")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
SARS-CoV-2 (COVID-19) Adhesion Site Protein Upregulation in Small Airways, Type 2 Pneumocytes, and Alveolar Macrophages of Smokers and COPD – Possible Implications for Interstitial Fibrosis
Authors Brake SJ, Eapen MS , McAlinden KD, Markos J, Haug G, Larby J, Chia C, Hardikar A, Singhera GK, Hackett TL, Lu W , Sohal SS
Received 21 July 2021
Accepted for publication 14 November 2021
Published 11 January 2022 Volume 2022:17 Pages 101—115
DOI https://doi.org/10.2147/COPD.S329783
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Min Zhang
Samuel James Brake,1 Mathew Suji Eapen,1 Kielan Darcy McAlinden,1 James Markos,1,2 Greg Haug,1,2 Josie Larby,1,2 Collin Chia,1,2 Ashutosh Hardikar,1,3 Gurpreet Kaur Singhera,4,5 Tillie L Hackett,4,5 Wenying Lu,1 Sukhwinder Singh Sohal1
1Respiratory Translational Research Group, Department of Laboratory Medicine, School of Health Sciences, College of Health and Medicine, University of Tasmania, Launceston, TAS, 7248, Australia; 2Department of Respiratory Medicine, Launceston General Hospital, Launceston, TAS, 7250, Australia; 3Department of Cardiothoracic Surgery, Royal Hobart Hospital, Hobart, TAS, 7000, Australia; 4Department of Anesthesiology, Pharmacology, and Therapeutics, University of British Columbia, Vancouver, BC, Canada; 5Department of Medicine, University of British Columbia Centre for Heart Lung Innovation, St. Paul’s Hospital, Vancouver, BC, Canada
Correspondence: Sukhwinder Singh Sohal
Respiratory Translational Research Group, Department of Laboratory Medicine, School of Health Sciences, College of Health and Medicine, University of Tasmania, Locked Bag – 1322, Newnham Drive, Launceston, TAS, 7248, Australia
Tel +61 3 6324 5434
Email [email protected]
Background: Smokers and patients with COPD are highly susceptible to SARS-CoV-2 infection, leading to severe COVID-19.
Methods: This cross-sectional study involved resected lung tissues from 16 patients with GOLD stage I or II COPD; of which 8 were current smokers COPD (COPD-CS), and 8 ex-smokers COPD (COPD-ES), 7 normal lung function smokers (NLFS), 9 patients with small airways disease (SAD), and 10 were never-smoking normal controls (NC). Immunostaining for ACE2, Furin, and TMPRSS2 was performed and analysed for percent expression in small airway epithelium (SAE) and counts for positively and negatively stained type 2 pneumocytes and alveolar macrophages (AMs) were done using Image ProPlus V7.0. Furthermore, primary small airway epithelial cells (pSAEC) were analysed by immunofluorescence after exposure to cigarette smoke extract (CSE).
Results: ACE2, Furin, and TMPRSS2 expression significantly increased in SAE and type 2 pneumocytes in all the subjects (except Furin for NLFS) compared to NC (p < 0.001). Similar significance was observed for ACE2 positive AM (p < 0.002), except COPD-ES, which decreased in ACE2 positive AMs (p < 0.003). Total type 2 pneumocytes and AMs significantly increased in the pathological groups compared to NC (p < 0.01), except SAD (p = 0.08). However, AMs are significantly reduced in COPD-ES (p < 0.003). Significant changes were observed for tissue co-expression of Furin and TMPRSS2 with ACE2 in SAE, type 2 pneumocytes and AMs. These markers also negatively correlated with lung function parameters, such as FEV1/FVC % predicted, FEF25-75%, DLCO% predicted. A strong co-localisation and expression for ACE2 (p < 0.0001), Furin (p < 0.01), and TMPRSS2 (p < 0.0001) was observed in pSAEC treated with 1% CSE than controls.
Discussion: The increased expression of ACE2, TMPRSS2 and Furin, in the SAE, type 2 pneumocytes and AMs of smokers and COPD are detrimental to lung function and proves that these patient groups could be more susceptible to severe COVID-19 infection. Increased type 2 pneumocytes suggest that these patients are vulnerable to developing post-COVID-19 interstitial pulmonary fibrosis or fibrosis in general. There could be a silently developing interstitial pathology in smokers and patients with COPD. This is the first comprehensive study to report such changes.
Keywords: COVID-19, SARS-CoV-2, smoking, COPD, ACE2, type 2 pneumocytes, alveolar macrophages, epithelium
Introduction
Since the emergence of the novel Coronavirus SARS-CoV-2 at the close of 2019, COVID-19 has spread rapidly around the globe and as of May 2021, the World Health Organisation (WHO) has reported over 169 million confirmed cases and 3.51 million deaths globally.1 SARS-CoV-2, the virus behind COVID-19, is one member of the large coronaviruses family.2 This family also includes the deadly MERS‐CoV, the causative virus of Middle East Respiratory Syndrome (MERS) and SARS-CoV, the causative virus of the previous severe acute respiratory syndrome (SARS).2–4 COVID-19 has a range of symptomatic presentations from respiratory distress, airway damage to death.5–7 In approximately 80% of COVID-19 infections, patients present with mild respiratory illness.8 Risk factors for more severe infections include age, comorbidities, hypoxia, and severe immune response.9
COPD is the fourth leading cause of death globally, a chronic lung disease characterised by partially reversible airflow obstruction, with increased exacerbations, dyspnoea, and decreased quality of life.10 Smoking primarily causes COPD, although air pollution through fossil fuel burning is a contributing factor too and is an increasingly likely comorbidity risk for severe COVID-19.11,12 COPD is partly characterised by the variable immune response, which sees increased macrophage recruitment in conjunction with other pathological inflammatory mediators.13,14 COPD patients are highly susceptible to respiratory infections, with a study showing 30% of COPD exacerbations related to respiratory viruses.15 Initially, based on observational epidemiological data, there was contention regarding the impact of cigarette smoke on COVID-19 infection severity and transmission rate.16 However, the WHO released a review finding that smokers are more likely to develop severe COVID-19 than non-smokers, urging caution about amplifying unproven claims that tobacco or nicotine could reduce COVID-19 risk.17 Aside from these weak and strongly disputed claims, it is inevitable that cigarette smoking, and COPD impacts the immune system, causing functional dysregulation.18,19 Therefore, both cigarette smoking and COPD are likely risk factors for the increased severity of COVID-19. Along with cigarette smoking and COPD being linked to dysfunctional immunity, they are also shown to relate to an increased expression of proteins linked to pathogen adhesion, driving smoking-induced pneumonia and other respiratory infections.20–25
Human angiotensin-converting enzyme 2 (ACE2), Furin and transmembrane serine protease 2 (TMPRSS2) are utilised by some respiratory viruses as a receptor for cell adhesion and entry.26 SARS-CoV-2 utilises the ACE2 enzyme as the critical entry point into human cells.26,27 Interestingly, the viral-binding protein, spike (S) protein, of SARS-CoV-2 has a significantly stronger affinity for ACE2 than the original SARS-CoV, with 10-to-20-fold greater binding efficiency.28 This increased specificity makes ACE2 a significant mediator in the severity and transmission of COVID-19. Our preliminary observations identified that smoking could upregulate ACE2 expression, with upregulation of ACE2 observed in limited patient tissue of COPD-current smokers (COPD-ES) and normal lung function smokers (NLFS) in comparison with normal controls.29
Furthermore, a small airway epithelia ACE2 expression study concluded that cigarette smoking and COPD upregulate ACE2 expression in small airway epithelium.30,31 Therefore, smoking-induced upregulation of ACE2 in the lung is a largely avoidable risk factor linked to an increased susceptibility of developing COVID-19. The cells targeted by SARS-CoV and now SARS-CoV-2 are predominantly type 2 pneumocytes and alveolar macrophages, with studies showing increases in these cell types attributed to smoking and COPD.32–37 New data is drawing attention to host mechanisms by which SARS-CoV-2 enters the cell.38 Furin is a proprotein convertase that is believed to be an essential protein in the configuration of the SARS-CoV-2 envelope, processing essential membrane proteins.39 SARS-CoV-2 utilises S protein, a granule shaped structural protein which aids in viral-cellular binding. During infection, host Furin cleaves the viral S protein into an N-terminal S1 extracellular domain, allowing recognition by cell surface receptors, and a C terminal S2 membrane anchor protein, which is involved in viral translocation into the cell.40,41 TMPRSS2, like Furin, is believed to cleave the viral S glycoprotein with a similar outcome. These processes are also seen in other coronaviruses, such as SARS-CoV, and viruses in the Orthomyxoviridae family, such as influenza and H1N1.42 Current data suggests that smoking may upregulate Furin expression in lung tissue; however, the effect of smoking on TMPRSS2 is debated, some reporting no effect while others showing increased expression.31,43
A recent two-centre pulmonary post-mortem of COVID-19 cases found that of the 38 post-mortems, all demonstrated type 2 pneumocyte hyperplasia, and the inflammatory infiltrate in 24 of the cases was primarily composed of alveolar macrophages.44 These findings further link the pathological changes and cell population changes associated with COPD and COVID-19. Our initial assessment was that type 2 pneumocyte and alveolar macrophages showed the most significant increase in ACE2 expression.29 This cross-sectional study aims to expand this understanding further using quantitative assessment tools and provide a vital link between smoking and ACE2, Furin and TMPRSS2 expression in the small airway epithelium, primary small airway epithelial cells, type 2 pneumocytes and alveolar macrophages. This paper also explores the levels of the SARS-CoV-2-related biomarkers in the small airways of normal controls and uses this as a baseline reference against smokers and patients with small airway disease and COPD.
Methods
Patient Demographics
Surgically resected small airway tissue from 42 patients was available from our biobank (ethics ID: H0012374). The approvals were obtained from the Tasmanian Health and Medical Human Research Ethics Committee. All subjects gave written, informed consent prior to participation. All subjects in the pathological cohorts were diagnosed with primary non-small cell lung cancer. Patient details are given in Table 1. The small airway resected tissue was taken well away from the primary tumour. Among the 42 patients, 16 patients were diagnosed with Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage I or stage II COPD (forced expiratory ratio <70%), of which 8 were current smokers (COPD-CS) and 8 were ex-smokers (COPD-ES) (>1-year smoking cessation); 9 patients had small airway disease (SAD) only, with expiratory limb scalloping and forced expiratory flow25–75 < 68% predicted, 7 were normal lung function smokers (NLFS); 10 were never-smoking normal controls (NC). The NC were kindly provided by James Hogg Lung Registry, the University of British Columbia, with approval from the Providence Health Care Research Ethics Board (H00-50110). The exclusion criterion for this study were as follows: subjects with other respiratory diseases and those on systemic or inhaled corticosteroids treatment. Written, informed consent was obtained prior to participation.
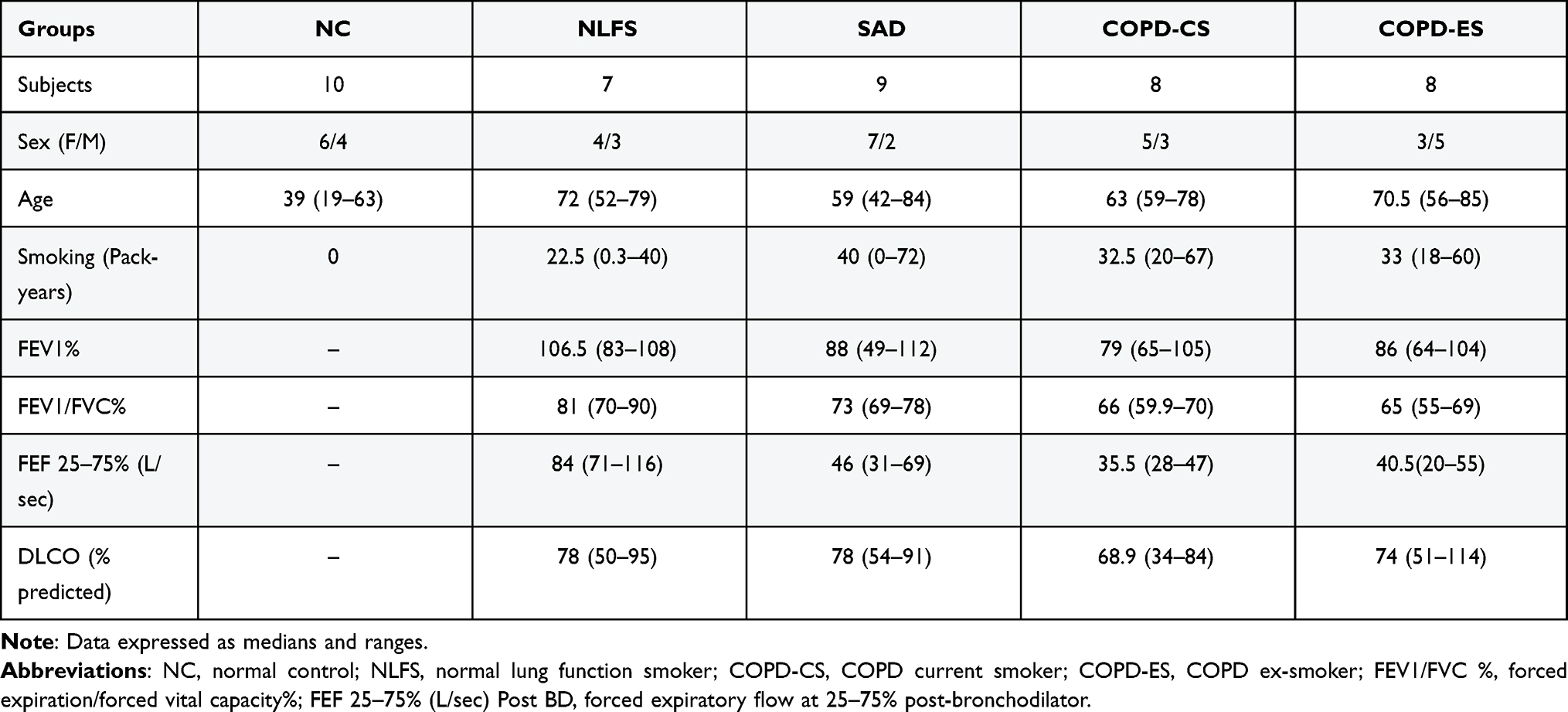 |
Table 1 Patient Demographics |
Immunohistochemical Staining and Analysis
Surgical resections were fixed in formalin within minutes of surgery.45 After processing, the resected small airway (<2 mm internal diameter) tissue blocks were separately embedded in paraffin wax for research analyses. The tissue was sectioned at 3.5 µm and processed with standard immunohistochemical staining procedures.45 Immunostaining was completed by using rabbit polyclonal anti-ACE2 antibody (Catalog No. Ab15348, Abcam, 1:800), Furin rabbit polyclonal antibody (bs-13228R; Bioss antibodies; 1:200) and TMPRSS2 rabbit polyclonal antibody (bs-6285R, Bioss antibodies, 1:250). Antibody binding was visualised by a substrate 3,3ʹ-diaminobenzidine (DAB) reaction producing a brown colour indicating positive staining. Nuclear counterstain was achieved with gills hematoxylin. All slides were coded and randomised to blind the analyst.
Tissue Analysis for ACE2, Furin and TMPRSS2
Images were taken of the small airway epithelium (airways less than 2 mm in diameter and lacking cartilaginous support) at x40 and of lung parenchyma at x20 using a Leica ICC50 W microscope camera mounted to a Leica DM 500.46 From the total images, eight images were randomly selected for measurements. Epithelial ACE2, Furin and TMPRSS2 staining were measured as a percentage positive staining in the small airway epithelium. In addition, Type-2 pneumocytes, both positive and negative for ACE2, Furin and TMPRSS2 for type-2 pneumocytes, were counted per parenchymal tissue area in the alveolar epithelium. Similarly, alveolar macrophages staining positive and negative for ACE2, Furin and TMPRSS2 were counted per parenchymal tissue area. All counts and measurements were performed using brightfield microscopy and computer-assisted Image ProPlus V7.0 software (Media Cybernetics, USA).
Immunofluorescence Staining and Analysis of ACE2, Furin and TMPRSS2 in Primary Small Airway Epithelial Cells
Primary small airway epithelial cells (20×103 cells per chamber) were seeded into 8-chamber culture (Millicell EZ SLIDE 8-well, Merck, North Ryde BC, New South Wales, Australia) slides and incubated at 37°C in 5% CO2 for 24 hours to attain at least 80% confluence. The following day, cells were treated with cigarette smoke extract and appropriate vehicle control VC (1%DMSO) and further incubated for 24 hours. The cells were rinsed with PBS (cold) and fixed and permeabilised with 4% paraformaldehyde containing 0.1% Triton X-100 in PBS for 10 min at room temperature. After blocking with a mix of 10% goat and donkey serum in PBS for a half-hour, the cells were stained with mouse monoclonal anti-human ACE2 antibody (ab89111, AbCam, Melbourne, Victoria, Australia) 1:100 and rabbit polyclonal anti-human Furin (Thermofisher Cat#bs13228R, Bioss antibodies, VIC, Australia) 1:100 or TMPRSS2 (Thermofisher Cat#bs13228R, Bioss antibodies, VIC, Australia) 1:100, incubated overnight at 4°C. The cells were washed PBS thrice for 3 min each, before incubating them with Alexa 488-labelled goat anti-mouse (Alexa Fluor 488, Invitrogen, Scoresby, Victoria, Australia) and Alexa 594-labelled donkey anti-rabbit secondary antibody (Thermofisher Cat#R37119, Life Technologies, VIC, Australia) (1:500 each) at room temperature for 1 hour. Following three washes with PBS, DAPI (Invitrogen, Scoresby, Victoria, Australia) was added to visualise nuclear staining. The cells were examined, and images were taken using an Olympus FV1200 confocal laser scanning microscope (Olympus Life Science Europe GmbH, Hamburg, Germany). Alexa Fluor-488 images were captured under excitation: 470–495 nm, dichroic beam splitter 505 nm, and 510–650 nm emission. Alexa Fluor-594 images were captured under excitation: 530–550 nm, dichroic beam splitter 570 nm, and emission of 575–670 nm. The images were acquired at 40× objectives and taken at a speed of 2 µs/pixel. With the aid of ImageJ software, averaged corrected total cell fluorescence was calculated by measuring the integrated density of individual cells minus the integrated density of the background. Colocalisation of immunofluorescent staining and Pearson’s coefficients were calculated with ImageJ software by splitting colour channels of the images and running the JACoP plugin with conserved thresholds.
Statistical Analysis
Following normal distribution check, the analysis is represented as median and range, non-parametric (Kruskal–Wallis) analysis of variance with multiple comparisons using Dunn’s test. Further comparison with the Mann Whitney U-test was performed where appropriate. Linear regression and Pearson r’ were used for correlation analysis. All statistical analysis was done using PRISM V9.2 software (GraphPad, La Jolla, CA, USA), p < 0.05 was considered statistically significant.
Results
ACE2, Furin and TMPRSS Tissue Protein Expression and Localisation in Smokers, SAD and COPD Patients
Overall, a significant increase in the ACE2, Furin and TMPRSS2 expression was observed in the tissue from smokers and patients with COPD compared to normal controls (Figure 1). An increase in protein expression of ACE2, Furin, and TMPRSS was also notable in type 2 pneumocytes and alveolar macrophages in all the pathological groups compared to normal controls. Total type 2 pneumocytes and alveolar macrophages also increased in the pathological groups compared to normal controls. These proteins also negatively correlated with lung function parameters. Comparatively, similar changes were observed for primary small airway epithelial cells on exposure to cigarette smoke extract.
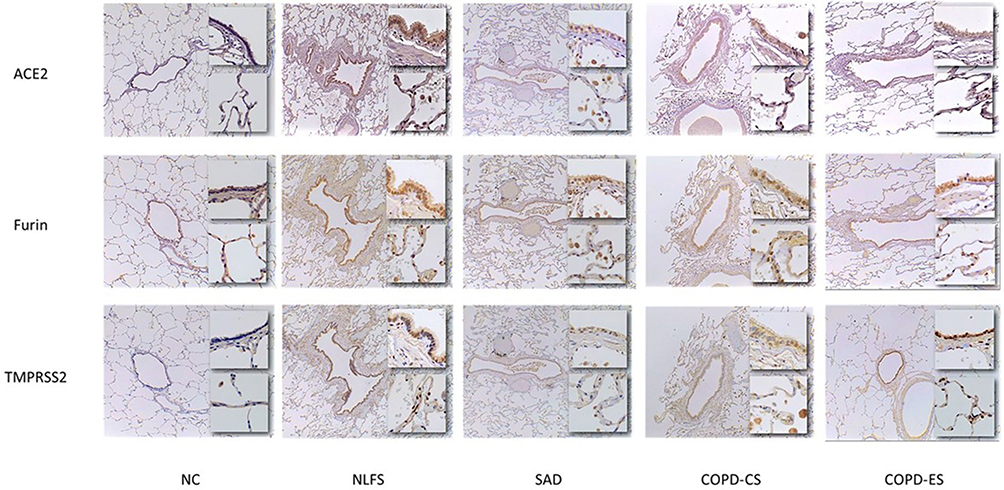 |
Figure 1 Micrographs of ACE2, Furin and TMPRSS2 protein expression in the small airways of smokers and COPD patients. The top inset images depict small airway epithelium taken at x40 magnification. The bottom inset images depict the parenchymal alveoli at x20 magnification. Abbreviations: NC, Normal control; NLFS, Normal Lung Function Smokers; SAD, Small Airways Disease; COPD-CS, Chronic Obstructive Pulmonary Disease – Current Smokers; COPD-ES; Chronic Obstructive Pulmonary Disease – Ex Smokers micrographs. |
Increase in Total Type 2 Pneumocytes and Macrophages in Parenchyma of Smokers and COPD Patients
Type 2 pneumocytes increased significantly across all the pathological groups compared to normal controls except SAD (Figure 2A). NLFS (p < 0.05), COPD-CS (p < 0.001), and COPD-ES (p < 0.05) had a significantly greater number of type 2 pneumocytes per parenchymal area compared with normal controls. In comparison, there was no significant difference in SAD patient total type 2 pneumocytes compared to normal controls (p = 0.08), which was probably a type 2 error. Similar changes were observed for alveolar macrophages except for COPD-ES compared to normal controls. NLFS (p < 0.01), SAD (p < 0.05), and COPD-CS (p < 0.0001) had a significantly greater total number of macrophages per parenchymal area compared to normal controls (Figure 2B). No significant difference between COPD-ES and NC (p = 0.46) for alveolar macrophages was observed. The highest significant increase in total alveolar macrophage numbers was seen in the COPD-CS group. A trend for smoking cessation exists here, with a statistically significant difference between the COPD-ES group and the COPD-CS group (p < 0.05), and essentially normalised when compared to normal controls (Figure 2B).
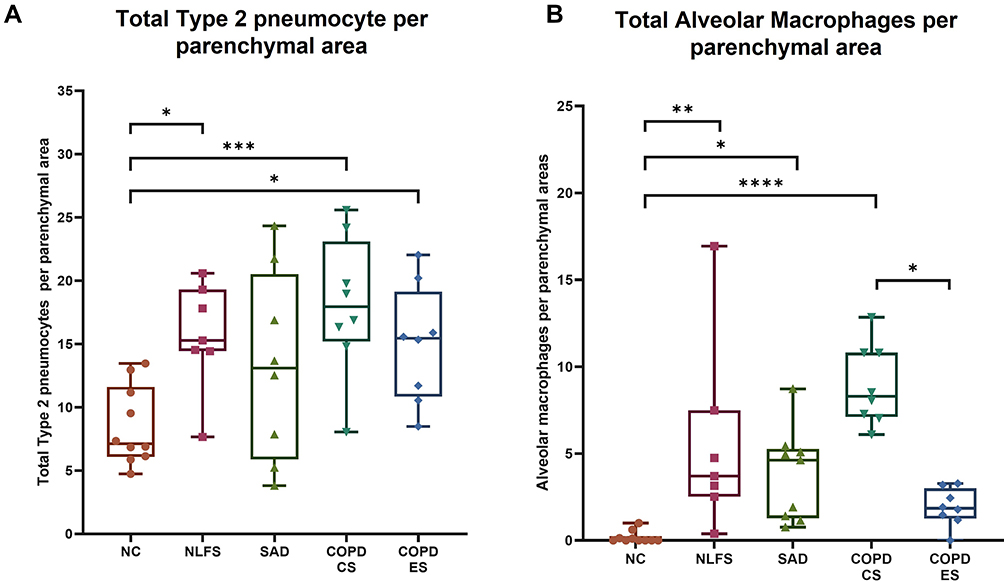 |
Figure 2 Comparison of total type 2 pneumocytes and alveolar macrophages in normal controls, normal lung function smokers, SAD, and COPD patients with normal controls. An increase in (A) the total number of type 2 pneumocytes per parenchymal area and (B) the total number of alveolar macrophages per parenchymal area in NLFS, SAD, COPD-CS, and COPD-ES compared to NC. Significance is indicated as: *p <0.05, **p< 0.01, ***p<0.001 and ****p<0.0001, the one-way ANOVA test was used for statistical analysis. |
Increased ACE2, Furin and TMPRSS2 Expression in the Small Airway Epithelium of Smokers, SAD, and COPD Patients
A significant increase in ACE2, Furin and TMPRSS2 protein expression was observed across the small airway epithelium for the pathological groups compared to normal controls (Figure 3A–C). Although the degree of such significance differed across the groups, the consistency in these vital SARS-CoV-2 adhesion and processing proteins suggested effects of both smoking and COPD. For ACE2 expression, compared to the COPD-CS group, the COPD-ES showed a decreasing trend, though this decline was not noticed for Furin and TMPRSS2 expression.
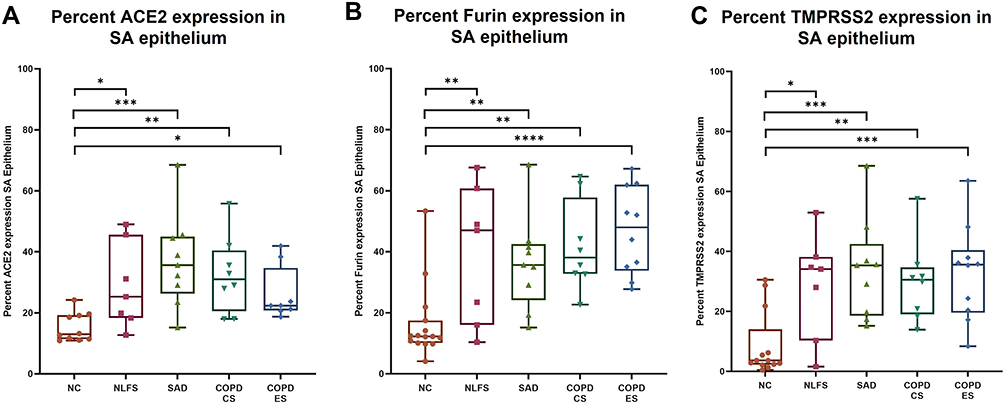 |
Figure 3 Percent small airway (SA) epithelial expression of ACE2, Furin and TMPRSS2 in normal controls, normal lung function smokers, SAD, and COPD patients. An increase in ACE2, Furin and TMPRSS2 expression was observed in all pathological groups NLFS, SAD, COPD-CS, and COPD-ES compared to NC for (A) ACE2, (B) Furin, and (C) TMPRSS2. Significance is indicated as: *p <0.05, **p< 0.01, ***p<0.001 and ****p<0.0001, the one-way ANOVA test was used for statistical analysis. |
Increased ACE2, Furin and TMPRSS2 Expression in Type 2 Pneumocytes and Alveolar Macrophages
Type 2 Pneumocytes
All the pathological groups showed increases in the percentage type 2 pneumocytes expressing ACE2, Furin and TMPRSS2 compared to NC (Figure 4). Notably, both the COPD cohorts, CS and ES, showed significant increases in type 2 pneumocytes, expressing ACE2, Furin and TMPRSS2. Further, in NLFS, the significant expression was seen with ACE2 (p < 0.05), TMPRSS2 (p < 0.05) was absent for Furin when compared to NC. In contrast, in SAD type 2 pneumocytes, we found significantly higher Furin (p < 0.01) upregulation than ACE2 (p < 0.05) and TMPRSS2 (p < 0.05). Further, in our intra-group type 2 pneumocytes analysis for the three proteins, we found that, in general, NCs had fewer positive protein-expressing type 2 pneumocytes than negative type 2 pneumocytes, with a very significant decline, especially in TMPRSS2 positive type 2 pneumocytes (p < 0.01). This changed in both COPD cohorts (p < 0.0001) and the SAD (p < 0.001), wherein ACE2, Furin and TMPRSS2 positive type 2 pneumocytes were significantly higher than their negative counterparts; however, in NLFS, these increases were insignificant. In Furin, this decline was significant across the groups when compared to the ACE2 and TMPRSS2. Overall, the fold change for each group showed a general positive trend and was greatest in the COPD cohorts.
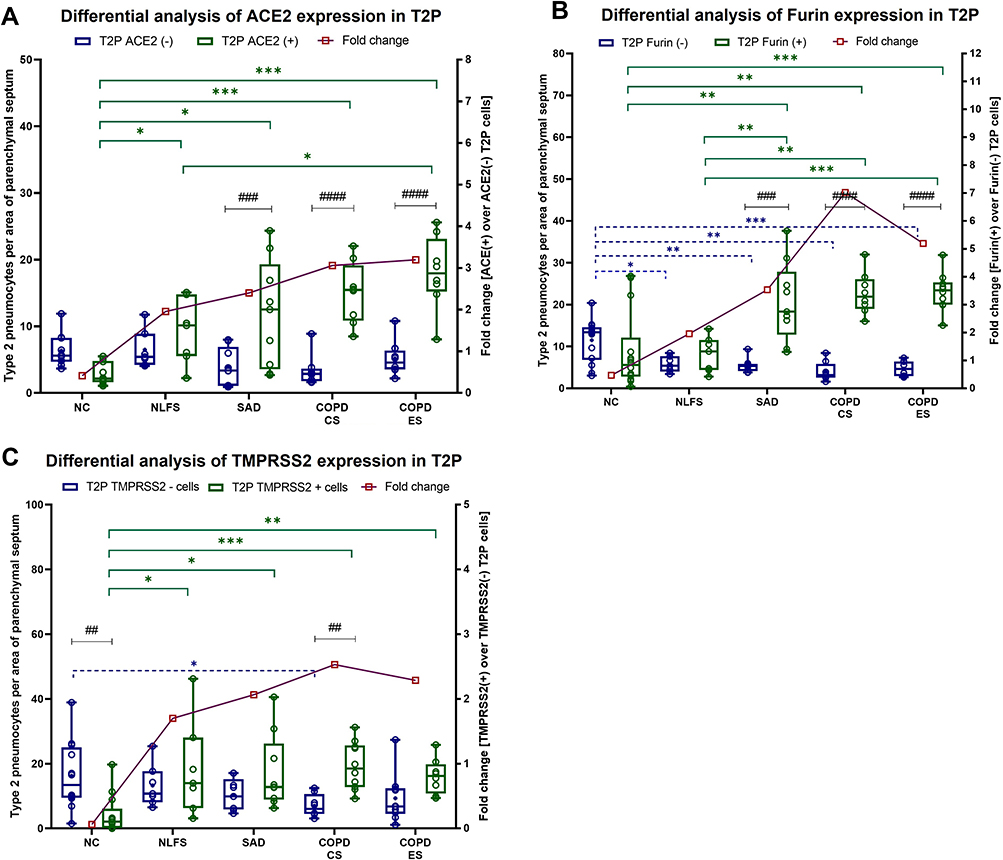 |
Figure 4 Differences in ACE2, Furin and TMPRSS2 positive and negative type 2 pneumocytes (T2P) in normal controls, normal lung function smokers, SAD, and COPD patients. (A) Differential inter and intragroup analysis of protein expression in type 2 pneumocytes as fold change for positive cells over negative cells in NC, NLFS, SAD, COPD-CS and COPD-ES for (A) ACE2, (B) Furin, and (C) TMPRSS2. Significance is indicated between intergroup *p <0.05, **p< 0.01, ***p<0.001 and intragroup ##p< 0.01, ###p<0.001 and ####p<0.0001 variability, the two-way ANOVA test was used for statistical analysis. |
Alveolar Macrophages
Similar to type 2 pneumocytes, we find a significant increase in alveolar macrophage (AMs) numbers expressing ACE2, Furin and TMPRSS2 across all pathological groups compared to NCs (Figure 5). Specifically, compared to NCs, ACE2 positive AMs showed significant increases in NLFS (p < 0.05), SAD (p < 0.01), COPD-CS (p < 0.01), and COPD-ES (p < 0.01) (Figure 5A). Furin, expression was more pronounced of the two markers, NLFS (p < 0.05), SAD (p < 0.001), COPD-CS (p < 0.0001), and COPD-ES (p < 0.01), while in TMPRSS2, except COPD-ES (p = 0.07), all other pathological cohort showed significant increases, NLFS (p < 0.01), SAD (p < 0.001), and COPD-CS (p < 0.01), than NCs. Notably, a significant downregulation of these proteins was observed in COPD-ES compared to SAD and COPD-CS, suggesting smoking cessation effects. Interestingly, unlike type 2 pneumocytes, in NCs, AMs positive for ACE2, Furin, and TMPRSS2 showed no significant difference from negative AMs. In NLFS groups, increased fold change was seen in ACE2, with significance in Furin (p < 0.01) and TMPRSS2 (p < 0.05) positive cells over that of the negative cells. Both COPD groups and the SAD cohort showed increased fold for the three proteins over their corresponding negative AMs, with the greatest significance seen in the COPD-CS. Among all pathological groups, fold differences in COPD-ES considerably declined across the three proteins.
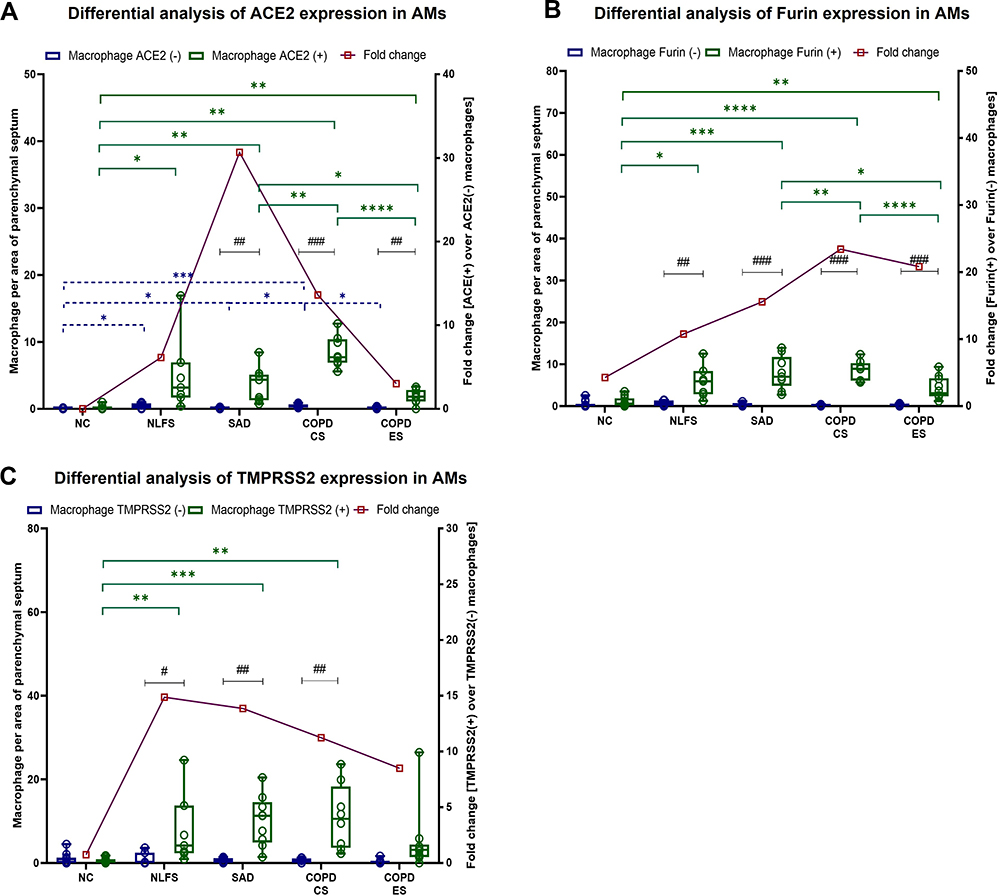 |
Figure 5 Differences in ACE2, Furin and TMPRSS2 positive and negative alveolar macrophages (AMs) in normal controls, normal lung function smokers, SAD, and COPD patients. (A) Differential inter and intra group analysis of protein expression in alveolar macrophages as fold change for positive cells over negative cells in NC, NLFS, SAD, COPD-CS and COPD-ES for (A) ACE2, (B) Furin, and (C) TMPRSS2. Significance is indicated as: *p <0.05, **p< 0.01, ***p<0.001 and ****p<0.0001 and intragroup #p <0.05, ##p< 0.01 and ###p<0.001 variability, the two-way ANOVA test was used for statistical analysis. |
Tissue Co-Expression of Furin and TMPRSS2 with ACE2 in Small Airway Epithelium, Type 2 Pneumocytes and Alveolar Macrophages
We further analysed the degree of co-expression of Furin and TMPRSS2 with ACE2 expression using Pearson r’ correlation coefficient (Figure 6). Interestingly, except for COPD-ES in the type 2 pneumocyte population, we found that in all the pathological groups Furin and TMPRSS2 showed positive Pearson r’ coefficient values with ACE2. In contrast, in NCs, both Furin and TMPRSS2 showed a negative correlation to ACE2 in SA epithelium and type 2 pneumocytes while showing minimal to no correlation in alveolar macrophages (AMs). Most notably, the significant positive correlation was observed in NLFS AMs in both Furin (r’=0.95; p < 0.001) and TMPRSS2 (r’=0.98; p < 0.001). Significance was also noticed in SAD TMPRSS2 levels (r’=0.78; p < 0.01) in SA epithelium and for Furin (r’=0.97; p < 0.001) in type 2 pneumocytes (Figure 6).
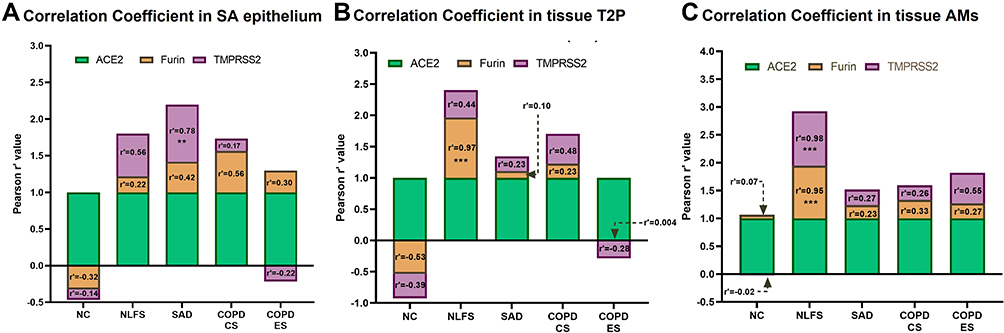 |
Figure 6 Correlation coefficient analysis for ACE2, Furin and TMPRSS2 in small airway epithelium, type 2 pneumocytes (T2P) and alveolar macrophages (AMs). Pearson r’ correlation analysis was performed to estimate the degree of co-expression of ACE2, Furin and TMPRSS2 in NLFS, SAD, COPD-CS and COPD-ES compared to NC in (A) small airway epithelium, (B) type 2 pneumocytes, and (C) alveolar macrophages. Significance is indicated as **p< 0.01 and ***p<0.001. |
ACE2, Furin, TMPRSS2 Expression and Correlation with Lung Function Parameters
Comparative correlation analysis across the four pathological groups was conducted with the data for ACE2, Furin and TMPRSS2 expression in SA epithelium, type 2 pneumocytes (T2Ps) and alveolar macrophages (AMs). Their association with physiological function such as lung function (FEV1/FVC %), small airway functionality (FEF25–75%) and gas exchange (DLCO %predicted) was determined (Table 2). In SA epithelium, negative correlation between Furin and TMPRSS2 expression with lung function parameters FEV1/FVC% [Furin (r’= −0.38; p < 0.05); TMPRSS2 (r’= −0.35; p < 0.05)] and FEF25–75% [Furin- (r’= −0.27; p = 0.06); TMPRSS2 (r’= −0.26; p = 0.07)] was observed, however no relation with ACE2 expression could be established. Also, no relationship with DLCO for the three makers was observed. In T2Ps, we found significant or near significant correlation for all three markers with the lung function parameters, for ACE2 [FEV1/FVC% (r’= −0.20; p = 0.1), and FEF25–75% (r’= −0.30; p = 0.053)], Furin [FEV1/FVC% (r’= −0.50; p < 0.01), and FEF25–75% (r’= −0.52; p < 0.001)] and TMPRSS2 [FEV1/FVC% (r’= −0.40; p < 0.01), and FEF25–75% (r’= −0.30; p < 0.05)]. As with SA epithelium, no noticeable relation between T2Ps and DLCO % predicted was observed. In contrast, AM expression for three markers showed significant or near significant correlation with DLCO % predicted, for ACE2 (r’= −0.40; p = 0.05), Furin (r’= −0.23; p = 0.09), and TMPRSS2 (r’= −0.27; p = 0.06). In addition, ACE2 expressing AMs were also observed to significantly affect the small airway functionality FEF25–75% (r’= −0.30; p < 0.05) and near significance was seen for lung function parameter, FEV1/FVC% (r’= −0.22; p = 0.1). We observed no significant relationship for AMs expressing Furin and TMPRSS2 with lung function parameters.
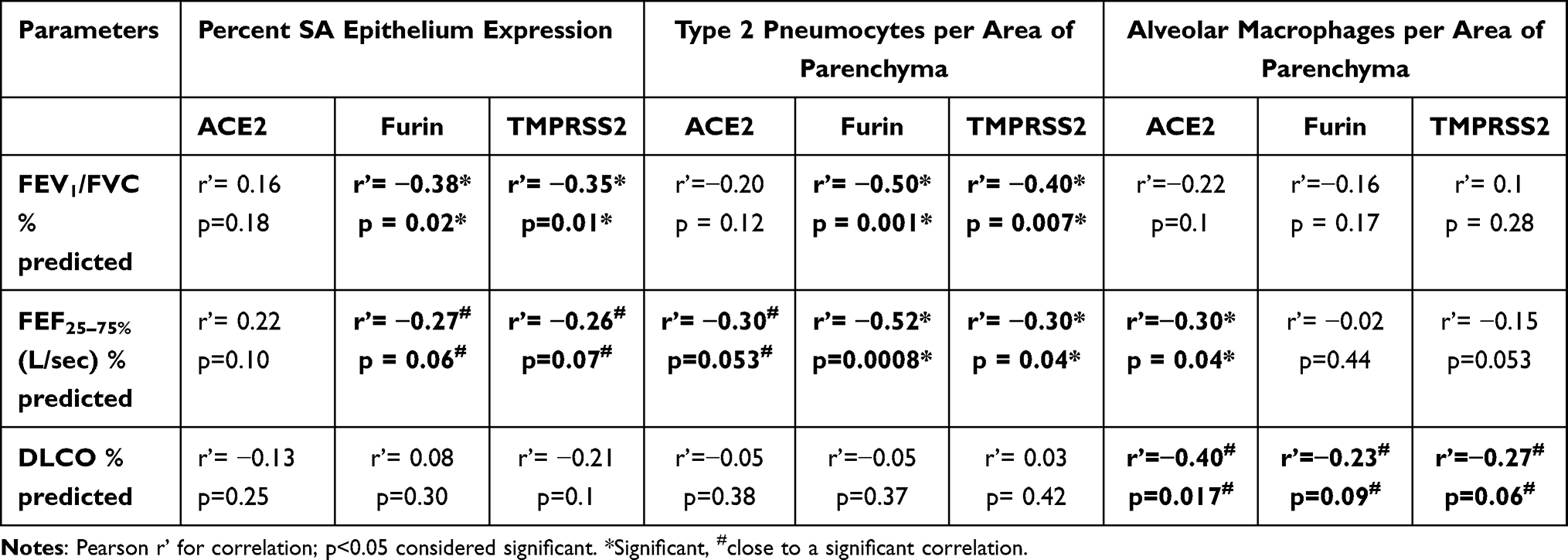 |
Table 2 Correlations of ACE2, Furin and TMPRSS2 with Physiological Lung Parameters |
Increased Co-Localisation of Furin and TMPRSS2 with ACE2 Was Observed in Primary Small Airway Epithelial Cells Treated with Cigarette Smoke Extract in vitro
Following our observation with immunohistochemical analysis of small airway tissue, we demonstrated these findings using in vitro primary small airway epithelial cells. We observed increased Furin, TMPRSS2, and ACE2 expression of primary small airway epithelial cells when treated with 1% cigarette smoke extract over vehicle control (Figure 7A). Furin and ACE2 both showed increased peripheral expression and co-expressed in similar areas within the cell membrane. Although similar ACE2-TMPRSS2 co-expression was observed, TMPRSS2 expression was found to be more within the cytoplasm than in the plasma membrane (Figure 7A). Corrected total cell fluorescence significantly increased for ACE-2 (p < 0.0001), Furin (p < 0.01), and TMPRSS2 (p < 0.0001) following treatment with 1% cigarette smoke extract compared to vehicle controls (Figure 7B). No significant difference between cigarette smoke extract-treated and VC was seen for co-localisations of Furin and ACE2 or TMPRSS2 and ACE2 (Figure 7C).
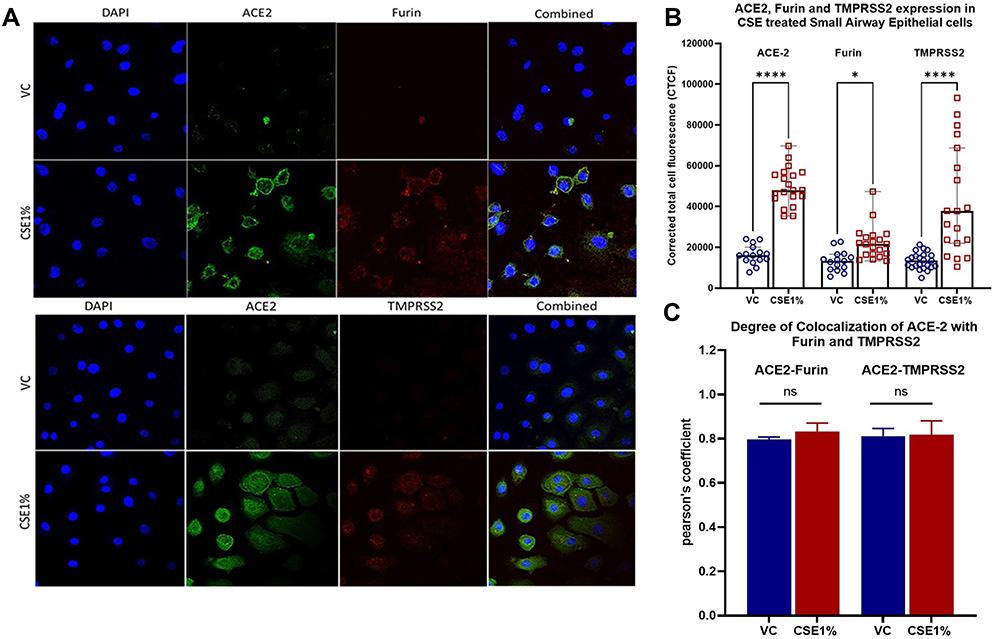 |
Figure 7 Immunofluorescence of proteins (ACE2, Furin, and TMPRSS2) associated with SARS-CoV-2 adhesion and infection. ACE2, Furin and TMPRSS2 protein expression of small airway epithelial cells following cigarette smoke exposure. Small airway epithelial cells were exposed to 1% cigarette smoke extract (CSE) or appropriate vehicle control (VC) (1% DMSO) and incubated for 24 hours. (A) Protein expression of ACE2 (green), Furin (red, top) and TMPRSS2 (red, bottom) with DAPI (blue) was assessed through immunofluorescence staining and confocal imaging. Images at 40×. (B) Cigarette smoke extract-treated cells had significantly greater corrected total cell fluorescence of ACE-2, Furin, and TMPRSS2. (C) Co-localisation does not vary between cigarette smoke extract-treated cells and vehicle controls (VC). Corrected total cell fluorescence and Pearson’s coefficient were calculated using ImageJ. Results are represented as medians ± range. Ns= non-significant; *p<0.05; ****p<0.0001. |
Discussion
This study demonstrates links between smoking, COPD and an increase in multiple biomarkers associated with SARS-CoV-2 viral adhesion and cellular entry. This study expands on our previous work, which first identified this link.29 To our knowledge, this study is the first to demonstrate a quantitative increase in expression of ACE2, Furin and TMPRSS2 in type 2 pneumocytes and alveolar macrophages in the small airways of NLFS and SAD and COPD patients. Further, we also identified an increase in both Furin and TMPRSS2 in the small airway epithelium, type 2 pneumocytes and alveolar macrophages, which corresponded with the areas of increased ACE2 expression. Total numbers of type 2 pneumocytes and alveolar macrophages per parenchymal area were increased in smokers and COPD patients, along with the percentage of ACE2, Furin and TMPRSS2 positive cells in all groups compared to normal controls. An increase in type 2 pneumocytes also possibly suggests active interstitial pathology like idiopathic pulmonary fibrosis (IPF).47–49 Interestingly, COPD-ES tissues showed a significant reduction in the total number of alveolar macrophages and the number of ACE2, Furin and TMPRSS2 positive alveolar macrophages, indicating a smoking cessation effect. Our findings are further strengthened with in-vitro experiments in which healthy primary small airway epithelial cells were observed to have increased fluorescent intensity of ACE2, Furin, and TMPRSS2 following cigarette smoke extract stimulation. An observed co-localisation of ACE2/Furin and ACE2/TMPRSS2 was unchanged between cigarette smoke extract 1% treated cells and vehicle controls. This unchanged co-localisation of these proteins highlights their likely complex synergy which is utilised for adhesion and engulfment of SARS-CoV-2.
The role of ACE2 as an adhesion site for coronaviruses was first identified in connection with the 2003 outbreak of SARS (SARS-CoV-1),50 but the emergence of SARS-CoV-2 has once again drawn the attention of the scientific community to the human ACE2 protein. Increased small airway epithelium expression of ACE2 has been previously reported, however, our finding shows that smoking, independent of respiratory disease, provides sufficient stimulus to elevate respiratory epithelial ACE2 expression significantly. This suggests an inherent risk of a more severe COVID-19 infection for smokers, irrespective of lung function. The increased expression of ACE2 was further exacerbated by the development of small airway disease and COPD. The binding affinity between SARS-CoV-2 and ACE2 is significantly higher, which is at the core of the strong infectious nature of this virus. The increased expression of Furin and TMPRSS2 in smokers is further evidence of the greater mechanism of possible infection for smokers and COPD patients, as viral adhesion and cell entry is facilitated. Our correlation coefficient analysis between ACE2, Furin and TMPRSS2 further indicates the degree of co-expression between these proteins, especially under pathological conditions. Interestingly, we find that smokers and patients with small airway dysfunction have greater co-dependency of these proteins than normal healthy controls, suggesting their vulnerability to early disease onset to a SARS infection. It is interesting to note that, in this context, the downregulation of cofactors Furin and TMRSS2 in normal controls compared to ACE2 suggests that ACE2 alone is not sufficient, but smokers, patients with COPD or with small airway disease, the three proteins are upregulated, thus enhancing the chance for SARS-CoV-2 infections, and further exacerbating COVID-19.
SARS-CoV-2 and other similar enveloped viruses use the host cell endocytic pathway for hijacked cellular entry before fusing it with lysosomal membranes.51 We have recently shown a concurrent upregulation of both endocytic machinery proteins and ACE2 with similar localisation in normal lung function smokers and COPD (current- and ex-smokers) patients compared with normal human control tissue, promoting the theory of endocytic facilitation of viral entry for SARS-CoV-2.52 This study further amplifies the increased susceptibility and risk for this demographic in relation to the COVID-19 pandemic.
Correlations with clinical data showed a significant negative relationship between ACE2, Furin and TMPRSS2 positive alveolar macrophages and DLCO % predicted. In addition, a strong correlation between ACE2 positive alveolar macrophage and small airway capacity FEF 25–75% was evident here. Furin and TMPRSS2 expression in small airway epithelium and type 2 pneumocytes interestingly showed a greater propensity for airway dysfunction than ACE2. This indicates that a link between the degree of small airway epithelium, type 2 pneumocytes and macrophages expressing ACE2, Furin and TMPRSS2 and poor lung function outcomes in smokers and COPD patients could indeed drive more severe COVID-19.
Studies have shown a significant increase in the number of macrophages in the small airways and lung parenchyma in patients with COPD compared to normal lung functions smokers, which is similarly observed in our study.53,54 This increased macrophage population has been established as part of the pathogenic mechanisms in COPD. Supported clinically, studies have demonstrated a positive correlation between increasing alveolar macrophage numbers and COPD severity.55 One study found that patients with COVID-19 related severe respiratory failure displayed features of macrophage activation syndrome.56 It has been suggested that alveolar macrophages play the role of the trojan horse in COVID-19, providing a mobile viral anchoring point specifically within the alveolar spaces and pulmonary parenchyma.35,57
We have previously shown an increase in the number of pro-inflammatory and cytotoxic M1 macrophages in the small airway wall of NLFS and COPD-CS, with a corresponding reduction in anti-inflammatory M2 macrophages, with phenotype reverting towards normal levels in COPD-ES.45 Interestingly, alveolar macrophage populations from bronchoalveolar lavage showed the inverse, with increased M2 populations seen in NLFS and COPD cohorts. Furthermore, the polarisation and increase in M1 macrophages in the small airways correlate directly with smoking pack-years.45 Correlating these earlier findings with those in this study suggests that COPD sees a pathological increase in small airway pro-inflammatory M1 macrophages, which would help drive acute respiratory disease syndrome (ARDS) commonly presenting in COVID-19 patients.58 Furthermore, an increased alveolar macrophage population that predominantly expresses ACE2 protein will facilitate inhaled viral binding and cellular entry. Currently, there is a limited understanding of the connection between alveolar macrophages and COVID-19; other than that, alveolar macrophages are a targeted binding site for SARS-CoV-2.57 However, recent studies have suggested that alveolar macrophages play a crucial role in the cytokine storm seen in severe COVID-19.59 On top of predominantly greater neutrophil to lymphocyte ratios in severe cases of COVID-19,60 hyperinflammation and highly elevated concentrations of cytokines and chemokines drive the severity of disease,61 of which macrophages constitute a significant source.59,62 IL-6 and the NF-κB pathway is heavily implicated and highly correlated with lethal complications from COVID-19,56,63 and given the role of IL-6 in chronic respiratory diseases,64,65 smoking and COPD could initiate and exacerbate the severity of COVID-19 cytokine storm.62
It has recently been suggested that the smoking-induced increase in ACE2 expression is nicotine-dependent and mediated by α7-subtype nicotinic receptors (α7-nAChR).66 Therefore, we have explored other nicotine delivery systems such as electronic cigarettes and the effect that their use may have on ACE2 expression and susceptibility to SARS-CoV-2 infection.67 In our preliminary studies, ACE2 expression was increased in large and small airway epithelial cells following exposure to electronic cigarette aerosol condensate and cigarette smoke extract with predominant nicotine dependence.68
When considering the findings from this study, it should be noted that patient tissue samples came from lung resection involving non-small cell lung cancers, but to mitigate the risk of interference with results, the tissue used in this analysis was significantly distant from any tumour. Altered ACE2, Furin, and TMPRSS2 expression in lung cancer has been reported69 but should have little to no impact on our measurements. Another limitation of this study is the low but precious number of samples from subjects available, contributing to the clinical groups. However, our robust statistics show that these changes will only further exaggerate with higher numbers. Therefore, we believe that the results presented are statistically robust. The variability in age across the clinical groups is also a minor and unavoidable limitation, with the potential correlation of results with age not being verified.70 Further work is required to confirm that increased ACE2 expression correlates with increased infectivity and severity of COVID-19 clinical outcomes.
Since the emergence of COVID-19, much work has been done to identify possible links between the virus and potential risk populations.71 It has been established that, like SAR-COV-1, the causative agent of SARS, SAR-COV-2 enters cells via the ACE2 receptor.72 Work conducted by our group and others has shown an increase in ACE2 and that this viral spike protein-binding site is linked to smoking and COPD.29,30,43 Increased small and large airway epithelial ACE2 expression has been observed in smokers and patients with COPD.73 Smoking has been demonstrated to upregulate the expression of ACE2 both in mouse models and in cross-sectional studies;74 similar reports have been published about the risk of vaping.67,68 This novel study provides further evidence supporting the observations of increased ACE2 expression in small airway epithelium, alveolar macrophages, and type 2 pneumocytes of smokers and patients with COPD.
Conclusion
This is the first comprehensive multigroup study to show ACE2, Furin and TMPRSS2 expression in the small airway epithelium, type 2 pneumocytes and alveolar macrophages. We also believe this is the first report on a significant increase in type 2 pneumocytes in smokers and patients with COPD, suggesting active interstitial pathology similar to IPF. This increase indicates that smokers and patients with COPD could be at a higher risk of developing post-COVID-19 interstitial pulmonary fibrosis or fibrosis in general. We have provided links between increased expression of ACE2 due to smoking, SAD, and COPD in both type 2 pneumocytes and macrophages, correlating this with previous information linking possible pathogenic phenotypes between COPD and COVID-19. We show that along with increased ACE2 expression, the cofactors Furin and TMPRSS2 also increase in smokers, SAD, COPD, and cigarette smoke extract treated small airway primary epithelial cells. The overexpression of these proteins links smokers and COPD patients, increased susceptibility to the virus, and the potential for severe manifestations of COVID-19. Mounting evidence supports that smoking and vaping are avoidable risk factors during the COVID-19 pandemic.75–77
Ethics
This study was conducted in accordance with the Declaration of Helsinki.
Funding
Grants were received from the Clifford Craig Foundation, Launceston General Hospital and the Rebecca L. Cooper Medical Research Foundation.
Disclosure
Dr Josie Larby reports personal fees from GSK outside the submitted work. Dr Sukhwinder Singh Sohal reports personal fees from Chiesi outside the submitted work. The aforementioned authors report no other potential conflicts of interest in this work and all the other authors report no conflicts of interest in this work.
References
1. World Health Organization. COVID-19 Weekly epidemiological update - 24 November 2020; 2020. Available from: https://www.who.int/publications/m/item/weekly-epidemiological-update---24-november-2020. Accessed March 3, 2021.
2. World Health Organisation. Coronavirus; 2020. Available from: https://www.who.int/health-topics/coronavirus.
3. Asrani P, Hasan GM, Sohal SS, Hassan MI. Molecular basis of pathogenesis of coronaviruses: a comparative genomics approach to planetary health to prevent zoonotic outbreaks in the 21st century. Omics. 2020;24(11):634–644. doi:10.1089/omi.2020.0131
4. Asrani P, Hussain A, Nasreen K, et al. Guidelines and safety considerations in the laboratory diagnosis of SARS-CoV-2 infection: a prerequisite study for health professionals. Risk Manag Healthc Policy. 2021;14:379–389. doi:10.2147/RMHP.S284473
5. Rodriguez-Morales AJ, Cardona-Ospina JA, Gutiérrez-Ocampo E, et al. Clinical, laboratory and imaging features of COVID-19: a systematic review and meta-analysis. Travel Med Infect Dis. 2020;34:101623. doi:10.1016/j.tmaid.2020.101623
6. Asrani P, Eapen MS, Chia C, et al. Diagnostic approaches in COVID-19: clinical updates. Expert Rev Respir Med. 2021;15(2):197–212. doi:10.1080/17476348.2021.1823833
7. Kumari P, Singh A, Ngasainao MR, et al. Potential diagnostics and therapeutic approaches in COVID-19. Clin Chim Acta. 2020;510:488–497. doi:10.1016/j.cca.2020.08.013
8. Thevarajan I, Buising K, Cowie B. Clinical presentation and management of COVID-19. Med J Aust. 2020;213(3):134–139. doi:10.5694/mja2.50698
9. Wei YY, Wang RR, Zhang DW, et al. Risk factors for severe COVID-19: evidence from 167 hospitalized patients in Anhui, China. J Infect. 2020;81(1):e89–e92. doi:10.1016/j.jinf.2020.04.010
10. World Health Organisation. Chronic obstructive pulmonary disease (COPD); 2020. Available from: https://www.who.int/respiratory/copd/en/.
11. Asrani P, Eapen MS, Hassan MI, Sohal SS. Implications of the second wave of COVID-19 in India. Lancet Respir Med. 2021;9(9):e93–e94. doi:10.1016/S2213-2600(21)00312-X
12. Bourdrel T, Annesi-Maesano I, Alahmad B, Maesano CN, Bind MA. The impact of outdoor air pollution on COVID-19: a review of evidence from in vitro, animal, and human studies. Eur Respir Rev. 2021;30(159):159. doi:10.1183/16000617.0242-2020
13. Wilkinson TMA. Immune checkpoints in chronic obstructive pulmonary disease. Eur Respir Rev. 2017;26(144):144. doi:10.1183/16000617.0045-2017
14. Marsland BJ, Konigshoff M, Saglani S, Eickelberg O. Immune system dysregulation in chronic lung disease. Eur Respir J. 2011;38(3):500–501. doi:10.1183/09031936.00103211
15. Atto B, Eapen MS, Sharma P, et al. New therapeutic targets for the prevention of infectious acute exacerbations of COPD: role of epithelial adhesion molecules and inflammatory pathways. Clin Sci (Lond). 2019;133(14):1663–1703. doi:10.1042/CS20181009
16. van Zyl-smit RN, Richards G, Leone FT. Tobacco smoking and COVID-19 infection. Lancet Respir Med. 2020;8(7):664–665. doi:10.1016/S2213-2600(20)30239-3
17. World Health Organization. WHO statement: tobacco use and COVID-19; 2020. Available from: https://www.who.int/news/item/11-05-2020-who-statement-tobacco-use-and-covid-19.
18. Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91(2):142–149. doi:10.1177/0022034511421200
19. Huttunen R, Heikkinen T, Syrjanen J. Smoking and the outcome of infection. J Intern Med. 2011;269(3):258–269. doi:10.1111/j.1365-2796.2010.02332.x
20. Sohal SS, Eapen MS, Naidu VGM, Sharma P. IQOS exposure impairs human airway cell homeostasis: direct comparison with traditional cigarette and e-cigarette. ERJ Open Res. 2019;5(1):00159–2018. doi:10.1183/23120541.00159-2018
21. Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164(20):2206–2216. doi:10.1001/archinte.164.20.2206
22. Grigg J, Walters H, Sohal SS, et al. Cigarette smoke and platelet-activating factor receptor dependent adhesion of Streptococcus pneumoniae to lower airway cells. Thorax. 2012;67(10):908–913. doi:10.1136/thoraxjnl-2011-200835
23. Eapen MS, Sharma P, Sohal SS. Mitochondrial dysfunction in macrophages: a key to defective bacterial phagocytosis in COPD. Eur Respir J. 2019;54(4):1901641. doi:10.1183/13993003.01641-2019
24. Eapen MS, Sharma P, Moodley YP, Hansbro PM, Sohal SS. Dysfunctional immunity and microbial adhesion molecules in smoking-induced pneumonia. Am J Respir Crit Care Med. 2019;199(2):250–251. doi:10.1164/rccm.201808-1553LE
25. Eapen MS, Sohal SS. Understanding novel mechanisms of microbial pathogenesis in chronic lung disease: implications for new therapeutic targets. Clin Sci. 2018;132(3):375–379. doi:10.1042/CS20171261
26. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi:10.1126/science.1116480
27. Xu X, Chen P, Wang J, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63(3):457–460. doi:10.1007/s11427-020-1637-5
28. Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi:10.1126/science.abb2507
29. Brake SJ, Barnsley K, Lu W, McAlinden KD, Eapen MS, Sohal SS. Smoking upregulates angiotensin-converting enzyme-2 receptor: a potential adhesion site for novel coronavirus SARS-CoV-2 (Covid-19). J Clin Med. 2020;9(3):841. doi:10.3390/jcm9030841
30. Leung JM, Yang CX, Tam A, et al. ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur Respir J. 2020;55(5):2000688. doi:10.1183/13993003.00688-2020
31. Saheb Sharif-Askari N, Saheb Sharif-Askari F, Alabed M, et al. Airways expression of SARS-CoV-2 receptor, ACE2, and TMPRSS2 is lower in children than adults and increases with smoking and COPD. Mol Ther Methods Clin Dev. 2020;18:1–6. doi:10.1016/j.omtm.2020.05.013
32. Mossel EC, Wang J, Jeffers S, et al. SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology. 2008;372(1):127–135. doi:10.1016/j.virol.2007.09.045
33. Rockx B, Kuiken T, Herfst S, et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science. 2020;368(6494):1012–1015. doi:10.1126/science.abb7314
34. Mason RJ. Pathogenesis of COVID-19 from a cell biology perspective. Eur Respir J. 2020;55(4):2000607. doi:10.1183/13993003.00607-2020
35. Park MD. Macrophages: a Trojan horse in COVID-19? Nat Rev Immunol. 2020;20(6):351. doi:10.1038/s41577-020-0317-2
36. Kadkhoda K, Rosenberg HF. COVID-19: an immunopathological view. mSphere. 2020;5(2). doi:10.1128/mSphere.00344-20
37. Kapellos TS, Bassler K, Aschenbrenner AC, Fujii W, Schultze JL. Dysregulated functions of lung macrophage populations in COPD. J Immunol Res. 2018;2018:2349045. doi:10.1155/2018/2349045
38. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. doi:10.1016/j.antiviral.2020.104742
39. Ming Y, Qiang L. Involvement of spike protein, furin, and ACE2 in SARS-CoV-2-related cardiovascular complications. SN Compr Clin Med. 2020:1–6. doi:10.1007/s42399-020-00400-2
40. Liu PP, Blet A, Smyth D, Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation. 2020;142(1):68–78. doi:10.1161/CIRCULATIONAHA.120.047549
41. He WT, Ji X, He W, et al. Genomic epidemiology, evolution, and transmission dynamics of porcine deltacoronavirus. Mol Biol Evol. 2020;37(9):2641–2654. doi:10.1093/molbev/msaa117
42. Mollica V, Rizzo A, Massari F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Fut Oncol. 2020;16(27):2029–2033. doi:10.2217/fon-2020-0571
43. Cai G, Bosse Y, Xiao F, Kheradmand F, Amos CI. Tobacco smoking increases the lung gene expression of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med. 2020;201(12):1557–1559. doi:10.1164/rccm.202003-0693LE
44. Carsana L, Sonzogni A, Nasr A, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. 2020;20(10):1135–1140. doi:10.1016/S1473-3099(20)30434-5
45. Eapen MS, Hansbro PM, McAlinden K, et al. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci Rep. 2017;7(1):13392. doi:10.1038/s41598-017-13888-x
46. Eapen MS, Lu W, Hackett TL, et al. Increased myofibroblasts in the small airways, and relationship to remodelling and functional changes in smokers and COPD patients: potential role of epithelial-mesenchymal transition. ERJ Open Res. 2021;7(2):00876–2020. doi:10.1183/23120541.00876-2020
47. Liu G, Philp AM, Corte T, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther. 2021;225:107839. doi:10.1016/j.pharmthera.2021.107839
48. Eapen MS, Lu W, Gaikwad AV, et al. Endothelial to mesenchymal transition: a precursor to post-COVID-19 interstitial pulmonary fibrosis and vascular obliteration? Eur Respir J. 2020;56(4):2003167. doi:10.1183/13993003.03167-2020
49. Aghaei M, Dastghaib S, Aftabi S, et al. The ER Stress/UPR axis in Chronic Obstructive Pulmonary Disease and idiopathic pulmonary fibrosis. Life. 2020;11(1). doi:10.3390/life11010001.
50. Jia HP, Look DC, Shi L, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. 2005;79(23):14614–14621. doi:10.1128/JVI.79.23.14614-14621.2005
51. Zheng Y, Shang J, Yang Y, et al. Lysosomal proteases are a determinant of coronavirus tropism. J Virol. 2018;92(24). doi:10.1128/JVI.01504-18.
52. Eapen MS, Lu W, Hackett TL, et al. Dysregulation of endocytic machinery and ACE2 in small airways of smokers and COPD patients can augment their susceptibility to SARS-CoV-2 (COVID-19) infections. Am J Physiol Lung Cell Mol Physiol. 2021;320(1):L158–L63. doi:10.1152/ajplung.00437.2020
53. Vlahos R, Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. 2014;5:435. doi:10.3389/fimmu.2014.00435
54. Retamales I, Elliott WM, Meshi B, et al. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med. 2001;164(3):469–473. doi:10.1164/ajrccm.164.3.2007149
55. Di Stefano A, Capelli A, Lusuardi M, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158(4):1277–1285. doi:10.1164/ajrccm.158.4.9802078
56. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992–1000 e3. doi:10.1016/j.chom.2020.04.009
57. Abassi Z, Knaney Y, Karram T, Heyman SN. The lung macrophage in SARS-CoV-2 infection: a friend or a foe? Front Immunol. 2020;11:1312. doi:10.3389/fimmu.2020.01312
58. Wark PAB, Pathinayake PS, Eapen MS, Sohal SS. Asthma, COPD and SARS-CoV-2 infection (COVID-19): potential mechanistic insights. Eur Respir J. 2021;58(2):2100920. doi:10.1183/13993003.00920-2021
59. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; What we know so far. Front Immunol. 2020;11:1446. doi:10.3389/fimmu.2020.01446
60. Sun S, Cai X, Wang H, et al. Abnormalities of peripheral blood system in patients with COVID-19 in Wenzhou, China. Clin Chim Acta. 2020;507:174–180. doi:10.1016/j.cca.2020.04.024
61. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. doi:10.1016/S0140-6736(20)30628-0
62. Hojyo S, Uchida M, Tanaka K, et al. How COVID-19 induces cytokine storm with high mortality. Inflamm Regen. 2020;40(1):37. doi:10.1186/s41232-020-00146-3
63. DeDiego ML, Nieto-Torres JL, Regla-Nava JA, et al. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J Virol. 2014;88(2):913–924. doi:10.1128/JVI.02576-13
64. Song W, Zhao J, Li Z. Interleukin-6 in bronchoalveolar lavage fluid from patients with COPD. Chin Med J. 2001;114(11):1140–1142.
65. Dawson RE, Jenkins BJ, Saad MI. IL-6 family cytokines in respiratory health and disease. Cytokine. 2021;143:155520. doi:10.1016/j.cyto.2021.155520
66. Russo P, Bonassi S, Giacconi R, Malavolta M, Tomino C, Maggi F. COVID-19 and smoking: is nicotine the hidden link? Eur Respir J. 2020;55(6):2001116. doi:10.1183/13993003.01116-2020
67. McAlinden KD, Eapen MS, Lu W, Chia C, Haug G, Sohal SS. COVID-19 and vaping: risk for increased susceptibility to SARS-CoV-2 infection? Eur Respir J. 2020;56(1):2001645. doi:10.1183/13993003.01645-2020
68. McAlinden KD, Lu W, Ferdowsi PV, et al. Electronic cigarette aerosol is cytotoxic and increases ACE2 expression on human airway epithelial cells: implications for SARS-CoV-2 (COVID-19). J Clin Med. 2021;10(5):1028. doi:10.3390/jcm10051028
69. Samad A, Jafar T, Rafi JH. Identification of angiotensin-converting enzyme 2 (ACE2) protein as the potential biomarker in SARS-CoV-2 infection-related lung cancer using computational analyses. Genomics. 2020;112(6):4912–4923. doi:10.1016/j.ygeno.2020.09.002
70. Wark PAB, Pathinayake PS, Kaiko G, et al. ACE2 expression is elevated in airway epithelial cells from older and male healthy individuals but reduced in asthma. Respirology. 2021;26(5):442–451. doi:10.1111/resp.14003
71. Haug G, Eapen MS, Sohal SS. Renin-angiotensin-aldosterone system inhibitors in Covid-19. N Engl J Med. 2020;382(24):e92.
72. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80 e8. doi:10.1016/j.cell.2020.02.052
73. Jacobs M, Van Eeckhoutte HP, Wijnant SRA, et al. Increased expression of ACE2, the SARS-CoV-2 entry receptor, in alveolar and bronchial epithelium of smokers and COPD subjects. Eur Respir J. 2020;56(2):2002378. doi:10.1183/13993003.02378-2020
74. Smith JC, Sausville EL, Girish V, et al. Cigarette smoke exposure and inflammatory signaling increase the expression of the SARS-CoV-2 receptor ACE2 in the respiratory tract. Dev Cell. 2020;53(5):514–29.e3. doi:10.1016/j.devcel.2020.05.012
75. The rise of electronic nicotine delivery systems and the emergence of electronic-cigarette-driven disease. McAlinden KD, Eapen MS, Lu W, Sharma P, Sohal SS. Am J Physiol Lung Cell Mol Physiol. 2020;319(4):L585–L595. doi:10.1152/ajplung.00160.2020
76. Electronic cigarettes: Modern instruments for toxic lung delivery and posing risk for the development of chronic disease. McAlinden KD, Lu W, Eapen MS, Sohal SS. Int J Biochem Cell Biol. 2021;137:106039. doi:10.1016/j.biocel.2021.106039
77. Cochrane review update leaves big questions unanswered regarding vaping: implications for medical practitioners. McAlinden KD, Barnsley K, Weber HC, Haug G, Chia C, Eapen MS, Sohal SS. Eur Respir J. 2021;57(5):2100022. doi:10.1183/13993003.00022-2021
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.