the other hand, transgenic mice overexpressing SOST exhibit low bone mass and decreased bone strength, as a result of reduced osteoblast activity, and also have reduced load-induced bone formation.40,41 Thus, based on human disorders of loss of function of sclerostin and mouse models of targeted gene inactivation and transgenic overexpression of SOST, inhibition of sclerostin was put forward as a potential strategy in the management of the human disorder of osteoporosis.
Sclerostin antibody in animal studies
Several animal studies have helped to further elucidate the effects of sclerostin inhibition on bone (Figure 3).
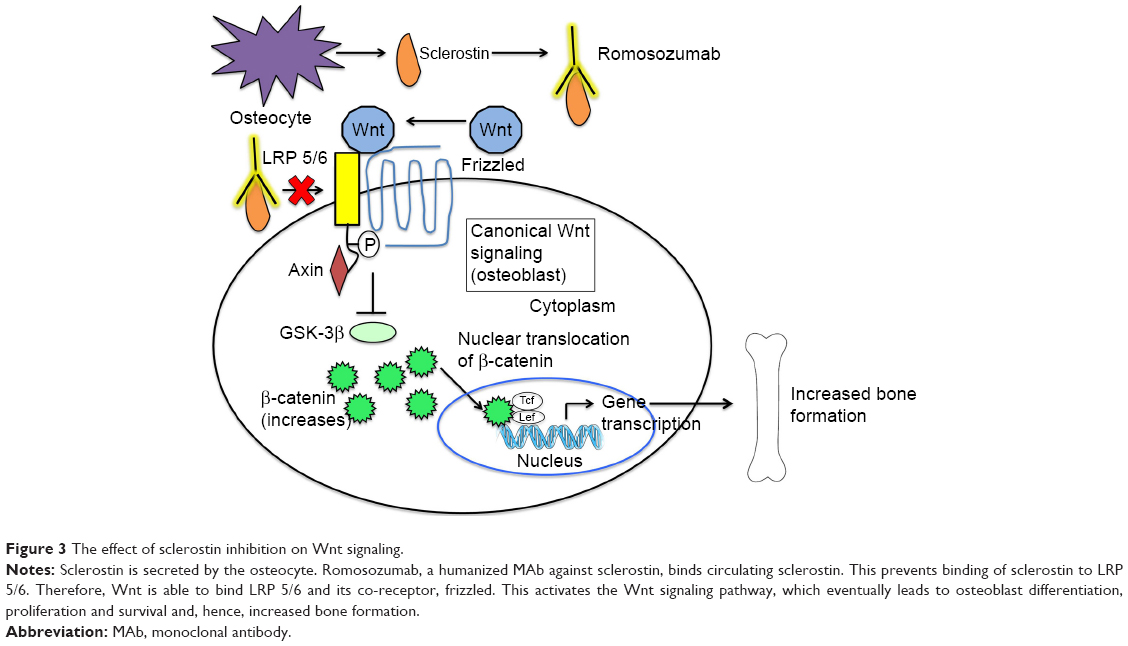 | Figure 3 The effect of sclerostin inhibition on Wnt signaling. |
Studies of osteoporosis in female rats
Of greatest clinical interest is the effect of sclerostin inhibition on osteoporosis due to estrogen deficiency. Li et al studied sclerostin inhibition in aged ovariectomized female rats, a standard model of postmenopausal osteoporosis.42 Six-month-old female rats were ovariectomized and left untreated for 1 year to allow for estrogen-deficiency induced bone loss to develop. Rats were then treated with an sclerostin neutralizing MAbs for 5 weeks.42 By dual energy X-ray absorptiometry (DXA), this treatment resulted in a 26% increase in BMD at the lumbar vertebrae and a 17% increase at the femur-tibia (defined as the entire femur in addition to the part of the tibia above the tibia-fibular junction) relative to baseline, while BMD in controls and in mice that underwent sham surgery remained at pre-treatment levels.42 This was accompanied by a significant increase in osteocalcin, a bone formation marker.42 Micro-CT analysis of the distal femur demonstrated that treatment with the sclerostin MAb increased trabecular thickness, trabecular volumetric BMD, and bone volume, restoring microarchitectural parameters to sham control levels.42 Histomorphometric analysis demonstrated that treatment with the sclerostin MAb led to an increased osteoblast surface, decreased osteoclast surface, and increases in mineralizing surface, mineral apposition rate, and bone formation rate in the proximal tibia.42 In the femoral midshaft, MAb treatment led to increased bone formation on periosteal and endocortical surfaces.42 Finally, results of mechanical testing showed that sclerostin MAb treatment led to greater bone strength in the lumbar vertebrae and femoral midshaft.42 In a rat model, Ominsky et al showed that treatment of ovariectomized rats with sclerostin MAb led to an increase in bone volume by increasing modeling-based bone formation on trabecular and cortical surfaces and prolonging the formation period at both modeling and remodeling sites while reducing bone resorption.43 Therefore, these pivotal studies underscored sclerostin inhibition as a promising therapeutic approach for the treatment of postmenopausal osteoporosis.
Additional studies have provided further insight into the effects of sclerostin inhibition on skeletal metabolism. In a study of ovariectomized 4-month-old Sprague-Dawley rats, treatment with sclerostin MAb for 6 or 26 weeks led to thicker trabeculae in the vertebrae and increased trabecular bone volume and increased cortical bone volume in the tibia, increased parameters of bone formation, increased bone formation rates, and reduced parameters of bone resorption at 6 weeks.44 At week 26, bone resorption markers were no longer significantly reduced, and bone formation markers and periosteal bone formation rates were no longer significantly increased, although endocortical and trabecular bone formation rates remained elevated but diminished compared to those reported at week 6.44 Bone accrual continued between weeks 6 and 26, and the antiresorptive effects of the sclerostin MAb remained histologically evident at 26 weeks.44 Interestingly, the sclerostin MAb-treated group had increased skeletal mRNA expression of several osteocyte genes, with SOST showing the greatest induction.44 This suggests that the increased osteocytic expression of sclerostin may serve to modulate gains in bone mass.44 Another study in which 6-month-old, osteopenic, ovariectomized rats were treated with sclerostin MAb for 6, 12 or 26 weeks showed similar findings.45 In this study, DXA analysis found that BMD at the lumbar vertebrae and femur-tibia increased progressively during the 26 weeks of treatment with sclerostin MAb, along with increases in trabecular and cortical bone mass and strength.45 Similar to other studies, trabecular, endocortical, and periosteal bone formation rates peaked at week 6 with sclerostin MAb treatment.45 At 26 weeks, periosteal bone formation rates had returned to control levels while endocortical and trabecular bone formation rates remained greater than controls, although both had declined compared to week 6.45 Finally, osteoclast surface and eroded surface were significantly lower in the sclerostin MAb-treated group compared to controls.45 This study suggests that early gains in bone mass with sclerostin inhibition are due to reduced osteoclast activity and increased trabecular and cortical bone formation, while later gains are due to residual endocortical and trabecular osteoblast stimulation and persistent low osteoclast activity.45
One concern is that pre-treatment or co-treatment with antiresorptive agents might blunt the anabolic effects of sclerostin inhibition, as seen with treatment with PTH. Therefore, Li et al46 examined the effect of pre-treatment and co-treatment with alendronate on the anabolic actions of sclerostin MAb in ovariectomized rats. Ten-month-old, osteopenic, ovariectomized rats were pre-treated with alendronate (or vehicle) for 6 weeks, after which they were switched to sclerostin MAb alone, a combination of sclerostin MAb and alendronate, or vehicle alone for 6 weeks.46 In rats pre-treated with alendronate or vehicle, sclerostin MAb treatment increased areal BMD both in the lumbar vertebrae and femur-tibia, volumetric BMD, trabecular and cortical bone mass, and bone strength.46 Furthermore, serum osteocalcin and bone formation on trabecular, endocortical and periosteal surfaces were similar between alendronate and vehicle pre-treated rats.46 Interestingly, co-treatment with alendronate did not have significant effects on the increased bone formation, bone mass, and bone strength seen with sclerostin MAb treatment.46 Therefore, it appears that the anabolic effect of sclerostin MAb treatment is not blunted by pre- or co-treatment with alendronate in ovariectomized osteopenic female rats. Such information is highly relevant to women with osteoporosis, since many have received prior bisphosphonate therapy.
Additional rat studies
The effect of sclerostin MAb treatment has also been investigated in aged male rats, which serve as a model for low bone mass in elderly men. Aged, 16-month-old, male Sprague-Dawley rats were treated with sclerostin MAb for 5 weeks.47 DXA analysis showed that there was an increase in areal BMD in the lumbar vertebrae and femur-tibia compared with controls.47 Micro-CT showed improved trabecular and cortical microarchitecture in the treated group compared with controls.47 Bone formation parameters, including mineralizing surface, mineral apposition rate, and bone formation rate, were greater on trabecular, periosteal, and endocortical surfaces in the treated group compared with controls.47 These findings suggest than sclerostin inhibition may also be a promising therapy for male osteoporosis.
There is also evidence from preclinical studies that sclerostin inhibition may enhance fracture healing. In a rat closed femoral fracture model, 7 weeks of treatment with sclerostin MAb led to greater increases in bone mass in the region of the fracture, callus area and volume, peak load, and stiffness compared to these parameters in controls.48 In addition, treatment with sclerostin MAb led to recovery in bone parameters such that they approached the strength seen in intact, unfractured bone of controls.48 Furthermore, treatment of osteoporotic rats with a femur fracture with a single chain antibody directed against sclerostin (scFv) led to indistinct fracture lines, callus formation that connected both fracture ends, and increased BMD on fracture ends, thereby enhancing fracture healing.49 Subsequently, two studies of rats with femoral defects found that treatment with sclerostin MAb can enhance bone repair in this bone defect model and in the surrounding host bone; however, only a small subset of rats experienced complete healing of the defects.50,51 Therefore, treatment with sclerostin MAb seems to be most effective in bone repair when there is cortical integrity.50
Sclerostin expression is upregulated in unloaded bone and downregulated by loading.52 Agholme et al52 investigated the effect of unloading of the right hind limb and fixation of a steel screw in the tibia in rats treated with sclerostin MAb and found that treatment improved fixation of the steel screw in both normal loaded and unloaded bone in male rats, which reflects an increase in pull-out force and bone strength, and hence, an enhanced fracture healing response. Therefore, sclerostin inhibition may be a useful therapeutic option to mitigate bone loss in settings where bone is unloaded.
Additional studies have demonstrated that treatment with sclerostin MAb increases bone formation and volume and decreases bone resorption at both red and yellow marrow skeletal sites.53 While sclerostin inhibition increases bone volume and bone mass, it does not negatively impact bone matrix quality, as determined by measures of mineralization, or bone strength, as determined by three-point bending and micro-finite element analysis.54,55 In fact, treatment with sclerostin MAb resulted in increased strength compared with controls.55
Additional findings from mouse studies
Studies in mice have also served to elucidate the effect of loss-of-function of sclerostin and sclerostin inhibition. As discussed above, loss-of-function in both LRP5 alleles in humans leads to osteoporosis-pseudoglioma syndrome; in mice this genetic manipulation leads to low bone mass. However, mice with both LRP5 and SOST genes knocked-out have larger, denser, and stronger bones compared to LRP5 KO mice.21 Furthermore, treatment of LRP5 KO mice with sclerostin MAb results in increased BMD, bone mineral content, and bone formation rates, suggesting that the anabolic effects of sclerostin inhibition may be mediated via receptors other than LRP5, such as LRP4 and LRP6.21
In a mouse fracture healing model, in which mice have undergone an osteotomy, treatment with an sclerostin MAb increased bone formation rates by histomorphometry at 2 weeks, BMD and bone volume by micro-CT at 4 weeks, and bone strength at the fracture site by mechanical testing at 6 weeks,56 suggesting that sclerostin inhibition in the early stage of fracture healing may promote fracture healing.
Primate studies
The role of sclerostin inhibition in nonhuman primates has also been investigated, although not to the same extent as in rodents. Administration of sclerostin MAb to gonad-intact female cynomolgus monkeys for 2 months resulted in increased bone formation on trabecular, periosteal, endocortical, and intracortical surfaces, as well as increased BMD at several skeletal sites.57 In addition, there were significant increases in trabecular thickness and bone strength at the lumbar vertebrae.57 This study further confirmed that sclerostin inhibition may be an appealing therapeutic approach for treating osteoporosis in humans. As in rats, treatment with sclerostin MAb increased total bone forming surface, modeling-based bone formation on trabecular and endocortical surfaces, and lifespan of bone formation on modeling surfaces. Together, these changes resulted in increased bone volume along with reduced bone resorption.43
In a fibular osteotomy model in cynomolgus monkeys, treatment with the sclerostin MAb for 10 weeks increased bone mass and strength at the site of the fracture.48 Furthermore, this strategy led to less callus cartilage and smaller fracture gaps, which had more bone and less fibrovascular tissue, consistent with enhanced fracture healing.48 As in rats, treatment of cynomolgus monkeys with sclerostin MAb did not negatively impact bone matrix quality, and more specifically, treatment did not affect the mineral-to-matrix ratio, crystallinity, or collagen cross-linking in the endocortical, intracortical, or trabecular compartments.54 The latter testing reflects measures of bone quality.
Human studies of sclerostin inhibition
Based on data in animal models, a sclerostin neutralizing MAb was developed by Amgen and has been investigated in Phase I and Phase II studies. Phase III studies are currently ongoing. Another neutralizing MAb, blosozumab, has been developed by Eli Lilly, and Phase I and II studies have been completed.
Phase I study
The first study with the humanized sclerostin MAb, romosozumab, included 72 healthy men and postmenopausal women (Table 1).58 In this double-blind, placebo-controlled, ascending, single-dose study, subjects received romosozumab or placebo in a 3:1 ratio.58 Romosozumab was given subcutaneously (SC) at doses of 0.1, 0.3, 1, 3, 5, or 10 mg/kg or intravenously (IV) at doses of 1 or 5 mg/kg; and subjects were followed for up to 85 days.58 Pharmacokinetics were non-linear with dose, with peak serum concentrations occurring within the first week after subcutaneous administration.58 There were dose-dependent increases in bone formation markers including P1NP, bone-specific alkaline phosphatase (BSAP), and osteocalcin with 184%, 126%, and 176% increases in these markers, respectively, in patients receiving the MAb at 10 mg/kg SC.58 There were 167%, 125%, and 143% increases in these markers, respectively, in patients receiving 5 mg/kg IV.58 There was also a dose-dependent decrease in the bone resorption marker, serum C-telopeptide (sCTX), of 54% (patients receiving 10 mg/kg SC) and of 49% (patients receiving 5 mg/kg IV).58 Furthermore, dose-dependent increases in BMD were noted on days 29, 57, and 85 with increases of 5.3% in the lumbar spine and 2.8% in the total hip on day 85 in patients receiving MAb at 10 mg/kg SC.58 BMD increased by 5.2% in the lumbar spine and by 1.1% in the total hip on day 85 in patients receiving 5 mg/kg dose IV.58 Romosozumab was generally well tolerated with one treatment-related serious adverse event of non-specific hepatitis in the 10 mg/kg SC group.58 In this subject, elevated liver function tests were seen 1 day after dosing, peaked 3 days after dosing with alanine aminotransferase and aspartate aminotransferase 13 times and six times the upper limit of normal and by day 26, resolution had occurred.58 Of note, hepatitis panels and abdominal ultrasound were normal.58 Other reported adverse effects in the SC group included injection-site erythema, injection-site hemorrhage, back pain, headache, constipation, arthralgia, and dizziness, all of which were considered mild.58 In the IV group, adverse effects included constipation, cough, headache, and hot flush, all of which were considered mild. Adverse events were reported in 60% of subjects who received romosozumab SC, 64% of subjects who received placebo SC, 50% of subjects who received placebo IV and 25% of subjects who received romosozumab IV.58 There were no deaths or study discontinuations.58 Mild, transient decreases in mean serum ionized calcium (4% from baseline) occurred after a single dose of the study drug associated with increases in PTH, but values returned to baseline during the study or follow-up period.58 However, these fluctuations in serum ionized calcium and in PTH were not associated with the reported adverse events.58 Of those who received romosozumab, six subjects developed anti-romosozumab antibodies, two of which were neutralizing – one in the 10 mg/kg SC group and one in the 5 mg/kg IV group.58 However, there was no apparent effect of these antibodies on pharmacokinetics or pharmacodynamics, although this was not determined.58 There were no adverse events or abnormalities in laboratory results, vital signs or electrocardiograms in these subjects.58 The data from this study suggested that single doses of romosozumab are generally well-tolerated with notable increases in bone formation markers, warranting further investigation of this compound.
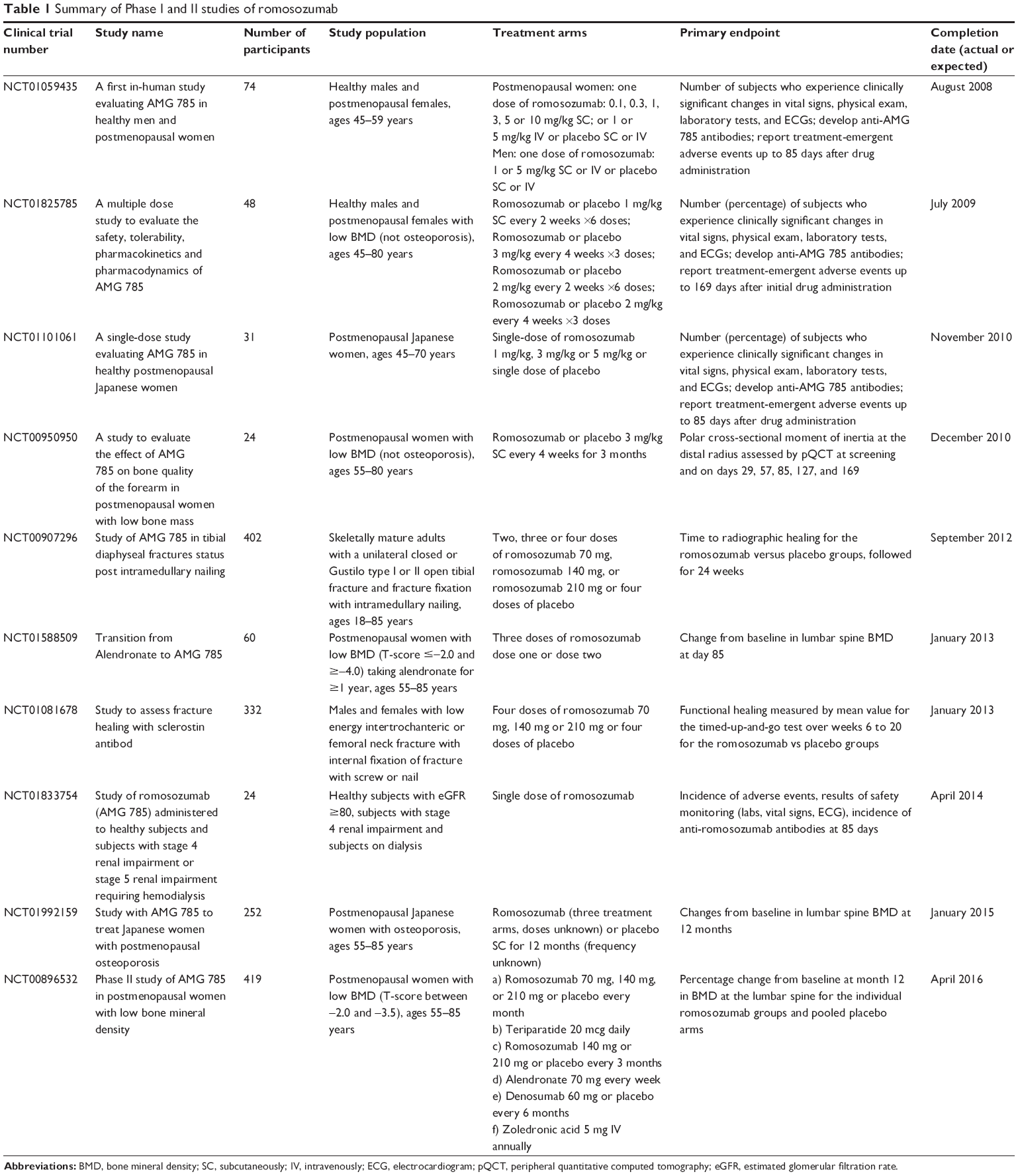 | Table 1 Summary of Phase I and II studies of romosozumab |
Phase II study
Subsequently, a Phase II, multicenter, international, randomized, placebo-controlled, parallel, eight-group study was conducted over a 12-month period in 419 postmenopausal women between the ages of 55 and 85 years with low BMD (T-score between −2.0 and −3.5 at the lumbar spine, total hip, or femoral neck) (Table 1).59 Participants were randomly assigned to one of eight groups: placebo every 3 months or monthly, romosozumab 140 mg SC every 3 months, romosozumab 210 mg SC every 3 months, romosozumab 70 mg SC monthly, romosozumab 140 mg SC monthly, romosozumab 210 mg SC monthly, alendronate 70 mg weekly, and teriparatide 20 mcg SC daily.59 Participants were followed for 12 months, with BMD assessed at baseline and 3, 6, and 12 months and labs done at baseline, week 1, and months 1, 2, 3, 6, 9, and 12.59 Of the 419 participants enrolled in the study, 383 completed the 12-month visit, while 36 withdrew from the study.59
The primary endpoint of the study was percentage change from baseline in BMD at the lumbar spine at month 12, and secondary endpoints included percentage change from baseline in BMD at the total hip, femoral neck, and distal third of the radius at month 12.59 Compared to placebo, all of the groups treated with romosozumab had significant increases in BMD at the lumbar spine, total hip, and femoral neck at 12 months.59 The largest gains were seen with monthly dosing of romosozumab (210 mg) with an 11.3% increase in lumbar spine BMD, 4.1% increase in total hip BMD, and 3.7% increase in femoral neck BMD at 12 months, which were greater than the increases seen in the alendronate- and teriparatide-treated groups.59 There were no significant differences in the distal third of the radius at 12 months in any group.59 Participants in the cohort treated with monthly romosozumab (140 mg) had greater increases in lumbar spine BMD at 12 months, compared to those in the alendronate- and teriparatide-treated groups.59
Changes in BMD at the lumbar spine, total hip, and femoral neck were also assessed at 6 months as secondary endpoints.59 Teriparatide was superior to romosozumab given every 3 months.59 At 6 months, lumbar spine and total hip BMD were significantly increased in all of the groups treated with romosozumab, compared to placebo treatment, while femoral neck BMD was greater in the groups that received romosozumab 140 mg monthly, 210 mg monthly or 210 mg every 3 months, compared to placebo treatment.59 In addition, increases in BMD at the lumbar spine, total hip, and femoral neck at month 6 were significantly greater in the groups that received the higher doses of romosozumab (140 mg or 210 mg monthly) than in the groups that received alendronate or teriparatide.59
Bone turnover markers were also measured as secondary endpoints at months 1, 3, 6, 9, and 12.59 In all cohorts receiving romosozumab, increases in bone formation markers (P1NP, osteocalcin, and BSAP) were transitory – with increases noted at 1 week and greatest at 1 month after the dose, after which levels returned to baseline or fell below baseline.59 In all of the romosozumab groups, the bone resorption marker, sCTX, fell from baseline, with the largest decrease in the 1st week, and in the groups receiving monthly doses of romosozumab, it remained below baseline at month 12.59
Study participants were closely observed for adverse events and changes in clinical and biochemical parameters. Treatment with romosozumab was associated with a dose-dependent decrease in calcium levels of 1.30–2.68% from baseline, with a nadir at month 1, and a compensatory increase in serum PTH.59 The serum calcium returned to baseline at follow-up visits, and there were no associated adverse events with that laboratory value change. There was no difference in the proportion of participants reporting adverse events between the placebo group (90%) and romosozumab groups (87%), and there was no apparent relationship between dose and adverse events.59 However, injection site reactions were more common with romosozumab than with placebo.59 There was also no difference in the proportion of participants experiencing serious adverse effects between the placebo group (14%) and the romosozumab groups (7%).59 Serious adverse events in the group receiving romosozumab (210 mg monthly) included: breast cancer, chronic obstructive pulmonary disease, non-cardiac chest pain, wrist fracture, and renal oncocytoma (benign).59 There were two deaths in the study – one in the placebo group (due to colon cancer) and one in the group receiving romosozumab (70 mg monthly), due to complications after aortobifemoral bypass surgery.59 In those who received romosozumab, 20% tested positive for binding antibodies, with neutralizing activity seen in vitro in 3%. The development of antibodies, however, did not have any effect on adverse events, pharmacokinetics, or pharmacodynamics.59
In this study, treatment with romosozumab was associated with increased BMD and bone formation markers and decreased bone resorption markers and was generally well-tolerated. The results are promising. The pattern of brief anabolic stimulation coupled with chronic suppression of bone resorption is unprecedented, compared to other therapies for osteoporosis.60
Ongoing studies
Based on the promising results seen in initial studies, there are several additional Phase I and II studies that have been completed or are in progress (Table 1),61 including a study evaluating the safety of romosozumab in patients with renal impairment and end-stage renal disease and studies assessing the effects of romosozumab on fracture healing. In addition, there are five ongoing Phase III studies (Table 2)61 – four investigating the use of romosozumab in postmenopausal women with osteoporosis and one investigating the use of romosozumab in men with osteoporosis.
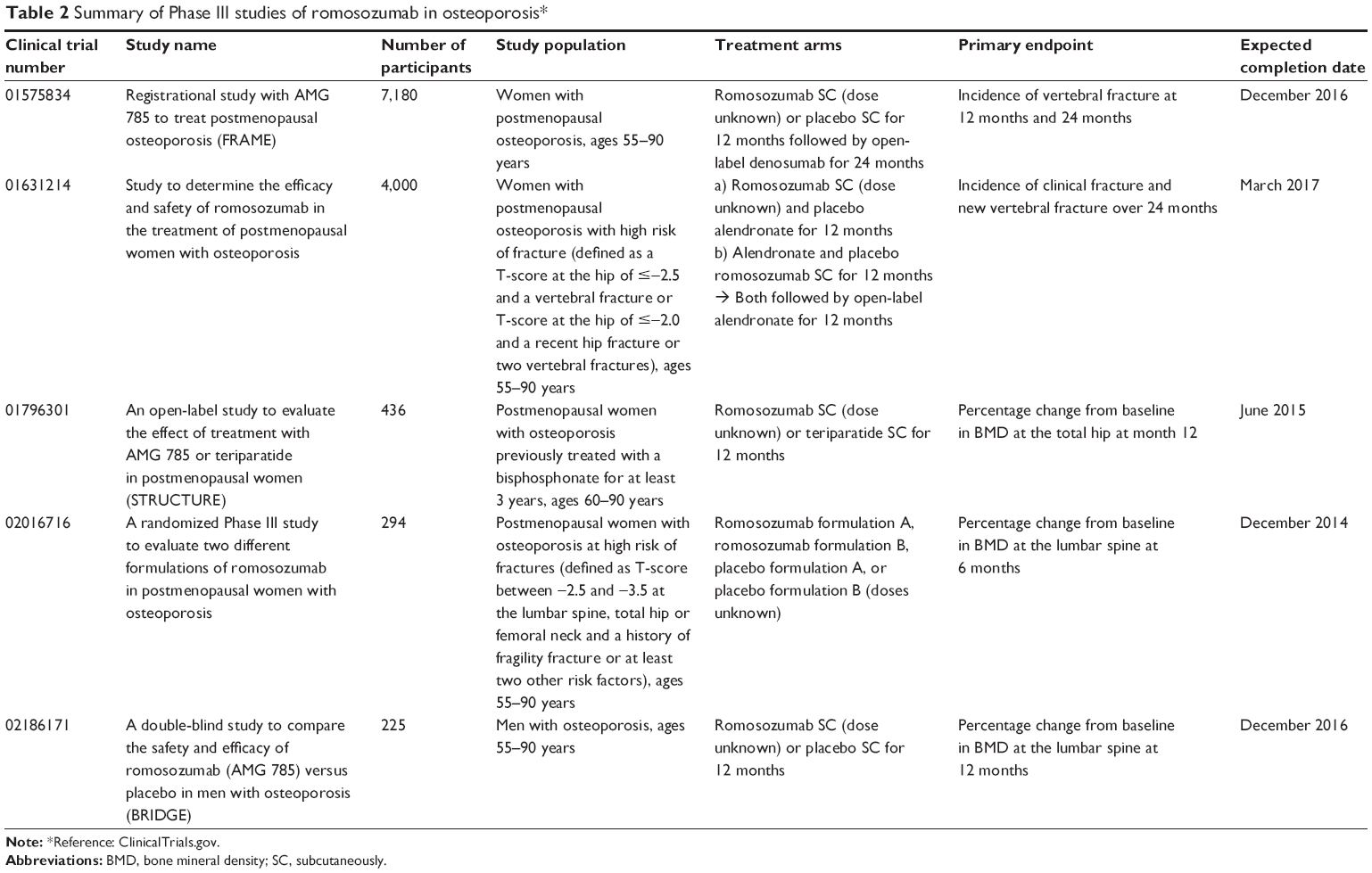 | Table 2 Summary of Phase III studies of romosozumab in osteoporosis* |
Studies of other sclerostin MAbs
While investigation of romosozumab continues, other humanized MAbs against sclerostin are being developed. More specifically, blosozumab has been investigated in Phase I and Phase II studies. In the Phase I study conducted in healthy postmenopausal women, investigators found that there were dose-dependent responses in sclerostin, P1NP, BSAP, osteocalcin, sCTX, and BMD with both single and multiple doses of blosozumab.62 At day 85, there was up to a 3.41% increase and up to a 7.71% increase from baseline in lumbar spine BMD in the single dose and multiple dose groups respectively.62 In the Phase II study, 120 postmenopausal women with low BMD received blosozumab or placebo SC at varying doses for12 months.63 At study end, BMD in the lumbar spine increased by 17.7% and BMD in the total hip increased by 6.2% in the highest dose group.63 In addition, markers of bone formation increased initially and then trended towards pre-treatment levels while markers of bone resorption remained reduced.63 Overall, the drug was well-tolerated.63 Therefore, blosozumab also shows promise as a treatment for osteoporosis.
Potential complications of sclerostin inhibition
Based on our understanding of the physiology of sclerostin and data from human studies, there are several potential complications and adverse effects to consider. First of all, patients with sclerosteosis and van Buchem disease develop thickening of the skull and facial bones, which leads to entrapment of cranial nerves; such patients are also at risk for the development of spinal stenosis. It might be speculated that similar complications could occur with long-term treatment with MAbs directed against sclerostin, due to excessive accumulation of bone in unwanted locations. However, upregulation of sclerostin may regulate gains in bone mass as suggested by Stolina et al,44 and this may prevent accumulation of excess bone mass. Such an effect, however, remains to be determined in humans. Phase I and II studies have shown that romosozumab is generally well tolerated with only mild adverse effects. However, in the Phase I study, one patient developed transient hepatitis. It is not known whether this was causally related to the drug. A decrease in serum calcium was seen with initial dosing. While patients had compensatory increases in serum PTH and return of serum calcium levels to baseline, these decreases in serum calcium could potentially become clinically significant in those with vitamin D deficiency or kidney disease. In both the Phase I and II studies, a few subjects developed neutralizing antibodies. While this did not seem to impact the pharmacokinetics or pharmacodynamics of drug action, with long-term treatment, it might impact the efficacy and potency of the drug. Finally, the other anabolic agents, teriparatide and rhPTH(1–84), come with a black box warning about osteosarcoma based on studies done in rats.64 While post-marketing surveillance in the US has not shown an increased rate of osteosarcoma in patients on teriparatide,65 osteosarcoma remains a theoretical risk in patients being treated with anabolic agents.
The past, the present, and the future
Current approved treatments for osteoporosis include bisphosphonates, denosumab, selective estrogen receptor modulators, teriparatide, and strontium ranelate; all of them are antiresorptive agents except for teriparatide, which has a regulatory limitation of 2 years lifetime use. Therefore, the availability of additional anabolic agents would be a welcome option for managing patients with osteoporosis who are at high risk of fracture.
By understanding the role of sclerostin in modulating Wnt signaling, sclerostin inhibition has emerged as a promising therapeutic approach in the treatment of osteoporosis. Furthermore, disorders of sclerostin, namely sclerosteosis and van Buchem disease, have given us insight into both the positive and negative effects of sclerostin inhibition. Animal studies have further elucidated the cellular mechanism underlying the effects of sclerostin inhibition and eventually led to the development of sclerostin neutralizing MAbs. Initial studies of one such MAb, romosozumab, in humans have been promising, showing striking increases in BMD and in bone formation markers with treatment. Phase III studies in postmenopausal women with osteoporosis will help us to understand whether the increased BMD seen with treatment in the Phase I and II studies will translate into anti-fracture efficacy. Furthermore, studies in men with osteoporosis and chronic kidney disease will also help us to understand the effects of romosozumab in other groups of patients with low BMD.
Additional studies will be needed to determine the optimal duration of treatment and to better understand the efficacy and safety of long-term romosozumab treatment. It also remains to be determined which patients are most likely to benefit from romosozumab and when such treatment should be instituted. Will this be first-line therapy for some patients? It might be speculated that cyclical use of romosozumab or combination with an antiresorptive could enhance the skeletal benefits. Should it be used in combination with an antiresorptive, or should it be followed with an antiresorptive, as is the current practice with teriparatide? Will this agent be effective in subsequent treatment courses? What determines when that timing is optimal? Further investigation will shed light and provide answers to these questions.
Conclusion
There is a clinical need for anabolic therapies in addition to PTH for the management of osteoporosis. Understanding the Wnt signaling pathway and specifically, the inhibitory effect of sclerostin, has spurred the development of a new class of anabolic agents, humanized MAbs against sclerostin, including romosozumab and blosozumab. Initial human studies of romosozumab are encouraging, and clinicians await the results of Phase III studies. Further investigation will shed light on where sclerostin inhibition fits into treatment strategies for osteoporosis. While many questions remain unanswered, sclerostin inhibition has emerged as a novel therapeutic approach in the treatment of osteoporosis and may add to our armamentarium of anabolic agents.
Disclosure
ADS and DS have no conflicts of interest to disclose. EML has received institutional grant/research support from Amgen, Merck, and Eli Lilly; he has served on scientific advisory boards for Amgen, Merck, Eli Lilly, Radius Health, AgNovos Healthcare, Alexion, NPS, and AbbVie.
References
NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285(6):785–795. | ||
Costa AG, Bilezikian JP, Lewiecki EM. Update on romosozumab: a humanized monoclonal antibody to sclerostin. Expert Opin Biol Ther. 2014;14(5):697–707. | ||
International Osteoporosis Foundation [homepage on the Internet]. Osteoporosis facts and statistics. International Osteoporosis Foundation Bone Health. Available from: http://www.iofbonehealth.org/facts-statistics. Accessed February 11, 2015. | ||
Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res. 2007;22(3):465–475. | ||
Kanis JA on behalf of the World Health Organization Scientific Group. Assessment of osteoporosis at the primary healthcare level. Technical Report. World Health Organization Collaborating Centre for Metabolic Bone Diseases, University of Sheffield, UK; 2007. Available from: http://www.shef.ac.uk/FRAX/pdfs/WHO_Technical_Report.pdf. Accessed February 11, 2015. | ||
Wade SW, Strader C, Fitzpatrick LA, Anthony MS, O’Malley CD. Estimating prevalence of osteoporosis: Examples from industrialized countries. Arch Osteoporos. 2014;9(1):182. | ||
National Osteoporosis Foundation [homepage on the Internet]. What women need to know. Available from: http://nof.org/articles/235. Accessed February 11, 2015. | ||
Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int. 2006;17(12):1726–1733. | ||
Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: Now and the future. Lancet. 2011;377(9773):1276–1287. | ||
Martin TJ. Bone biology and anabolic therapies for bone: Current status and future prospects. J Bone Metab. 2014;21(1):8–20. | ||
Tella SH, Gallagher JC. Biological agents in management of osteoporosis. Eur J Clin Pharmacol. 2014;70(11):1291–1301. | ||
Bernabei R, Martone AM, Ortolani E, Landi F, Marzetti E. Screening, diagnosis and treatment of osteoporosis: A brief review. Clin Cases Miner Bone Metab. 2014;11(3):201–207. | ||
Augustine M, Horwitz MJ. Parathyroid hormone and parathyroid hormone-related protein analogs as therapies for osteoporosis. Curr Osteoporos Rep. 2013;11(4):400–406. | ||
Hoeppner LH, Secreto FJ, Westendorf JJ. Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets. 2009;13(4):485–496. | ||
Veverka V, Henry AJ, Slocombe PM, et al. Characterization of the structural features and interactions of sclerostin: Molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. 2009;284(16):10890–10900. | ||
Burgers TA, Williams BO. Regulation of Wnt/beta-catenin signaling within and from osteocytes. Bone. 2013;54(2):244–249. | ||
Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat Med. 2013;19(2):179–192. | ||
Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148(6):2635–2643. | ||
Baron R, Rawadi G. Wnt signaling and the regulation of bone mass. Curr Osteoporos Rep. 2007;5(2):73–80. | ||
Ai M, Heeger S, Bartels CF, Schelling DK; Osteoporosis-Pseudoglioma Collaborative Group. Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am J Hum Genet. 2005;77(5):741–753. | ||
Kedlaya R, Veera S, Horan DJ, et al. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med. 2013;5(211):211ra158. | ||
Keupp K, Beleggia F, Kayserili H, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92(4):565–574. | ||
van Bezooijen RL, Svensson JP, Eefting D, et al. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res. 2007;22(1):19–28. | ||
van Dinther M, Zhang J, Weidauer SE, et al. Anti-sclerostin antibody inhibits internalization of sclerostin and sclerostin-mediated antagonism of Wnt/LRP6 signaling. PLoS One. 2013;8(4):e62295. | ||
Poole KE, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19(13):1842–1844. | ||
Atkins GJ, Rowe PS, Lim HP, et al. Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J Bone Miner Res. 2011;26(7):1425–1436. | ||
Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10(5):537–543. | ||
Van Hul W, Balemans W, Van Hul E, et al. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am J Hum Genet. 1998;62(2):391–399. | ||
van Lierop AH, Hamdy NA, van Egmond ME, Bakker E, Dikkers FG, Papapoulos SE. Van Buchem disease: Clinical, biochemical, and densitometric features of patients and disease carriers. J Bone Miner Res. 2013;28(4):848–854. | ||
Brunkow ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68(3):577–589. | ||
Staehling-Hampton K, Proll S, Paeper BW, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110(2):144–152. | ||
Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–97. | ||
Wergedal JE, Veskovic K, Hellan M, et al. Patients with van Buchem disease, an osteosclerotic genetic disease, have elevated bone formation markers, higher bone density, and greater derived polar moment of inertia than normal. J Clin Endocrinol Metab. 2003;88(12):5778–5783. | ||
van Lierop AH, Hamdy NA, Hamersma H, et al. Patients with sclerosteosis and disease carriers: Human models of the effect of sclerostin on bone turnover. J Bone Miner Res. 2011;26(12):2804–2811. | ||
Gardner JC, van Bezooijen RL, Mervis B, et al. Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab. 2005;90(12):6392–6395. | ||
Costa AG, Bilezikian JP. Sclerostin: Therapeutic horizons based upon its actions. Curr Osteoporos Rep. 2012;10(1):64–72. | ||
Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–869. | ||
Niziolek PJ, Farmer TL, Cui Y, Turner CH, Warman ML, Robling AG. High-bone-mass-producing mutations in the Wnt signaling pathway result in distinct skeletal phenotypes. Bone. 2011;49(5):1010–1019. | ||
Lin C, Jiang X, Dai Z, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24(10):1651–1661. | ||
Winkler DG, Sutherland MK, Geoghegan JC, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22(23):6267–6276. | ||
Tu X, Rhee Y, Condon KW, et al. SOST downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50(1):209–217. | ||
Li X, Ominsky MS, Warmington KS, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24(4):578–588. | ||
Ominsky MS, Niu QT, Li C, Li X, Ke HZ. Tissue-level mechanisms responsible for the increase in bone formation and bone volume by sclerostin antibody. J Bone Miner Res. 2014;29(6):1424–1430. | ||
Stolina M, Dwyer D, Niu QT, et al. Temporal changes in systemic and local expression of bone turnover markers during six months of sclerostin antibody administration to ovariectomized rats. Bone. 2014;67:305–313. | ||
Li X, Niu QT, Warmington KS, et al. Progressive increases in bone mass and bone strength in an ovariectomized rat model of osteoporosis after 26 weeks of treatment with a sclerostin antibody. Endocrinology. 2014;155(12):4785–4797. | ||
Li X, Ominsky MS, Warmington KS, et al. Increased bone formation and bone mass induced by sclerostin antibody is not affected by pretreatment or cotreatment with alendronate in osteopenic, ovariectomized rats. Endocrinology. 2011;152(9):3312–3322. | ||
Li X, Warmington KS, Niu QT, et al. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res. 2010;25(12):2647–2656. | ||
Ominsky MS, Li C, Li X, et al. Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res. 2011;26(5):1012–1021. | ||
Yao Q, Ni J, Hou Y, Ding L, Zhang L, Jiang H. Expression of sclerostin scFv and the effect of sclerostin scFv on healing of osteoporotic femur fracture in rats. Cell Biochem Biophys. 2014;69(2):229–235. | ||
Alaee F, Virk MS, Tang H, et al. Evaluation of the effects of systemic treatment with a sclerostin neutralizing antibody on bone repair in a rat femoral defect model. J Orthop Res. 2014;32(2):197–203. | ||
Virk MS, Alaee F, Tang H, Ominsky MS, Ke HZ, Lieberman JR. Systemic administration of sclerostin antibody enhances bone repair in a critical-sized femoral defect in a rat model. J Bone Joint Surg Am. 2013;95(8):694–701. | ||
Agholme F, Isaksson H, Li X, Ke HZ, Aspenberg P. Anti-sclerostin antibody and mechanical loading appear to influence metaphyseal bone independently in rats. Acta Orthop. 2011;82(5):628–632. | ||
Tian X, Setterberg RB, Li X, Paszty C, Ke HZ, Jee WS. Treatment with a sclerostin antibody increases cancellous bone formation and bone mass regardless of marrow composition in adult female rats. Bone. 2010;47(3):529–533. | ||
Ross RD, Edwards LH, Acerbo AS, et al. Bone matrix quality after sclerostin antibody treatment. J Bone Miner Res. 2014;29(7):1597–1607. | ||
Spatz JM, Ellman R, Cloutier AM, et al. Sclerostin antibody inhibits skeletal deterioration due to reduced mechanical loading. J Bone Miner Res. 2013;28(4):865–874. | ||
Cui L, Cheng H, Song C, et al. Time-dependent effects of sclerostin antibody on a mouse fracture healing model. J Musculoskelet Neuronal Interact. 2013;13(2):178–184. | ||
Ominsky MS, Vlasseros F, Jolette J, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25(5):948–959. | ||
Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26(1):19–26. | ||
McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370(5):412–420. | ||
Becker CB. Sclerostin inhibition for osteoporosis – a new approach. N Engl J Med. 2014;370(5):476–477. | ||
ClinicalTrials.gov [homepage on the Internet]. Romosozumab. ClinicalTrials.gov A service of the US National Institutes of Health. Available from: http://clinicaltrials.gov/ct2/results?term=romosozumab&Search=Search. Accessed February 11, 2015. | ||
McColm J, Hu L, Womack T, Tang CC, Chiang AY. Single- and multiple-dose randomized studies of blosozumab, a monoclonal antibody against sclerostin, in healthy postmenopausal women. J Bone Miner Res. 2014;29(4):935–943. | ||
Recker R, Benson C, Matsumoto T, et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res. 2015;30(2):216–224. | ||
Cipriani C, Irani D, Bilezikian JP. Safety of osteoanabolic therapy: A decade of experience. J Bone Miner Res. 2012;27(12):2419–2428. | ||
Andrews EB, Gilsenan AW, Midkiff K, et al. The US postmarketing surveillance study of adult osteosarcoma and teriparatide: Study design and findings from the first 7 years. J Bone Miner Res. 2012;27(12):2429–2437. | ||
Balemans W, Van Den Ende J, Freire Paes-Alves A, et al. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am J Hum Genet. 1999;64(6):1661–1669. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.