Back to Journals » Journal of Inflammation Research » Volume 18
SCAP/SREBP2-Mediated Cholesterol Biosynthetic Pathway Suppresses Particulate Matter-Induced Macrophage Activation and Airway Inflammation
Authors Chen WS ![]() , Lou JF, Li JY, Wu YP, Gao SW, Zheng K, Huang JQ, Li M, Zhu C, Zhang SY, Chen ZH
, Lou JF, Li JY, Wu YP, Gao SW, Zheng K, Huang JQ, Li M, Zhu C, Zhang SY, Chen ZH ![]() , Wu YF, Li W
, Wu YF, Li W ![]()
Received 3 April 2025
Accepted for publication 10 October 2025
Published 29 October 2025 Volume 2025:18 Pages 15117—15133
DOI https://doi.org/10.2147/JIR.S527083
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Yuhan Xing
Wei-Song Chen,1,2,* Jia-Fei Lou,1,* Jie-Yu Li,1 Yan-Ping Wu,1 Shen-Wei Gao,1 Kua Zheng,1 Jia-Qi Huang,1 Miao Li,1 Chen Zhu,1 Shu-Yang Zhang,3 Zhi-Hua Chen,1 Yin-Fang Wu,1 Wen Li1
1Key Laboratory of Respiratory Disease of Zhejiang Province, Department of Respiratory and Critical Care Medicine, Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China; 2Department of respiratory medicine, Affiliated Jinhua Hospital Zhejiang University School of Medicine, Jinhua, Zhejiang, People’s Republic of China; 3The First Clinical Medical College, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wen Li, Email [email protected] Yin-Fang Wu, Email [email protected]
Objective: To investigate the role and mechanism of the SREBP cleavage activating protein/Sterol-regulatory element binding proteins 2(SCAP/SREBP2)-mediated cholesterol synthesis pathway in particulate matter (PM)-induced airway inflammation in macrophages.
Methods: Bone marrow-derived macrophages (BMDMs) were cultured in vitro, and after intervention with PM, the macrophage phagocytic activity and expression of related inflammatory factors were assessed. RNA sequencing (RNA-seq) was performed on BMDMs before and after PM exposure to identify significantly altered pathways. The change of these pathways was validated. In vitro, the SCAP/SREBP2-mediated cholesterol synthesis pathway in macrophages was inhibited using LysMCre-Scapf/f mice, inhibitors, and siRNAs, and the effects on PM-induced macrophage activation were detected. The specific mechanism was further explored. In vivo, PM-induced airway inflammation models were established using LysMCre-Scapf/f mice and their littermate controls (Scapf/f) to examine the role of the SCAP/SREBP2-mediated cholesterol synthesis pathway in PM-induced airway inflammation.
Results: Macrophages were able to phagocytose PM, which led to the expression of inflammatory factors. After phagocytosis of PM, the cholesterol levels in macrophages decreased, which in turn triggered the maturation of SREBP2 and activated the cholesterol synthesis pathway. Both LysMCre-Scapf/f mice and siRNA-mediated knockdown of SREBP2 exhibited an enhanced expression of inflammatory factors after PM exposure. Further studies revealed that inhibition of cholesterol biosynthesis also promoted the expression of inflammatory factors induced by PM, while cholesterol supplementation suppressed the PM-induced inflammatory response. Additionally, myeloid-specific knockout of SCAP exacerbated PM-induced airway inflammation and mucus secretion.
Conclusion: The SCAP/SREBP2-mediated cholesterol synthesis pathway inhibits macrophage activation and airway inflammation induced by PM through the production of cholesterol.
Keywords: SCAP, SREBP2, cholesterol, macrophage, PM
Introduction
Air pollution is a major public health challenge faced by cities worldwide. It is characterized by the presence of particulate matter (PM2.5 and PM10), ozone, nitrogen dioxide, sulfur dioxide, and carbon monoxide, and has emerged as a leading environmental risk factor contributing to respiratory and cardiovascular diseases.1 PM refers to the mixture of solid and liquid particles suspended in the atmosphere. Based on particle size, PM can be classified as coarse particles (2.5–10 μm, PM10), fine particles (0.1–2.5 μm, PM2.5), and ultrafine particles (<0.1 μm).2 Among these pollutants, fine particulate matter (PM2.5) is the major contributor to particulate pollution and poses substantial health risks.3 Because of its small aerodynamic diameter, PM2.5 can remain suspended in the air for extended periods and, once inhaled, penetrate deep into the lungs. These particles are able to cross the air–blood barrier, enter the circulatory system, and reach multiple organs and systems, thereby causing adverse health effects.4 Epidemiological studies have shown that exposure to PM is associated with increased mortality rates and the development of cardiovascular and respiratory diseases, such as asthma, chronic obstructive pulmonary disease (COPD), myocardial infarction, and coronary artery disease,5–8 Therefore, elucidating the mechanisms by which PM particles induce pulmonary injury may provide new and effective strategies for disease prevention and treatment, as well as theoretical guidance and experimental evidence for the development of novel therapeutics.
Pulmonary damage caused by PM has been reported to involve multiple mechanisms, such as oxidative stress, activation of inflammatory pathways, DNA damage, and epigenetic changes.9,10 Among these, activation of inflammatory responses represents a particularly critical pathway. Macrophages, one of the most abundant immune cell populations in the lung, play essential roles in maintaining pulmonary immune homeostasis through diverse biological functions, such as antigen presentation, immune regulation, secretion of cytokines, and production of bioactive mediators.11 Lung macrophages consist of tissue-resident populations—including alveolar macrophages (AMs) and interstitial macrophages (IMs)—as well as monocyte-derived macrophages (MDMs) that are recruited under inflammatory conditions.12,13 Under steady-state conditions, the AM pool is maintained primarily through self-renewal, with little contribution from bone marrow–derived monocytes. However, during inflammation, circulating monocytes are robustly recruited to inflamed alveoli and differentiate into recruited AMs, which substantially reshape the pulmonary immune landscape.12,13 Given that AMs engulf inhaled particles and play a central role in clearing deposited PM from the alveolar space, understanding how PM exposure regulates AM function and activation is essential for elucidating the mechanisms underlying PM-induced pulmonary injury. Macrophage activation is divided into M1 activation and M2 activation. M1 macrophages dominate during the early stages of inflammation, releasing pro-inflammatory cytokines (eg, TNF-α, IL-6, IL-1β) and generating reactive oxygen species and nitric oxide to enhance bactericidal activity. By contrast, M2 macrophages primarily contribute to antiparasitic defense, tissue repair, and the resolution of inflammation by secreting anti-inflammatory mediators such as TGF-β and IL-10.14 Once PM is phagocytosed by macrophages, it initiates macrophage activation, induces M1 polarization, and leads to the release of large amounts of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) regulating the immune response.15 Increasing evidence suggests that PM stimulates macrophages to produce large amounts of pro-inflammatory cytokines,16 thereby amplifying lung inflammation and contributing to disease progression.
Recent studies have shown that metabolic pathways regulate macrophage activation and, consequently, inflammatory responses.11 For instance, serine metabolism, as a key regulator of glycolysis, impacts fatty acid synthesis, amino acid metabolism, and nucleotide metabolism, all of which are critical for macrophage polarization and the immune response to pathogens.12 Additionally, glutamine has been shown to play an important role in macrophage phenotyping through its involvement in the tricarboxylic acid cycle.13 Furthermore, pyruvate metabolism influences macrophage polarization, mitochondrial dynamics, and mitochondria-associated membrane formation, all of which are essential for macrophage function and immune responses.14
Studies indicate that lipids and their metabolites play a key role in regulating macrophage phagocytosis and inflammatory responses via intricate signaling networks.17,18 The role of cholesterol metabolism in lung diseases has recently attracted growing attention, particularly in conditions such as asthma,17 pulmonary fibrosis, and chronic obstructive pulmonary disease (COPD). Among the multiple facets of cholesterol metabolism, cholesterol biosynthesis represents a fundamental pathway, as nearly all cells possess the intrinsic capacity to synthesize cholesterol de novo.18,19 The SCAP–SREBPs pathway acts as a central regulatory axis controlling cellular lipid and cholesterol homeostasis.18,20 SREBPs (SREBP1 and SREBP2) are synthesized as inactive precursors anchored to the endoplasmic reticulum (ER), where they form a complex with SREBP cleavage-activating protein(SCAP).21 When lipid or cholesterol demand increases, SCAP escorts SREBPs to the Golgi apparatus, where sequential cleavage by Site-1 protease (S1P) and Site-2 protease (S2P) produces mature, transcriptionally active SREBPs. Functionally, SREBP1 primarily regulates fatty acid and triglyceride synthesis, whereas SREBP2 predominantly controls cholesterol biosynthesis; together with SCAP, they constitute an integrated lipid-sensing and transcriptional execution module.22
Accumulating evidence underscores the importance of this pathway in pulmonary biology and disease. In the lung, SCAP/SREBP signaling, particularly SREBP1, plays a critical role in surfactant lipid synthesis and normal lung maturation.23 In cancer biology, SREBP2 activation promotes the progression of non-small cell lung cancer (NSCLC) through the mevalonate–Akt pathway.24 Moreover, our previous work demonstrated that the SCAP–SREBP1–S1P/S2P axis regulates inflammatory responses in acute lung injury by modulating NF-κB activation.25
Although lipid metabolism has been implicated in lung physiology and disease, the specific contribution of the SCAP–SREBP2 axis to PM-induced airway inflammation has not been elucidated. Elucidating this mechanism may provide novel perspectives on the interplay between lipid metabolism and PM-driven airway inflammation.
In this study, a combination of molecular and functional approaches was used to demonstrate that PM exposure leads to alterations in cholesterol synthesis enzymes and associated cytokines in macrophages. Key regulatory factors contributing to these changes were identified. By inhibiting the SREBP2–cholesterol synthesis pathway or supplementing with exogenous cholesterol, the role and underlying mechanisms of this pathway in regulating PM-induced macrophage inflammatory responses were elucidated. Finally, in vivo experiments were performed to further examine the effects of macrophage-specific disruption of the SREBP2–cholesterol synthesis pathway on PM-induced airway inflammation. These findings provide novel insights into the crosstalk between lipid metabolism and PM-induced airway inflammation, offering potential therapeutic implications.
Materials and Methods
Mice
C57BL/6 mice (Shanghai SLAC Laboratory Animal, China) were used as wild-type (WT) mice. Scapf/f mice (Jackson Laboratory, stock No. 004162) were kindly provided by Prof. Bao-Liang Song. This strain carries loxP sites flanking exon 1 of the Scap gene and is viable, fertile, and phenotypically normal, enabling conditional knockout when crossed with Cre-expressing lines. These mice were crossed with LysMCre mice to generate myeloid cell-specific Scap conditional knockout mice (LysMCre-Scapf/f). Age- and sex-matched Scapf/f mice were used as controls. For the in vitro BMDM experiments, 4 to 6-week-old mice of the same sex were used. For in vivo PM exposure models, 6 to 8-week-old male mice were used. All mice were maintained in a specific pathogen-free facility. All experimental manipulations were approved by the Zhejiang University Medical Laboratory Animal Care and Use Committee and Ethics Committee for Animal Studies at Zhejiang University. The primer sequences for genotyping were ordered from Hangzhou Youkang Biotechnology and are listed in Table 1.
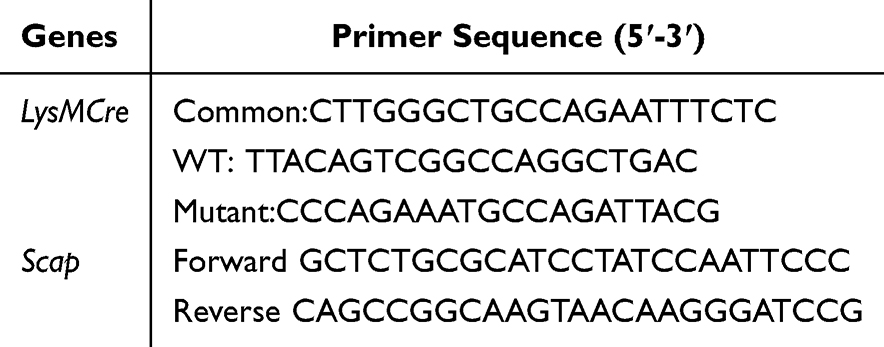 |
Table 1 The Primer Sequences for Gene Identification |
PM Preparation
PM used in this study was purchased from the National Institute of Standards and Technology (USA). The particle diameter was approximately 2.5–10 µm. PM was suspended in sterile phosphate-buffered saline (PBS) and sonicated for 1 h to obtain a stock solution at 2 mg/mL, which was used in both in vivo and in vitro experiments.
Animal Models
In vivo, PM was suspended and sonicated in sterile saline (0.9% NaCl) to a final concentration of 2 mg/mL. Mice were administered 100 µg PM (suspended in 50 µL saline) via intratracheal instillation for four consecutive days to establish an acute airway inflammation model. Control mice received an equal volume of saline.
Cell Culture: BMDMs
Bone marrow cells were isolated from the tibia and femur of 4 to 6-week-old C57BL/6 mice, and erythrocytes were lysed using red blood cell (RBC) lysis buffer (Sigma-Aldrich, USA). The remaining cells were seeded in plates and cultured in RPMI-1640 medium (Biochannel, China) supplemented with 15% heat-inactivated fetal bovine serum (Dcell, China) and 1% penicillin–streptomycin (Beyotime, China). Macrophage differentiation was induced by adding 20 ng/mL M-CSF (Novoprotein, China). Cells were incubated at 37 °C with 5% CO2, and on day 3 the medium was replaced with fresh complete medium. By day 5, BMDMs had matured. BMDMs were then exposed to PM at a concentration of 120 µg/mL for 2 h, 4 h, or 8 h.
Transmission Electron Microscope
BMDMs were fixed in 2.5% glutaraldehyde in PBS for 24 h following standard procedures. Samples were washed, post-fixed in 1% osmium tetroxide, and stained with 4% uranyl acetate. They were dehydrated in graded ethanol/acetone, embedded in epoxy resin, and sectioned (85–90 nm). Sections were stained with uranyl acetate and lead citrate. Images were acquired with a Tecnai G2 Spirit 120-kV TEM (FEI, USA). Cytoplasmic area was measured with Image-Pro Plus 6.0, and data were expressed as PM particles per 100 µm².
Real-Time Quantitative PCR (RT-qPCR)
Total RNA was extracted from lung tissue or cells using Trizol reagent (Accurate Biotechnology, China) according to the manufacturer’s instructions. RNA quality was assessed using a NanoDrop One spectrophotometer (Thermo Fisher Scientific, USA). Complementary DNA (cDNA) was synthesized from total RNA using a reverse transcription kit (Accurate Biotechnology, China). Real-time quantitative PCR was performed using SYBR Green Master Mix (Accurate Biotechnology, China) on a StepOne Real-Time PCR System (Applied Biosystems, USA). Each assay was performed with three biological replicates and two technical replicates per sample. Primer sequences used for RT-qPCR are listed in Table 2.
 |
Table 2 The Primer Sequences for RT-PCR |
Enzyme-Linked Immunosorbent Assay (ELISA)
Supernatants from BMDMs and bronchoalveolar lavage fluid (BALF) from animal models were collected. IL-6, CXCL1, and TNF concentrations were measured with ELISA kits (MultiSciences, China) according to the manufacturer’s protocol.
RNA-Seq Detection and Analysis
Total RNA was extracted from BMDMs using Trizol reagent according to the manufacturer’s instructions. RNA quality and quantity were assessed with an Agilent 2100 Bioanalyzer (Agilent, USA). Library preparation was performed using the Optimal Dual-Mode mRNA Library Prep Kit (BGI-Shenzhen, China). The library was amplified by polymerase chain reaction (PCR) with primers annealing to universal adapter sequences to generate DNA nanoballs (DNBs). Paired-end 150 bp sequencing (PE150) was conducted on the T7 sequencer (BGI-Shenzhen, China) using the combined probe-anchoring polymerization technique. The reads were mapped to reference gene sets using Bowtie2 (v2.4.5), and differential gene expression between groups was analyzed with DESeq2 (v1.40.2). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of differentially expressed genes was performed with Phyper in R (R Foundation for Statistical Computing, Austria; v3.5.3) using the hypergeometric test.
Isolation and Culture of Alveolar Macrophages
After anesthesia and euthanasia, BALF was collected by gently lavaging the lungs four times with 1 mL sterile PBS. The recovered lavage fluid was centrifuged at 1600 rpm for 5 min, and the supernatant was discarded. The resulting cell pellet was resuspended in complete medium consisting of RPMI-1640 (Biochannel, China) with 15% fetal bovine serum (Dcell, China), 1% penicillin–streptomycin (Beyotime, China), and 20 ng/mL GM-CSF (Novoprotein, China). Cells were plated (1.5 × 10^5/well, 12-well plates). After 3 h, the medium was replaced to remove non-adherent cells. After 24 h, cells were exposed to PM (80 µg/mL) for 6 h.
Immunofluorescence Staining
Cells were fixed and blocked with 5% BSA, followed by incubation with anti-HMGCR antibody (Abcam, UK; 1:200) at 4 °C overnight. After washing with PBS, cells were incubated with Alexa Fluor 555–conjugated secondary antibody (Thermo Fisher Scientific, USA; 1:500) at room temperature for 1 h, followed by DAPI counterstain (Beyotime, China; 1:1000). Fluorescence images were acquired using a super-resolution confocal microscope (Olympus IX83-FV3000-OSR, Japan). Quantification was performed with Image J.
Apoptosis Assay
To assess apoptosis, the Annexin V-APC/PI Apoptosis Kit (MultiSciences, China) was used. BMDMs were harvested from one well of a 12-well plate per sample. Cells were resuspended in binding buffer and stained with 5 µL Annexin V–APC and 10 µL PI for 5 min at room temperature. Flow cytometry was performed on a CytoFLEX (Beckman Coulter, USA). Gating strategy: (1) FSC vs SSC to exclude debris; (2) FSC-height vs FSC-area to remove doublets. Annexin V−/PI− cells were considered viable.
Western Blot
After PM exposure, cells were lysed in SDS-PAGE sample loading buffer (Beyotime, China). Protein lysates were separated by SDS-PAGE (Epizyme, China) and transferred to membranes for immunoblotting with the indicated antibodies using standard methods. The primary antibodies used were anti-ACTB (Abclonal, China; 1:10,000), anti-SREBP2 (Abclonal, China; 1:500), anti-SREBP1 (BD Biosciences, USA; 1:1000), and anti-HMGCR (Abcam, UK; 1:1000). ACTB served as the loading control. Fluorescently labeled secondary antibodies were used for detection, and imaging was performed with the Odyssey two-color infrared laser imaging system (LI-COR, USA). Final results were presented as grayscale images.
Cholesterol Measurement
Total cholesterol was measured with the Cholesterol Quantitation Kit (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions on a Varioskan Flash multimode plate reader (Thermo Fisher Scientific, USA). Lipid droplets were stained with BODIPY™ 493/503 (Invitrogen, USA), and lipid rafts were detected with cholera toxin subunit B (Sigma-Aldrich, USA). These two readouts were quantified by flow cytometry as mean fluorescence intensity (MFI) on a CytoFLEX instrument (Beckman Coulter, USA).
Small Interfering RNA (siRNA) Transfection
BMDMs (1 × 10^6) were transfected with 100 nM siRNA using a commercial transfection reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.
BALF Cell Collection
Twenty-four hours after the last instillation, BALF was collected by three lavages with 0.4 mL PBS. The total number of BALF cells was counted, and the remaining cells were centrifuged at 3000 × g for 10 min at 4 °C to collect cells for Wright–Giemsa staining (Baso, China) to determine differential cell counts. Differential counts were assessed by counting 200 cells. Supernatant was stored at −80 °C for cytokine detection.
Histologic Analysis
After treatment with PM, the lungs of mice were isolated and fixed in formalin for 24 h. Paraffin-embedded sections were stained with hematoxylin and eosin (H&E) and with Periodic acid–Schiff (PAS). Inflammation scores were assessed according to published guidelines.26 Periodic Acid-Schiff (PAS) stained goblet cells in airway epithelium were scored as described previously.27
Statistical Analysis
GraphPad Prism 8.0.2 (GraphPad Software, USA) was used for statistical analysis and data presentation. Quantitative data are expressed as mean ± SD. Differences between two groups were analyzed using an unpaired Student’s t-test, and multiple group comparisons were performed by one-way analysis of variance (ANOVA). Statistical significance was set at *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant.
Results
Macrophages Phagocytose PM Particles and Produce Inflammatory Factors
To elucidate the mechanism of PM-induced airway inflammation, both in vivo and in vitro approaches were employed. We first established a mouse model of PM-induced airway inflammation by intratracheal instillation of PM, following previously published protocols.28 The results showed that PM exposure led to an increase in inflammatory cell infiltration around the airways (Figure 1A). In lung tissue sections, we observed that alveolar macrophages were able to phagocytose PM (Figure 1B). Because monocyte-derived alveolar macrophages play a central role in airway responses to PM, bone marrow–derived macrophages (BMDMs) were used in vitro as a surrogate model. BMDMs were cultured and reached maturity after 5 days, as indicated by CD11b⁺F4/80⁺ expression29 (Figure S1A). Cell viability assays showed no significant changes after exposure to PM for 2, 4, or 8 hours (Figure S2A). In vitro experiments also showed that macrophages internalized PM (Figure 1C and D). To further investigate the macrophage response to PM, we examined the expression level of inflammatory cytokines. As shown in Figure 1E, G, and I, PM exposure resulted in a significant increase in the mRNA transcription of IL-6, CXCL1, and TNF in macrophages, respectively. Similarly, protein levels of IL-6 and CXCL1 were also significantly increased in the supernatant (Figure 1F and H). Collectively, these results indicate that PM can be phagocytosed by macrophages, thereby triggering their activation.
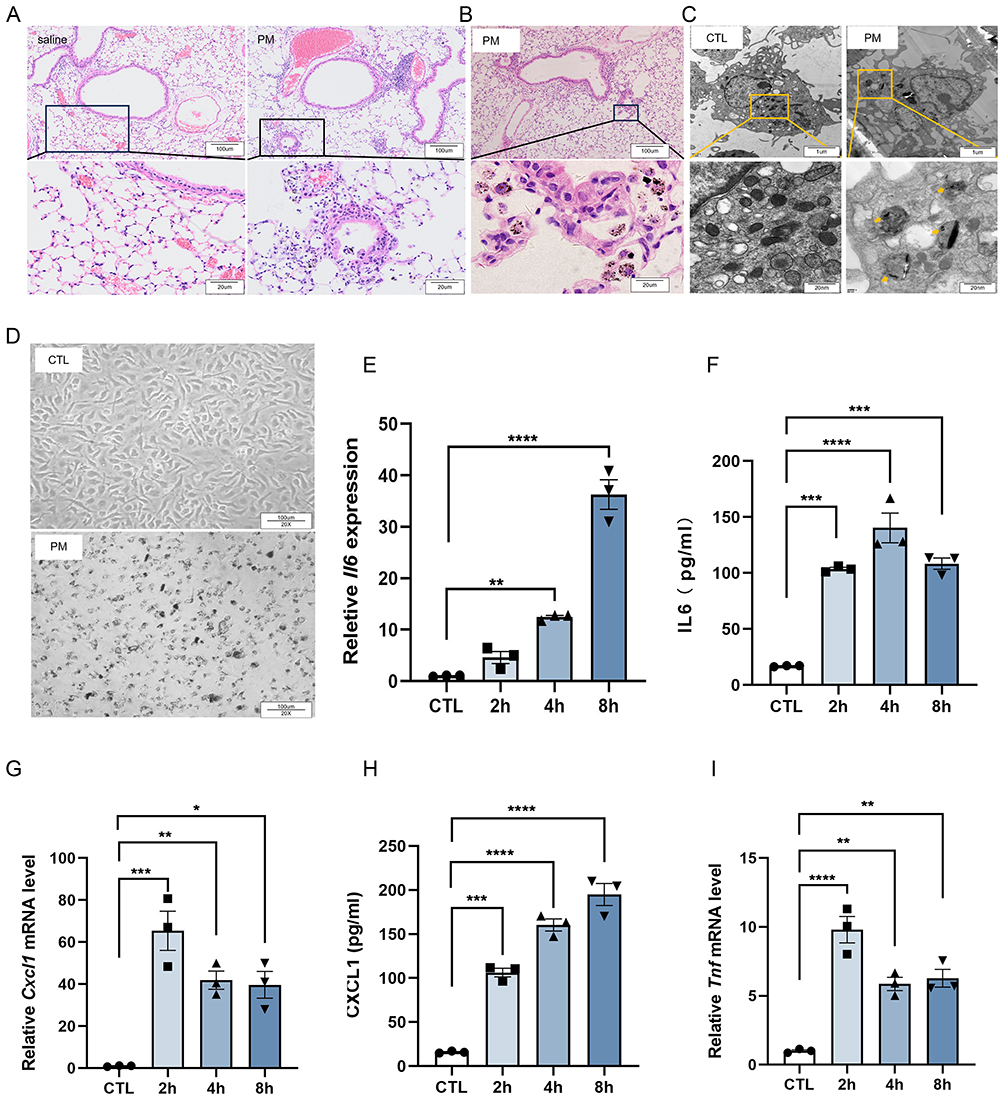 |
Figure 1 Macrophages phagocytose PM and produce inflammatory cytokines. C57BL/6 mice were treated with 100 µg PM via intratracheal instillation for four consecutive days. Lung tissues were harvested on day 5 for H&E staining. (A) Representative images showing inflammatory cell infiltration around the airways. (B) Lung macrophages phagocytosing PM particles. BMDMs were exposed to PM (120 µg/mL) for 8 h. (C) Electron microscopy images of macrophages phagocytosing PM. (D) Microscopic views of macrophages in control (CTL) and PM-exposed groups. BMDMs were collected after PM exposure for 2 h, 4 h, and 8 h to assess inflammatory cytokines by qPCR and ELISA. (E) qPCR analysis of Il6 mRNA (n = 3). (F) ELISA analysis of IL-6 levels in culture supernatants (n = 3). (G) qPCR analysis of Cxcl1 mRNA transcription (n = 3). (H) ELISA analysis of CXCL1 levels in culture supernatants (n = 3). (I) qPCR analysis of Tnf mRNA transcription (n = 3). All results were confirmed in at least three independent experiments, and data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
PM Reduces Cholesterol Levels in Macrophages, Promoting SREBP2 Maturation and Activation of the Cholesterol Synthesis Pathway
To explore the mechanism behind PM-induced inflammatory cytokine expression, we performed RNA sequencing on both CTL and PM-exposed BMDMs. KEGG pathway enrichment analysis revealed significant alterations in the Cholesterol metabolism pathway (Figure S3A; Gene List). Then we found that PM exposure reduced intracellular cholesterol levels (Figure 2A). Within macrophages, cholesterol is stored by esterifying free cholesterol and packaging it into lipid droplets. Flow cytometry analysis showed no significant difference in cytoplasmic lipid droplet levels between groups (Figure 2B and C). However, membrane lipid raft levels were significantly lower in PM-exposed macrophages compared with controls (Figure 2D and E). We speculate that the reduction in total cholesterol content may be attributable to the decrease in cholesterol incorporated into membrane lipid rafts. This raises the question of what functional consequences reduced cholesterol levels may exert. Previous studies have reported that a decline in cholesterol content can trigger compensatory activation of the cholesterol synthesis pathway. To test this possibility, we next examined the expression of cholesterol synthesis–related genes and performed Western blotting of key enzymes. In our study, PM exposure significantly increased the mRNA levels of cholesterol synthesis enzymes compared to the control group (Figure 2F). Western blot analysis of selected enzymes (eg, HMGCS1, CYP51) showed a corresponding increase in protein levels (Figure 2G). To further validate these findings, primary alveolar macrophages (AMs) were examined. Consistently, the key enzyme HMGCR was found to be upregulated at both the transcriptional and protein levels in PM-exposed AMs (Figure S4A–C). SREBP2 is a pivotal transcription factor that mediates the transcription of enzymes involved in cholesterol synthesis. In PM-exposed macrophages, we observed enhanced maturation of SREBP2 (Figure 2H). To validate this, macrophages were treated with the cholesterol derivative 25-HC. 25-HC treatment effectively suppressed transcription of cholesterol synthesis–related enzymes (Figure 2I). Western blotting confirmed that 25-HC reduced the elevated protein levels of HMGCR, HMGCS1, and CYP51 induced by PM (Figure 2J and K). Furthermore, we examined the activation of the SREBP1-mediated fatty acid synthesis pathway and found that PM exposure did not significantly affect the maturation of SREBP1 or the expression of key enzymes in this pathway (Figure S5A–E).
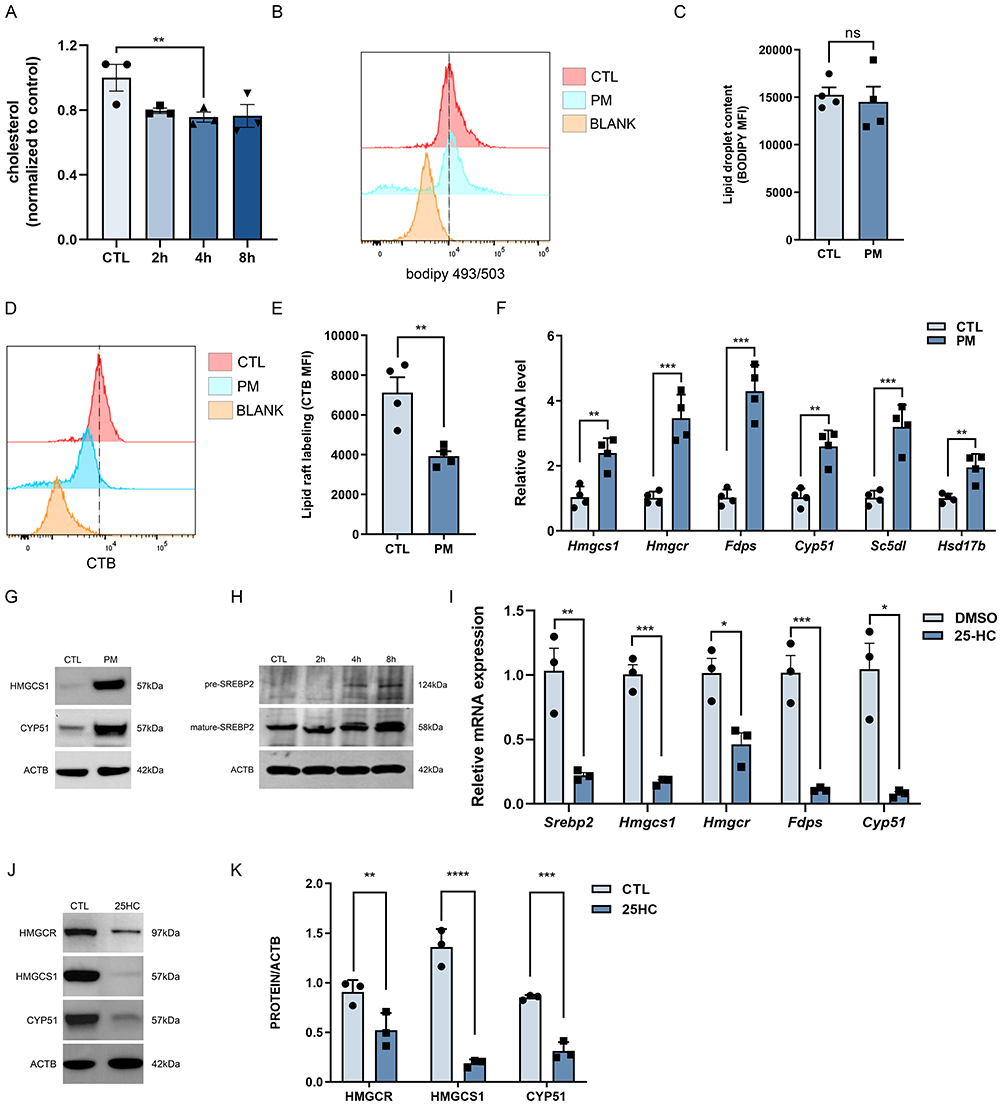 |
Figure 2 PM reduces cholesterol levels, promoting SREBP2 maturation and activating the cholesterol biosynthetic pathway in macrophages. BMDMs were exposed to PM (120 µg/mL) for 8 h. (A) Cholesterol content in CTL and PM groups at 2 h, 4 h, and 8 h (n = 3). (B) Flow cytometry histogram showing BODIPY staining to assess lipid droplet content. (C) Quantification of lipid droplet content as mean fluorescence intensity (MFI) (n = 4). (D) Flow cytometry histogram showing CT-B staining to assess lipid raft labeling. (E) Quantification of lipid raft labeling as mean fluorescence intensity (MFI) (n = 4). (F) qPCR analysis of cholesterol synthesis–related genes (Hmgcs1, Hmgcr, Fdps, Cyp51, Sc5dl, Hsd17b) in the CTL and PM-treated groups (n = 4). (G) WB analysis of CYP51 and HMGCS1 proteins. (H) WB analysis of precursor (pre-) and mature SREBP2 in CTL and PM-exposed groups at 2 h, 4 h, and 8 h. (I) qPCR analysis of Srebp2, Hmgcs1, Hmgcr, Fdps, and Cyp51 after treatment with 25-hydroxycholesterol (25-HC, 10 µM) for 36 h vs DMSO groups (n = 3). (J) WB of HMGCR, HMGCS1, and CYP51 after 25-HC treatment. (K) Quantification of HMGCR, HMGCS1, and CYP51 normalized to ACTB (n = 3). All results were confirmed in at least three independent experiments, and data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant. |
Inhibition of SREBP2 Maturation Promotes PM-Induced Inflammatory Cytokine Expression
SCAP is a key regulator of SREBP2 maturation. When cholesterol levels are low, SCAP facilitates the transfer of SREBP2 from the endoplasmic reticulum to the Golgi apparatus, where it undergoes proteolytic cleavage to form its mature active form. To further elucidate the role of SCAP/SREBP2 in macrophage activation, LysMCre-Scapf/f mice, which specifically lack SCAP in myeloid cells, were used. Bone marrow-derived macrophages (BMDMs) from these mice were exposed to PM. The results showed that SCAP-deficient BMDMs exhibited significantly enhanced expression of inflammatory cytokines, including IL-6 and CXCL1 at both mRNA and protein levels, and TNF at the mRNA level (Figure 3A–E). Because SCAP also promotes SREBP1 maturation, siRNAs targeting SREBF1 or SREBF2 were employed. Knockdown efficiency was shown in Figure S6A and B. Knockdown of SREBP1 had no effect, whereas SREBP2 knockdown significantly increased cytokine expression in response to PM (Figure 3F–H).
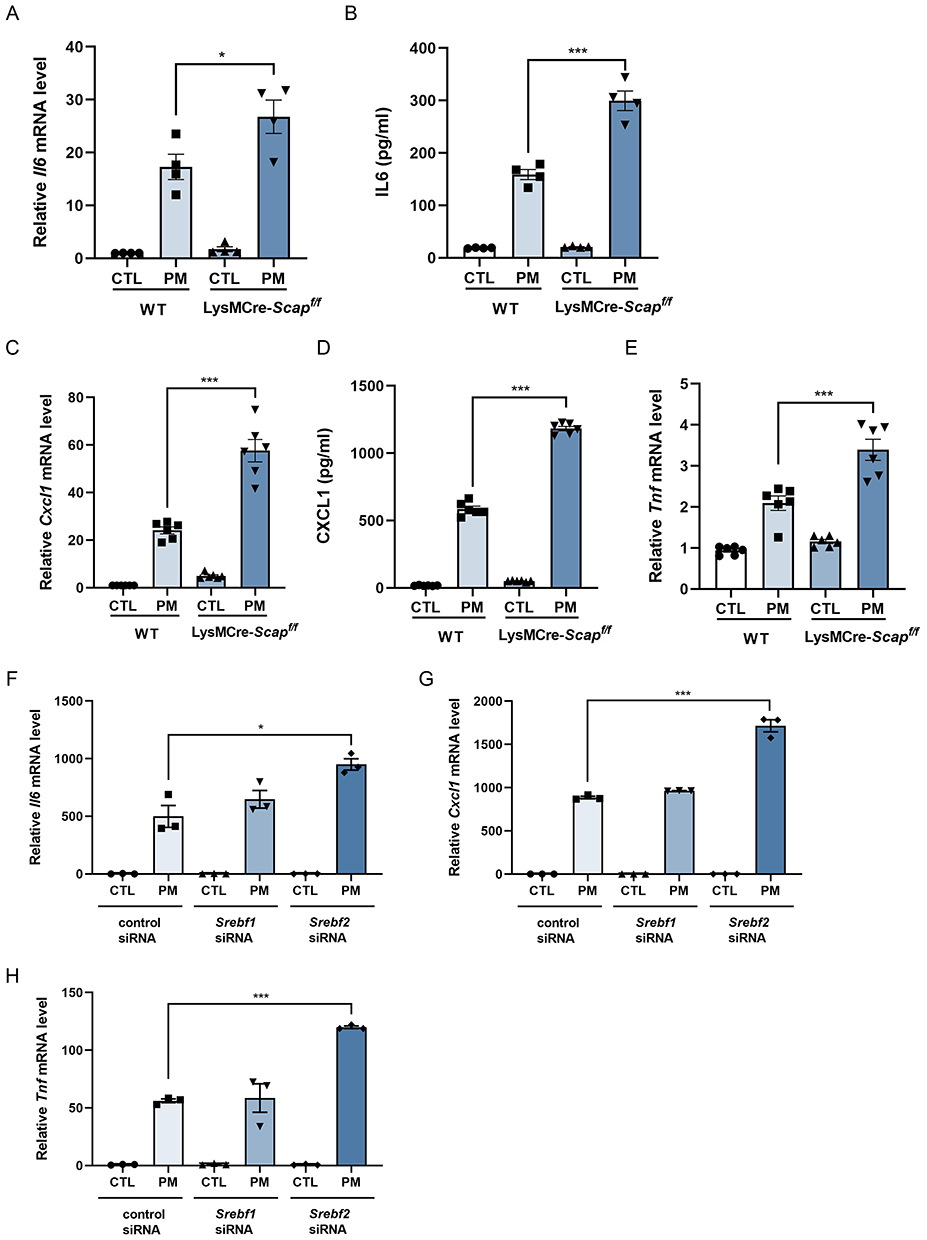 |
Figure 3 Inhibition of SREBP2 maturation upregulates PM-induced inflammatory cytokines expression in macrophages. BMDMs were exposed to PM (120 µg/mL) for 8 h. (A and B) qPCR analysis of Il6 mRNA (A) and ELISA analysis of IL-6 protein (B) in Scapf/f and LysMCre-Scapf/f mice (n = 3). (C and D) qPCR analysis of Cxcl1 mRNA (C) and ELISA analysis of CXCL1 protein (D) in Scapf/f and LysMCre-Scapf/f mice (n = 6). (E) qPCR analysis of Tnf mRNA in Scapf/f and LysMCre-Scapf/f mice (n = 6). (F–H) qPCR analysis of Il6 (F), Cxcl1 (G), and Tnf (H) mRNA following siRNA-mediated knockdown of Srebf1 or Srebf2 versus control siRNA (n = 3). All results were confirmed in at least three independent experiments, and data are presented as mean ± SD. *p < 0.05, ***p < 0.001. |
Inhibition of Cholesterol Synthesis Promotes PM-Induced Inflammatory Cytokine Expression
Mature SREBP2 activates expression of cholesterol synthesis enzymes. To examine this process, HMGCR, a key rate-limiting enzyme, was evaluated. PM exposure significantly increased HMGCR mRNA and protein levels (Figure 4A and B). Quantitative analysis of protein expression also supported the increase in HMGCR levels (Figure S7A). Treatment with lovastatin or atorvastatin suppressed HMGCR induction (Figure S8A–D), supporting a role for HMGCR activity in macrophage activation. Following PM exposure, lovastatin increased the expression of IL-6, CXCL1, and TNF, as demonstrated by qPCR (Figure 4C–E) and ELISA (Figure 4F and G) analysis. Similarly, atorvastatin treatment also increased expression of these inflammatory cytokines (Figure 4H–J). These findings indicate that inhibition of cholesterol synthesis exacerbates PM-induced macrophage activation.
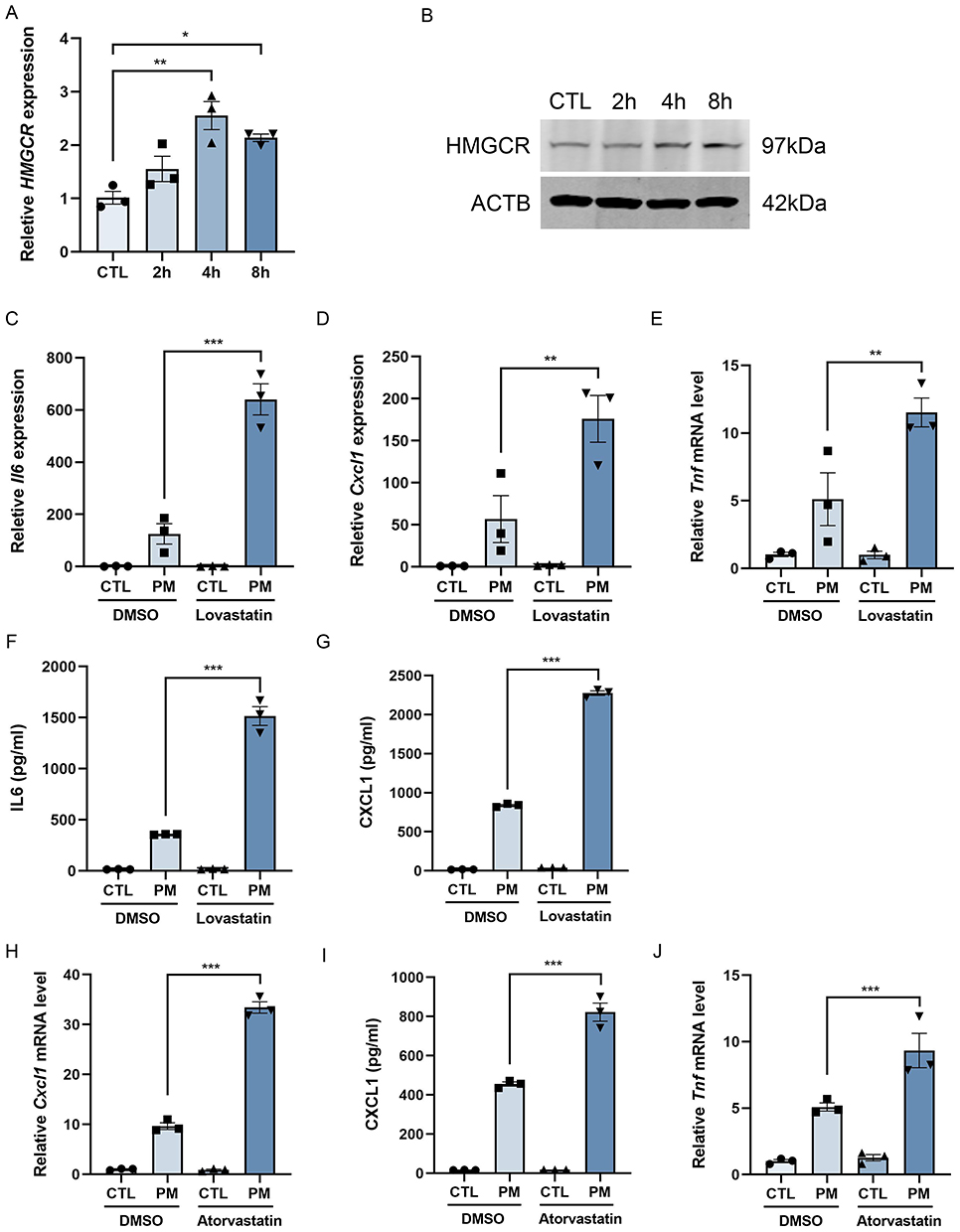 |
Figure 4 Inhibition of the rate-limiting enzyme HMGCR in cholesterol synthesis promotes the expression of PM-induced inflammatory cytokines in macrophages. BMDMs were exposed to PM (120 µg/mL) for 2, 4, and 8 h. (A) qPCR analysis of Hmgcr mRNA (n = 3). (B) WB analysis of HMGCR protein. (C–E) qPCR analysis of Il6 (C), Cxcl1 (D), and Tnf (E) mRNA after PM exposure with or without lovastatin (20 µM) (n = 3). (F and G) ELISA analysis of IL-6 (F) and CXCL1 (G) in culture supernatants with or without lovastatin (20 µM) (n = 3). (H and I) qPCR analysis of Cxcl1 mRNA (H) and ELISA analysis of CXCL1 protein (I) with or without atorvastatin (20 µM) (n = 3). (J) qPCR analysis of Tnf mRNA with or without atorvastatin (20 µM) (n = 3). All results were confirmed in at least three independent experiments, and data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. |
Cholesterol Suppresses PM-Induced Inflammatory Cytokine Expression in Macrophages
Given the aforementioned findings, we further investigated the role of cholesterol in PM-induced macrophage activation. 25-HC, an endogenous cholesterol derivative, was used to treat macrophages exposed to PM. Our results demonstrated that 25-HC significantly attenuated the expression of IL-6, CXCL1, and TNF-α both in cells (Figure 5A–C) and in the culture supernatant (Figure 5D and E) compared to the PM exposed-only group, indicating that cholesterol exerts a suppressive effect on PM-induced macrophage activation.
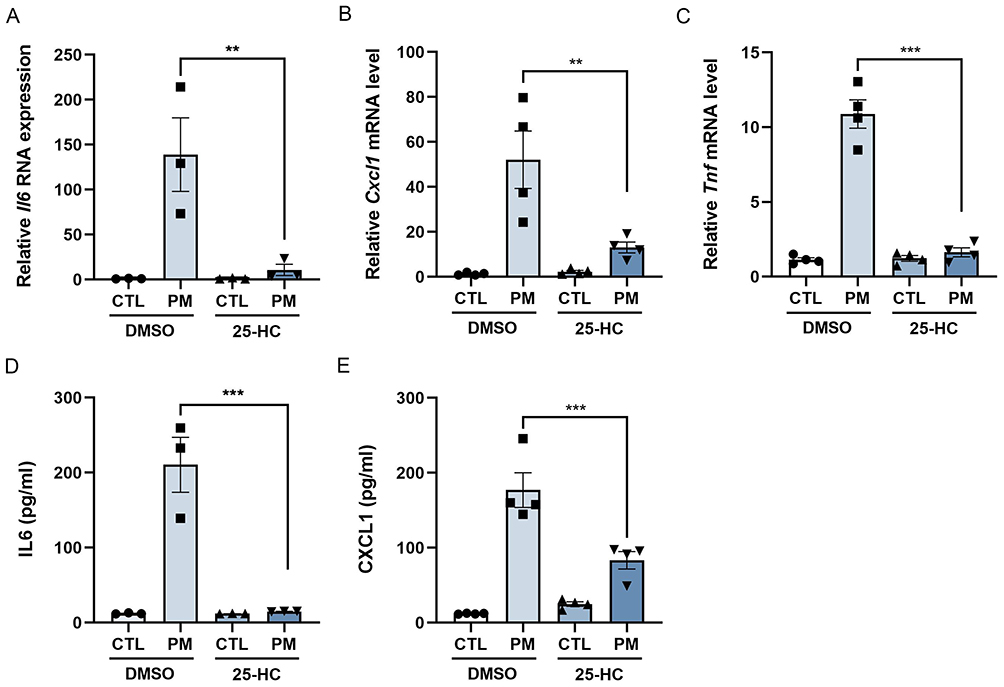 |
Figure 5 Cholesterol suppresses the level of PM-induced inflammatory cytokines in macrophages. BMDMs were pretreated with 25-hydroxycholesterol (25-HC, 10 µM) for 24 h and then exposed to PM (120 µg/mL) for 8 h. (A) qPCR analysis of Il6 mRNA (n = 3). (B) qPCR analysis of Cxcl1 mRNA (n = 4). (C) qPCR analysis of Tnf mRNA (n = 4). (D) ELISA analysis of IL-6 in culture supernatants (n = 3). (E) ELISA analysis of CXCL1 levels in culture supernatants (n = 4). All results were confirmed in at least three independent experiments, and data are presented as mean ± SD. **p < 0.01, ***p < 0.001. |
Myeloid-Specific SCAP Knockout Exacerbates PM-Induced Airway Inflammation and Mucus Secretion
The in vitro findings demonstrated that the SCAP/SREBP2 pathway regulates PM-induced macrophage activation by modulating cholesterol synthesis. Enhanced cholesterol synthesis mitigated inflammatory cytokine expression, whereas reduced cholesterol synthesis exacerbated this expression. Finally, to assess the in vivo role of SCAP/SREBP2, LysMCre-Scapf/f and control mice were subjected to PM-induced airway inflammation.
In vivo analysis demonstrated that myeloid-specific SCAP knockout exacerbated PM-induced airway inflammation. H&E and PAS staining revealed enhanced inflammatory infiltration and mucus secretion (Figure 6A–C). Consistently, both the total cell counts and the number of neutrophils in BALF were significantly increased in SCAP-deficient mice (Figure 6D and E). Moreover, levels of CXCL1 and IL-6 in BALF were markedly increased compared with controls (Figure 6F and G).
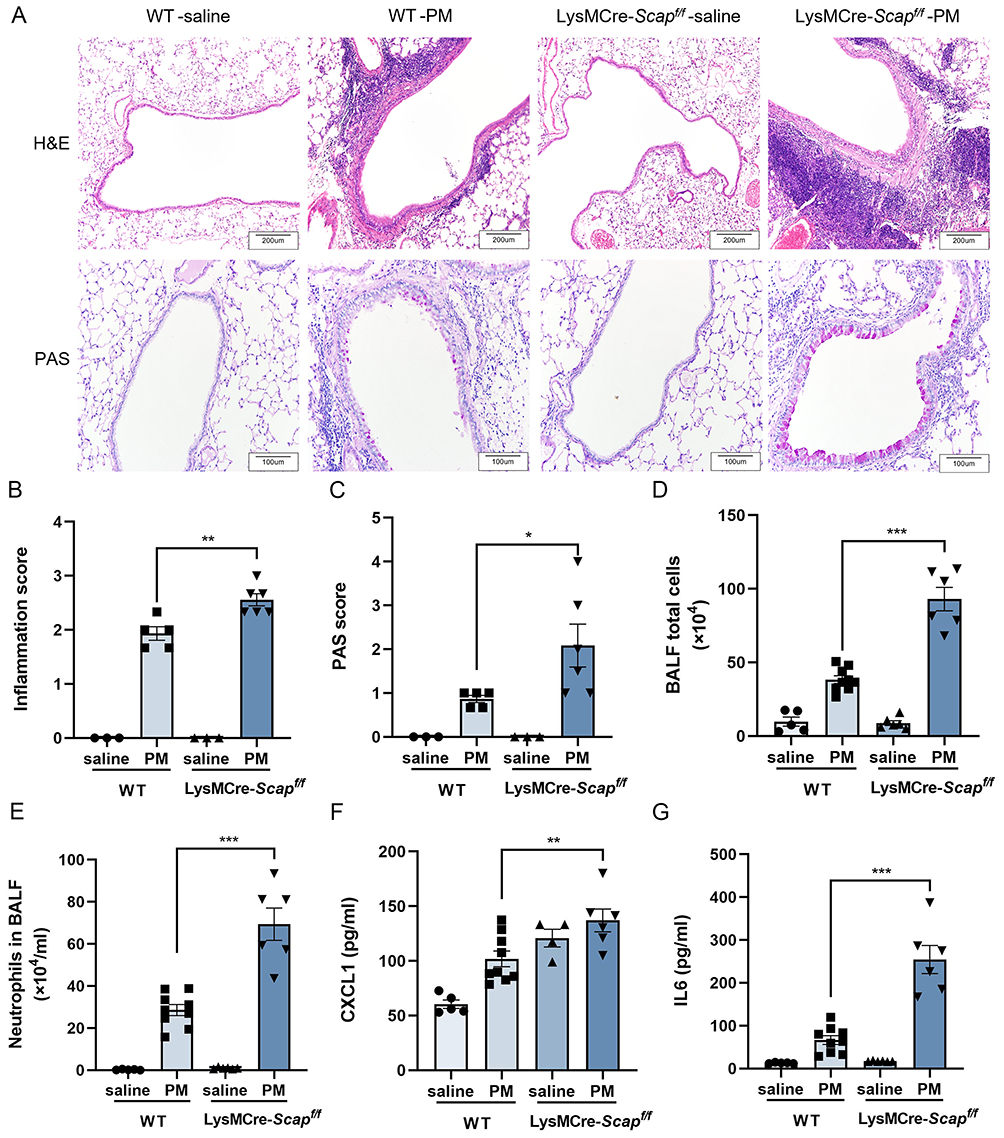 |
Figure 6 Myeloid-specific SCAP knockout exacerbates PM-induced airway inflammation and mucus secretion. (A) H&E and PAS staining of lung tissue from Scapf/f and LysMCre-Scapf/f mice after exposure to saline or PM. (B) Quantitative analysis of HE staining (n = 3–6). (C) Quantitative analysis of PAS staining (n = 3–5). (D) Total cell counts in BALF (n = 5–9). (E) Neutrophil counts in BALF (n = 5–9). (F) ELISA analysis of CXCL1 levels in BALF (n = 4–9). (G) ELISA analysis of IL-6 levels in BALF (n = 5–9). All results were confirmed in at least three independent experiments, and data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. |
Discussion
This study demonstrates that when macrophages are exposed to PM, they can phagocytose the particles and induce the expression of intracellular inflammatory factors. The cholesterol levels in the PM exposure group were significantly lower than those in the control group. Through RNA-seq analysis and KEGG pathway enrichment, we identified that PM exposure altered the cholesterol metabolism pathway. Further investigation revealed that PM exposure reduced intracellular cholesterol levels, while simultaneously increasing the maturation of SREBP2, the expression of enzymes related to the cholesterol synthesis pathway, and activating cholesterol synthesis. Furthermore, cholesterol intervention was found to suppress the expression of these cholesterol synthesis-related enzymes. These findings suggest that PM causes a reduction in intracellular cholesterol levels, which in turn feeds back to activate the cholesterol synthesis pathway and promote cholesterol production in macrophages. We also found that specific knockout of SCAP or knockdown of SREBP2 in macrophages inhibits SREBP2 maturation and promotes the expression of inflammatory factors induced by PM exposure. Moreover, inhibition of HMGCR, a key enzyme in cholesterol synthesis, also enhanced PM-induced inflammatory cytokine expression, whereas cholesterol supplementation significantly suppressed macrophage inflammation. These results collectively indicate that the SCAP/SREBP2 pathway inhibits PM-induced macrophage activation by activating cholesterol synthesis. Consistent with our in vitro results, in vivo experiments using myeloid-specific SCAP knockout mice demonstrated that SCAP knockout in macrophages exacerbated PM-induced airway inflammation by inhibiting SREBP2 maturation.
Moreover, our previous work demonstrated that PM could induce the formation of macrophage extracellular traps (METs) both in vitro and in vivo through peptidylarginine deiminase type 4 (PAD4)-mediated histone citrullination. Mechanistically, METs played a crucial role in enhancing macrophage phagocytosis and facilitating the clearance of PM, thereby suppressing airway inflammation.30 Further elucidating the complex mechanisms underlying PM-induced inflammatory cytokine production following its cellular internalization continues to be a critical and ongoing focus of our research.
Endoplasmic reticulum (ER) stress refers to a cellular stress response triggered by the accumulation of unfolded or misfolded proteins due to ER dysfunction. PM exposure activates the ER stress response, leading to the secretion of inflammatory factors such as TNF-α and IL-6. These inflammatory cytokines not only exacerbate systemic inflammation but also further intensify ER stress. Our study, through RNA-seq analysis, also observed that PM exposure induces ER protein processing.
Previous studies have reported the specific mechanisms linking PM exposure to ER stress. The literature suggests that PM triggers cellular responses by activating ER stress pathways such as inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6) and protein kinase RNA-like ER kinase (PERK), which in turn leads to the exacerbation of inflammatory responses.31
The ER also plays a crucial role in lipid metabolism. The literature suggests that the ER maintains cellular lipid homeostasis by regulating processes such as fatty acid synthesis and storage.32 Interestingly, although our study did not focus on ER stress, we observed that PM exposure significantly altered lipid metabolism. Specifically, we noted an increase in the nuclear translocation of SREBP2, indicating that PM exposure may exacerbate the ER’s burden by influencing lipid metabolic pathways. Therefore, while the current study primarily focused on lipid metabolism changes, ER stress remains an important direction for our future research.
In recent years, several studies have indicated that PM exposure is closely associated with changes in cholesterol levels. A longitudinal study conducted in China found that exposure to PM2.5 and PM10 was linked to elevated levels of triglycerides, total cholesterol, and low-density lipoprotein cholesterol, while high-density lipoprotein cholesterol levels were reduced.33 Another study in China found that higher levels of PM1 (particles with a diameter ≤ 1.0 μm), which make up 80% of the total PM2.5 mass,34 were associated with dyslipidemia. This was indicated by increased total cholesterol and low-density lipoprotein cholesterol, along with decreased triglyceride and high-density lipoprotein cholesterol levels.35 In Mexico City, exposure to PM2.5 during the later stages of pregnancy was associated with increased total cholesterol and low-density lipoprotein cholesterol levels, but with lower triglyceride and high-density lipoprotein cholesterol levels in children.36 In an in vitro three-dimensional skin model, exposure to PM2.5 significantly increased cholesterol levels, and this cholesterol accumulation promoted the onset of inflammation. Additionally, a study on dry eye disease showed that SD rats injected with PM2.5 in both eyes exhibited significantly higher serum total cholesterol (TC) levels compared with the control group, and the inflammatory response in the lacrimal glands was also enhanced.37 Another study on PM-induced liver damage demonstrated that rats exposed to PM had significantly higher serum cholesterol levels compared with the control group, and the expression of inflammatory cytokines in both serum and liver was also increased.38 However, studies focusing on the lungs have yet to provide clear conclusions regarding the impact of PM airway exposure on cholesterol content.
Interestingly, a recent transcriptomic analysis of primary keratinocytes exposed to PM2.5 found that PM2.5 exposure significantly upregulated the transcription of cholesterol metabolism-related genes, many of which encode key enzymes involved in cholesterol synthesis from acetyl-CoA.39 This finding is consistent with the results of our study. We found that PM exposure decreased cholesterol levels in macrophages, which, in turn, feed back to promote the activation of the SCAP-SREBP2 pathway involved in cholesterol synthesis. Notably, inhibiting the SCAP/SREBP2-mediated cholesterol synthesis pathway enhanced PM-induced macrophage activation and the expression of inflammatory cytokines.
These findings suggest that the changes in cholesterol metabolism induced by PM exposure may vary across different tissues or cell types, further influencing the inflammatory response. Therefore, we believe that the dynamic changes in metabolic processes triggered by PM may lead to distinct inflammatory responses and metabolic alterations in different diseases.
Several studies have shown that statins play an important role in pulmonary inflammation, particularly in type 1 and type 2 inflammation. Pulmonary inflammation is primarily divided into two types: Type 1 inflammation is typically driven by neutrophils and is commonly seen in acute responses such as bacterial infections, while Type 2 inflammation is predominantly driven by eosinophils and is common in allergic diseases like asthma. Existing research has indicated that statins have significant anti-inflammatory effects in Type 1 inflammation. For example, high doses of simvastatin have been shown to reduce the production of IL-6 and IL-1β in LPS-induced monocytes.40 Moreover, atorvastatin has demonstrated similar anti-inflammatory effects by reducing levels of various pro-inflammatory cytokines, such as TNF, IL-1, and IL-6.41
In Type 2 inflammation, statins have also shown anti-inflammatory effects, especially in models of allergic diseases like asthma. For instance, simvastatin in an OVA-induced asthma mouse model was able to reduce the infiltration of inflammatory cells in BALF and lower the levels of IFN-γ, IL-4, and IL-5, which are associated with Type 2 immune responses.42 Additionally, elevated cholesterol levels in the blood are closely linked to enhanced Type 2 inflammation. Studies suggest that high cholesterol exacerbates allergic inflammation by promoting the secretion of pro-inflammatory cytokines.
Although existing studies indicate that statins have the potential to suppress both Type 1 and Type 2 inflammation, our research has found that, under conditions of PM exposure, statins may influence inflammation through different mechanisms. Therefore, this finding suggests that further exploration of the mechanisms by which statins affect different types of pulmonary inflammation is necessary, with the aim of providing new insights for treatment strategies.
There are two limitations. One is availability of alveolar macrophages: It was difficult to obtain a sufficient number of alveolar macrophages. Approximately 20–30 mice were required to yield enough cells to fill a single well of a 12-well plate. This limitation impacted the quantity of cells available for further experiments and consequently limited the scope of our investigation into the detailed functions of alveolar macrophages. The other is potential off-target effects of LysM knockout on neutrophils: This limitation arises from the use of LysM-Cre mice for gene knockout, although they are widely recognized as a standard tool for studying macrophages. While LysM-driven gene deletion is primarily intended to target macrophages, it may also affect neutrophils. This potential off-target effect could complicate the interpretation of our results and represents an area for future refinement in experimental design.
Conclusion
In conclusion, our study demonstrates that PM can be phagocytosed by macrophages, thereby inducing cellular activation. During this process, PM exposure decreases intracellular cholesterol levels, which in turn activates the SCAP/SREBP2 pathway through a feedback mechanism. This leads to SREBP2 maturation and the upregulation of enzymes involved in cholesterol synthesis. Consequently, increased cholesterol synthesis mitigates PM-induced macrophage activation and airway inflammation. To our knowledge, this is the first study to identify the cholesterol synthesis pathway as a protective regulator of macrophage responses to PM.
Data Sharing Statement
The raw data supporting the conclusions of this article will be available from the corresponding author on request.
Ethics Approval and Informed Consent
This study complied with the regulations of Animal Research Ethics Committee of Zhejiang University. The care of experimental animals follows the standard of experimental animal welfare of Zhejiang University.
Consent for Publication
This manuscript has never been published or accepted anywhere else. It is approved by all authors for the publication of this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (82270023, U22A20265, 82200026 and 82300102), the Science and Technology Department of Zhejiang Province (2023C03067), Major Project of Guangzhou National Laboratory (GZNL2024A02002) and Jinhua Municipal Science and Technology Program Project (2023-4-091).
Disclosure
The authors disclose no relevant financial or non-financial interests in this work.
References
1. WHO Guidelines Approved by the Guidelines Review Committee. WHO Global Air Quality Guidelines: Particulate Matter (PM(2.5) and PM(10)), Ozone, Nitrogen Dioxide, Sulfur Dioxide and Carbon Monoxide. Geneva: World Health Organization © World Health Organization; 2021.
2. Madureira J, Slezakova K, Costa C, Pereira MC, Teixeira JP. Assessment of indoor air exposure among newborns and their mothers: levels and sources of PM(10), PM(2.5) and ultrafine particles at 65 home environments. Environ Pollut. 2020;264:114746. doi:10.1016/j.envpol.2020.114746
3. Yu W, Xu R, Ye T, et al. Estimates of global mortality burden associated with short-term exposure to fine particulate matter (PM(2·5)). Lancet Planet Health. 2024;8(3):e146–e155. doi:10.1016/s2542-5196(24)00003-2
4. Li D, Li Y, Li G, Zhang Y, Li J, Chen H. Fluorescent reconstitution on deposition of PM(2.5) in lung and extrapulmonary organs. Proc Natl Acad Sci U S A. 2019;116(7):2488–2493. doi:10.1073/pnas.1818134116
5. Künzli N, Kaiser R, Medina S, et al. Public-health impact of outdoor and traffic-related air pollution: a European assessment. Lancet. 2000;356(9232):795–801. doi:10.1016/s0140-6736(00)02653-2
6. Ancona C, Badaloni C, Mataloni F, et al. Mortality and morbidity in a population exposed to multiple sources of air pollution: a retrospective cohort study using air dispersion models. Environ Res. 2015;137:467–474. doi:10.1016/j.envres.2014.10.036
7. Hu G, Zhong N, Ran P. Air pollution and COPD in China. J Thorac Dis. 2015;7(1):59–66. doi:10.3978/j.issn.2072-1439.2014.12.47
8. Lu F, Xu D, Cheng Y, et al. Systematic review and meta-analysis of the adverse health effects of ambient PM2.5 and PM10 pollution in the Chinese population. Environ Res. 2015;136:196–204. doi:10.1016/j.envres.2014.06.029
9. Duan S, Zhang M, Sun Y, et al. Mechanism of PM(2.5)-induced human bronchial epithelial cell toxicity in central China. J Hazard Mater. 2020;396:122747. doi:10.1016/j.jhazmat.2020.122747
10. Tao F, Gonzalez-Flecha B, Kobzik L. Reactive oxygen species in pulmonary inflammation by ambient particulates. Free Radic Biol Med. 2003;35(4):327–340. doi:10.1016/s0891-5849(03)00280-6
11. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. 2014;14(2):81–93. doi:10.1038/nri3600
12. Shi T, Denney L, An H, Ho LP, Zheng Y. Alveolar and lung interstitial macrophages: definitions, functions, and roles in lung fibrosis. J Leukoc Biol. 2021;110(1):107–114. doi:10.1002/jlb.3ru0720-418r
13. Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022;55(9):1564–1580. doi:10.1016/j.immuni.2022.08.010
14. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084. doi:10.3389/fimmu.2019.01084
15. Zhong Y, Liao J, Hu Y, et al. PM(2.5) upregulates MicroRNA-146a-3p and induces M1 polarization in RAW264.7 cells by targeting Sirtuin1. Int J Med Sci. 2019;16(3):384–393. doi:10.7150/ijms.30084
16. Mitschik S, Schierl R, Nowak D, Jörres RA. Effects of particulate matter on cytokine production in vitro: a comparative analysis of published studies. Inhal Toxicol. 2008;20(4):399–414. doi:10.1080/08958370801903784
17. Britt RD Jr, Porter N, Grayson MH, et al. Sterols and immune mechanisms in asthma. J Allergy Clin Immunol. 2023;151(1):47–59. doi:10.1016/j.jaci.2022.09.025
18. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2020;21(4):225–245. doi:10.1038/s41580-019-0190-7
19. Lee MS, Bensinger SJ. Reprogramming cholesterol metabolism in macrophages and its role in host defense against cholesterol-dependent cytolysins. Cell Mol Immunol. 2022;19(3):327–336. doi:10.1038/s41423-021-00827-0
20. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–340. doi:10.1016/s0092-8674(00)80213-5
21. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. 2017;13(12):710–730. doi:10.1038/nrendo.2017.91
22. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. doi:10.1172/jci15593
23. Bridges JP, Schehr A, Wang Y, et al. Epithelial SCAP/INSIG/SREBP signaling regulates multiple biological processes during perinatal lung maturation. PLoS One. 2014;9(5):e91376. doi:10.1371/journal.pone.0091376
24. You W, Su L, Weng S, et al. SREBP-2 promotes cancer progression through the mevalonate-Akt pathway in non-small cell lung cancer. Sci Rep. 2025;15(1):23103. doi:10.1038/s41598-025-07437-0
25. Fei X, Huang J, Li F, et al. The Scap-SREBP1-S1P/S2P lipogenesis signal orchestrates the homeostasis and spatiotemporal activation of NF-κB. Cell Rep. 2023;42(6):112586. doi:10.1016/j.celrep.2023.112586
26. Lee KS, Lee HK, Hayflick JS, Lee YC, Puri KD. Inhibition of phosphoinositide 3--kinase δ attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB Journal. 2006;20(3):455–465. doi:10.1096/fj.05-5045com
27. McMillan SJ, Bishop B, Townsend MJ, McKenzie AN, Lloyd CM. The absence of interleukin 9 does not affect the development of allergen-induced pulmonary inflammation nor airway hyperreactivity. J Exp Med. 2002;195(1):51–57. doi:10.1084/jem.20011732
28. Wu YF, Li ZY, Dong LL, et al. Inactivation of MTOR promotes autophagy-mediated epithelial injury in particulate matter-induced airway inflammation. Autophagy. 2020;16(3):435–450. doi:10.1080/15548627.2019.1628536
29. Harrison AG, Yang D, Cahoon JG, et al. UBXN9 governs GLUT4-mediated spatial confinement of RIG-I-like receptors and signaling. Nat Immunol. 2024;25(12):2234–2246. doi:10.1038/s41590-024-02004-7
30. Gao S, Zheng K, Lou J, et al. Macrophage extracellular traps suppress particulate matter-induced airway Inflammation. Am J Pathol. 2024;194(9):1622–1635. doi:10.1016/j.ajpath.2024.05.008
31. Mu W, Zhi Y, Zhou J, et al. Endoplasmic reticulum stress and quality control in relation to cisplatin resistance in tumor cells. Front Pharmacol. 2024;15:1419468. doi:10.3389/fphar.2024.1419468
32. Zhou X, Li Z, Ren F, et al. Endoplasmic reticulum stress and unfolded protein response in renal lipid metabolism. Exp Cell Res. 2025;446(1):114463. doi:10.1016/j.yexcr.2025.114463
33. Zhang K, Wang H, He W, et al. The association between ambient air pollution and blood lipids: a longitudinal study in Shijiazhuang, China. Sci Total Environ. 2021;752:141648. doi:10.1016/j.scitotenv.2020.141648
34. Chen G, Knibbs LD, Zhang W, et al. Estimating spatiotemporal distribution of PM(1) concentrations in China with satellite remote sensing, meteorology, and land use information. Environ Pollut. 2018;233:1086–1094. doi:10.1016/j.envpol.2017.10.011
35. Mao S, Li S, Wang C, et al. Is long-term PM(1) exposure associated with blood lipids and dyslipidemias in a Chinese rural population? Environ Int. 2020;138:105637. doi:10.1016/j.envint.2020.105637
36. McGuinn LA, Coull BA, Kloog I, et al. Fine particulate matter exposure and lipid levels among children in Mexico city. Environ Epidemiol. 2020;4(2):e088. doi:10.1097/ee9.0000000000000088
37. Lee H, Kim MY, Ji SY, et al. The protective effect of oral application of Corni fructus on the disorders of the cornea, conjunctiva, lacrimal gland and retina by topical particulate matter 2.5. Nutrients. 2021;13(9). doi:10.3390/nu13092986
38. Moradi M, Mard SA, Farbood Y, et al. The protective effect of p-Coumaric acid on hepatic injury caused by particulate matter in the rat and determining the role of long noncoding RNAs MEG3 and HOTAIR. J Biochem Mol Toxicol. 2023;37(7):e23364. doi:10.1002/jbt.23364
39. Liao Z, Nie J, Sun P. The impact of particulate matter (PM2.5) on skin barrier revealed by transcriptome analysis: focusing on cholesterol metabolism. Toxicol Rep. 2020;7:1–9. doi:10.1016/j.toxrep.2019.11.014
40. Moutzouri E, Tellis CC, Rousouli K, et al. Effect of simvastatin or its combination with ezetimibe on Toll-like receptor expression and lipopolysaccharide - induced cytokine production in monocytes of hypercholesterolemic patients. Atherosclerosis. 2012;225(2):381–387. doi:10.1016/j.atherosclerosis.2012.08.037
41. Ascer E, Bertolami MC, Venturinelli ML, et al. Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis. 2004;177(1):161–166. doi:10.1016/j.atherosclerosis.2004.07.003
42. McKay A, Leung BP, McInnes IB, Thomson NC, Liew FY. A novel anti-inflammatory role of simvastatin in a murine model of allergic asthma. J Immunol. 2004;172(5):2903–2908. doi:10.4049/jimmunol.172.5.2903
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.