Back to Journals » Drug Design, Development and Therapy » Volume 11
Salidroside pretreatment attenuates apoptosis and autophagy during hepatic ischemia–reperfusion injury by inhibiting the mitogen-activated protein kinase pathway in mice
Authors Feng J ![]() , Zhang Q, Mo WH
, Zhang Q, Mo WH ![]() , Wu L
, Wu L ![]() , Li S, Li J, Liu T, Xu S, Fan X, Guo C
, Li S, Li J, Liu T, Xu S, Fan X, Guo C ![]()
Received 11 March 2017
Accepted for publication 2 June 2017
Published 3 July 2017 Volume 2017:11 Pages 1989—2006
DOI https://doi.org/10.2147/DDDT.S136792
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Tuo Deng
Jiao Feng,1,* Qinghui Zhang,2,* Wenhui Mo,3,* Liwei Wu,1 Sainan Li,1 Jingjing Li,1 Tong Liu,1 Shizan Xu,4 Xiaoming Fan,5 Chuanyong Guo1
1Department of Gastroenterology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai, 2Department of Clinical Laboratory, Kunshan First People’s Hospital Affiliated to Jiangsu University, Kunshan, JiangSu, 3Department of Gastroenterology, Minhang Hospital, Fudan University, Shanghai, 4Department of Gastroenterology, Shanghai Tenth People’s Hospital, School of Clinical Medicine of Nanjing Medical University, Shanghai, 5Department of Gastroenterology, Jinshan Hospital of Fudan University, Jinshan, Shanghai, China
*These authors contributed equally to this work
Abstract: Ischemia–reperfusion injury (IRI) contributes to liver damage in many clinical situations, such as liver resection and liver transplantation. In the present study, we investigated the effects of the antioxidant, anti-inflammatory, and anticancer agent salidroside (Sal) on hepatic IRI in mice. The mice were randomly divided into six groups: normal control, Sham, Sal (20 mg/kg), IRI, IRI + Sal (10 mg/kg), and IRI + Sal (20 mg/kg). We measured liver enzymes, proinflammatory cytokines, TNF-α and interleukin-6, and apoptosis- and autophagy-related marker proteins at 2, 8, and 24 hours after reperfusion. Components of mitogen-activated protein kinase (MAPK) signaling, including P-38, jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK), were also measured using an MAPK activator anisomycin to deduce their roles in hepatic IRI. Our results show that Sal safely protects hepatocytes from IRI by reducing levels of liver enzymes in the serum. These findings were confirmed by histopathology. We concluded that Sal protects hepatocytes from IRI partly by inhibiting the activation of MAPK signaling, including the phosphorylation of P38, JNK, and ERK. This ameliorates inflammatory reactions, apoptosis, and autophagy in the mouse liver.
Keywords: liver injury, salidroside, apoptosis, autophagy, JNK, P38
Introduction
Recently, liver surgery has been widely adopted in clinical medicine for the treatment of liver diseases. Hepatic ischemia–reperfusion injury (IRI) is inevitable following hemorrhagic shock, liver resection, liver transplantation, and other medical conditions.1,2 This leads to hepatocyte necrosis, liver dysfunction, organ injury, and even death. Hepatic IRI includes warm and cold IRI. Although they occur in different situations, cold and warm IRI share similar pathophysiological processes. The mechanisms of hepatic IRI remain unclear, but involve anerobic metabolism and acidosis, mitochondria damage, oxidative stress, calcium overload, liver Kupffer cells, and inflammatory cytokines.3
Hepatic IRI is initiated by the production of reactive oxygen species (ROS), including peroxides, superoxide, hydroxyl radical, and singlet oxygen.4 After the release of ROS, a cascade of mediators are activated, which injure different cells, including hepatocytes and endothelial cells.3–6 Inflammatory reactions and inflammatory cytokines are key factors in hepatic IRI. Kupffer cells, or hepatic macrophages, are activated by ischemia and synthesize proinflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6, which act on other cells to induce hepatocyte damage. Kupffer cells also promote the accumulation and activation of neutrophils, which generate more proinflammatory cytokines. As a result, Kupffer cells are persistently active and liver injury is induced.3,5 As a consequence, a number of liver cells die during IRI.
Apoptosis is programmed cell death induced by endogenous or exogenous factors. It eliminates abnormal or dead cells to maintain homeostasis. Apoptosis and necrosis are the two main types of cell death during hepatic IRI and more than half of dead cells die by apoptosis during the first 24 hours of reperfusion.5–7 Antiapoptotic measures, including the overexpression of Bcl-2, Bax knockout, and caspase inhibition, can protect the liver from IRI.8–10 This strongly supports the theory that apoptotic cell death plays a role during hepatic IRI. Autophagy, or type II programmed cell death, is a self-digesting process that removes dysfunctional organelles, malformed proteins, and surplus or unnecessary cytoplasmic contents through lysosomal digestion.11 It promotes cell survival, but is considered as a double-edged sword because it leads to cell death when overactivated.12 In 1976, Sybers et al reported an increase in autophagic vacuoles following IRI.13 Decker et al also found that autophagy was induced by cardiac ischemia and reperfusion injury in rabbits.14 Recent studies have shown that autophagy plays a vital role in liver damage during IRI.6,15–17 Hence, it is important to elucidate the exact mechanisms underlying hepatic IRI, apoptosis, and autophagy.
Mitogen-activated protein kinases (MAPKs) are important cellular signaling proteins and include extracellular signal-regulated kinase (ERK), p38 MAPK (P38), and c-jun N-terminal kinase (JNK).18,19 They are activated by many factors, such as oxidative stress, proinflammatory cytokines, ultraviolet radiation, and heat shock.20 In 2004, Koike et al reduced IRI following heart, lung, and liver transplantations using P38 MAPK inhibitors.21 The ROS/JNK pathway is also activated by TNF-α and induces apoptosis and necrosis.22
Salidroside (Sal) is the main active component of Rhodiola Rosea L. It is a phenylpropanoid glycoside with antioxidant, anti-inflammatory, anticancer, antidepressive, neuroprotective, and cardioprotective properties.23 However, the protective effects of Sal on hepatic IRI have been poorly investigated. Studies have shown that Sal pretreatment effectively reduces cerebral IRI by decreasing TNF-α expression.23 What is more, Sal protects myocardia from IRI in rats by inhibiting apoptosis and inflammation.24 In addition, Sal inhibits MAPK signal transduction to protect organs from damaging factors, such as cardiac exhaustive injury, cadmium-induced hepatotoxicity, and cigarette smoke-induced lung diseases.18,25 Therefore, it is reasonable to infer that Sal could protect hepatic IRI through the downregulation of MAPKs to inhibit inflammation, apoptosis, and autophagy.
There are no effective prevention strategies or treatments for hepatic IRI. This places restrictions on liver surgery and the preservation of liver function. Thus, it is crucial to investigate the mechanism of hepatic IRI to develop new treatments and preventive strategies. The aim of this study was to explore the effects of Sal on hepatic IRI in mice and to deduce the underlying mechanisms. We hypothesized that Sal pretreatment ameliorates hepatic IRI by inhibiting inflammation, apoptosis, and autophagy through the MAPK pathway.
Materials and methods
Reagents
Sal and anisomycin (Ani) were purchased from Sigma-Aldrich (Saint Louis, MO, USA) and was diluted in normal saline and stored away from light at −20°C. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) microplate test kits were acquired from the Jiancheng Bioengineering Institute (Jiancheng Biotech, Nanjing, China). IL-6 and TNF-α enzyme-linked immunosorbent assay (ELISA) kits were obtained from Anogen (Ontario, Canada). Oligonucleotide primers were synthesized by Generay (Shanghai, China). The PrimeScript RT Reagent Kit and SYBR Premix Ex Taq were purchased from TaKaRa Biotechnology (Dalian, China). The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) apoptosis assay kit was obtained from Roche (Roche Ltd., Basel, Switzerland).
The primary antibodies used in this study were anti-Bax, anti-caspase 3, anti-caspase 9, anti-Beclin-1, anti-P62, anti-JNK, anti-P38, anti-p-P38 (Proteintech, Chicago, IL, USA), anti-TNF-α, anti-Bcl-2, antimicrotubule-associated protein 1 light chain 3 (LC3), antimammalian target of rapamycin (mTOR), anti-p-mTOR, anti-Akt, anti-p-Akt, anti-S6, anti-p-S6 (Cell Signaling Technology, Danvers, MA, USA), anti-IL-6, anti-p-JNK (Antibody Revolution, San Diego, CA, USA), anti-ERK, and anti-p-ERK (Signalway Antibody, College Park, MD, USA).
Animal preparation
This study was approved by the Animal Care and Use Committee of Shanghai Tongji University. Animal handling and care were performed according to the National Institutes of Health Guidelines. We made all our efforts to minimize the pain and suffering of mice throughout the study. Male Balb/c mice (6–8 weeks old, 23±2 g) were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The mice were raised in a clean and temperature-controlled room at 24°C±2°C under a 12-hour:12-hour light:dark cycle. They had free access to standard laboratory chow and water.
Experimental design
Eighty-four mice were randomly divided into six groups as follows:
- Normal control (NC): six mice were injected with vehicle (saline) only.
- Sham: eighteen mice underwent laparotomy after anesthesia. The abdominal cavity was closed without IRI.
- Sal (20 mg/kg): six mice were injected with 20 mg/kg Sal only.
- IRI: eighteen mice underwent ischemia and reperfusion.
- IRI + Sal (10 mg/kg): eighteen mice were injected with 10 mg/kg Sal 2 h before IRI.
- IRI + Sal (20 mg/kg): eighteen mice were injected with 20 mg/kg Sal 2 h before IRI.
To further investigate whether the protective effect of Sal was associated with MAPKs, we used Ani, a specific activator of P38 and JNK, to create another group of experiment. Thirty mice were randomly divided into five groups: 1) Sham group, 2) IRI group, 3) IRI + Sal (20 mg/kg), 4) IRI + Ani (20 mg/kg), and 5) IRI + Sal (20 mg/kg) + Ani (20 mg/kg). Six mice in each group were randomly sacrificed at 8 hours after IRI.
According to the pharmacokinetics and preliminary experiment of Sal, Sal and Ani were injected intraperitoneally 2 hours before IRI (Figure S1).23 The doses of Sal and Ani were determined according to a previous study and preliminary experiments. Six mice in each group were randomly sacrificed at 2, 8, or 24 h after IRI. Blood and liver tissues were collected for further experiments (Figure 1).
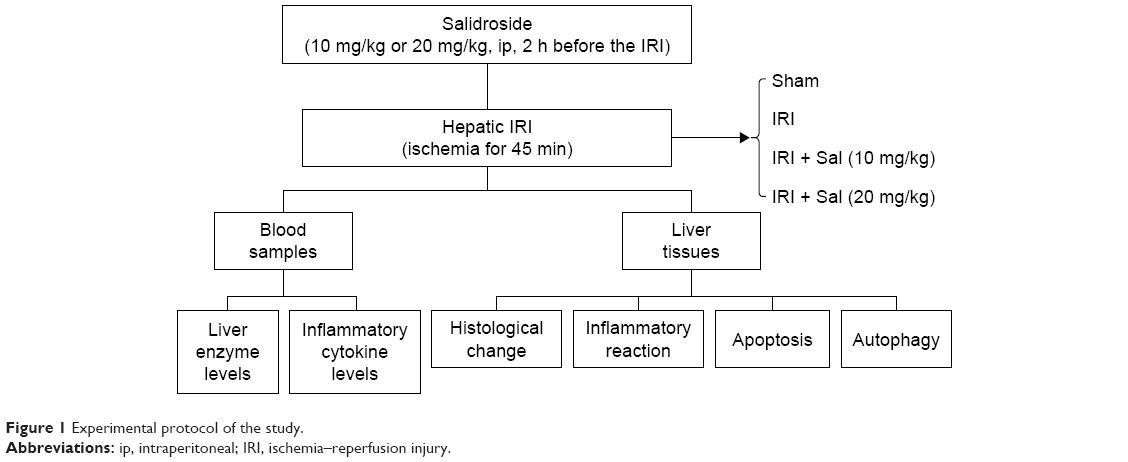 | Figure 1 Experimental protocol of the study. |
Establishment of a hepatic IRI mouse model
A warm hepatic IRI animal model was established in this study. Mice were fasted for 12 hours before surgery, but had free access to water. The mice were anesthetized by an intraperitoneal injection of sodium pentobarbital (40 mg/kg, 1.25%) (Nembutal, St Louis, MO, USA). After disinfection by alcohol, we entered the abdominal cavity by an incision in the skin and linea alba. The abdominal viscera were identified and then the hepatic lobes were turned over by wet cotton swabs to visualize the first porta hepatis. The portal vein, hepatic artery, and common bile duct were clipped by vascular clamps to induce hepatic ischemia. The liver lobes changed from crimson to light red immediately after the occlusion. The incision was covered with humid saline gauze to prevent visceral dehydration and mice were put on an electric blanket to maintain body temperature. The hepatic ischemic process lasted for 45 min. After this, the clamps were removed and the reperfusion process was initiated. The abdominal cavity was closed step by step and mice were put on electric blankets to recover from anesthesia.
Biochemical assays
Blood samples were collected by removing the eyes. Serum was separated from the blood by centrifugation at 2,000× g at 4°C for 10 min and stored at −80°C. Serum levels of ALT and AST were measured in an automated chemical analyzer (Olympus AU1000, Olympus, Tokyo, Japan) to evaluate liver IRI. Serum levels of IL-6 and TNF-α were measured using ELISA kits according to the manufacturers’ protocols.
Histopathological evaluation
A part of the liver tissue was excised, fixed in 4% paraformaldehyde for 24 hours, and dehydrated using different concentrations of ethanol. Then, tissues were embedded with paraffin and cut into 3-μm-thick sections. The sections were stained with hematoxylin and eosin (H&E) to observe the degree of injury.
Immunohistochemistry
We collected liver tissues 8 hours after IRI for immunohistochemistry. Paraffin-embedded 3-μm liver sections were dewaxed in a baking oven at 60°C for 20 min and then rehydrated with xylene and various concentrations of ethanol. Antigen retrieval was performed by heating to 95°C for 10 min and then cooling to room temperature. Hydrogen peroxide (H2O2) solution (3%) was added to the sections for 20 min at 37°C to block endogenous peroxidase activity and then nonspecific binding sites were blocked in 5% bovine serum albumin. The liver sections were incubated overnight with primary antibodies against Bcl-2 (1:500), Bax (1:500), Beclin-1 (1:500), and LC3 (1:500) at 4°C. Sections were incubated with a secondary antibody for 1 hour at 37°C. A diaminobenzidine kit was used to detect antibody binding. Sections were then counterstained with hematoxylin, dehydrated through a graded ethanol series and xylene, and mounted with entellan. Sections were examined under an optical microscope. The ratios of brown-stained areas versus total areas were calculated using Image-Pro Plus software 6.0.
Reverse transcription polymerase chain reaction (RT-PCR) and quantitative real-time PCR (qRT-PCR)
We extracted total RNA from liver tissues using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) and then reverse-transcribed it into cDNA. qRT-PCR was performed with SYBR Premix EX Taq according to the manufacturer’s instructions and products were detected with a 7900HT Fast PCR System (Applied Biosystems, Foster City, CA, USA). The primer details are provided in Table 1.
 | Table 1 Primers used for qRT-PCR in this study |
Western blot analysis
We extracted total protein from liver tissue using radioimmunoprecipitation assay buffer (Kaiji Biology, Nanjing, China). The protein concentration was measured with a bicinchoninic acid assay (Kaiji Biology). Protein samples were stored at −20°C in a loading buffer. A certain amount of protein samples was added to 10% or 12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis to separate proteins. After electrophoresis, proteins were transferred onto polyvinylidene fluoride membranes (Hybond™; Escondido, CA, USA) using the wet transfer method. Nonspecific binding sites were blocked with 5% nonfat milk dissolved in phosphate-buffered saline (PBS) for at least 1 hour. Membranes were incubated overnight at 4°C with the following primary antibodies: anti-β-actin (1:1,000), anti-IL-6 (1:500), anti-TNF-α (1:500), anti-Bcl-2 (1:1,000), anti-Bax (1:1,000), anticaspase 3 (1:500), anticaspase 9 (1:500), anti-Beclin-1 (1:1,000), anti-LC3 (1:1,000), anti-p62 (1:1,000), anti-mTOR (1:1,000), anti-p-mTOR (1:500), anti-S6 (1:1,000), anti-p-S6 (1:500), anti-Akt (1:1,000), anti-p-Akt (1:1,000), anti-P38 (1:1,000), anti-p-P38 (1:500), anti-ERK (1:500), anti-p-ERK (1:1,000), anti-JNK (1:1,000), and anti-p-JNK (1:500). The next day, the membranes were washed with PBS containing 0.1% Tween (PBST) three times and then incubated with peroxidase-conjugated secondary goat antimouse or antirabbit antibodies (1:2,000) for 1 hour at 37°C. After three PBST washes, the chromogenic results were visualized using an Odyssey two-color infrared laser imaging system (Licor, Lincoln, NE, USA).
TUNEL assay
We assessed the apoptosis of liver tissues using the TUNEL assay. Paraffin-embedded 3-μm liver sections were dewaxed with xylene, dehydrated with ethanol, and digested with 20 μg/mL proteinase K for 15 min at 37°C. After four washes, the sections were incubated with 2% H2O2 for 5 min at 37°C, washed twice, and then immersed in TUNEL reaction mixture at 37°C in a humidified atmosphere for 1 hour. The sections were observed by optical microscopy and positive areas with dark brown nuclei were quantified using Image-Pro Plus software 6.0.
Transmission electron microscopy (TEM)
Liver tissue was fixed in 2% glutaraldehyde buffer and then fixed in osmium tetroxide. The sections were observed by TEM (JEM 1230, JEOL, Tokyo, Japan).
Statistical analysis
Data were expressed as mean ± SD, and all experiments were repeated at least three times. The serum levels of ALT and AST, Western blot (Figure S2), immunohistochemistry, and TUNEL assay results were analyzed by Student’s t-test. The ELISA and qRT-PCR data were analyzed by one-way analysis of variance. P-values of <0.05 were considered statistically significant. All statistical graphs were drawn by GraphPad Prism 6 (GraphPad Software, Inc., San Diego, CA, USA).
Results
Sal and laparotomy do not affect liver function and kidney function
To determine whether Sal and laparotomy injured liver function and kidney function, we compared mice from the NC group with mice from the sham group and Sal (20 mg/kg) groups. The serum creatinine level was detected using an automated biochemical analyzer (Siemens Dimension RxL Analyser, Siemens AG, Health Care Division, Munich, Germany) at the Clinical Laboratory of Shanghai Tenth People’s Hospital. Serum ALT/AST levels, serum creatinine level, and H&E stained sections were not significantly different between these three groups (Figures 2 and S3). Sham and Sal (20 mg/kg) mice were as healthy as NC mice. Therefore, we used Sham mice as controls in our study. These results reveal that Sal and laparotomy do not harm liver function or cause liver injury in mice.
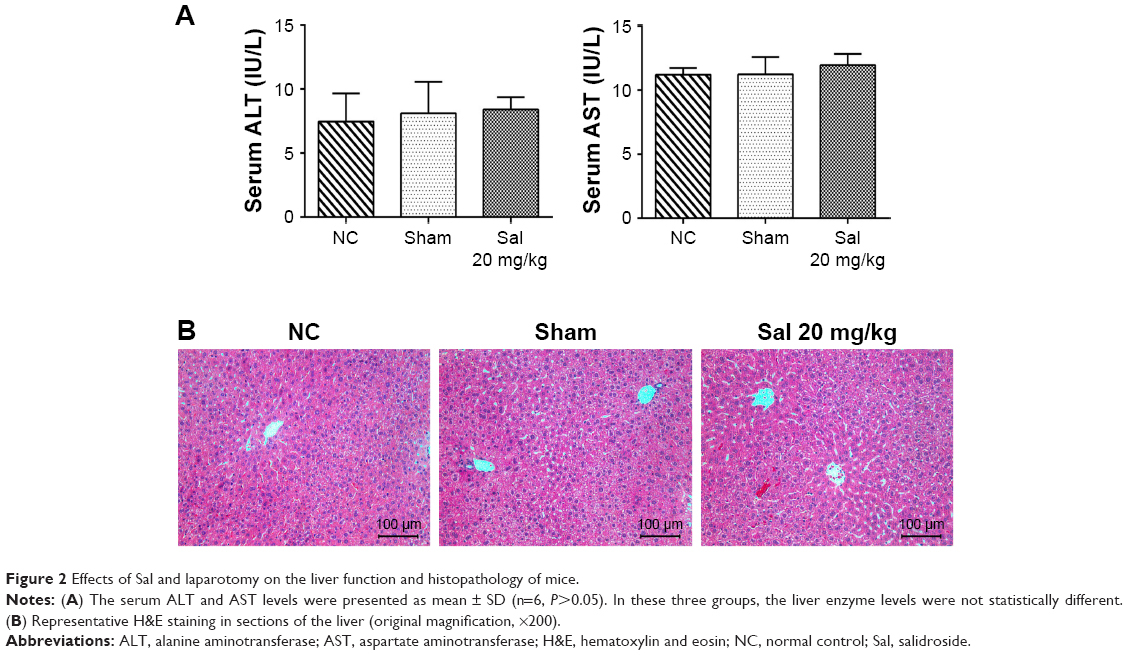 | Figure 2 Effects of Sal and laparotomy on the liver function and histopathology of mice. |
Sal pretreatment ameliorates hepatic IRI in mice
ALT and AST are sensitive markers of liver dysfunction. To assess the effects of Sal pretreatment on hepatic IRI, we measured ALT and AST serum levels and the histopathological appearance. As shown in Figure 3A, the serum levels of ALT and AST increased dramatically in IRI mice at three time points, especially 8 hours after reperfusion. Sal pretreatment (10 or 20 mg/kg) reduced the increase in ALT and AST levels after IRI. Furthermore, 20 mg/kg Sal had a better protective effect than 10 mg/kg. These findings suggest that Sal protects hepatocytes from IRI in a dose-dependent manner. We used H&E staining to further investigate the effects of Sal on hepatic IRI. In Sham group, the liver tissues were well structured and without necrosis. In the IRI group, we observed structural disruption, sinusoidal congestion, hepatocyte ballooning, areas of necrosis, and polymorphonuclear cells enrichment in liver tissues. Sal (10 or 20 mg/kg) pretreatment significantly alleviated hepatocyte necrosis and inflammation after IRI, and 20 mg/kg Sal had a stronger protective effect than the lower dose (Figure 3B). These results indicate that Sal pretreatment can effectively reduce hepatic IRI in mice in a dose-dependent manner.
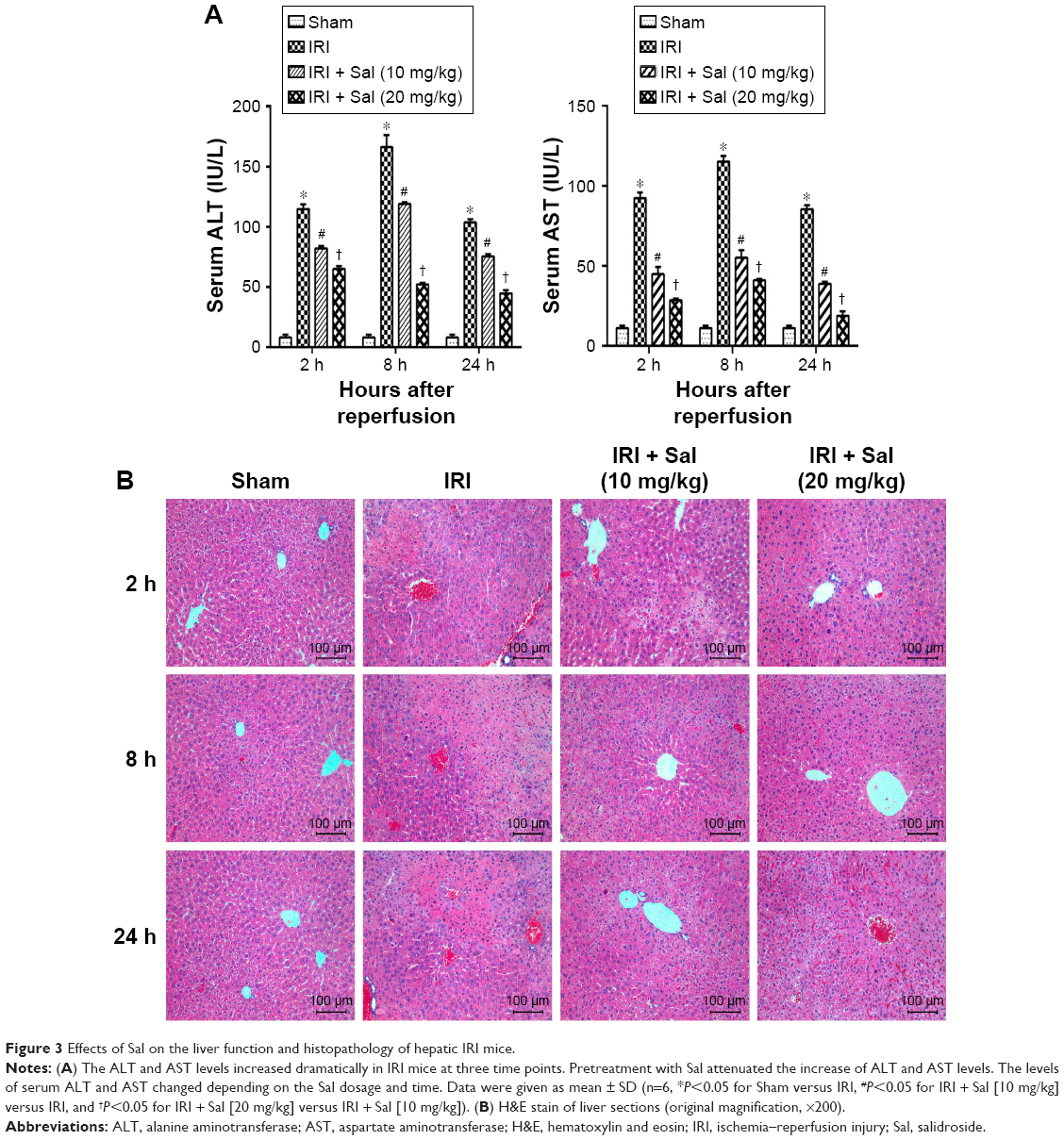 | Figure 3 Effects of Sal on the liver function and histopathology of hepatic IRI mice. |
Since serum liver enzyme levels peaked 8 hours after reperfusion, we used liver tissues collected at this time point for immunohistochemistry and TUNEL assay experiments.
Sal pretreatment inhibits the release of inflammatory cytokines including IL-6 and TNF-α
Inflammatory reactions are involved in the pathogenesis of hepatic IRI. Therefore, we investigated changes in IL-6 and TNF-α levels in liver tissues by Western blotting and immunohistochemistry. ELISA results demonstrated that IL-6 and TNF-α serum levels were elevated and peaked 8 hours after reperfusion in IRI mice. Pretreatment with Sal significantly decreased this elevation at all three time points (Figure 4A). These results were confirmed by qRT-PCR and Western blot (Figure 4B and C). These results suggest that Sal pretreatment inhibits the release of inflammatory cytokines, including IL-6 and TNF-α, during hepatic IRI in mice.
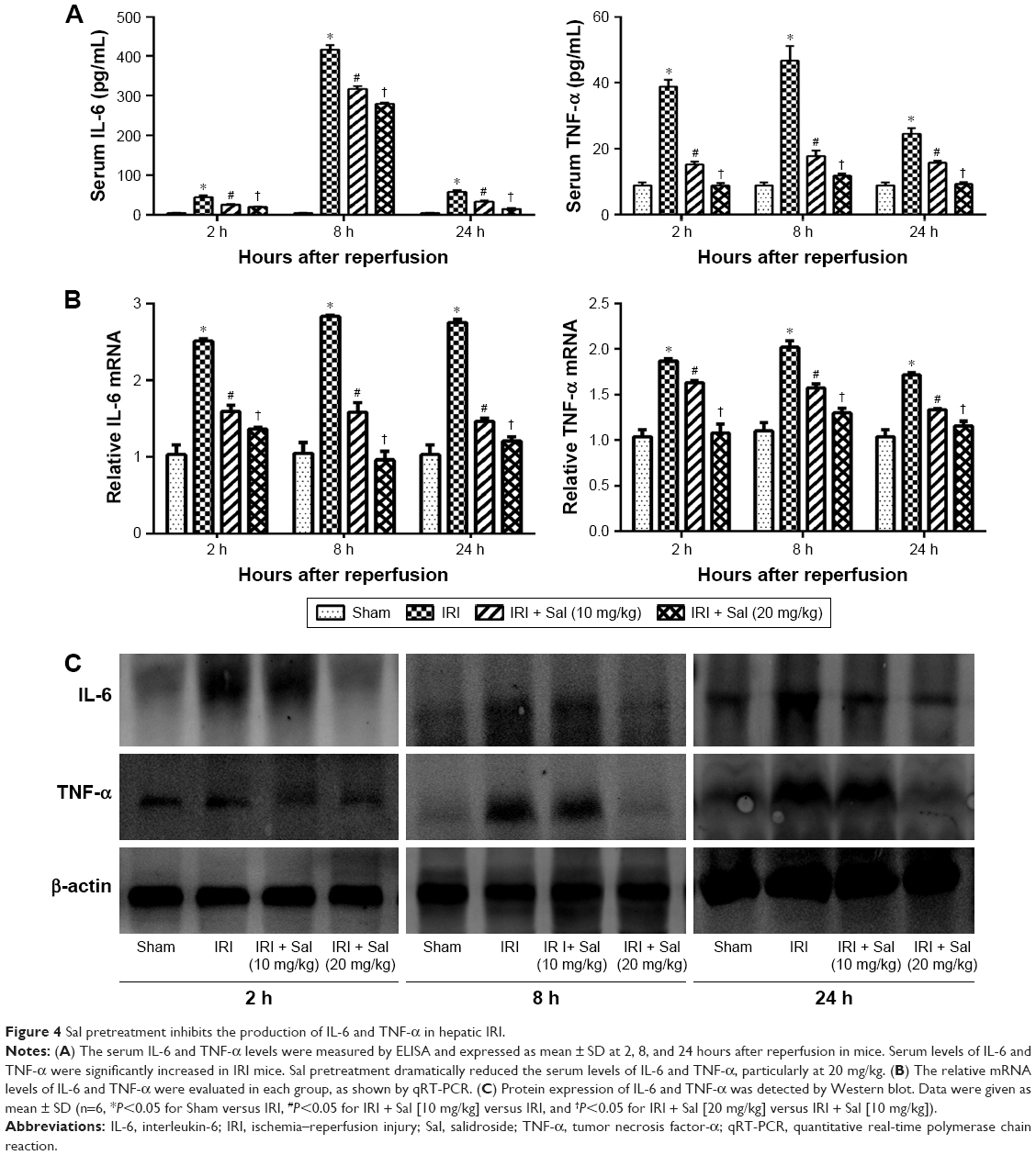 | Figure 4 Sal pretreatment inhibits the production of IL-6 and TNF-α in hepatic IRI. |
Sal pretreatment attenuates hepatocyte apoptosis and autophagy during hepatic IRI in mice
Apoptosis and autophagy are two types of cell death in hepatic IRI. To explore the protective effects of Sal against apoptosis and autophagy, we detected apoptosis and autophagy markers. Bcl-2 is an antiapoptotic protein, while Bax, caspase 3, and caspase 9 are known to promote apoptosis. Beclin-1, LC3, P62, Akt, mTOR, and S6 play an important role in autophagy. We measured the expression of these markers by qRT-PCR, Western blotting, and immunohistochemistry. The protein and cDNA expression of Bax, caspase 3, caspase 9, Beclin-1, and LC3 was elevated in IRI mice at all three time points, while the expression of Bcl-2, P62, p-Akt, p-mTOR, and p-S6 was decreased. This indicated that apoptosis and autophagy were enhanced in IRI mice. The opposite expression patterns were observed after pretreatment with Sal and the stronger effect was observed with 20 mg/kg Sal (Figure 5A–C). We used TUNEL assays to stain apoptotic cells in liver tissues. TUNEL-positive cells were cells with nuclear chromatin concentration and edge accumulation, and their nuclear chromatins were dyed dark brown. These cells were indicated by red arrows in Figure 5D. We found that fewer TUNEL-positive cells were observed in Sal pretreated groups (Figure 5D). We further used the TEM to observe the autophagosomes formation in 8 hours in liver tissues. The results showed that autophagosomes formation was more in the IRI group than in the Sal-treated group (Figure 5E). These results demonstrate that Sal can attenuate hepatocyte apoptosis and autophagy during IRI in mice.
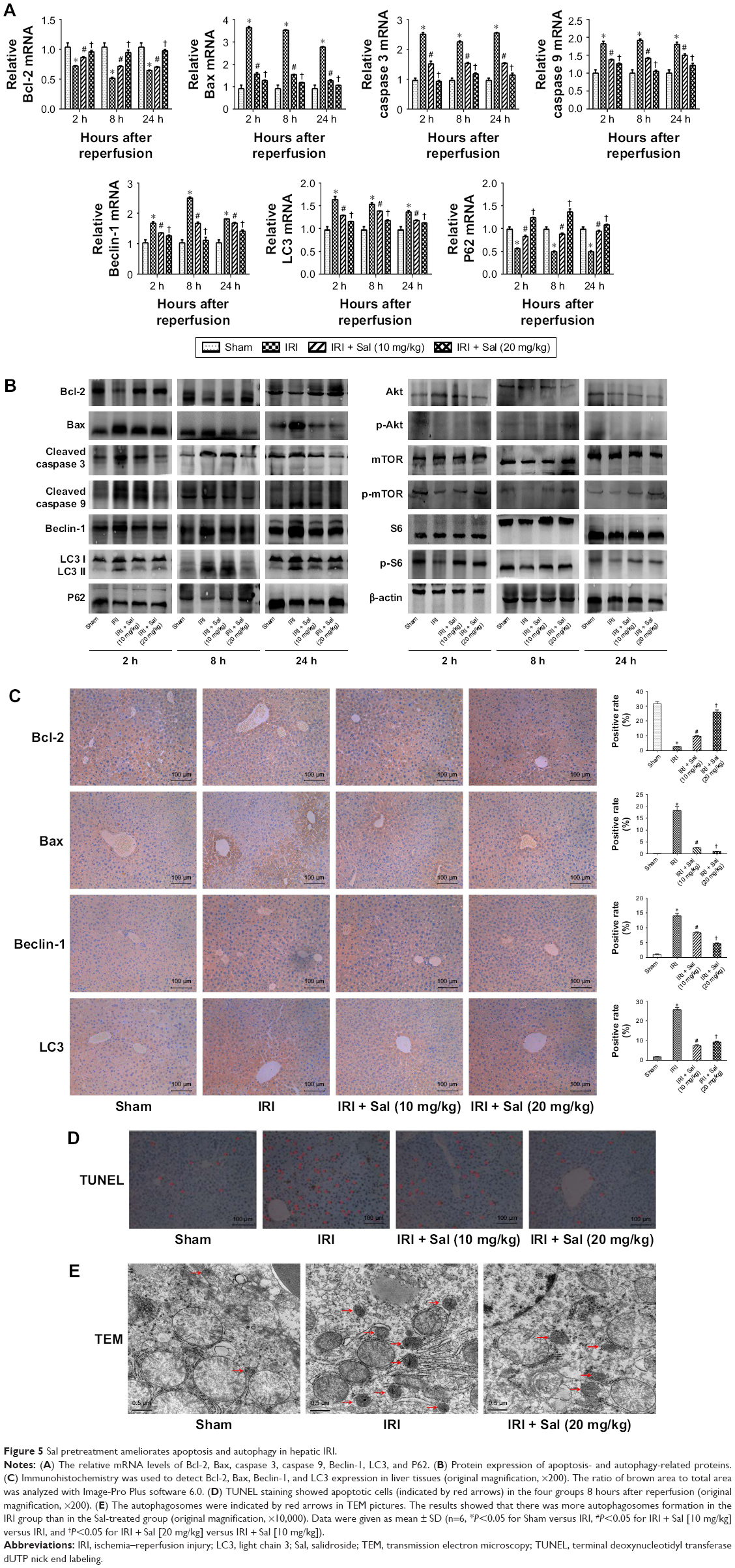 | Figure 5 Sal pretreatment ameliorates apoptosis and autophagy in hepatic IRI. |
Sal inhibits the activation of MAPK signaling in hepatic IRI
We quantified levels of the main components of the MAPK pathway, including P38, ERK, and JNK, and their phosphorylated proteins p-P38, p-ERK, and p-JNK in liver tissues by Western blotting. The expression of P38, ERK, and JNK was similar in the four groups. However, the levels of p-P38, p-ERK, and p-JNK were all upregulated in IRI mice. This was reversed by Sal pretreatment (both 10 and 20 mg/kg; stronger effect with 20 mg/kg) (Figure 6). These results suggest that MAPK activation is inhibited by Sal during hepatic IRI.
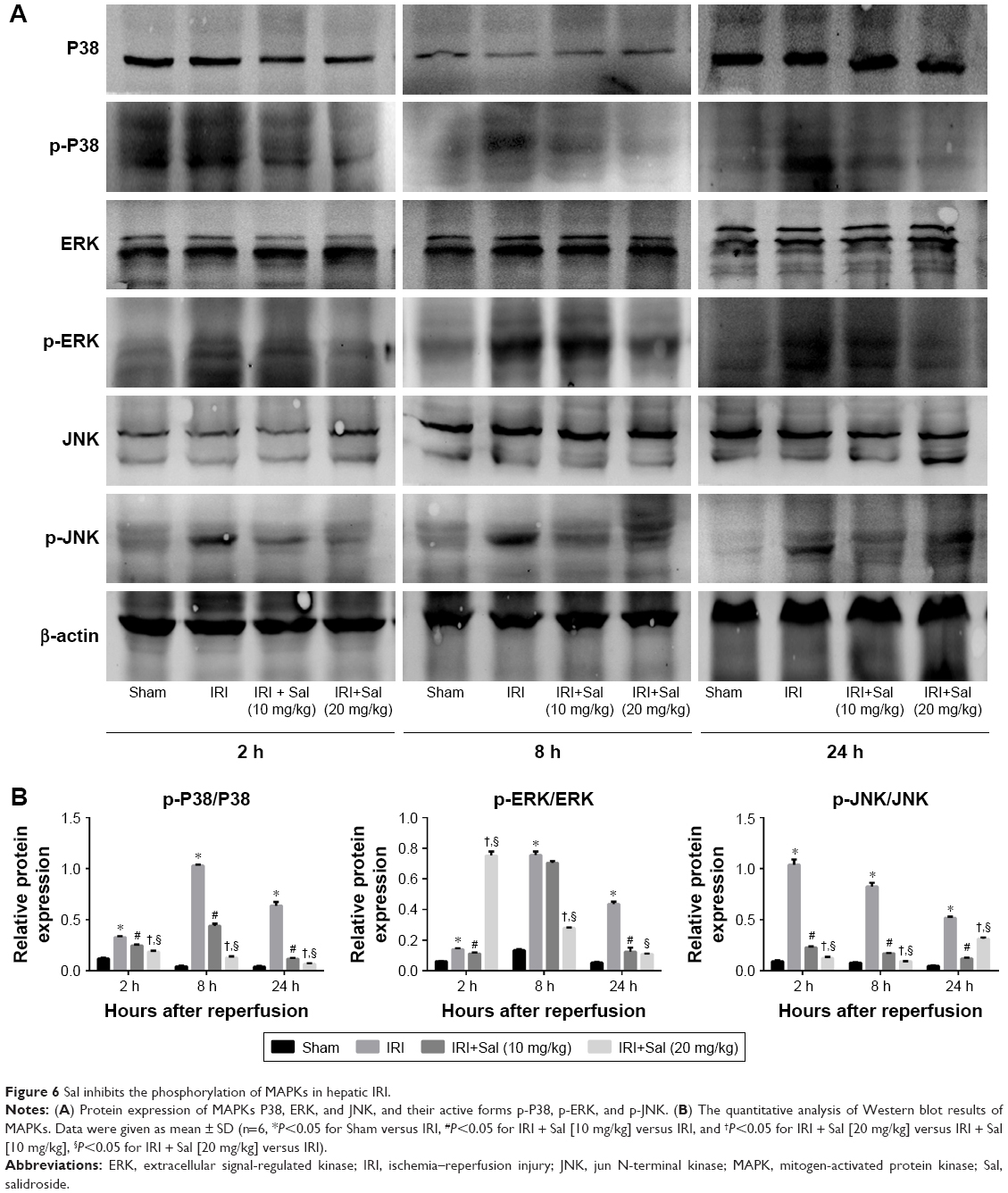 | Figure 6 Sal inhibits the phosphorylation of MAPKs in hepatic IRI. |
The protective effects of Sal on hepatic IRI are associated with MAPKs
We used anisomycin, a specific activator of P38 and JNK, to investigate whether MAPKs are involved in the protective effect of Sal against IRI. As shown in Figure 7A, the serum levels of ALT and AST were significantly higher in the IRI + Ani (20 mg/kg) group than in the IRI group. However, cotreatment with Sal ameliorated this increase. The mRNA expression levels of IL-6, TNF-α, Bax, Beclin-1, and LC3 were higher in the IRI + Ani (20 mg/kg) group than in the IRI groups, and the level of Bcl-2 was lower (Figure 7B). However, Sal treatment reversed these effects. These results were consistent with Western blot analysis (Figure 7C). These results indicated that liver injury following hepatic IRI is enhanced by anisomycin-induced MAPK overactivation and that this can be reversed by Sal treatment. These results support our speculation that Sal can relieve hepatic IRI by inhibiting inflammation, apoptosis, and autophagy, partly through the MAPK pathway.
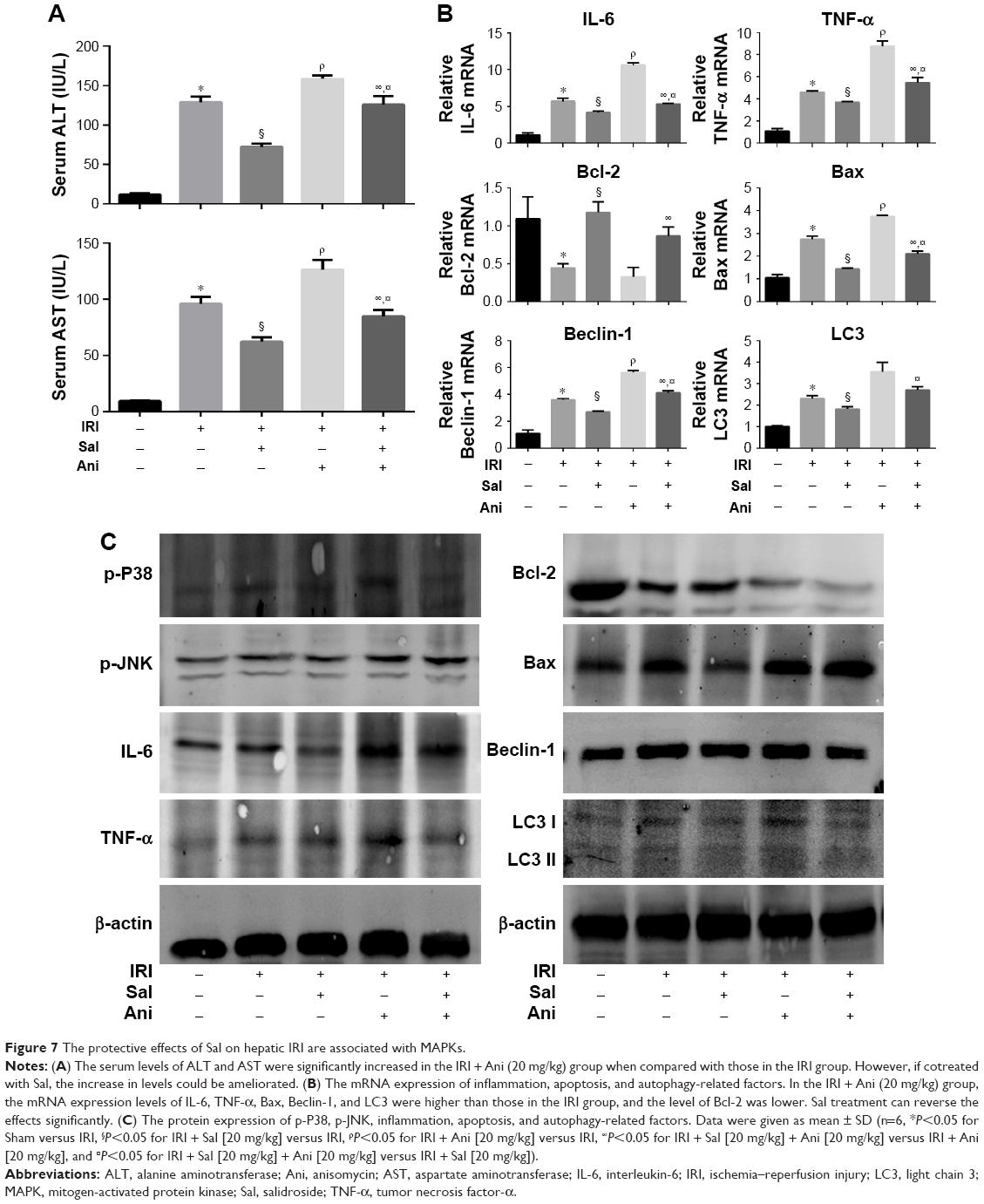 | Figure 7 The protective effects of Sal on hepatic IRI are associated with MAPKs. |
Discussion
Hepatic IRI is an important pathogenesis in clinical practice. It can be divided into ischemia and reperfusion phases.4,26,27 During the ischemia phase, liver tissues are exposed to ischemic anoxia, which disrupts mitochondria, Na+–K+ ATPase activity, and the production of reperfusion molecules such as ROS, inflammatory cytokines, chemokines, and adhesion molecules.4,5 The reperfusion phase is itself composed of two phases. In the early phase (0–6 hours after reperfusion), hepatocytes are swollen, ROS are released, and Kupffer cells are activated. The activation of Kupffer cells produces TNF-α, IL-6, IL-1β, interferon-γ (IFN-γ), and so on, leading to the activation and accumulation of neutrophils and T cells.27,28 In the late phase, neutrophils kill hepatocytes and promote hepatic IRI.3,5 T cells, especially CD4+ T cells, further activate Kupffer cells and cytokine release by producing granulocyte macrophage colony-stimulating factor, IFN-γ and TNF-β.29 These cascade reactions are cyclic processes that continuously injure liver tissue after hepatic IRI.
In this study, we investigated the effects of Sal, which does not negatively affect serum liver enzymes levels, on early- and late-stage hepatic IRI. Our results demonstrate that Sal pretreatment can reduce liver injury by downregulating the liver enzymes ALT and AST and reducing pathological manifestations. As discussed above, inflammatory reactions play a role in the initial phase of hepatocyte injury, so we measured IL-6 and TNF-α levels in the serum and liver tissues. Sal pretreatment dramatically decreased IL-6 and TNF-α levels in serum and liver tissues. Therefore, inflammatory reactions are inhibited by Sal in hepatic IRI. Next, we investigated the mechanisms underlying Sal-mediated protection.
MAPKs are widely expressed in vertebrate animals. They transmit extracellular stimuli to intracellular targets to regulate growth, differentiation, stress, inflammation, and apoptosis.19,30,31 MAPKs, including P38, JNK, and ERK, are activated during hepatic IRI and play a role in the necrosis, apoptosis, and autophagy of hepatocytes.6,32 In our study, we used the anisomycin, a specific activator of P38 and JNK, to further investigate whether the protective effect of Sal was associated with MAPKs. Our results showed that Sal can relieve hepatic IRI enhanced by Ani, including the inhibition of inflammation, apoptosis, and autophagy. These revealed that the Sal protects hepatocytes from IRI partly through the MAPK pathway.
MAPKs can be activated by phosphorylation cascades in response to oxidative stress, proinflammatory cytokines, DNA damage, heat injury, ultraviolet radiation, and so on.33,34 In hepatic IRI, ROS may directly act on cell membrane receptors to phosphorylate P38.6,27 P-P38 promotes translocation of Bax, which is a proapoptotic factor, from the cytoplasm to mitochondria.35 Mitochondria control apoptosis by altering their membrane permeability.36 The translocation of Bax to the mitochondria increases the permeability of the mitochondrial membrane, leading to the release of cytochrome c and the activation of caspase 9 and caspase 3.37 Then, apoptosis is activated by the phosphorylation of P38. P-P38 can also act on monocyte macrophages or promote the transcription of NF-KB, resulting in the release of more TNF-α, IL-6, and IL-1β (Figure 8).6
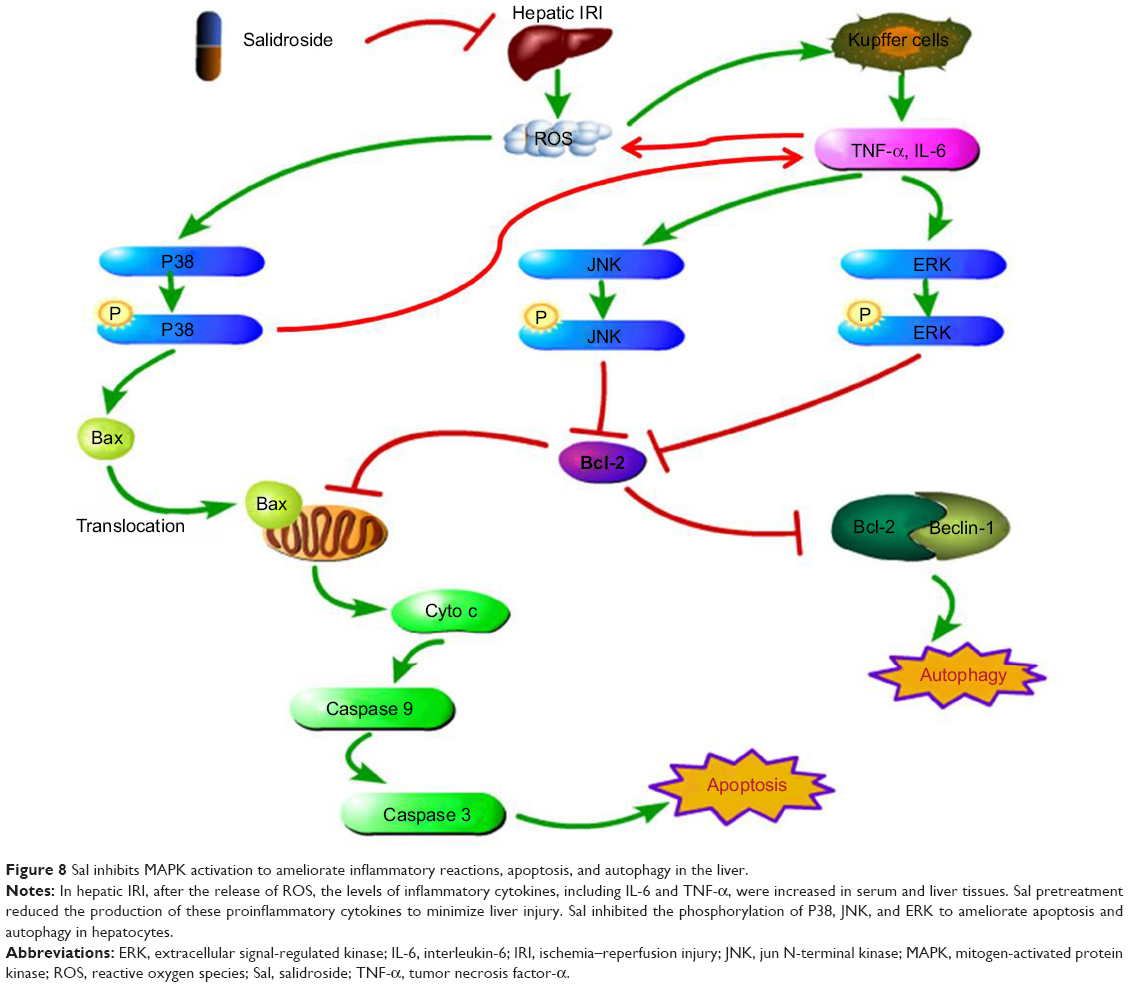 | Figure 8 Sal inhibits MAPK activation to ameliorate inflammatory reactions, apoptosis, and autophagy in the liver. |
TNF-α and other inflammatory cytokines activate JNK and ERK.6,22 TNF-α is generated by Kupffer cells, endothelial cells, and T cells, among others.38,39 It functions not only as a proinflammatory factor but also as a proapoptotic factor.40 TNF-α can strongly activate JNK and ERK, followed by the phosphorylation of Bcl-2. Bcl-2 is an antiapoptotic factor that reduces permeability of the mitochondria membrane and the release of cytochrome c. Phosphorylation of Bcl-2 inactivates it and facilitates apoptosis.41,42 Activated JNK also translocates into the nucleus, activating the transcription of proapoptotic genes, such as Fas-L, Bak, and p53.43 In addition, activated ERK can also translocate to the nucleus, promoting the transcription of genes involved in the proliferation of Kupffer cells.44 Hence, hepatocyte injury and apoptosis are accelerated by JNK and ERK activation.45
Our study showed that Bax, caspase 3, and caspase 9 were overexpressed in IRI mice, while Bcl-2 levels were decreased. These findings were consistent with previous reports that apoptosis is enhanced in hepatic IRI mice.6,15–17,22,27 We found that Sal pretreatment reduced these effects and inhibited the activation of apoptosis. p-P38, p-JNK, and p-ERK levels were all decreased in Sal pretreated groups, suggesting that Sal protects hepatocytes from IRI by inhibiting the activation of MAPK signaling to attenuate apoptosis.
Autophagy is characterized by the formation of autophagosomes and contributes to the pathogenesis of IRI.2,26,46,47 Sometimes, autophagy promotes survival under stress conditions. However, overactivation of autophagy results in cell death.43 Previous studies have found that autophagy is excessive during hepatic IRI in mice. Autophagy can be regulated by two pathways. The mTOR pathway is the negatively regulated one. The activation of mTOR can phosphorylate ULK1/2 and Atg13 to prevent the formation of mTOR complex1, which is crucial in autophagosome formation, to downregulate autophagy. What is more, mTOR can also phosphorylate and activate ribosomal protein S6 kinase, which can further activate ribosomal protein S6 and result in the protein synthesis, the adhesion of ribosome to endoplasmic reticulum, and the inhibition of autophagy.48 The positively regulated pathway is the Beclin-1 and phosphatidyl inositol 3-kinase class III (PI3K3C), or vesicular protein sorting 34 (VPS34). Autophagy and apoptosis are closely connected. Bcl-2 is a crucial protein in the crosstalk. In normal cells, Bcl-2 forms a complex with Beclin-1. This complex disrupts the interaction between Beclin-1 and PI3K3C, which participates in the initiation of autophagy.48 Besides, the LC3, which undergoes transformation from LC3 I to LC3 II during autophagy, conjugates to the autophagosomal membrane and plays a role in the extension of the autophagosome membrane and the eventual closure of the autophagic vesicle.12 The P62 is also degraded during the formation of autophagosomes.
In our study, we showed that Beclin-1 and LC3 were increased, and P62, p-Akt, p-mTOR, and p-S6 were decreased in IRI mice. This was reversed by Sal pretreatment to inhibit autophagy. These results were consistent with previous studies showing that autophagy is enhanced in hepatic IRI. Our results suggest that Sal is a putative antiautophagy candidate.
Taken together, our study has shown that Sal safely protects hepatocytes from IRI partly by inhibiting the activation of MAPK signaling, including the phosphorylation of P38, JNK, and ERK. This ameliorates inflammatory reactions, apoptosis, and autophagy in the liver. Our findings indicate that Sal can be used clinically as a preconditioning drug to reduce hepatic IRI during liver resection, liver transplantation, and other medical interventions.
However, there are some limitations to our study. For example, further investigation is required to clarify the structure–activity relationship between Sal and hepatic IRI. The comorbidities can also interfere with the protection effect of Sal. Whether Sal is safe for clinical use also remains unclear. Furthermore, the therapeutic effects of Sal need to be compared with existing clinical drugs.
Conclusions
The present study has demonstrated that Sal is a safe agent for the prevention of hepatic IRI in mice. We confirmed that Sal protects hepatocytes from IRI partly by inhibiting the activation of MAPK signaling, including the phosphorylation of P38, JNK, and ERK, to ameliorate the inflammatory reactions, apoptosis, and autophagy in the liver.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 81670472). We are grateful to Dr Wenwen Wang, Xiya Lu, Qiang Yu, Kan Chen, Yujing Xia, Jie Lu, Ling Xu, and Yingqun Zhou. They helped us a lot in the experiment process and data collection. Jiao Feng, Qinghui Zhang and Wenhui Mo shared first authorship for this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Li Y, Yang Y, Feng Y, et al. A review of melatonin in hepatic ischemia/reperfusion injury and clinical liver disease. Ann Med. 2014;46(7):503–511. | ||
Theodoraki K, Tympa A, Karmaniolou I, Tsaroucha A, Arkadopoulos N, Smyrniotis V. Ischemia/reperfusion injury in liver resection: a review of preconditioning methods. Surg Today. 2011;41(5):620–629. | ||
Guan LY, Fu PY, Li PD, et al. Mechanisms of hepatic ischemia-reperfusion injury and protective effects of nitric oxide. World J Gastrointest Surg. 2014;6(7):122–128. | ||
Datta G, Fuller BJ, Davidson BR. Molecular mechanisms of liver ischemia reperfusion injury: insights from transgenic knockout models. World J Gastroenterol. 2013;19(11):1683–1698. | ||
Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284(1):G15–G26. | ||
Li J, Wang F, Xia Y, et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar Drugs. 2015;13(6):3368–3387. | ||
Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33(2):397–405. | ||
Natori S, Selzner M, Valentino KL, et al. Apoptosis of sinusoidal endothelial cells occurs during liver preservation injury by a caspase-dependent mechanism. Transplantation. 1999;68(1):89–96. | ||
Bilbao G, Contreras JL, Eckhoff DE, et al. Reduction of ischemia-reperfusion injury of the liver by in vivo adenovirus-mediated gene transfer of the antiapoptotic Bcl-2 gene. Ann Surg. 1999;230(2):185–193. | ||
Ben-Ari Z, Pappo O, Cheporko Y, et al. Bax ablation protects against hepatic ischemia/reperfusion injury in transgenic mice. Liver Transpl. 2007;13(8):1181–1188. | ||
Go KL, Lee S, Zendejas I, Behrns KE, Kim JS. Mitochondrial dysfunction and autophagy in hepatic ischemia/reperfusion injury. BioMed Res Int. 2015;2015:183469. | ||
Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47(5):1773–1785. | ||
Sybers HD, Ingwall J, DeLuca M. Autophagy in cardiac myocytes. Recent Adv Stud Cardiac Struct Metab. 1976;12:453–463. | ||
Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98(2):425–444. | ||
Shen M, Lu J, Dai W, et al. Ethyl pyruvate ameliorates hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm. 2013;2013:461536. | ||
Wang C, Chen K, Xia Y, et al. N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS One. 2014;9(9):e108855. | ||
Cheng P, Wang F, Chen K, et al. Hydrogen sulfide ameliorates ischemia/reperfusion-induced hepatitis by inhibiting apoptosis and autophagy pathways. Mediators Inflamm. 2014;2014:935251. | ||
Wang Y, Xu P, Wang Y, Liu H, Zhou Y, Cao X. The protection of salidroside of the heart against acute exhaustive injury and molecular mechanism in rat. Oxid Med Cell Longev. 2013;2013:507832. | ||
Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15(1):11–18. | ||
Nakagawa H, Maeda S. Molecular mechanisms of liver injury and hepatocarcinogenesis: focusing on the role of stress-activated MAPK. Pathol Res Int. 2012;2012:172894. | ||
Koike N, Takeyoshi I, Ohki S, Tokumine M, Matsumoto K, Morishita Y. Effects of adding P38 mitogen-activated protein-kinase inhibitor to celsior solution in canine heart transplantation from non-heart-beating donors. Transplantation. 2004;77(2):286–292. | ||
Schwabe RF, Brenner DA. Mechanisms of liver injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290(4):G583–G589. | ||
Zhang Y, Li L, Lin L, et al. Pharmacokinetics, tissue distribution, and excretion of salidroside in rats. Planta Med. 2013;79(15):1429–1433. | ||
Zhu L, Wei T, Gao J, et al. The cardioprotective effect of salidroside against myocardial ischemia reperfusion injury in rats by inhibiting apoptosis and inflammation. Apoptosis. 2015;20(11):1433–1443. | ||
Zou H, Liu X, Han T, et al. Salidroside protects against cadmium-induced hepatotoxicity in rats via GJIC and MAPK pathways. PLoS One. 2015;10(6):e0129788. | ||
Ruan W, Liu Q, Chen C, Li S, Xu J. [Limb remote ischemic preconditioning attenuates liver ischemia reperfusion injury by activating autophagy via modulating PPAR-gamma pathway]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2016;41(9):918–928. | ||
Nastos C, Kalimeris K, Papoutsidakis N, et al. Global consequences of liver ischemia/reperfusion injury. Oxidative Med Cell Longevity. 2014;2014:906965. | ||
Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA Jr. Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest. 1990;85(6):1936–1943. | ||
Mendes-Braz M, Elias-Miro M, Jimenez-Castro MB, Casillas-Ramirez A, Ramalho FS, Peralta C. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J Biomed Biotechnol. 2012;2012:298657. | ||
Dufresne SD, Bjorbaek C, El-Haschimi K, et al. Altered extracellular signal-regulated kinase signaling and glycogen metabolism in skeletal muscle from p90 ribosomal S6 kinase 2 knockout mice. Mol Cell Biol. 2001;21(1):81–87. | ||
Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002;3(7):REVIEWS3009. | ||
Jiang Y, Gram H, Zhao M, et al. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem. 1997;272(48):30122–30128. | ||
Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265(5173):808–811. | ||
Sanchez-Fidalgo S, Villegas I, Rosillo MA, Aparicio-Soto M, de la Lastra CA. Dietary squalene supplementation improves DSS-induced acute colitis by downregulating p38 MAPK and NFkB signaling pathways. Mol Nutr Food Res. 2015;59(2):284–292. | ||
Park GB, Choi Y, Kim YS, Lee HK, Kim D, Hur DY. ROS-mediated JNK/p38-MAPK activation regulates Bax translocation in sorafenib-induced apoptosis of EBV-transformed B cells. Int J Oncol. 2014;44(3):977–985. | ||
Marchetti P, Hirsch T, Zamzami N, et al. Mitochondrial permeability transition triggers lymphocyte apoptosis. J Immunol. 1996;157(11):4830–4836. | ||
Gibson EM, Henson ES, Haney N, Villanueva J, Gibson SB. Epidermal growth factor protects epithelial-derived cells from tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by inhibiting cytochrome c release. Cancer Res. 2002;62(2):488–496. | ||
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65. | ||
Ibrahim MA, Abdel-Gaber SA, Amin EF, Ibrahim SA, Mohammed RK, Abdelrahman AM. Molecular mechanisms contributing to the protective effect of levosimendan in liver ischemia-reperfusion injury. Eur J Pharmacol. 2014;741:64–73. | ||
Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001;15(11):1419–1426. | ||
Chinnaiyan AM, Orth K, O’Rourke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996;271(9):4573–4576. | ||
Armstrong RC, Aja T, Xiang J, et al. Fas-induced activation of the cell death-related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;271(28):16850–16855. | ||
Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48. | ||
Lloberas J, Valverde-Estrella L, Tur J, Vico T, Celada A. Mitogen-activated protein kinases and mitogen kinase phosphatase 1: a critical interplay in macrophage biology. Front Mol Biosci. 2016;3:28. | ||
Malik S, Suchal K, Gamad N, Dinda AK, Arya DS, Bhatia J. Telmisartan ameliorates cisplatin-induced nephrotoxicity by inhibiting MAPK mediated inflammation and apoptosis. Eur J Pharmacol. 2015;748: 54–60. | ||
Dong Y, Undyala VV, Gottlieb RA, Mentzer RM Jr, Przyklenk K. Autophagy: definition, molecular machinery, and potential role in myocardial ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther. 2010;15(3):220–230. | ||
Hong JM, Kim SJ, Lee SM. Role of necroptosis in autophagy signaling during hepatic ischemia and reperfusion. Toxicol Appl Pharmacol. 2016;308:1–10. | ||
Baranyai T, Nagy CT, Koncsos G, et al. Acute hyperglycemia abolishes cardioprotection by remote ischemic perconditioning. Cardiovasc Diabetol. 2015;14:151. |
Supplementary materials
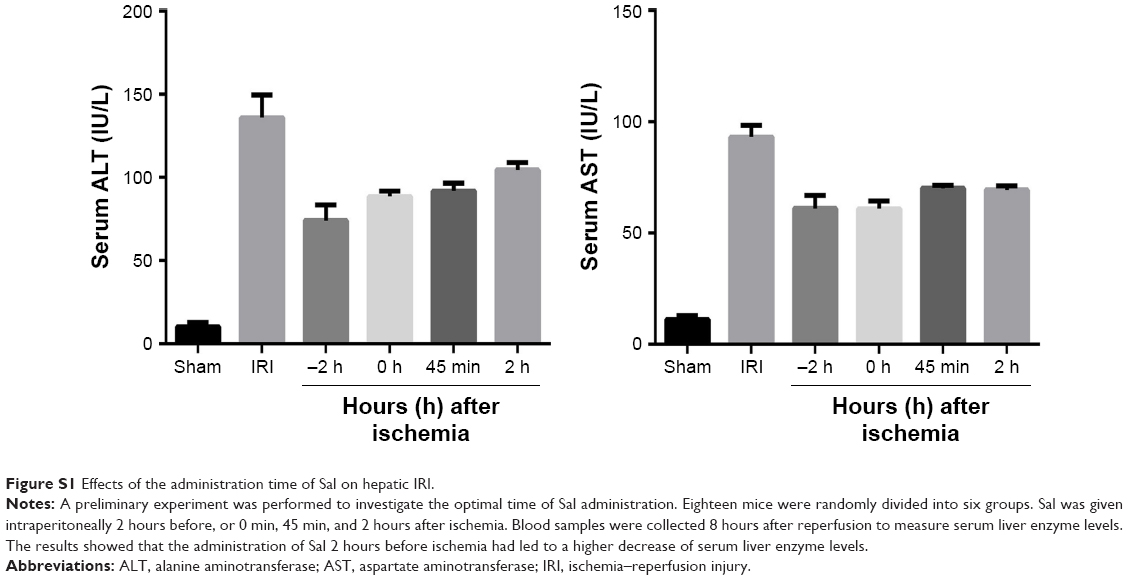 | Figure S1 Effects of the administration time of Sal on hepatic IRI. |
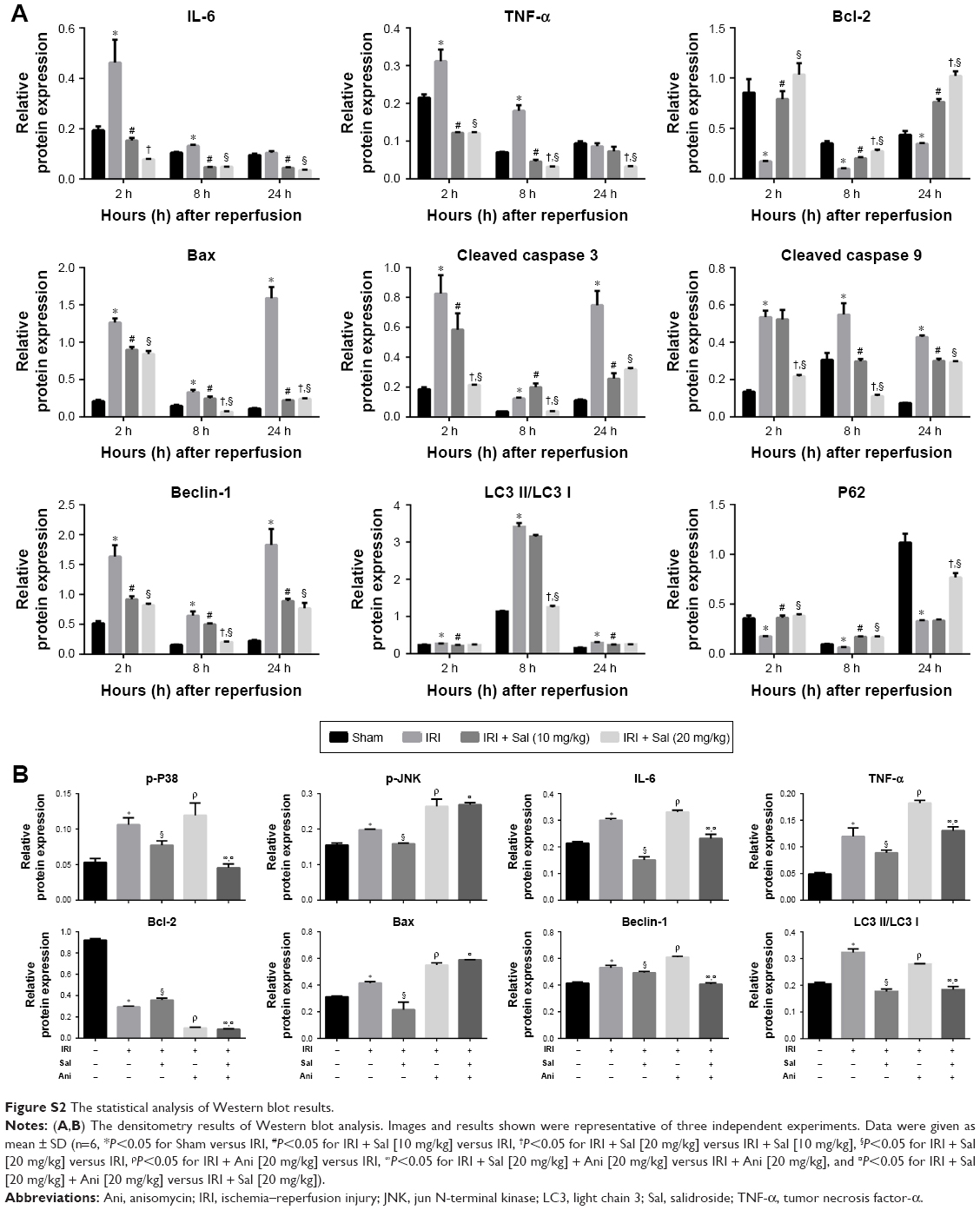 | Figure S2 The statistical analysis of Western blot results. |
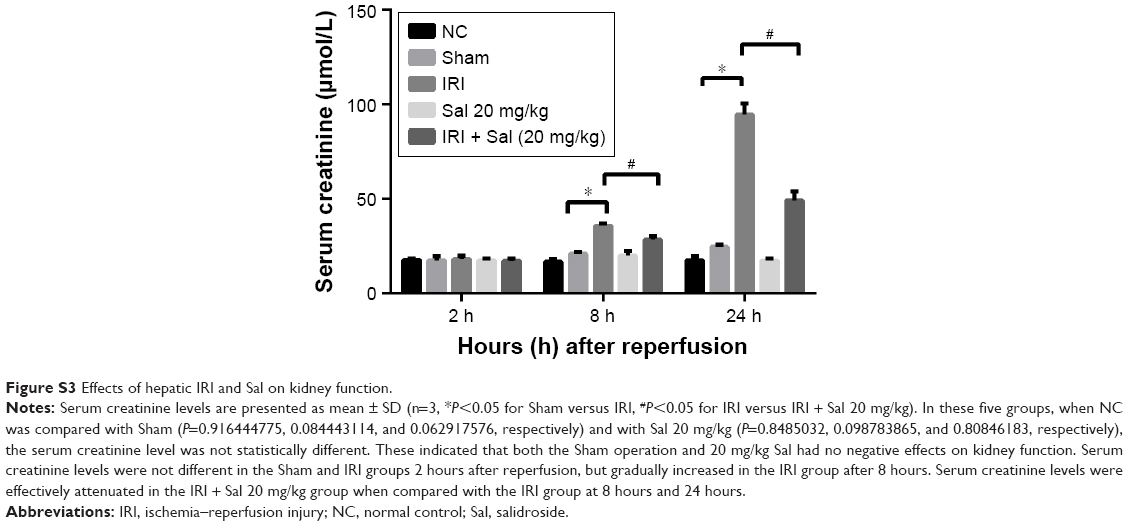 | Figure S3 Effects of hepatic IRI and Sal on kidney function. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.