")
Back to Journals » Journal of Inflammation Research » Volume 13
Saturated Fatty Acids in Obesity-Associated Inflammation
Authors Zhou H , Urso CJ , Jadeja V
Received 4 September 2019
Accepted for publication 11 November 2019
Published 6 January 2020 Volume 2020:13 Pages 1—14
DOI https://doi.org/10.2147/JIR.S229691
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Heping Zhou, CJ Urso, Viren Jadeja
Department of Biological Sciences, Seton Hall University, South Orange, NJ 07079, USA
Correspondence: Heping Zhou Email [email protected]
Abstract: Obesity is a major risk factor for the development of various pathological conditions including insulin resistance, diabetes, cardiovascular diseases, and non-alcoholic fatty liver disease (NAFLD). Central to these conditions is obesity-associated chronic low-grade inflammation in many tissues including adipose, liver, muscle, kidney, pancreas, and brain. There is increasing evidence that saturated fatty acids (SFAs) increase the phosphorylation of MAPKs, enhance the activation of transcription factors such as nuclear factor (NF)-κB, and elevate the expression of inflammatory genes. This paper focuses on the mechanisms by which SFAs induce inflammation. SFAs may induce the expression inflammatory genes via different pathways including toll-like receptor (TLR), protein kinase C (PKC), reactive oxygen species (ROS), NOD-like receptors (NLRs), and endoplasmic reticulum (ER) stress. These findings suggest that SFAs act as an important link between obesity and inflammation.
Keywords: saturated fatty acids, obesity, inflammation, Toll-like receptor, reactive oxygen species, lipid rafts, protein kinase C
Introduction
Obesity is an increasingly prevalent global issue. According to the 2018 World Health Organization (WHO) fact sheet, the number of people with obesity worldwide has nearly tripled since 1975, and more than 650 million adults were obese in 2016 (http://www.who.int/mediacentre/factsheets/fs311/en/). There is significant evidence that obesity is associated with the development of a range of pathological conditions including cardiovascular diseases, insulin resistance, diabetes, and non-alcoholic fatty liver disease (NAFLD).1 Chronic low-grade inflammation has been reported in the adipose tissue,2 liver,3 muscle,4 kidney,5 and hypothalamus6 of obese human subjects. Circulating levels of TNF-α and C-reactive protein (CRP) are also increased in obese children and adolescents.7 Elevated circulating IL-6 and higher levels of IL-1β, monocyte chemoattractant protein (MCP)-1, and IL-8 have been reported in the placenta of obese pregnant women.8
Inflammation is also detected in various tissues of both genetic and dietary animal models of obesity. For example, production of inflammatory mediators is increased in the liver, muscle, adipose tissue of ob/ob and db/db mice compared to control mice.9–11 Mice fed with palmitic acid-supplemented high-fat diet (HFD) also exhibit inflammation in the adipose tissue, liver, muscle, kidney, and hypothalamus compared to control animals.9,12–16
There is increasing evidence that chronic inflammation is an important underlying cause of various obesity-associated conditions.17 For example, tumor necrosis factor (TNF)-α, a proinflammatory cytokine, has been shown to induce insulin resistance when increased and improve insulin resistance when neutralized18 while decreased expression of adiponectin, an anti-inflammatory adipokine, has been implicated in the development of obesity-associated cardiovascular diseases.19
A significant number of studies have been conducted to identify the cause of obesity-associated inflammation with many focused on free fatty acids (FFAs). Circulating fatty acids are generally transported either free (nonesterified) or bound to cholesterol and other protein molecules. The circulating levels of FFAs may be increased in obesity and its associated conditions as a result of increased amount of adipose tissue, reduced response to insulin’s antilipolytic effect of obese adipose tissue, and decreased re-esterification of FFAs by obese adipocytes.20–22 Circulating levels of FFAs have been reported to be increased in obese subjects,22 morbidly obese subjects,23 overweight/obese subjects with diabetes mellitus,24 patients with severe non-insulin-dependent diabetes mellitus,25 and obese NAFLD patients.24,26 Karpe et al conducted a literature search on non-esterified fatty acids (NEFA) or FFA as well as obesity on PubMed in July 2009 and found 43 original reports on 953 nonobese (control) subjects and 1410 overweight/obese subjects with most studies reporting greater FFA level in the obese/overweight group even though the average difference is modest, and concluded that FFA concentration is undeniably higher in certain groups of obese individuals.27
Circulating FFAs may vary in the degree of saturation with saturated fatty acids (SFAs), monounsaturated fatty acids (MUFA) and polyunsaturated fatty acids (PUFA). They may also vary in the number of carbons with short-chain, medium-chain, and long-chain FFAs. Considering that the effects of different FFAs on innate immunity are quite complex depending on the number of carbons, degree of saturation, and location of the C=C double bond in the hydrocarbon chain, this paper is focused on examining how long-chain SFAs may contribute to inflammation.
Long-Chain SFAs Increase the Production of Inflammatory Mediators
Palmitic acid (C16:0) has been reported to increase the phosphorylation of mitogen-activated protein kinases (MAPKs) including p38, JNK, and extracellular-signal-regulated kinases (ERKs), enhance the activation of transcription factors including activator protein (AP)-1 and nuclear factor (NF)-κB, and induce the mRNA expression of cyclooxygenase (COX)-2, IL-1β, IL-6, and TNF-α in macrophages, monocytes, and monocyte-derived dendritic cells.28–34 Stearic acid (C18:0) has been reported to trigger the release of TNF-α, IL-1β, and IL-6 from astrocytes.35 Both stearic acid and palmitic acid induce the activation of NF-κB and stimulate the secretion of pro-inflammatory mediators in trophoblast cells isolated from human placentas,36,37 microglial cells,38 and prostate epithelial cells.39 Similarly, palmitic acid significantly activates JNK in HEPG2 cells;40 increases the expression of MCP-1 in mesangial cells;15 induces the expression of IL-6, IL-8, and MCP-1 in smooth muscle cells;41,42 increases the activation of p38, JNK, and NF-κB with enhanced expression of TNF-α in C2C12 skeletal muscle cells;43,44 enhances the activation of NF-κB with increased production of IL-6 and TNF-α in adipocytes;23,45 and induces the activation of p38, ERK, and JNK with increased expression of COX-2, IL-6 and MCP-1 in fibroblast cells.46,47
Dietary supplementation with high-fat diet (HFD) or infusion of Liposyn II (a 20% triglyceride nonpyrogenic emulsion), lipid/heparin, soybean oil, lard oil, or fatty acids has been used to increase circulating FFA level in human and animal model studies.48–51 Infusion of ethyl palmitic acid increases the expression of chemokines including MCP-1 and keratinocyte chemoattractant (KC) in β cells and the recruitment of monocytes/macrophages into the mouse islets.52 Intracerebroventricular administration of arachidic acid (C20:0) induces mRNA expression of TNF-α, IL-1β, IL-6, and IL-10 in the hypothalamus of rats.53 In addition, Dumas et al reported that subjects on high palmitic acid diet exhibit elevated circulating levels of IL-6 and IL-1β than subjects on low palmitic acid/high oleic acid diet.54 These studies suggest that increased level of SFAs may represent a key link between obesity and inflammation.
Involvement of TLR2 and/or TLR4 in SFA-Induced Inflammation
Toll-like receptors (TLRs) are pattern recognition receptors (PRRs) that recognize different pathogen-associated molecular pattern (PAMP) molecules. TLR4 is a TLR family member well known to recognize lipopolysaccharide (LPS), a main component of Gram-negative bacterial cell wall. Upon being brought to TLR4 and MD-2, LPS promotes the formation of TLR4-MD2-LPS complex, which, in turn, recruits myeloid differentiation primary response 88 (MyD88) and Toll–IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF), initiating a MyD88-dependent and MyD88-independent signaling pathways respectively.55,56 Activation of MyD88-dependent pathway leads to the activation of MAPKs and transcription factors such as NF-κB and AP-1, ultimately increasing the expression of inflammatory markers such as cytokines and chemokines.57,58 Activation of MyD88-independent pathway leads to the activation of IFN-regulatory factor (IRF)-3, ultimately increasing the expression of inflammatory genes such as IP-10.59
TLR2, another member of the TLR family, has been shown to recognize a broad range of ligands by forming a heterodimer with other TLRs. Triacyl lipopeptide (LP) from Gram-positive bacterial cell wall promotes the formation of TLR2/1 heterodimer and diacyl LP promotes the formation of TLR2/6 heterodimer,60–62 which, in turn, initiates the MyD88-dependent signaling pathway, ultimately increasing the expression of inflammatory markers.63,64
A number of studies have examined the role of TLR in SFA-induced inflammation. Knockdown of TLR4 has been found to markedly attenuate palmitic acid-induced NF-κB activation and IL-8 expression in human aortic vascular smooth muscle cells,42 and significantly reduce palmitic acid-induced increase of NF-κB activation and release of IL-1β and MCP-1 in THP-1 cells.65 Inhibition of TLR4 reduces palmitic acid-induced production of TNF-α and IL-6 in astrocytes35 and trophoblast cells.36 Dominant-negative TLR4 inhibits lauric acid-induced activation of NF-κB and expression of COX-2 in RAW 264.7 cells.32 Pretreatment with TLR4 antibody also inhibits palmitic acid-induced mRNA expression of TNF-α, IL-6, and IL-1β in microglial cells38 (Table 1). These in vitro studies suggest the necessary involvement of TLR4 in SFA-induced expression of inflammatory genes.
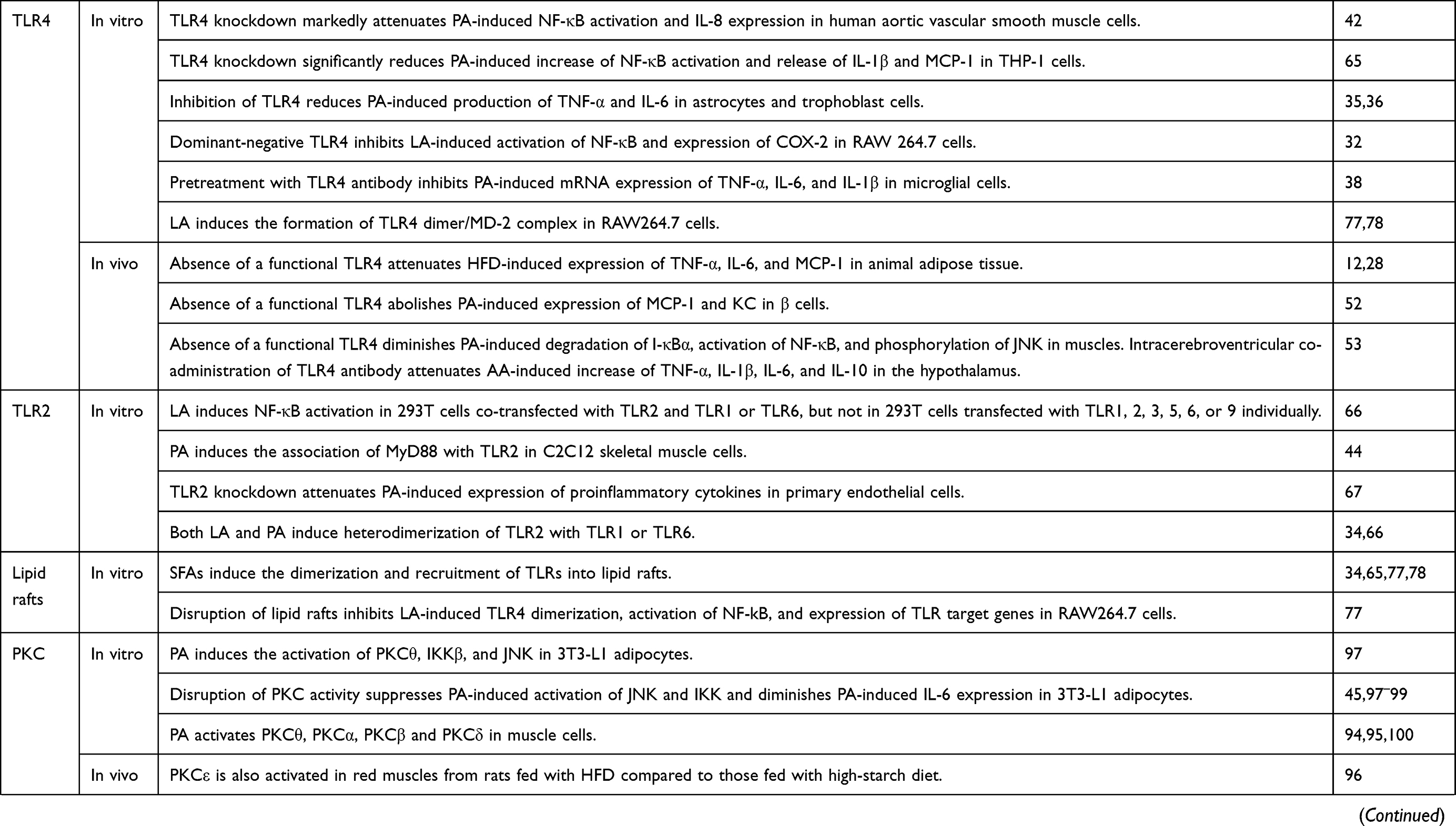 | 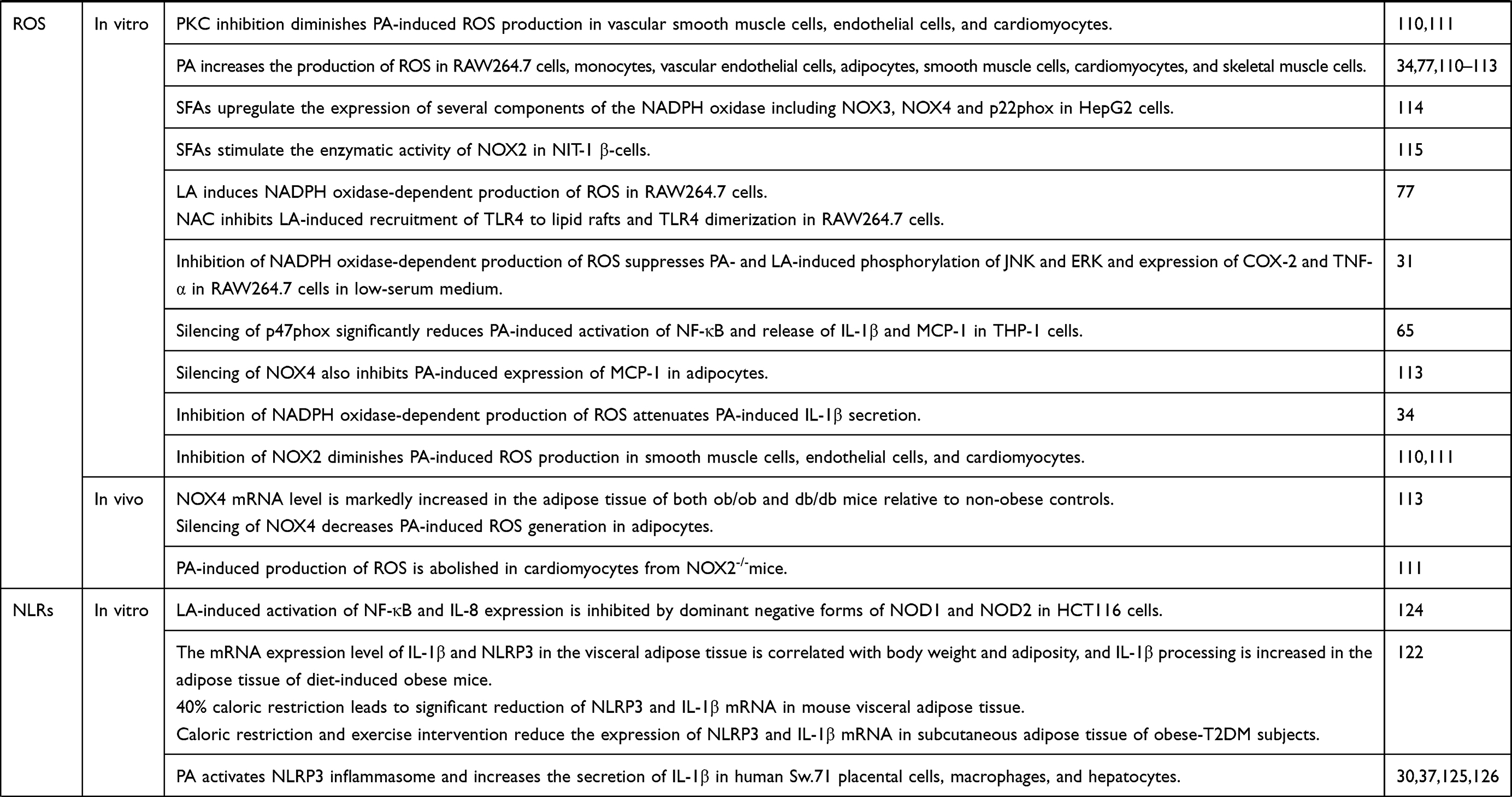 |  |
Table 1 Studies on the Mechanisms of SFA-Induced Inflammation |
Moreover, the absence of a functional TLR4 protects mice from the obesogenic effects of HFD with lard and palmitic acid,12 attenuates HFD-induced expression of TNF-α, IL-6, and MCP-1 in the adipose tissue,12,28 abolishes palmitic acid-induced expression of MCP-1 and KC in β cells,52 and diminishes palmitic acid-induced degradation of I-κBα, activation of NF-κB, and phosphorylation of JNK in isolated muscles compared to control mice.53 Furthermore, intracerebroventricular co-administration of TLR4 antibody attenuates arachidic acid (C20:0)-induced increase of TNF-α, IL-1β, IL-6, and IL-10 in the hypothalamus53 (Table 1). Taken together, both in vitro and in vivo studies suggest that TLR4 is clearly implicated in SFA-induced inflammation (Figure 1).
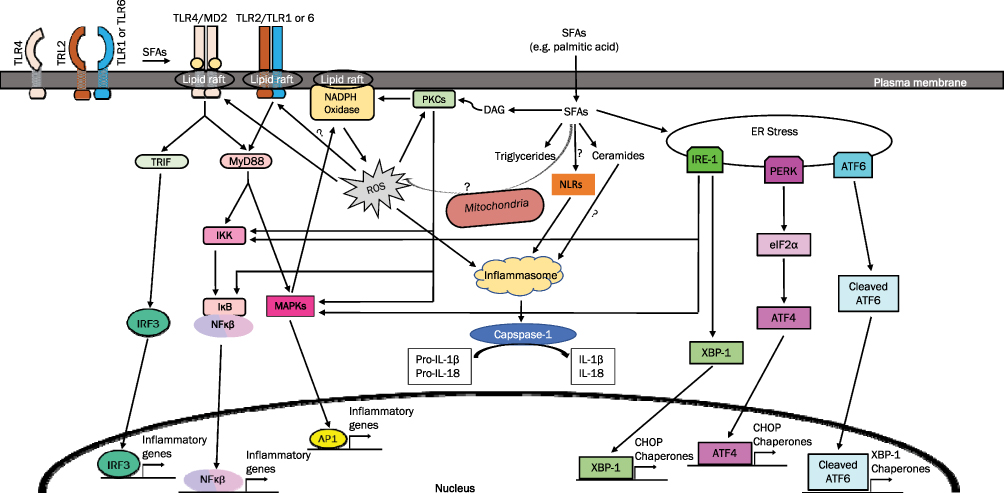 |
Figure 1 SFAs induce inflammation via several potential mechanisms. |
There is also evidence that SFA-induced expression of inflammatory markers may involve TLR2. Lauric acid is able to induce NF-κB activation in 293T cells co-transfected with TLR2 and TLR1 or TLR6, but not in 293T cells transfected with TLR1, 2, 3, 5, 6, or 9 individually,66 suggesting that lauric acid-induced NF-κB activation involves TLR2 with TLR1 or TLR6. Palmitic acid has also been found to induce the association of MyD88 with TLR2 in C2C12 skeletal muscle cells.44 Furthermore, knockdown of TLR2 attenuates palmitic acid-induced expression of proinflammatory cytokines in primary endothelial cells67 (Table 1). Taken together, TLR2 may also be involved in SFA-induced activation of NF-κB and expression of inflammatory markers (Figure 1).
Do SFAs Act as Ligands for TLR4 and TLR2?
While there is much evidence for the involvement of TLR2 and TLR4 in SFA-induced inflammation, questions remain as to how SFAs activate the TLR signaling. The lipid portion of LPS and LP critical for their binding to TLRs56,68–72 is structurally similar to SFAs,73,74 therefore SFAs have been postulated as ligands for TLRs. Nicholas et al also demonstrated the possibility of fitting 5 molecules of palmitic acid into the hydrophobic binding pocket in MD-2 using a theoretical approach.33 However, stearic acid is unable to compete with LPS in its binding to LPS-Trap fusion protein consisting of FLAG-tagged extracellular part of TLR4 fused to full-length MD-2.75 Lancaster et al also reported that multiple palmitic acid molecules would make the TLR4/MD2 active complex unstable using molecular simulations,76 suggesting the unlikelihood of palmitic acid to act as a TLR4 ligand.
Despite insufficient evidence for the physical binding of SFAs to TLRs, SFAs have been shown to induce dimerization of TLRs. For example, lauric acid induces the formation of TLR4 dimer/MD-2 complex in RAW264.7 cells.77,78 Panter et al showed that the chimeric protein consisting of the transmembrane and cytoplasmic domains of TLR4 and the MD2 or CD14 forms a constitutively active dimer that is able to activate the expression of luciferase reporter under the control of NF-κB promoter,79 suggesting that TLR4 dimerization is sufficient to activate TLR4 signaling. Furthermore, expression of LPS-induced inflammatory genes is significantly reduced when LPS-induced TLR4 dimerization is inhibited,80 suggesting that dimerization is necessary for the activation of TLR4. In terms of TLR2, both lauric acid and palmitic acid have been shown to induce heterodimerization of TLR2 with TLR1 or TLR6.34,66 Several studies further demonstrated that TLR2 exists as inactive, loosely bound heterodimer with TLR1 or TLR6 without the ligand and that the binding of LP to the preformed heterodimer brings the TLRs closer in proximity and stabilizes the heterodimer.62,81–83 These studies suggest that SFAs induce the dimerization of TLRs and thereby activate TLR signaling. Questions still remain as to how SFAs induce the dimerization of TLR4 or stabilize TLR2 dimers with TLR1 or TLR6.
Involvement of Lipid Rafts in SFA-Induced TLR Activation
Lipid rafts are specialized microdomains of plasma membrane that are insoluble in non-ionic detergents and highly enriched with cholesterol, sphingolipids, and glycolipids.84 Lipid rafts serve as a platform for bringing receptor molecules close together, formation of receptor complexes, receptor activation, assembly of various signaling proteins, and downstream signaling.85–89 TLR4 without ligands is generally believed to reside outside of lipid rafts.87 Ligand binding induces TLR4 recruitment to lipid rafts coupled to TLR4 dimerization and subsequent downstream signaling.77,83,90,91 TLR2 without ligands is believed to exist in pre-formed weakly bound dimers with TLR1 or TLR6,92 which are brought to closer proximity upon ligand binding, initiating the recruitment of downstream signaling molecules.81,86,92
SFAs have been reported to induce the dimerization and recruitment of TLRs into lipid rafts34,65,77,78 (Figure 1). Disruption of lipid rafts inhibits lauric acid-induced TLR4 dimerization, activation of NF-kB, and expression of TLR target genes in RAW264.7 cells77 (Table 1). Therefore, even though SFAs may not act as ligands that physically bind to TLRs, SFAs may alternatively modulate TLR activity by affecting their recruitment into the lipid rafts, which may in turn impact the formation of TLR receptor complexes and recruitment of downstream signaling mediators. It is currently unclear how SFAs induce the recruitment of TLRs to lipid rafts and the dimerization of TLRs. It has been suggested that SFAs may facilitate the recruitment of TLRs to the lipid rafts and TLR dimerization by affecting the physical composition of lipid rafts and the reorganization and remodeling of these microdomains.78 Another possible scenario may involve the association of TLR2/6 with CD36, a resident membrane protein in caveolae-lipid rafts, which may facilitate SFA-induced recruitment of TLR2/6 into lipid rafts.83 It remains to be determined how CD36 may help to recruit specific TLRs to lipid rafts in response to elevated levels of SFAs.
Involvement of PKC in SFA-Induced Inflammation
Once taken up intracellularly, fatty acids may be catabolized via β-oxidation, incorporated into phospholipids, turned into triglycerides and diacylglycerol (DAG), or used in the synthesis of many other molecules. Intracellular level of DAG is increased with elevated levels of circulating FFAs.49,93 Palmitic acid increases DAG level in C2C12 skeletal muscle cells94 and human muscle primary cultures.95 DAG level is also increased in the red muscles of rats fed with high-fat diet compared with those fed with isocaloric high-starch diet.96
DAG is a known activator for classical PKCs, such as PKCα, βI, βII, and γ, and novel PKCs, such as PKCδ, ε, η, and θ. When activated, PKCs are translocated from the cytosol to membranes, increasing the ratio of particulate to cytosolic PKCs. Palmitic acid treatment has been reported to induce the activation of PKCθ, IKKβ, and JNK in 3T3-L1 adipocytes97 (Table 1). Disrupting PKC activity suppresses palmitic acid-induced activation of JNK and IKK and diminishes palmitic acid-induced IL-6 expression in 3T3-L1 adipocytes,45,97–99 suggesting that activation of PKC may contribute to palmitic acid-induced activation of IKK and JNK and expression of inflammatory genes in adipocytes (Figure 1). PKCε is also activated in red muscles from rats fed with high-fat diet compared to those fed with high-starch diet.96 Palmitic acid exposure activates PKCθ,94,95 PKCα, PKCβ and PKCδ100 in association with enhanced activation of NF-κB,94 increased activation of JNK, p38MAPK and ERK1/2,100 and elevated expression of IL-6 and TNF-α in C2C12 skeletal muscle cells94 (Figure 1). Kadotani et al reported that palmitic acid and stearic acid-induced COX-2 expression in myotubes requires activation of p38 MAPK and NF-κB but may not involve PKCθ even though PKCθ phosphorylation is strongly augmented following treatment with saturated fatty acids.101 It should be noted that Kadotani et al used rottlerin as an inhibitor for PKCθ in their study. Rottlerin is a protein kinase inhibitor with some reported specificity for PKCδ and PKCθ and its efficacy has been questioned.102 More studies are needed to investigate whether and how various PKCs contribute to SFA-induced expression of inflammatory genes.
Involvement of ROS in SFA-Induced Inflammation
Reactive oxygen species (ROS) may affect various cellular processes including immunity, cell signaling pathways, and gene expression regulation.103 ROS may be produced from different pathways including reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, nitric oxide synthase, and mitochondria. NADPH oxidase is a multi-subunit protein with NOX as the catalytic subunit. NOX1-4 exists in stable complexes with membrane-bound p22phox. While NOX4-p22phox complex is constitutively active, activation of NOX1-3 requires further assembly with cytosolic components (p47phox, p67phox, p40phox and a GTPase Rac1 or Rac2).104–106 The assembly may take place at the lipid rafts107 and is tightly regulated by protein–protein interactions and phosphorylation of p47phox.108,109 In response to stimulation, p47phox may become phosphorylated by a number of kinases including PKC‐α, PKC‐βII, PKCδ, PKC‐σ, PKC‐ζ, p38 MAPK, and ERK1/2, which leads to changes in its conformation, enabling p47phox to interact with other cytosolic and membrane components, and subsequent activation of the oxidase.108,109 In support, inhibition of PKC diminishes palmitic acid-induced ROS production in vascular smooth muscle cells, endothelial cells, and cardiomyocytes.110,111
Palmitic acid has been shown to increase the production of ROS in a variety of cells including RAW264.7 cells, monocytes, vascular endothelial cells, adipocytes, smooth muscle cells, cardiomyocytes, and skeletal muscle cells.34,77,110–113 SFA-induced production of ROS may be mediated by the activation of NADPH oxidase. Firstly, SFAs have been reported to upregulate the expression of several components of the NADPH oxidase including NOX3, NOX4 and p22phox in HepG2 hepatocytes.114 Secondly, SFAs have been shown to stimulate the enzymatic activity of NOX2 in NIT-1 β-cells.115 Thirdly, NOX4 mRNA level is markedly increased in the adipose tissue of both ob/ob and db/db mice relative to non-obese controls.113 Fourthly, decreased activity of NADPH oxidase reduces SFA-induced production of ROS. For example, silencing of NOX4 decreases palmitic acid-induced ROS generation in adipocytes.113 Inhibition of NOX2 diminishes palmitic acid-induced ROS production in smooth muscle cells, endothelial cells, and cardiomyocytes.110,111 Palmitic acid-induced production of ROS is also abolished in cardiomyocytes from NOX2-/-mice111 (Table 1). These studies suggest that NADPH oxidase may be critical for and represent a key source of SFA-induced ROS (Figure 1). Consistently, lauric acid has also been shown to induce NADPH oxidase-dependent production of ROS in RAW264.7 cells.77
It is not clear whether fatty acid oxidation in mitochondria may also contribute to the production of ROS in cells exposed to increased levels of fatty acids. While Frayn et al reported that after entering adipocytes, FFAs are predominantly rapidly converted to fatty acyl-coA and stored as triglycerides without significant mitochondrial oxidation,116 Joseph et al showed that palmitic acid-induced increase of NOX2 activity is prevented by the inhibition of mitochondrial uptake of fatty acids,111 and proposed that the mitochondrial uptake of palmitic acid may cause a small initial increase in mitochondrial ROS, which then activates PKC and NADPH oxidase to feed forward the production of ROS in the cytosol111,117 (Figure 1).
Once generated, ROS may be involved in the production of inflammatory genes in different ways. For example, ROS has been shown to induce TLR4 recruitment into hepatic lipid rafts, and p47phox-deficient mice exhibit significantly less recruitment of TLR4 into lipid rafts in the liver.118 N-acetyl cysteine, an antioxidant, also inhibits lauric acid-induced recruitment of TLR4 to lipid rafts and TLR4 dimerization in RAW264.7 cells77 (Table 1). These studies suggest that SFA treatment induces NADPH oxidase-dependent generation of ROS, which, in turn, helps to recruit TLR4 to the lipid rafts and contribute to subsequent TLR4 dimerization and activation (Figure 1).
Inhibition of NADPH oxidase-dependent production of ROS suppresses palmitic acid- and lauric acid-induced phosphorylation of JNK and ERK and expression of COX-2 and TNF-α in RAW264.7 cells in low-serum medium,31 and attenuates palmitic acid-induced IL-1β secretion.34 Silencing of p47phox significantly reduces palmitic acid-induced activation of NF-κB and release of IL-1β and MCP-1 in THP-1 cells.65 Silencing of NOX4 also inhibits palmitic acid-induced expression of MCP-1 in adipocytes113 (Table 1) These studies suggest that NADPH oxidase-dependent generation of ROS may contribute to the recruitment of TLR to the lipid rafts and increase the production of inflammatory markers.
Involvement of NLRs in SFA-Induced Inflammation
A family of intracellular PRRs called nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) may also be implicated in SFA-induced inflammation. NLRs have been found to recognize both pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs).119 Among the NLR family members, activation of NOD1 and NOD2 has been shown to orchestrate NF-kB and MAPK signaling,120,121 and activation of NLRP3, NLRP1B, and NLRC4 recruits the apoptotic speck protein (ASC) and pro-caspase I. The activated caspase 1, in turn, regulates the cleavage of pro-IL-1β and pro-IL-18 to form active IL-1β and IL-18, respectively.119,122,123
Lauric acid-induced activation of NF-κB and IL-8 expression is inhibited by dominant negative forms of NOD1 and NOD2 in HCT116 cells.124 The mRNA expression level of IL-1β and NLRP3 in the visceral adipose tissue is correlated with body weight and adiposity, and IL-1β processing is increased in the adipose tissue of diet-induced obese mice.122 Furthermore, 40% caloric restriction leads to significant reduction of NLRP3 and IL-1β mRNA in mouse visceral adipose tissue.122 Vandanmagsar et al also reported that the caloric restriction and exercise intervention leads to reduced expression of NLRP3 and IL-1β mRNA in subcutaneous adipose tissue of obese-T2DM subjects122 (Table 1).
Palmitic acid has been shown to activate NLRP3 inflammasome and increase the secretion of IL-1β in human Sw.71 placental cells, macrophages, and hepatocytes.30,37,125,126 ROS and NF-κB have been suggested to mediate palmitic acid-induced activation of NLRP3-ASC inflammasome may be mediated by30,126 (Figure 1). Further studies are needed to determine whether SFAs directly interact with any of the intracellular NLRs.
Involvement of ER Stress in SFA-Induced Inflammation
SFAs have been shown to induce significant endoplasmic reticulum (ER) stress in different cells due to accumulation of unfolded or misfolded proteins. As shown in Figure 1, there are three major ER stress pathways: protein kinase RNA-like endoplasmic reticulum kinase (PERK) undergoes autophosphorylation, which, in turn, phosphorylates α-subunit of eukaryotic translation initiation factor-2 (eIF2α), slowing the rate of translation initiation of many mRNAs while increasing the translation of ATF4 and the expression of C/EBP-homologous protein (CHOP);127 inositol-requiring kinase 1 (IRE1) undergoes autophosphorylation, which, in turn, leads to the activation of inflammatory pathways including JNK and NF-κB, and the splicing of Xbox protein-1 (XBP1); transcription factor 6 (ATF6) is cleaved and activated, inducing the expression of XBP1 mRNA. ATF6 and XBP1 also regulate the expression of the 78-kDa glucose-regulated protein (GRP78), a major ER chaperone.98
Palmitic acid increases the levels of ER stress markers in different cells including skeletal muscle cells, monocytes, and hypothalamic neurons.34,128,129 Animals fed with high-fat diet exhibit increased phosphorylation of eIF2α and PERK and/or elevated expression of GRP78 in the liver and adipose tissue compared to control animals.51,130 Rats infused with lard express markers of ER stress and expression of inflammatory markers in liver and adipose tissue compared to control50,131 (Table 1).
Moreover, ER stress may be implicated in obesity-induced inflammatory response. Inhibition of palmitic acid-induced ER stress attenuates IL-1β production in monocytes,34 and suppresses the expression of TNF-α and IL-6 in the adipose tissue of animals fed with high-fat diet.51 Oral administration of chemical chaperones to alleviate ER stress has also been reported to improve chronic inflammation in adipose tissue of obese mice.132 These studies suggest that ER stress may be implicated in SFA-induced inflammation. Consistently, mice deficient in XBP-1 decease the activation of JNK in the liver.130
Conclusions and Future Directions
Chronic low-grade inflammation is central to the development of various obesity-associated pathologies including insulin resistance, diabetes, cardiovascular diseases, and NAFLD. This paper examined the mechanisms by which SFAs induce the expression of inflammatory mediators and showed that SFAs may increase the activity of NF-κB and AP-1, enhance the activation of MAPKs, and elevate the expression of inflammatory markers via the activation of several interacting pathways.
There is significant evidence that TLR4 and TLR2 pathways are involved in SFA-induced expression of inflammatory genes. While there is no sufficient evidence for the physical binding of SFAs to TLR2 or TLR4 receptor complexes as ligands, SFAs do induce the recruitment of TLRs to the lipid rafts and their dimerization (Figure 1). Further studies need to address how SFAs specifically recruit TLR4 or TLR2 to the lipid rafts and induce their activation.
There is also evidence suggesting the involvement of TLR-independent pathways in SFA-induced inflammation. Snodgrass et al reported that silencing of TLR2 suppresses palmitic acid-induced IL-1β by 15% while suppressing Pam3CSK4-TLR2-induced IL1β by 63%, and that TLR4 inhibition reduces palmitic acid-induced IL1β by 19% while inhibiting LPS-TLR4-induced IL-1β by 80%.34 Furthermore, palmitic acid-induced gene expression pattern is different from that of LPS-TLR4 activation. For example, LPS abolishes palmitic acid-induced mRNA expression of arginase 1 while palmitic acid abolishes LPS-induced mRNA expression of C/EBPδ.133 Palmitic acid potentiates LPS-induced IL-1β but reduces LPS-induced IL6 mRNA expression.133 These studies suggest that SFAs may not act only through activation of TLR2 or TLR4 pathways.
Indeed, SFAs have been shown to increase the activation of PKCs which may, in turn, activate IKK, MAPK, and NADPH oxidase. Activation of IKK-NF-κB and MAPKs leads to increased expression of inflammatory genes while activation of NADPH oxidase increases the production of ROS. ROS enhances the recruitment of TLRs to the lipid rafts, increases the activity of PKCs, and activates the NLRP3 inflammasome (Figure 1). Further studies are needed to address whether SFAs induce a rise in mitochondrial ROS and whether the initial increase in mitochondrial ROS stimulates NADPH-mediated production of ROS in the cytosol.
The intracellular NLRs may also be involved in SFA-induced activation of inflammatory genes (Figure 1). NOD1 and NOD2 have been shown to be critical for lauric acid-induced activation of NF-κB and IL-8,124 and NLRP3 inflammasome has been reported to be activated following treatment with palmitic acid and increase the secretion of IL-1β in human Sw.71 placental cells, macrophages, and hepatocytes.30,37,125,126 Further studies are needed to determine how SFAs activate NLR pathways. There is also evidence suggesting that SFAs may increase the ER stress which, in turn, induces the expression of inflammatory genes and ER chaperones including GRP78 (Figure 1).
Besides the mechanistic pathways described above, SFAs may also induce inflammation via binding to G-protein-coupled receptors (GPRs)23 or production of ceramides.29,134,135 The increased production of ceramides may contribute to the activation of NLRP3 inflammasome.122 Further studies are needed to delineate how ceramides activate NLRP3, what GPRs are activated by SFAs, and how different GPR pathways may interact to affect inflammation. There are also studies suggesting that high-fat diet may affect the gut microbiota136 and increase the diffusion of LPS from the gut to the circulatory system and/or the absorption by enterocytes during chylomicron secretion137,138 while Dalby et al (2018) showed that changes in the composition of caecal microbiota is not consistent within genotypes following high-fat diet consumption.139 The focus of this paper is on the mechanisms of long-chain saturated fatty-acids-induced inflammation. Many studies examining how unsaturated fatty acids affect inflammation140–142 are out of the scope of this paper. Considering the complexity of our diet, the effects our diet may have on gut microbiota, and the intricate effects different fatty acids have on multiple pathways involved in inflammation, a wholistic approach needs to be taken when evaluating preventative and therapeutic strategies to target inflammation.
Acknowledgment
This work was supported by the Research Fund from the Department of Biological Sciences, Seton Hall University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Pi-Sunyer X. The medical risks of obesity. Postgrad Med. 2009;121(6):21–33. doi:10.3810/pgm.2009.11.2074
2. Unamuno X, Gomez-Ambrosi J, Rodriguez A, Becerril S, Fruhbeck G, Catalan V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur J Clin Invest. 2018;48(9):e12997. doi:10.1111/eci.2018.48.issue-9
3. Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol. 2012;56(3):704–713. doi:10.1016/j.jhep.2011.09.020
4. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest. 2017;127(1):43–54. doi:10.1172/JCI88880
5. Ramkumar N, Cheung AK, Pappas LM, Roberts WL, Beddhu S. Association of obesity with inflammation in chronic kidney disease: a cross-sectional study. J Ren Nutr. 2004;14(4):201–207. doi:10.1016/S1051-2276(04)00133-5
6. Kreutzer C, Peters S, Schulte DM, et al. Hypothalamic inflammation in human obesity is mediated by environmental and genetic factors. Diabetes. 2017;66(9):2407–2415. doi:10.2337/db17-0067
7. Assuncao SNF, Sorte N, Alves CAD, Mendes PSA, Alves CRB, Silva LR. Inflammatory cytokines and non-alcoholic fatty liver disease (NAFLD) in obese children and adolescents. Nutr Hosp. 2018;35(1):78–83. doi:10.20960/nh.1317
8. Roberts KA, Riley SC, Reynolds RM, et al. Placental structure and inflammation in pregnancies associated with obesity. Placenta. 2011;32(3):247–254. doi:10.1016/j.placenta.2010.12.023
9. Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi:10.1038/nature01137
10. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi:10.1126/science.7678183
11. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi:10.1172/JCI200319451
12. Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring). 2008;16(6):1248–1255. doi:10.1038/oby.2008.210
13. Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi:10.1016/j.cell.2009.12.052
14. Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11(2):183–190. doi:10.1038/nm1166
15. Decleves AE, Mathew AV, Cunard R, Sharma K. AMPK mediates the initiation of kidney disease induced by a high-fat diet. J Am Soc Nephrol. 2011;22(10):1846–1855. doi:10.1681/ASN.2011010026
16. Wang X, Ge A, Cheng M, et al. Increased hypothalamic inflammation associated with the susceptibility to obesity in rats exposed to high-fat diet. Exp Diabetes Res. 2012;2012:847246.
17. Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132(6):2087–2102. doi:10.1053/j.gastro.2007.03.052
18. Moller DE. Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2000;11(6):212–217. doi:10.1016/S1043-2760(00)00272-1
19. Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96(9):939–949. doi:10.1161/01.RES.0000163635.62927.34
20. Frohnert BI, Jacobs DR
21. Zhao L, Ni Y, Ma X, et al. A panel of free fatty acid ratios to predict the development of metabolic abnormalities in healthy obese individuals. Sci Rep. 2016;6:28418. doi:10.1038/srep28418
22. Arner P, Ryden M. Fatty acids, obesity and insulin resistance. Obes Facts. 2015;8(2):147–155. doi:10.1159/000381224
23. Rodriguez-Pacheco F, Gutierrez-Repiso C, Garcia-Serrano S, et al. The pro-/anti-inflammatory effects of different fatty acids on visceral adipocytes are partially mediated by GPR120. Eur J Nutr. 2016;56(4):1743–1752.
24. Zhang J, Zhao Y, Xu C, et al. Association between serum free fatty acid levels and nonalcoholic fatty liver disease: a cross-sectional study. Sci Rep. 2014;4:5832. doi:10.1038/srep05832
25. Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37(8):1020–1024. doi:10.2337/diab.37.8.1020
26. Feng R, Luo C, Li C, et al. Free fatty acids profile among lean, overweight and obese non-alcoholic fatty liver disease patients: a case-control study. Lipids Health Dis. 2017;16(1):165. doi:10.1186/s12944-017-0551-1
27. Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60(10):2441–2449. doi:10.2337/db11-0425
28. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi:10.1172/JCI28898
29. Haversen L, Danielsson KN, Fogelstrand L, Wiklund O. Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis. 2009;202(2):382–393. doi:10.1016/j.atherosclerosis.2008.05.033
30. Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–415. doi:10.1038/ni.2022
31. Huang S, Rutkowsky JM, Snodgrass RG, et al. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res. 2012;53(9):2002–2013. doi:10.1194/jlr.D029546
32. Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276(20):16683–16689. doi:10.1074/jbc.M011695200
33. Nicholas DA, Zhang K, Hung C, et al. Palmitic acid is a toll-like receptor 4 ligand that induces human dendritic cell secretion of IL-1beta. PLoS One. 2017;12(5):e0176793. doi:10.1371/journal.pone.0176793
34. Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1beta in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. 2013;191(8):4337–4347. doi:10.4049/jimmunol.1300298
35. Gupta S, Knight AG, Keller JN, Bruce-Keller AJ. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J Neurochem. 2012;120(6):1060–1071. doi:10.1111/j.1471-4159.2012.07660.x
36. Yang X, Haghiac M, Glazebrook P, Minium J, Catalano PM, Hauguel-de Mouzon S. Saturated fatty acids enhance TLR4 immune pathways in human trophoblasts. Hum Reprod. 2015;30(9):2152–2159. doi:10.1093/humrep/dev173
37. Shirasuna K, Takano H, Seno K, et al. Palmitic acid induces interleukin-1beta secretion via NLRP3 inflammasomes and inflammatory responses through ROS production in human placental cells. J Reprod Immunol. 2016;116:104–112. doi:10.1016/j.jri.2016.06.001
38. Wang Z, Liu D, Wang F, et al. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-kappaB signalling. Br J Nutr. 2012;107(2):229–241. doi:10.1017/S0007114511002868
39. Liu J, Hu S, Cui Y, et al. Saturated fatty acids up-regulate COX-2 expression in prostate epithelial cells via toll-like receptor 4/NF-kappaB signaling. Inflammation. 2014;37(2):467–477. doi:10.1007/s10753-013-9760-6
40. Lee JY, Cho HK, Kwon YH. Palmitate induces insulin resistance without significant intracellular triglyceride accumulation in HepG2 cells. Metabolism. 2010;59(7):927–934. doi:10.1016/j.metabol.2009.10.012
41. Oberbach A, Schlichting N, Bluher M, et al. Palmitate induced IL-6 and MCP-1 expression in human bladder smooth muscle cells provides a link between diabetes and urinary tract infections. PLoS One. 2010;5(5):e10882. doi:10.1371/journal.pone.0010882
42. Quan J, Liu J, Gao X, et al. Palmitate induces interleukin-8 expression in human aortic vascular smooth muscle cells via Toll-like receptor 4/nuclear factor-kappaB pathway (TLR4/NF-kappaB-8). J Diabetes. 2014;6(1):33–41. doi:10.1111/1753-0407.12073
43. Jove M, Planavila A, Sanchez RM, Merlos M, Laguna JC, Vazquez-Carrera M. Palmitate induces tumor necrosis factor-alpha expression in C2C12 skeletal muscle cells by a mechanism involving protein kinase C and nuclear factor-kappaB activation. Endocrinology. 2006;147(1):552–561. doi:10.1210/en.2005-0440
44. Senn JJ. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem. 2006;281(37):26865–26875. doi:10.1074/jbc.M513304200
45. Ajuwon KM, Spurlock ME. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J Nutr. 2005;135(8):1841–1846. doi:10.1093/jn/135.8.1841
46. Paik JS, Cho WK, Oh EH, Lee SB, Yang SW. Palmitate induced secretion of IL-6 and MCP-1 in orbital fibroblasts derived from patients with thyroid-associated ophthalmopathy. Mol Vis. 2012;18:1467–1477.
47. Oh E, Yun M, Kim SK, et al. Palmitate induces COX-2 expression via the sphingolipid pathway-mediated activation of NF-kappaB, p38, and ERK in human dermal fibroblasts. Arch Dermatol Res. 2014;306(4):339–345. doi:10.1007/s00403-013-1434-6
48. Boden G, Chen X, Rosner J, Barton M. Effects of a 48-h fat infusion on insulin secretion and glucose utilization. Diabetes. 1995;44(10):1239–1242. doi:10.2337/diab.44.10.1239
49. Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51(7):2005–2011. doi:10.2337/diabetes.51.7.2005
50. Nivala AM, Reese L, Frye M, Gentile CL, Pagliassotti MJ. Fatty acid-mediated endoplasmic reticulum stress in vivo: differential response to the infusion of soybean and lard oil in rats. Metabolism. 2013;62(5):753–760. doi:10.1016/j.metabol.2012.12.001
51. Li X. Endoplasmic reticulum stress regulates inflammation in adipocyte of obese rats via toll-like receptors 4 signaling. Iran J Basic Med Sci. 2018;21(5):502–507. doi:10.22038/IJBMS.2018.27346.6674
52. Eguchi K, Manabe I, Oishi-Tanaka Y, et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012;15(4):518–533. doi:10.1016/j.cmet.2012.01.023
53. Milanski M, Degasperi G, Coope A, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci. 2009;29(2):359–370. doi:10.1523/JNEUROSCI.2760-08.2009
54. Dumas JA, Bunn JY, Nickerson J, et al. Dietary saturated fat and monounsaturated fat have reversible effects on brain function and the secretion of pro-inflammatory cytokines in young women. Metabolism. 2016;65(10):1582–1588. doi:10.1016/j.metabol.2016.08.003
55. Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013;45:e66. doi:10.1038/emm.2013.97
56. Kim HM, Park BS, Kim JI, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell. 2007;130(5):906–917. doi:10.1016/j.cell.2007.08.002
57. Yin J, Peng Y, Wu J, Wang Y, Yao L. Toll-like receptor 2/4 links to free fatty acid-induced inflammation and beta-cell dysfunction. J Leukoc Biol. 2014;95(1):47–52. doi:10.1189/jlb.0313143
58. Jialal I, Kaur H, Devaraj S. Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J Clin Endocrinol Metab. 2014;99(1):39–48. doi:10.1210/jc.2013-3092
59. Kawai T, Takeuchi O, Fujita T, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167(10):5887–5894. doi:10.4049/jimmunol.167.10.5887
60. Oliveira-Nascimento L, Massari P, Wetzler LM. The Role of TLR2 in infection and immunity. Front Immunol. 2012;3:79. doi:10.3389/fimmu.2012.00079
61. Hanzelmann D, Joo HS, Franz-Wachtel M, et al. Toll-like receptor 2 activation depends on lipopeptide shedding by bacterial surfactants. Nat Commun. 2016;7:12304. doi:10.1038/ncomms12304
62. Kang JY, Nan X, Jin MS, et al. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity. 2009;31(6):873–884. doi:10.1016/j.immuni.2009.09.018
63. Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond). 2011;121(9):367–387. doi:10.1042/CS20110164
64. Nilsen NJ, Vladimer GI, Stenvik J, et al. A role for the adaptor proteins TRAM and TRIF in toll-like receptor 2 signaling. J Biol Chem. 2015;290(6):3209–3222. doi:10.1074/jbc.M114.593426
65. Dasu MR, Jialal I. Free fatty acids in the presence of high glucose amplify monocyte inflammation via Toll-like receptors. Am J Physiol Endocrinol Metab. 2011;300(1):E145–E154. doi:10.1152/ajpendo.00490.2010
66. Lee JY, Zhao L, Youn HS, et al. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279(17):16971–16979. doi:10.1074/jbc.M312990200
67. Jang HJ, Kim HS, Hwang DH, Quon MJ, Kim JA. Toll-like receptor 2 mediates high-fat diet-induced impairment of vasodilator actions of insulin. Am J Physiol Endocrinol Metab. 2013;304(10):E1077–E1088. doi:10.1152/ajpendo.00578.2012
68. Kitchens RL, Ulevitch RJ, Munford RS. Lipopolysaccharide (LPS) partial structures inhibit responses to LPS in a human macrophage cell line without inhibiting LPS uptake by a CD14-mediated pathway. J Exp Med. 1992;176(2):485–494. doi:10.1084/jem.176.2.485
69. Kumazawa Y, Nakatsuka M, Takimoto H, et al. Importance of fatty acid substituents of chemically synthesized lipid A-subunit analogs in the expression of immunopharmacological activity. Infect Immun. 1988;56(1):149–155.
70. Schletter J, Heine H, Ulmer AJ, Rietschel ET. Molecular mechanisms of endotoxin activity. Arch Microbiol. 1995;164(6):383–389. doi:10.1007/BF02529735
71. Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi:10.1146/annurev.biochem.76.010307.145803
72. Maeshima N, Fernandez RC. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front Cell Infect Microbiol. 2013;3:3. doi:10.3389/fcimb.2013.00003
73. Kato H, Haishima Y, Iida T, Tanaka A, Tanamoto K. Chemical structure of lipid A isolated from flavobacterium meningosepticum lipopolysaccharide. J Bacteriol. 1998;180(15):3891–3899.
74. Zhao H, Shao D, Jiang C, et al. Biological activity of lipopeptides from bacillus. Appl Microbiol Biotechnol. 2017;101(15):5951–5960. doi:10.1007/s00253-017-8396-0
75. Schaeffler A, Gross P, Buettner R, et al. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology. 2009;126(2):233–245. doi:10.1111/j.1365-2567.2008.02892.x
76. Lancaster GI, Langley KG, Berglund NA, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. 2018;27(5):1096–1110 e1095. doi:10.1016/j.cmet.2018.03.014
77. Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009;284(40):27384–27392. doi:10.1074/jbc.M109.044065
78. Hwang DH, Kim JA, Lee JY. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur J Pharmacol. 2016;785:24–35. doi:10.1016/j.ejphar.2016.04.024
79. Panter G, Jerala R. The ectodomain of the Toll-like receptor 4 prevents constitutive receptor activation. J Biol Chem. 2011;286(26):23334–23344. doi:10.1074/jbc.M110.205419
80. Park SJ, Youn HS. Suppression of homodimerization of toll-like receptor 4 by isoliquiritigenin. Phytochemistry. 2010;71(14–15):1736–1740. doi:10.1016/j.phytochem.2010.07.008
81. Jin MS, Kim SE, Heo JY, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130(6):1071–1082. doi:10.1016/j.cell.2007.09.008
82. Tapping RI, Tobias PS. Mycobacterial lipoarabinomannan mediates physical interactions between TLR1 and TLR2 to induce signaling. J Endotoxin Res. 2003;9(4):264–268. doi:10.1177/09680519030090040801
83. Triantafilou M, Gamper FG, Haston RM, et al. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem. 2006;281(41):31002–31011. doi:10.1074/jbc.M602794200
84. Anderson RG, Jacobson K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 2002;296(5574):1821–1825. doi:10.1126/science.1068886
85. Lee JY, Zhao L, Hwang DH. Modulation of pattern recognition receptor-mediated inflammation and risk of chronic diseases by dietary fatty acids. Nutr Rev. 2010;68(1):38–61. doi:10.1111/nure.2010.68.issue-1
86. Soong G, Reddy B, Sokol S, Adamo R, Prince A. TLR2 is mobilized into an apical lipid raft receptor complex to signal infection in airway epithelial cells. J Clin Invest. 2004;113(10):1482–1489. doi:10.1172/JCI200420773
87. Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115(Pt 12):2603–2611.
88. Olsson S, Sundler R. The role of lipid rafts in LPS-induced signaling in a macrophage cell line. Mol Immunol. 2006;43(6):607–612. doi:10.1016/j.molimm.2005.04.011
89. Li PL, Zhang Y, Yi F. Lipid raft redox signaling platforms in endothelial dysfunction. Antioxid Redox Signal. 2007;9(9):1457–1470. doi:10.1089/ars.2007.1667
90. Schoeniger A, Fuhrmann H, Schumann J. LPS- or pseudomonas aeruginosa-mediated activation of the macrophage TLR4 signaling cascade depends on membrane lipid composition. PeerJ. 2016;4:e1663. doi:10.7717/peerj.1663
91. Nakahira K, Kim HP, Geng XH, et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med. 2006;203(10):2377–2389. doi:10.1084/jem.20060845
92. Hellwing C, Schoeniger A, Roessler C, Leimert A, Schumann J. Lipid raft localization of TLR2 and its co-receptors is independent of membrane lipid composition. PeerJ. 2018;6:e4212. doi:10.7717/peerj.4212
93. Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am. 2008;37(3):
94. Coll T, Eyre E, Rodriguez-Calvo R, et al. Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J Biol Chem. 2008;283(17):11107–11116. doi:10.1074/jbc.M708700200
95. Montell E, Turini M, Marotta M, et al. DAG accumulation from saturated fatty acids desensitizes insulin stimulation of glucose uptake in muscle cells. Am J Physiol Endocrinol Metab. 2001;280(2):E229–E237. doi:10.1152/ajpendo.2001.280.2.E229
96. Schmitz-Peiffer C, Browne CL, Oakes ND, et al. Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes. 1997;46(2):169–178. doi:10.2337/diab.46.2.169
97. Yang L, Qian Z, Ji H, et al. Inhibitory effect on protein kinase ctheta by crocetin attenuates palmitate-induced insulin insensitivity in 3T3-L1 adipocytes. Eur J Pharmacol. 2010;642(1–3):47–55. doi:10.1016/j.ejphar.2010.05.061
98. So JS. Roles of endoplasmic reticulum stress in immune responses. Mol Cells. 2018;41(8):705–716. doi:10.14348/molcells.2018.0241
99. Gao Z, Zhang X, Zuberi A, et al. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18(8):2024–2034. doi:10.1210/me.2003-0383
100. Ragheb R, Shanab GM, Medhat AM, Seoudi DM, Adeli K, Fantus IG. Free fatty acid-induced muscle insulin resistance and glucose uptake dysfunction: evidence for PKC activation and oxidative stress-activated signaling pathways. Biochem Biophys Res Commun. 2009;389(2):211–216. doi:10.1016/j.bbrc.2009.08.106
101. Kadotani A, Tsuchiya Y, Hatakeyama H, Katagiri H, Kanzaki M. Different impacts of saturated and unsaturated free fatty acids on COX-2 expression in C(2)C(12) myotubes. Am J Physiol Endocrinol Metab. 2009;297(6):E1291–E1303. doi:10.1152/ajpendo.00293.2009
102. Soltoff SP. Rottlerin: an inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol Sci. 2007;28(9):453–458. doi:10.1016/j.tips.2007.07.003
103. Zhang J, Wang X, Vikash V, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. 2016;(2016:4350965.
104. Meitzler JL, Antony S, Wu Y, et al. NADPH oxidases: a perspective on reactive oxygen species production in tumor biology. Antioxid Redox Signal. 2014;20(17):2873–2889. doi:10.1089/ars.2013.5603
105. Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43(3):319–331. doi:10.1016/j.freeradbiomed.2007.03.028
106. Brandes RP, Weissmann N, Schroder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med. 2014;76:208–226. doi:10.1016/j.freeradbiomed.2014.07.046
107. Vilhardt F, van Deurs B. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J. 2004;23(4):739–748. doi:10.1038/sj.emboj.7600066
108. Quinn MT, Gauss KA. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol. 2004;76(4):760–781.
109. El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med. 2009;41(4):217–225. doi:10.3858/emm.2009.41.4.058
110. Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi:10.2337/diabetes.49.11.1939
111. Joseph LC, Barca E, Subramanyam P, et al. Inhibition of NAPDH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS One. 2016;11(1):e0145750. doi:10.1371/journal.pone.0145750
112. Yuzefovych L, Wilson G, Rachek L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am J Physiol Endocrinol Metab. 2010;299(6):E1096–E1105. doi:10.1152/ajpendo.00238.2010
113. Han CY, Umemoto T, Omer M, et al. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J Biol Chem. 2012;287(13):10379–10393. doi:10.1074/jbc.M111.304998
114. Cremonini E, Oteiza PI. (-)-Epicatechin and its metabolites prevent palmitate-induced NADPH oxidase upregulation, oxidative stress and insulin resistance in HepG2 cells. Arch Biochem Biophys. 2018;646:55–63. doi:10.1016/j.abb.2018.03.027
115. Yuan H, Zhang X, Huang X, et al. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of beta-cells via JNK, p38 MAPK and p53 pathways. PLoS One. 2010;5(12):e15726. doi:10.1371/journal.pone.0015726
116. Frayn KN, Langin D, Karpe F. Fatty acid-induced mitochondrial uncoupling in adipocytes is not a promising target for treatment of insulin resistance unless adipocyte oxidative capacity is increased. Diabetologia. 2008;51(3):394–397. doi:10.1007/s00125-007-0901-z
117. Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51(7):1289–1301. doi:10.1016/j.freeradbiomed.2011.06.033
118. Das S, Alhasson F, Dattaroy D, et al. NADPH oxidase-derived peroxynitrite drives inflammation in mice and human nonalcoholic steatohepatitis via TLR4-lipid raft recruitment. Am J Pathol. 2015;185(7):1944–1957. doi:10.1016/j.ajpath.2015.03.024
119. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
120. Moreira LO, Zamboni DS. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol. 2012;3:328. doi:10.3389/fimmu.2012.00328
121. Saxena M, Yeretssian G. NOD-like receptors: master regulators of inflammation and cancer. Front Immunol. 2014;5:327. doi:10.3389/fimmu.2014.00327
122. Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188. doi:10.1038/nm.2279
123. Lukens JR, Dixit VD, Kanneganti TD. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discov Med. 2011;12(62):65–74.
124. Zhao L, Kwon MJ, Huang S, et al. Differential modulation of Nods signaling pathways by fatty acids in human colonic epithelial HCT116 cells. J Biol Chem. 2007;282(16):11618–11628. doi:10.1074/jbc.M608644200
125. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54(1):133–144. doi:10.1002/hep.v54.1
126. Sui YH, Luo WJ, Xu QY, Hua J. Dietary saturated fatty acid and polyunsaturated fatty acid oppositely affect hepatic NOD-like receptor protein 3 inflammasome through regulating nuclear factor-kappa B activation. World J Gastroenterol. 2016;22(8):2533–2544. doi:10.3748/wjg.v22.i8.2533
127. B’Chir W, Maurin AC, Carraro V, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41(16):7683–7699. doi:10.1093/nar/gkt563
128. Salvado L, Coll T, Gomez-Foix AM, et al. Oleate prevents saturated-fatty-acid-induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia. 2013;56(6):1372–1382. doi:10.1007/s00125-013-2867-3
129. Mayer CM, Belsham DD. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5ʹ monophosphate-activated protein kinase activation. Endocrinology. 2010;151(2):576–585. doi:10.1210/en.2009-1122
130. Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi:10.1126/science.1103160
131. Zhao M, Zang B, Cheng M, Ma Y, Yang Y, Yang N. Differential responses of hepatic endoplasmic reticulum stress and inflammation in diet-induced obese rats with high-fat diet rich in lard oil or soybean oil. PLoS One. 2013;8(11):e78620. doi:10.1371/journal.pone.0078620
132. Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799. doi:10.1038/srep00799
133. Tracy LM, Bergqvist F, Ivanova EV, Jacobsen KT, Iverfeldt K. Exposure to the saturated free fatty acid palmitate alters BV-2 microglia inflammatory response. J Mol Neurosci. 2013;51(3):805–812. doi:10.1007/s12031-013-0068-7
134. Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem. 1999;274(34):24202–24210. doi:10.1074/jbc.274.34.24202
135. Schilling JD, Machkovech HM, He L, et al. Palmitate and lipopolysaccharide trigger synergistic ceramide production in primary macrophages. J Biol Chem. 2013;288(5):2923–2932. doi:10.1074/jbc.M112.419978
136. Teixeira TF, Collado MC, Ferreira CL, Bressan J, Peluzio Mdo C. Potential mechanisms for the emerging link between obesity and increased intestinal permeability. Nutr Res. 2012;32(9):637–647.
137. Moreira AP, Texeira TF, Ferreira AB, Peluzio Mdo C, Alfenas Rde C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr. 2012;108(5):801–809. doi:10.1017/S0007114512001213
138. Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology. 2012;142(5):1100–1101 e1102. doi:10.1053/j.gastro.2012.01.034
139. Dalby MJ, Aviello G, Ross AW, Walker AW, Barrett P, Morgan PJ. Diet induced obesity is independent of metabolic endotoxemia and TLR4 signalling, but markedly increases hypothalamic expression of the acute phase protein, SerpinA3N. Sci Rep. 2018;8(1):15648. doi:10.1038/s41598-018-33928-4
140. Fedor D, Kelley DS. Prevention of insulin resistance by n-3 polyunsaturated fatty acids. Curr Opin Clin Nutr Metab Care. 2009;12(2):138–146. doi:10.1097/MCO.0b013e3283218299
141. Orr SK, Trepanier MO, Bazinet RP. n-3 polyunsaturated fatty acids in animal models with neuroinflammation. Prostaglandins Leukot Essent Fatty Acids. 2013;88(1):97–103. doi:10.1016/j.plefa.2012.05.008
142. Rogero MM, Calder PC. Obesity, inflammation, toll-like receptor 4 and fatty acids. Nutrients. 2018;10(4):432. doi:10.3390/nu10040432
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.