Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 8
Saroglitazar for the treatment of hypertriglyceridemia in patients with type 2 diabetes: current evidence
Authors Sosale A ![]() , Saboo B, Sosale B
, Saboo B, Sosale B
Received 22 July 2014
Accepted for publication 19 August 2014
Published 15 April 2015 Volume 2015:8 Pages 189—196
DOI https://doi.org/10.2147/DMSO.S49592
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Aravind Sosale,1 Banshi Saboo,2 Bhavana Sosale1
1Diacon Hospital, Bangalore, 2Dia Care (Diabetes Care and Hormone Clinic), Ahmedabad, India
Abstract: Diabetes mellitus (DM) is one of the most dreaded metabolic disorders in the world today. It is the leading cause of morbidity and mortality, and plays a cardinal role in quality of life and health economics. DM is associated with a high prevalence of microvascular and macrovascular complications. DM is a very important cardiovascular (CV) risk factor. Cardiovascular disease (CVD) has been implicated as the prime cause of mortality and morbidity in patients with DM. Hence, treatment of DM goes beyond glycemic control, and demands a multidisciplinary approach that comprehensively targets risk factors inherent in CV events. Lipid abnormalities are undoubtedly common in patients with DM, and they contribute to an increased risk of CVD. A high-risk lipid profile, termed atherogenic dyslipidemia of diabetes (ADD), is known to occur in patients with DM. The use of lipid-lowering agents, a quintessential part of the multifactorial risk factor approach, is a crucial intervention to minimize diabetes-related complications. In this article, we discuss the role of peroxisome proliferator activator receptor (PPAR) alpha/gamma (α/γ) agonist, saroglitazar, in the management of ADD. While statins are irrefutably the first line of drugs for dyslipidemia management in patients with residual CV risk while on a statin, PPAR α/γ agonists have been found to be of substantial benefit. Data from the PRESS I–VI clinical trials testify to the fact that saroglitazar and fibrates have similar efficacy in reducing triglycerides and improving high-density lipoprotein. The ancillary benefit of improved glycemic control, without the weight gain of PPAR γ agonists, is an added advantage. Reduction in ADD, improved glycemic control, efficacy at par with fibrates, and an acceptable safety profile form the grounds on which this group of PPAR α/γ agonists, with their novel mechanism, holds a promising future in the management of diabetic dyslipidemia.
Keywords: diabetes mellitus, dyslipidemia, cardiovascular disease, atherosclerosis, PPAR α/γ agonists
Introduction
Diabetes mellitus (DM) is one of the most common metabolic disorders in the world. DM is a very important cardiovascular risk factor, and most diabetic patients die due to cardiovascular disease (CVD).1 DM and dyslipidemia commonly coexistent in many patients. The most common type of dyslipidemia in type 2 DM (T2DM) is atherogenic dyslipidemia of diabetes (ADD). Many different modalities are available for management of ADD. Here we discuss the use of saroglitazar, a newly approved (in India) dual peroxisome proliferator activator receptor (PPAR) alpha/gamma (α/γ) agonist for the management of ADD.
Atherogenic dyslipidemia of diabetes
The term “atherogenic dyslipidemia” was coined in 1990 by Austin et al, who described a high-risk lipid profile that comprises a higher proportion of small, dense, low-density lipoprotein cholesterol (sdLDL-C) particles, reduced high-density lipoprotein cholesterol (HDL-C), and hypertriglyceridemia.2 Strong clinical evidence from multiple studies has demonstrated that all components of ADD are important risk factors for development of CVD.3–4 ADD is commonly observed in patients with T2DM.5
Pathogenesis of ADD
As demonstrated in Figure 1, insulin resistance (IR) is primarily responsible for the pathogenesis of ADD in T2DM. IR at the adipose tissue results in disinhibition of hormone-sensitive lipase, leading to lipolysis and increased release of free fatty acids (FFA) into the circulation. Increased FFA flux and increased apolipoprotein B (Apo B) production in the liver stimulate increased assembly and secretion of very low-density lipoprotein cholesterol (VLDL-C), resulting in hypertriglyceridemia. In addition, cholesterol ester transport protein (CETP) stimulates the exchange of cholesteryl esters from both HDL-C and low-density lipoprotein cholesterol (LDL-C) to VLDL-C, and opposite movements of triglycerides (TG). TG-enriched HDL-C is easily metabolized by lipases, and Apo A is separated. Free Apo A-I is cleared rapidly from plasma, in part by excretion through the kidney, thus reducing the availability of HDL-C for reverse cholesterol transport. TG-enriched, low-density lipoprotein cholesterol (LDL-C) can also undergo lipolysis and become smaller and denser, known as sdLDL-C.6 Low levels of HDL-C and the presence of sdLDL-C are each independent risk factors for CVD.7
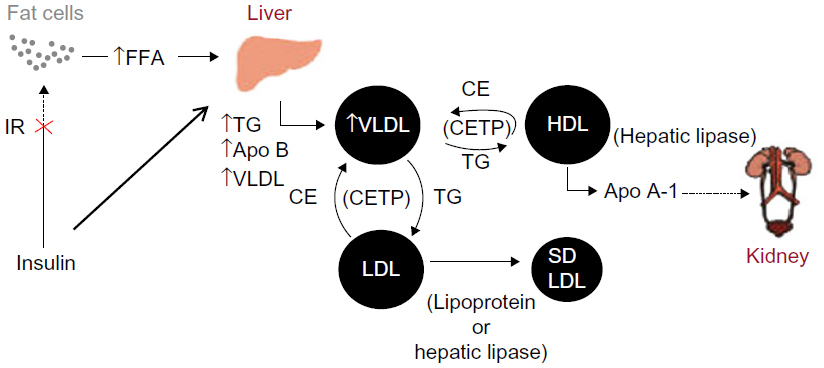 | Figure 1 Pathogenesis of ADD. |
Current options for ADD management and their limitations
Statins
Statins are very effective for reducing LDL-C, but they are only modestly effective in controlling the TGs (10%–15% decrease) and HDL levels (5%–10% increase), which are major abnormalities in ADD. So a significant residual risk remains even after the use of a maximal dose of statins (50% of CV events continue to occur after using the highest doses of statins)8,9 in T2DM patients. Also, many patients develop muscle-related adverse effects (myopathy, myositis, and, rarely, rhabdomyolysis) with statin therapy.
Fibrates
Fibrates reduce TG levels and increase HDL-C levels. Fibrates are known to worsen renal function by reducing glomerular filtration rate, especially when patients have established chronic kidney disease.10 This is especially important in DM patients, as DM is a very common cause of chronic kidney disease.11 When fibrates are used with statins, there is an increase in the risk of muscle-related adverse events, specifically rhabdomyolysis.12 There is also an increased risk of cholelithiasis and pancreatitis13 with fibrate therapy. Fibrates are contraindicated in patients with pre-existing gall bladder disease or with an unexplained increase in liver enzymes.14
Niacin
Niacin was widely used for management of hypertriglyceridemia and low HDL-C. But, in the current statin era, niacin has failed to provide additional cardiovascular benefits in two landmark trials: the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH)15 and the Heart Protection Study 2: Treatment of HDL to Reduce the Incidence of Vascular Events (HPS-2 THRIVE).16 In addition to this, serious adverse events were more common in niacin-treated patients in both these studies. As niacin itself is known to increase insulin resistance, its use in ADD should be limited.
Omega 3 fatty acids
These are also commonly used in Western countries, but their effects on residual risk in statin-treated diabetic patients are not clear.17 Moreover, high dose (2–4 g/day) and the prevalence of strict vegetarian diets in India preclude the use of these drugs.
Benefits of lowering TG levels in T2DM patients
Many recent studies have shown that lowering of TG levels by PPAR α agonist agents in T2DM patients with ADD can reduce CV events. In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study, 9,795 T2DM patients were randomized to fenofibrate or placebo. The follow-up was for 5 years. Serum TG levels were significantly reduced by fenofibrate (by 22%). Fenofibrate reduced CVD events by 27% in the subgroup of ADD patients (baseline: TG >200 mg/dL and HDL-C <40 mg/dL).18 In the lipid arm of the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial, the use of fenofibrate (in addition to simvastatin) was examined in 5,518 T2DM patients who were at high risk for CVD. The mean follow-up was 4.7 years.19 Fenofibrate reduced TG levels significantly (by 26%). In the subgroup analysis of patients with ADD (baseline: TG >204 mg/dL and HDL-C <34 mg/dL), the primary end-point (the first occurrence of nonfatal myocardial infarction, nonfatal stroke, or death from CV causes) was significantly reduced by fenofibrate (by 31%, P<0.03).20 European Society of Cardiology guidelines for dyslipidemia recommend use of drugs to lower TG in subjects with TG >200 mg/dL who cannot lower them by lifestyle measures, and if the subject is at high total CV risk.21 As we have discussed, DM patients are at high CV risk.1
Role of dual PPAR α/γ agonists in ADD
Dual PPAR α/γ agonists are newer agents to control all different pathogenetic factors in ADD simultaneously. Their PPAR α agonist action results in increased lipoprotein lipase activity (causing catabolism of TG in VLDL and chylomicrons), reduced secretion of VLDL, inhibition of Apo CIII expression, and increased production of apolipoproteins Apo AI and Apo AII. PPAR γ agonist actions increase insulin sensitivity in peripheral tissues, increasing glucose uptake, and reduce blood glucose levels. Their overall actions are summarized in Table 1.22
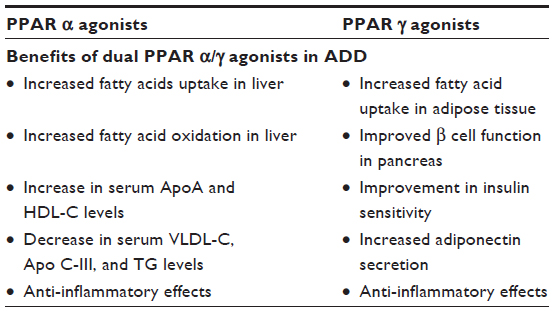 | Table 1 Effects of dual PPAR alpha and gamma agonists in ADD |
Many such dual PPAR agonists have been developed in the past and have failed in clinical trials, due to lack of efficacy or safety concerns. Table 2 outlines the details of such molecules.23
 | Table 2 Dual PPAR α/γ agonists that failed during clinical development |
Saroglitazar: the first and only approved dual PPAR α/γ agonist
Pharmacology and clinical studies
Saroglitazar (Lipaglyn™, Zydus Cadila, Ahmedabad, India) is a dual PPAR α/γ agonist drug approved in India by Drug Controller General of India for the treatment of diabetic dyslipidemia and hypertriglyceridemia with T2DM not controlled by statin therapy.24 It is the first drug in this novel group of dual PPAR α/γ agonists (glitazars) to be approved and clinically used, anywhere in the world. The recommended dose of saroglitazar is 4 mg per day.
Pharmacodynamics
PPAR α activation by saroglitazar increases the hepatic oxidation of fatty acids (FA) and reduces the synthesis and secretion of TG. This, in turn, increases diversion of FA from peripheral tissues (eg, skeletal muscle and fat tissue) to the liver, thereby decreasing both FA synthesis and delivery of TG to peripheral tissues. Saroglitazar also causes increased lipolysis and elimination of TG-rich particles from plasma by activating lipoprotein lipase (LPL) and reducing production of Apo C-III, an inhibitor of LPL activity. Saroglitazar was also found to reduce plasma LDL-C. PPAR activation by saroglitazar also induces an increase in the synthesis of apolipoproteins A-I and A-II, and HDL-C.24
Although saroglitazar is predominantly a PPAR α agonist, it also causes activation of PPAR γ and regulates the transcription of insulin-responsive genes involved in the control of glucose production, transport, and utilization. Saroglitazar increases the expression of numerous PPAR γ-responsive genes involved in carbohydrate and lipid metabolism, including adiponectin, adipocyte fatty acid-binding protein, LPL, fatty acid transport protein, and fatty acid translocase (CD36). By increasing the expression of these genes, saroglitazar decreases the postprandial rise of plasma FFA, improves postabsorptive, insulin-mediated suppression of hepatic glucose output, reduces the metabolic burden on liver and muscle, and promotes glucose utilization. Robust antidiabetic and insulin-sensitizing effects of saroglitazar were observed in preclinical models, in which hyperglycemia and/or impaired glucose tolerance is a consequence of insulin resistance in target tissues.24
As discussed, saroglitazar is predominantly a PPAR α agonist, with modest PPAR γ agonist actions. This is in contrast to previously developed dual PPAR α/γ agonists, like muraglitazar, aleglitazar, and tesaglitazar, which had either predominantly PPAR γ agonistic activity or equivocal agonistic activity on both PPAR α and PPAR γ receptors.
Pharmacokinetics
In its initial clinical trials, peak plasma levels of saroglitazar occurred at approximately 1 hour post-dosing in healthy volunteers. Its absorption is not affected by food. After a single oral dose of 4 mg saroglitazar in healthy volunteers, maximum serum concentration (Cmax) of 337.1±91.0 ng/mL (mean ± standard deviation) was observed.25 It is extensively protein-bound (around 96%) in human plasma. The mean plasma half-life of saroglitazar following a single dose of 4 mg saroglitazar is 2.9±0.9 hours. Multiple-dose studies in humans have not shown any accumulation of saroglitazar on repeat dosing once daily for 10 days.24 In vitro studies using pooled human liver microsomes showed that saroglitazar is metabolically stable. Saroglitazar was found to be metabolized into three minor oxidative metabolites. The exposure of the most abundant oxidative metabolite was found to be less than 10% of the exposure of saroglitazar. Saroglitazar is excreted primarily through the hepatobiliary route.
Preclinical studies
Saroglitazar did not show any adverse effects on mating or fertility in male rats up to 125 mg/kg (more than 250 times the approved human dose, on body surface area basis). In female rats, no adverse effects on fertility were observed up to 3 mg/kg (7 times the approved human dose on body surface area basis). A two-year carcinogenicity study of saroglitazar was conducted in Wistar rats. No potential carcinogenic concern for humans was identified, which was further confirmed by a mechanistic study in nonhuman primates, employing molecular biomarkers. Saroglitazar was found to be nonmutagenic and nongenotoxic, as evident in the Ames bacterial mutagenicity test, a chromosomal aberration assay using peripheral human blood lymphocytes and the mouse micronucleus assay.24
Clinical trials
Phase I clinical trial
Ninety-six healthy volunteers were administered saroglitazar (from 0.125 mg to 128 mg) in its phase I clinical trial program. Single-dose and multiple ascending dose studies were carried out. The pharmacokinetics of saroglitazar has demonstrated dose-dependent linearity in both single and multiple doses. Saroglitazar was safe and well-tolerated up to 128 mg, as a single dose, and up to 8 mg once per day for up to 10 days.25 Saroglitazar did not show any clinically relevant abnormal findings in the clinical laboratory investigations, physical examination, vital signs, and electrocardiogram. No serious adverse events were reported.
Phase II and III clinical trials of saroglitazar were identified as Prospective Randomized Efficacy and Safety study of Saroglitazar (PRESS).
Phase III clinical trials
PRESS V trial
This was a phase III, multicenter, randomized, double-blind study to evaluate the safety and efficacy of saroglitazar 2 mg and 4 mg, compared to pioglitazone 45 mg, in patients with diabetic dyslipidemia.26 A total of 122 patients were randomized into three groups: saroglitazar 2 mg/day, saroglitazar 4 mg/day, and pioglitazone 45 mg/day. Follow-up was done for 24 weeks. The primary end-point of the study was the percent change in serum TG levels after a 24-week treatment period, as compared against baseline.
There was 45% reduction in serum TG levels with saroglitazar 4 mg, which was statistically significant, compared against baseline and also compared against pioglitazone 45 mg (15.5%) (Figure 2). The maximum effect of saroglitazar on TG was achieved by week 12 and was sustained at week 24. Saroglitazar reduced VLDL-C (45.5%) and total cholesterol (7.7%) significantly more, compared to pioglitazone and/or baseline (Table 3). Both glycemic parameters, fasting plasma glucose and glycosylated hemoglobin, were significantly reduced at week 24, compared against baseline, in the saroglitazar and pioglitazone arms (Table 3 and Figure 3). An antiglycemic effect of saroglitazar was comparable to pioglitazone, and there was no significant difference between the saroglitazar 4 mg and pioglitazone 45 mg arms at the end of the study period.
 | Figure 2 Primary end point of PRESS V study. |
 | Table 3 Efficacy parameters at week 24 in PRESS V study (mITT population, LOCF method) |
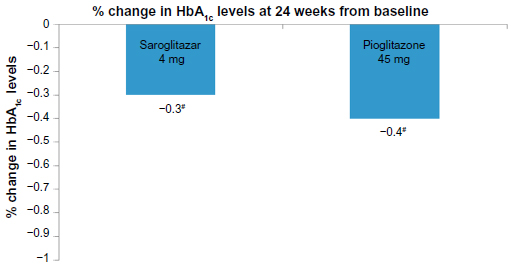 | Figure 3 Change in HbA1c levels in PRESS V study. |
Saroglitazar 2 mg and 4 mg doses were well-tolerated throughout the study. The most frequently reported adverse events were asthenia, gastritis, chest discomfort, peripheral edema, dizziness, and tremors. Most of the adverse events were of mild intensity, and were considered unrelated to treatment. Saroglitazar did not change body weight, while there was a modest increase in body weight in the pioglitazone group (Table 4). Hypoglycemia was not seen in any patient in any arm.
 | Table 4 Change in body weight at week 24 in PRESS V trial |
PRESS VI trial
This was a multicenter, prospective, randomized, double-blind study to evaluate safety and efficacy of saroglitazar 2 mg and 4 mg, compared to placebo, in hypertriglyceridemia with T2DM not controlled with atorvastatin therapy.27 The study consisted of a 4-week run-in period, involving discontinuation of any antidyslipidemic drugs other than atorvastatin 10 mg. Also, patients were put on a dietary and lifestyle modification program at this time. A total of 302 subjects were randomized to receive one of the treatments, saroglitazar 2 mg (n=101 subjects) or saroglitazar 4 mg (n=99 subjects), or matching placebo (n=102 subjects). The primary end-point was change in serum TG level from the baseline at 12 weeks of treatment.
The efficacy results of the PRESS VI study are shown in Table 5. Saroglitazar 2 mg and 4 mg decreased TG levels by 45.5% and 46.7%, respectively (Figure 4); non-HDL cholesterol was reduced by 29.2% and 32.5%, respectively. These decreases in TG and non-HDL levels were statistically significant, compared against baseline and placebo (P<0.05). Both doses of saroglitazar showed significant increases in HDL-C, as compared to placebo. Patients in the 4 mg saroglitazar arm achieved better reduction in LDL-C (31.3%), VLDL-C (46%), Apo B (32%), and total cholesterol (26.1%) than those in the placebo arm. There was also a statistically significant decrease in fasting plasma glucose level after 12 weeks of treatment with saroglitazar 2 mg and 4 mg, as compared to the placebo arm.
 | Table 5 Efficacy parameters at week 12 in PRESS VI study (mITT population) |
 | Figure 4 Primary end point of PRESS VI study. |
Both doses of saroglitazar were well tolerated throughout duration of the study. There were similar numbers of adverse events in the saroglitazar and placebo arms. Dyspepsia and gastritis were the most common adverse effects in the saroglitazar arms. Most of the adverse events were not related to treatment and were mild-to-moderate in intensity. No significant changes in hepatic function tests (transaminases and billirubin), renal function tests (blood urea nitrogen, serum creatinine), creatine phosphokinase, hemogram, or body weight were observed in patients in the saroglitazar arms.
In the PRESS VI trial, subjects were also evaluated for cardiac function and events. Electrocardiogram and 2-D echocardiography were carried out at the start of the study, at 12 weeks, and at 24 weeks after the last dose of the study. No adverse findings were reported, as far as cardiac safety is concerned.
Thus, in both phase II and phase III clinical trials, saroglitazar was effective in controlling both dyslipidemia and glycemic parameters. It was found to be devoid of conventional adverse events of typical PPAR α agonist agents (like reduced glomerular filtration rate, increased myopathy with statins, and hepatotoxicity), as well as of PPAR γ agonist agents (like pedal edema, weight gain, and congestive heart failure).
Conclusion
ADD is a complex metabolic abnormality seen in most DM patients. Its presence increases CV risk in DM, which is itself considered equivalent to CVD. Traditional drugs like fibrate and niacin can control lipid parameters (hypertriglyceridemia and low HDL-C) in ADD, but do not provide any glycemic benefits; niacin, rather, increases blood glucose levels. Saroglitazar is a novel drug, with unique mechanism of action, which has established its efficacy and safety in diabetic dyslipidemia in clinical trials. It not only corrects lipid abnormalities, but also helps to achieve glycemic targets in T2DM. Further studies should be carried out to evaluate saroglitazar’s exact role in management of ADD. Use of saroglitazar will help clinicians to better manage metabolic abnormalities in T2DM patients.
Disclosure
The authors report no conflicts of interest in this work.
References
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–3421. | |
Austin MA, King MC, Vranizan KM, Krauss RM. Atherogenic lipoprotein phenotype. A proposed genetic marker for coronary heart disease risk. Circulation. 1990;82(2):495–506. | |
Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375(9726):1634–1639. | |
Li C, Ford ES, Tsai J, Zhao G, Balluz LS, Gidding SS. Serum non-high-density lipoprotein cholesterol concentration and risk of death from cardiovascular diseases among US adults with diagnosed diabetes: the Third National Health and Nutrition Examination Survey linked mortality study. Cardiovasc Diabetol. 2011;10:46. | |
Kathiresan S, Otvos JD, Sullivan LM, et al. Increased small low-density lipoprotein particle number: a prominent feature of the metabolic syndrome in the Framingham Heart Study. Circulation. 2006;113(1):20–29. | |
Manoria PC, Chopra HK, Parashar SK, et al. The nuances of atherogenic dyslipidemia in diabetes: focus on triglycerides and current management strategies. Indian Heart J. 2013;65(6):683–690. | |
Toft-Petersen AP1, Tilsted HH, Aarøe J, et al. Small dense LDL particles – a predictor of coronary artery disease evaluated by invasive and CT-based techniques: a case-control study. Lipids Health Dis. 2011;10:21. | |
Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364(9435):685–696. | |
Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 5963 people with diabetes: a randomised placebo controlled trial Lancet. 2003;361:2005–2016. | |
Jun M, Zhu B, Tonelli M, et al, Effects of fibrates in kidney disease: a systematic review and meta-analysis. J Am Coll Cardiol. 2012;60(20):2061–2071. | |
Collins AJ, Kasiske B, Herzog C, et al. Excerpts from the United States Renal Data System 2006 Annual Data Report. Am J Kidney Dis. 2007;49(1 Suppl 1):A6–A7, S1–S296. | |
Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292(21):2585–2590. | |
Preiss D, Tikkanen MJ, Welsh P, et al. Lipid-modifying therapies and risk of pancreatitis: a meta-analysis. JAMA. 2012;308(8):804–811. | |
Lofibra (Micronized fenofibrate) [prescribing information]. North Wales, PA, USA: Teva Pharmaceuticals; 2010. | |
AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267. | |
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34(17):1279–1291. | |
Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369(9567):1090–1098. | |
Scott R, O’Brien R, Fulcher G, et al. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetes Care. 2009;32(3):493–498. | |
ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, et al, Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362(17):1563–1564. | |
Tenenbaum A, Fisman EZ. Fibrates are an essential part of modern anti-dyslipidemic arsenal: spotlight on atherogenic dyslipidemia and residual risk reduction. Cardiovasc Diabetol. 2012;11:125. | |
Catapano AL, Reiner Z, De Backer G, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32(14):1769–1818. | |
Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54(8):2460–2470. | |
Balakumar P, Rose M, Ganti SS, Krishan P, Singh M. PPAR dual agonists: are they opening Pandora’s Box? Pharmacol Res. 2007;56(2):91–98. | |
Lipaglyn (Saroglitazar) [prescribing information]. Ahmedabad, India: Cadila Healthcare; 2013. | |
Jani RH, Kansagra K, Jain MR, Patel H. Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects. Clin Drug Investig. 2013;33(11):809–816. | |
Pai V, Paneerselvam A, Mukhopadhyay S, et al. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of saroglitazar 2 and 4 mg compared to pioglitazone 45 mg in diabetic dyslipidemia (PRESS V). J Diabetes Sci Technol. 2014;8(1):132–141. | |
Jani RH, Pai V, Jha P, et al. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI). Diabetes Technol Ther. 2014;16(2):63–71. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.