")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Safety, Tolerability, and Pharmacokinetics of Ibuprofenamine Hydrochloride Spray (NSAIDs), a New Drug for Rheumatoid Arthritis and Osteoarthritis, in Healthy Chinese Subjects
Authors Xie P, Xue W, Qi W, Li Y, Yang L, Yang Z, Shi A
Received 2 December 2020
Accepted for publication 23 January 2021
Published 16 February 2021 Volume 2021:15 Pages 629—638
DOI https://doi.org/10.2147/DDDT.S294849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Panpan Xie,1 Wei Xue,1 Wenyuan Qi,1 Yang Li,1 Lei Yang,1 Zhaojun Yang,2 Aixin Shi1
1Clinical Trial Center, Beijing Hospital, National Center of Gerontology; Institute of Geriatric Medicine, Chinese Academy of Medical Science; Beijing Key Laboratory of Drug Clinical Risk and Personalized Medication Evaluation, Beijing, 100730, People’s Republic of China; 2Research and Development Department, Xin Chen Taifei Medical Technology Co. LTD, Tianjin, 300221, People’s Republic of China
Correspondence: Aixin Shi No. 1 Dahua Road, Dongcheng District, Beijing, People’s Republic of China
Email [email protected]
Background: Ibuprofenamine hydrochloride spray is novel transdermal nonsteroidal anti-inflammatory drugs (NSAIDs), under clinical development for the treatment of Rheumatoid Arthritis and Osteoarthritis as a novel transdermal drug.
Methods: A single and multiple ascending dose study investigated the safety, tolerability and pharmacokinetics of ibuprofenamine hydrochloride in healthy Chinese subjects. A total of 34 subjects (single-dose study: 34 subjects and multiple-dose study: 20 subjects) were involved in the trial. In the single-dose study, subjects were assigned to one of the four groups received 35, 70, 140, 280 mg. In the 70 mg and 140 mg treatment groups, subjects received one dose on the first day and twice a day from day 6 to 12. The starting dose was determined considering the no observed adverse effect level based on preclinical studies, and the dose escalations in subsequent cohorts were decided based on safety, tolerability, and pharmacokinetic data from previous dose cohorts.
Results: After a single dose, both ibuprofenamine and ibuprofen plasma exposure showed a more than dose-proportional increase across a dose range of 35– 280 mg. After multiple dosing, both ibuprofenamine and ibuprofen steady-state exposure increased obviously more than dose-proportional manner across the evaluated dose range (twice a day for 7 days) resulted in obvious accumulation. Single or multiple doses of ibuprofenamine hydrochloride were generally well tolerated and no obvious skin irritation was observed.
Conclusion: Ibuprofenamine hydrochloride exhibited a safety and pharmacokinetic profile that supports its future investigation as a potential therapeutic for Rheumatoid Arthritis and Osteoarthritis.
Keywords: ibuprofenamine hydrochloride spray, tolerability, safety, pharmacokinetics
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used to relieve symptoms of rheumatoid arthritis, osteoarthritis and other inflammations, but also cause many side effects. The most common side effects are gastrointestinal reactions, symptoms include indigestion, heartburn, vomiting, gastrointestinal bleeding, stomach ulcers and gastritis. Gastric and duodenal bleeding caused by NSAIDs is usually painless but can cause blood in the stool, leading to long-term iron deficiency anemia and even life-threatening.1,2
Transdermal administration can avoid direct damage to the gastrointestinal tract, which is safer than oral administration. Due to the low transdermal rate of nonsteroidal anti-inflammatory drugs, there are several insufficient transdermal preparations for NSAIDs.3,4 First, drugs are absorbed slowly through the skin, resulting in low bioavailability about 3–5%. Second, a high concentration of the drug in the skin area inhibits the synthesis of prostaglandins required for skin regeneration, thereby causing damage to the skin. Third, the effective dose is very high, generally requires 3–4 times a day. Finally, NSAIDs cannot effectively penetrate the articular cartilage tissue, it is difficult to achieve the purpose of treatment.
Ibuprofenamine hydrochloride (2-(Diethylamino) ethyl 2-(4-isobutylphenyl) propionate hydrochloride, structure shown in Figure 1) spray is new transdermal NSAIDs, developed by Xin Chen Taifei Medical Technology Co. Ltd., Tianjin, China with good transdermal properties and can penetrate the skin and biological barrier into the lesion tissue after administration. Ibuprofenamine hydrochloride is the prodrug of ibuprofen, which will be converted into therapeutic ibuprofen soon after absorption. Based on experimental data obtained in animals, the concentration of ibuprofen in the blood exceeds 95% of the total drug. The bioavailability of ibuprofenamine hydrochloride is 75%, reaching the level of many oral drugs, while the bioavailability of ibuprofen transdermal absorption is only 1%. Although ibuprofenamine hydrochloride is quickly converted into Ibuprofen in the blood, but very little conversion in the skin, indicating that the damage to the skin is small.
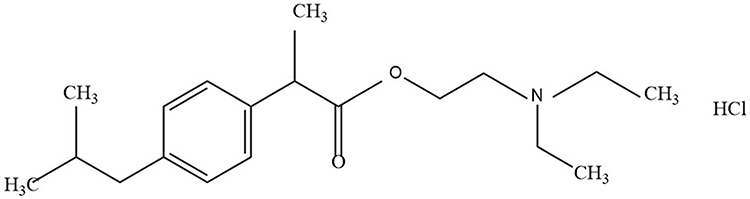 |
Figure 1 The chemical structure of ibuprofenamine hydrochloride. |
The safety study results of the single and multiple doses in healthy male and female subjects receiving ibuprofenamine hydrochloride 17.5 mg to 280 mg show that the subjects are well tolerated in the USA. This study aimed to evaluate the safety, tolerability and pharmacokinetics (PKs) of ibuprofenamine hydrochloride after single and multiple administrations in healthy Chinese subjects.
Materials and Methods
Subjects
All the subjects were given detailed written and oral information about the study and written informed consent was obtained before screening for eligibility. The eligible subjects were healthy subjects aged between 18 and 45 years whose body mass index (BMI) was in the range of 19.0–28.0 kg/m2. All the subjects were deemed healthy by a physical examination, medical history, 12-lead electrocardiograms (ECGs), vital signs, and laboratory tests (hematology, blood chemistry, and urinalysis). Subjects were excluded if there was a history or evidence of any of the following: significant disease of the vital organs; skin diseases wounds or other symptoms in the medication area; non-steroidal anti-inflammatory drug allergy; alcoholism or drug abuse or smoker; the use of any prescription drug within 4 weeks before the scheduled administration of study drug; the use of any over-the-counter medication or herbal medication or foods that affect drug-metabolizing enzymes within 2 weeks before the scheduled administration of study drug; and participation in any other clinical trial within 3 months before the scheduled administration of study drug. Subjects who had the following vital signs were also excluded: systolic blood pressure ≤89 mmHg or ≥140 mmHg and diastolic blood pressure ≤59 mmHg or ≥90 mmHg.
Study Design
This study was performed at the clinical trial research center of Beijing hospital, Republic of China in accordance with the Declaration of Helsinki and principles of the Declaration of China Good Clinical Practice. The study was approved by China Food and Drug Administration, the protocol was reviewed and approved by the Ethics Review Committee of Beijing Hospital. This study was registered at ClinicalTrials.gov (https://ClinicalTrial.gov, identifier: NCT02794740).
The study was designed as a double-blinded, randomized, placebo-controlled, dose-escalation study. The study consisted of two parts: a single ascending dose study and a multiple dose study, as shown in Figure 2. A total of 34 subjects (single-dose study: 34 subjects and multiple-dose study: 20 subjects) were involved in the trial. In the single-dose study, subjects were assigned to one of the four groups that received 35, 70, 140, 280 mg ibuprofenamine hydrochloride. In the 70 mg and 140 mg treatment groups, subjects received one dose on the first day and twice a day from day 6 to 12. Each treatment group consisted of 10 subjects (study drug-to-placebo ratio was 8:2), except for the 35 mg group, there are only 4 subjects without placebo. All subjects were randomized to treatment, and a randomization code was generated by an independent external provider. An escalation to the next dose level was decided only after the safety and tolerability data for all subjects in the cohort from the previously administered dose cohort were reviewed.
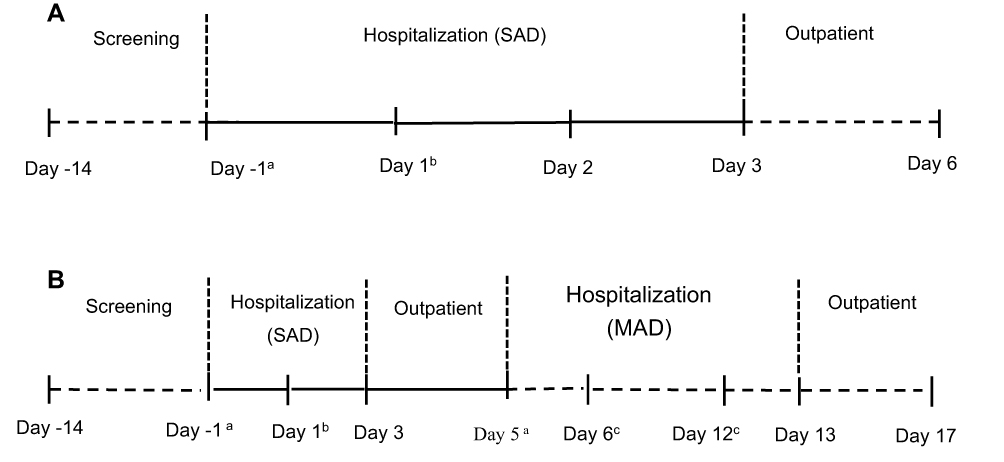 |
Figure 2 Study design of the single ascending dose (SAD) study (A) and multiple ascending dose (MAD) study (B). Notes: aBaseline; bRandomization and study drug administration; cStudy drug administration; A: 35 mg, 280 mg; B: 70 mg, 140 mg. |
PK Assessments
In the SAD study, venous blood samples (4 mL) for the PK study of ibuprofenamine and ibuprofen were collected before administration and 1, 3, 6, 9, 12, 16, 24, 32, 36, 40, 48, 72, 96 and 120 hours after administration. In the MAD study, venous blood samples for the PK study of ibuprofenamine and ibuprofen were collected before administration and 1, 3, 6, 9, 12, 16, 24, 32, 36, 40, 48, 72, 96 and 120 hours after administration on day 1. On day 11, venous blood samples were taken before administration. On day 12, venous blood samples were taken before administration and 1, 3, 6, 9, 12, 16, 24, 32, 36, 40, 48, 72, 96 and 120 hours after administration.
The blood samples were centrifuged at 3,000 rpm for 10 min at 4°C, and the obtained plasma samples were transferred into 4 polypropylene tubes (1.5 mL). All the tubes were immediately stored in a freezer at −70°C until analysis.
The plasma concentration of ibuprofenamine and ibuprofen were analyzed using high-performance liquid and mass spectrometry (HPLC-MS/MS). The system comprised an LC-20AD™ (Shimadzu System, Shimadzu, Japan) coupled with a mass spectrometry (Qtrap 5500, AB SCIEX, USA) to determine the ibuprofenamine concentration in plasma. The system comprised an LC-30AD™ (Shimadzu System, Shimadzu, Japan) coupled with a mass spectrometry (API 5000, AB SCIEX, USA) to determine the ibuprofen concentration in plasma. Briefly, 40 μL of plasma was mixed with 40 μL buffer solution, added 320 μL acetonitrile with 1% formic acid, vortexed for 3 min, and centrifuged for 5 min at 13,000 rpm. After transferring 150 μL of supernatant to another EP tube, 150 μL of acetonitrile and water (10:90) with 0.1% formic acid was added, vortexed for 1 min, and centrifuged for 5 min at 13,000 rpm. Then, 20 μL of the aliquot was injected into the HPLC-MS/MS system for analysis of ibuprofenamine. 100 μL of plasma was mixed with 100 μL acetonitrile and water (10:90) with 0.1% formic acid, added 1000 μL ethyl acetate, vortexed for 3 min, and centrifuged for 5 min at 13,000 rpm. After transferring 700 μL of supernatant to another EP tube, concentrated under reduced pressure apparatus evaporated to dryness, 300 μL of acetonitrile and water (10:90) was added, vortexed for 1 min, and centrifuged for 10 min at 13,000 rpm. Then, a 15 μL of the aliquot was injected into the HPLC-MS/MS system for analysis of ibuprofen. The quantification was performed using multiple reaction monitoring of the transitions m/z 306.2→161.4 and m/z 205.1→161.1 for ibuprofenamine and ibuprofen, respectively. The calibration curves were linear over the range of 40–20,000 pg mL−1 for ibuprofenamine and 1–500 ng mL−1 for ibuprofen. The limits of quantification for ibuprofenamine and ibuprofen were 40 pg mL−1 and 1 ng mL−1, respectively.
The PK parameters of ibuprofennamine and ibuprofen were estimated using non-compartment model. The maximum plasma concentration (Cmax) after a single dose, time to Cmax (Tmax), and the area under the plasma concentration–time curve (AUC) from zero extrapolated to infinity (AUCinf), the AUC from time 0 hour to the last quantifiable concentration (AUClast), the maximum plasma concentration at steady-state (Cmax,ss) after multi-dose, time to Cmax,ss (Tmax) and the AUC over the dosing interval at steady-state (AUCss) were calculated using the linear trapezoidal rule. Any BLQ (below the limit of quantification) values occurring prior to Cmax were assigned a value of zero; BLQ values after the last quantifiable concentration were treated as missing. The accumulation index was calculated as the ratio of the AUClast and Cmax,ss on day 12 to the AUClast and Cmax on day 1.
Safety and Tolerability Assessments
Safety and tolerability assessments were conducted in all subjects who received one or more doses of the study drug. Safety and tolerability were evaluated based on data from physical examination, including vital signs such as systolic and diastolic blood pressures, pulse rate and temperature, ECG, laboratory test results (hematology, clinical chemistry and urinalysis) and skin irritation score obtained throughout the study. Adverse events were reported by the subjects or observed by the medical investigators. All adverse events were coded using MedDRA version 20.1.
The administration site includes knees, thighs, calves and back, the area was marked by 5 cm × 5 cm as a unit. Each unit is sprayed once, while the volume of each spray is 0.07 mL. According to the calculation of the dose, the 35 mg dose group is 8 sprays, the 70 mg dose group is 16 sprays, the 140 mg dose group is 32 sprays, and the 280 mg dose group is 64 sprays. Skin irritation was assessed using a skin irritation assessment system (7 points) and performed on each received site (back, back of thigh, calf and knee) before administration and 30 min after administration. Skin irritation assessment system (0–7 points) were used to assess skin irritation, erythema, edema, or other skin irritation levels after visual inspection: 0 points (no irritation), 1 point (minimal erythema, almost hard to detect), 2 points (obvious erythema, mild edema or papule reaction), 3 points (erythema and pimples), 4 points (significant edema), 5 points (erythema, edema, papules), 6 points (blisters), 7 points (strong reaction beyond the site of administration).
Statistical Analyses
All demographic data, PK parameters, and safety data were summarized using descriptive statistics for continuous variables and using the number and percentage of subjects for categorical variables.
The dose proportionality was assessed for Cmax, AUClast, and AUCinf in the SAD study and for Cmax, AUClast (on day 1), Cmax,ss, Cmin,ss, and AUCss (on day 12) in the MAD study, using a power model with the following equation:
ln (PK parameter) = β0 +β1 × ln (Dose)
The estimate of the slope of the regression line (β1) and the corresponding 95% CI were calculated. All statistical analyses were performed in the SAS® version 9.3 software.
Results
Subjects
A total of 233 subjects were screened, 34 subjects were enrolled, 33 subjects received ibuprofenamine hydrochloride spray (33 in the SAD study and 19 in the MAD study), and 6 subjects received placebo. One subject withdrew from the trial due to high blood pressure before administration, and the remaining 33 subjects completed the study. The safety analysis included 33 subjects in the SAD (N=33) or MAD (N=19) studies. The demographic characteristics of the enrolled subjects are enumerated in Table 1.
 |
Table 1 Demographic Characteristics of the Study Subjects |
Safety and Tolerability
In total, 17 adverse events (AEs) were reported from 11 of the 33 subjects in the SAD study (Table 2), 4 AEs of these were considered to be related to the ibuprofenamine hydrochloride by the investigator. No treatment- or dose-related trend in the incidence of AEs was observed. There were no serious adverse events (SAEs) reported, no subjects were withdrawn from the SAD study due to AEs, and there were no AEs leading to death.
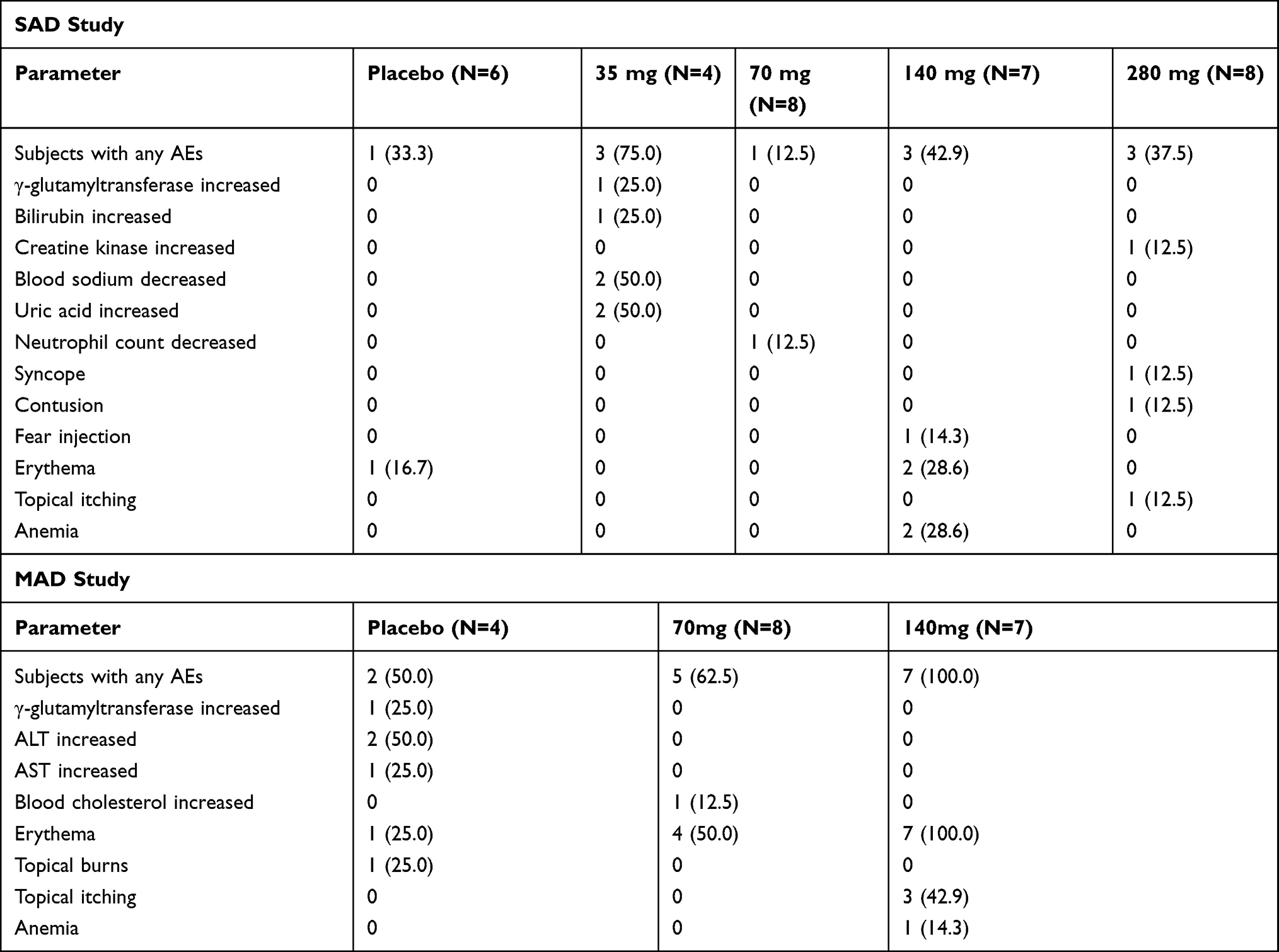 |
Table 2 Adverse Events in the SAD and MAD Studies |
In total, 22 AEs were reported from 47 of the 19 subjects in the MAD study (Table 2). Ninety-one percent of the 47 reported AEs were considered to be related to study treatment by the investigator. No treatment-related trend in the incidence of AEs was founded, and the AEs in the 140 mg treatment group where adverse events occurred in all subjects focused on the assessment of skin irritation. An increase in ALT and AST were showed in the placebo group, the severity was evaluated as mild by the investigator in the MAD study. There were no AEs leading to death.
In the SAD study for the first dose group (35 mg), an increasing trend of bilirubin and uric acid levels were apparent on day 6, with bilirubin and uric acid levels returning to normal after 3 days. An increasing trend of γ-glutamyltransferase levels was apparent on day 6, with γ-glutamyltransferase levels returning to normal after 14 days. A decreasing trend of blood sodium was apparent on day 6, with blood sodium levels returning to normal after 3 days. In the SAD study for 70 mg group, a decreasing trend of neutrophil count was apparent on day 6, with neutrophil count levels returning to normal on day 7. In the SAD study for 280 mg group, a creatine kinase increasing was apparent on day 6, but the subject gave up the follow-up. No other treatment- or dose-related or clinically relevant trends were observed for the clinical laboratory parameters, vital signs measurements, 12-lead ECG, or physical examination evaluations during the study. Overall, no significant safety concerns were observed from the safety data in the study.
Skin Irritation Assessment
In the SAD study, the knee joint and calf skin irritation assessment scores for all subjects were 0 after a single dose, including 35 mg, 70 mg, 140 mg and 280 mg. In the SAD study for 140 mg group, the back skin irritation assessment score is 0.3±0.8 in the placebo group, while the score was 0.1±0.4 in the treatment group. Skin irritation assessment score of the back was 0 in other treatment groups.
In the MAD study, the knee joint and calf skin irritation assessment scores for all subjects were 0 after administration. The evaluation scores of back skin irritation were between 0 and 2, compared with the baseline results, no significant changes observed in the placebo group and treatment groups.
Skin irritation mainly occurs at the site of administration. The main symptoms were erythema, pruritus and most of them disappeared within 30 minutes after administration, which may be related to 25% alcohol in the preparation.
Pharmacokinetics
The mean plasma concentration-time profiles of ibuprofenamine and ibuprofen, the metabolite of ibuprofenamine, in the SAD and MAD studies are presented in Figures 3 and 4. In the SAD study, the pharmacokinetics parameters of the ibuprofenamine and ibuprofen are presented in Tables 3 and 4. The plasma concentrations of ibuprofenamine were low, reflecting the rapid conversion to the Ibuprofen. According to the results of exposure, more than 99% of the ibuprofenamine were converted to Ibuprofen. In the single-dose studies, the Cmax and AUClast of ibuprofenamine and ibuprofen increased with increasing dose of ibuprofenamine hydrochloride, but the Cmax and AUClast were not in the 90% CIs range from 0.893 to 1.107 in the power model. The assessment of dose proportionality of ibuprofenamine and ibuprofen pharmacokinetic parameters (power model) in the SAD study are presented in Table 5. Based on the power model, the AUClast and Cmax of ibuprofenamine showed a less than dose-proportional increase after a single dose of ibuprofenamine hydrochloride except for 70 mg, the AUClast and Cmax of ibuprofen showed a less than dose-proportional increase after a single dose of ibuprofenamine hydrochloride except for 280 mg.
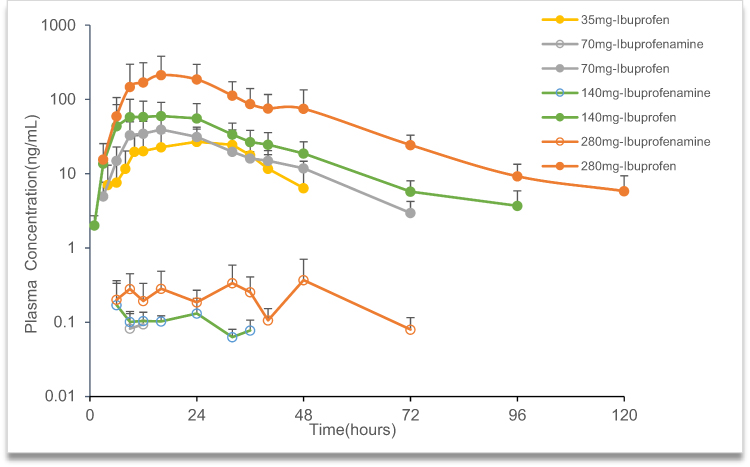 |
Figure 3 Mean plasma concentration-time profile of ibuprofenamine and ibuprofen in the single ascending dose (SAD) studies. |
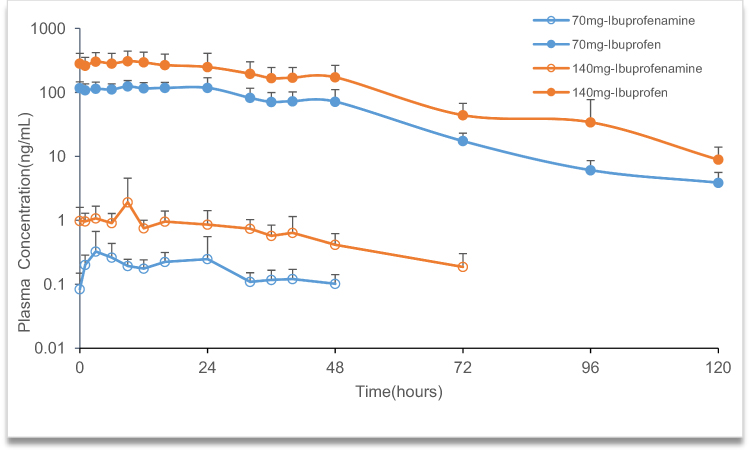 |
Figure 4 Mean plasma concentration-time profile of ibuprofenamine and ibuprofen in the multiple ascending dose (MAD) studies. |
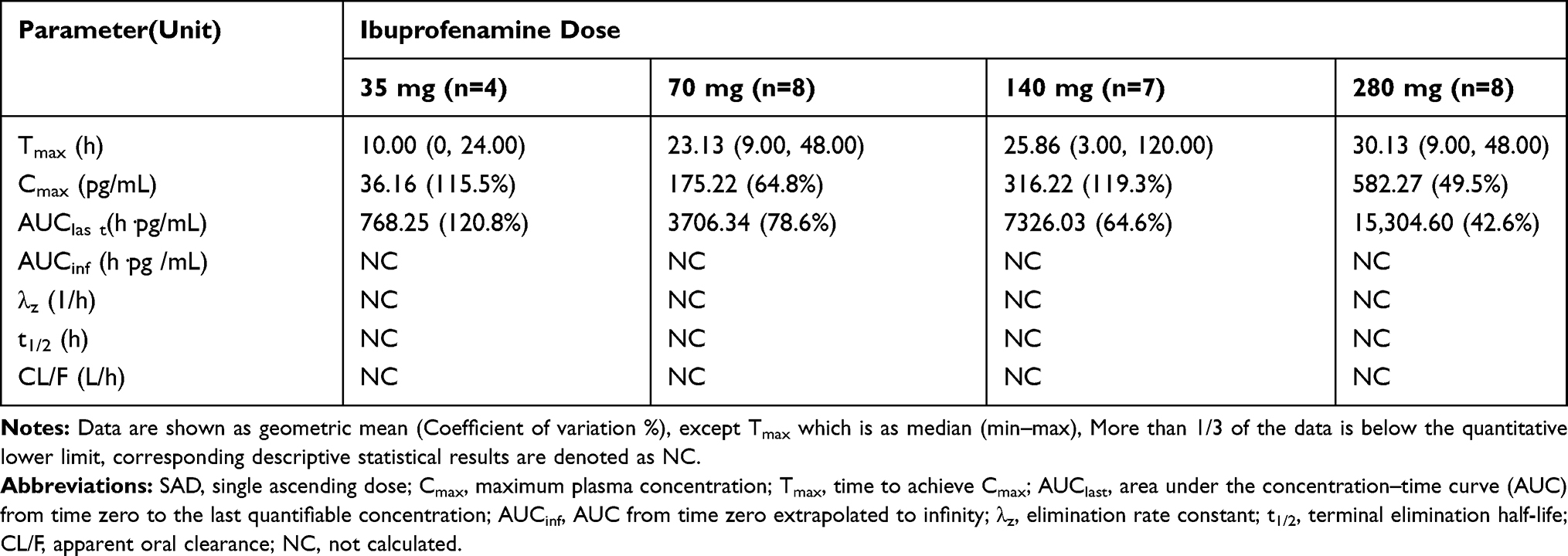 |
Table 3 Pharmacokinetic Parameters of Ibuprofenamine in the SAD Study |
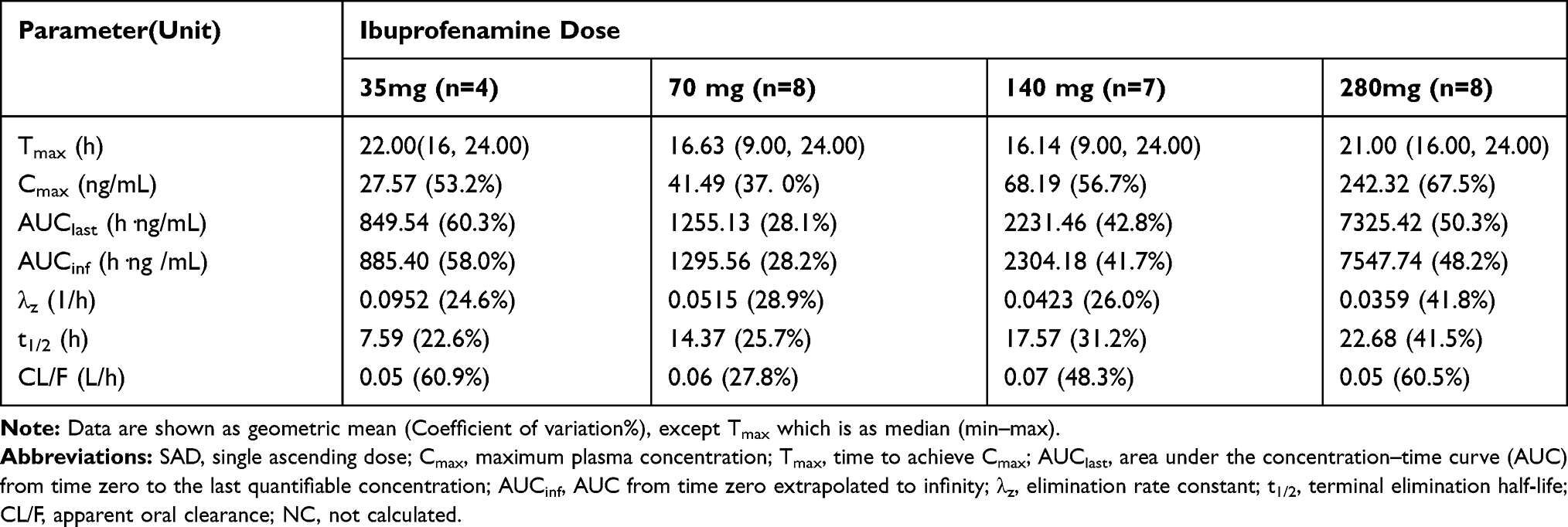 |
Table 4 Pharmacokinetic Parameters of Ibuprofen in the SAD Study |
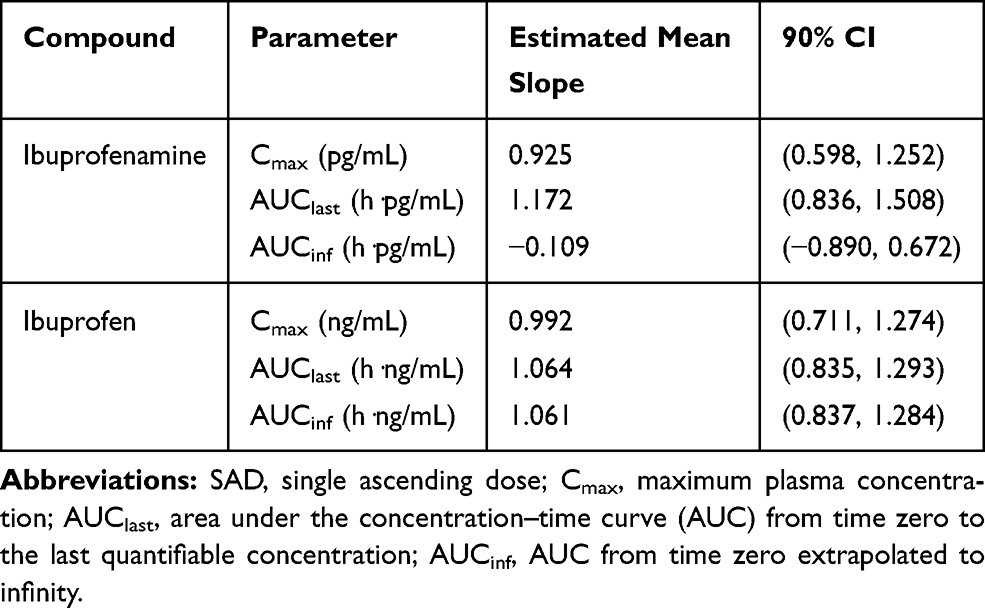 |
Table 5 Assessment of Dose Proportionality of Ibuprofenamine and Ibuprofen Pharmacokinetic Parameters (Power Model) in the SAD Study |
In the MAD study, the pharmacokinetic parameters of ibuprofenamine and ibuprofen are presented in Tables 6 and 7. From the mean accumulation ratio results, both ibuprofenamine and ibuprofen show significant accumulation, the mean accumulation ratio increased significantly more than the dose-proportional increase.
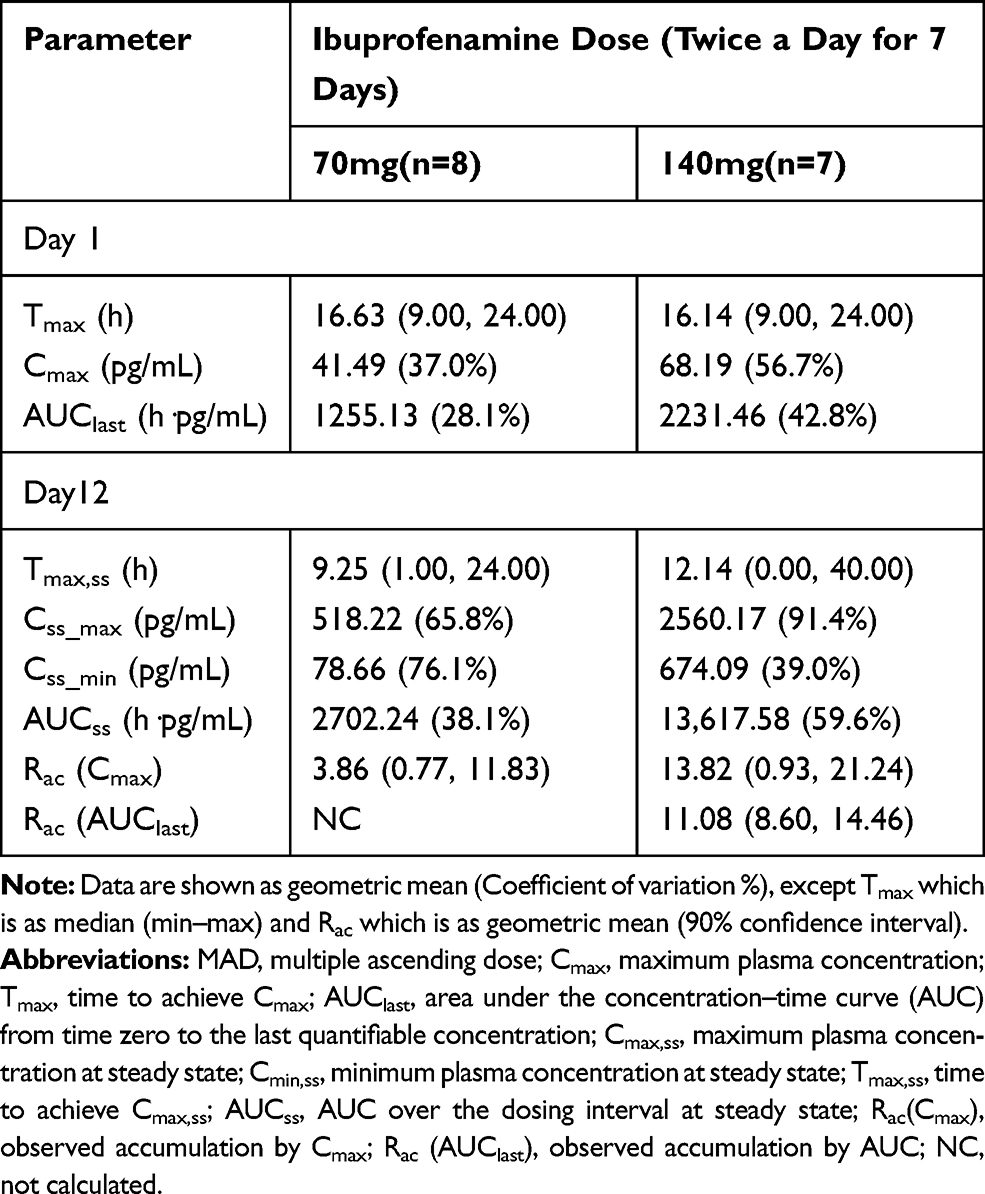 |
Table 6 Pharmacokinetic Parameters of Ibuprofenamine in the MAD Study |
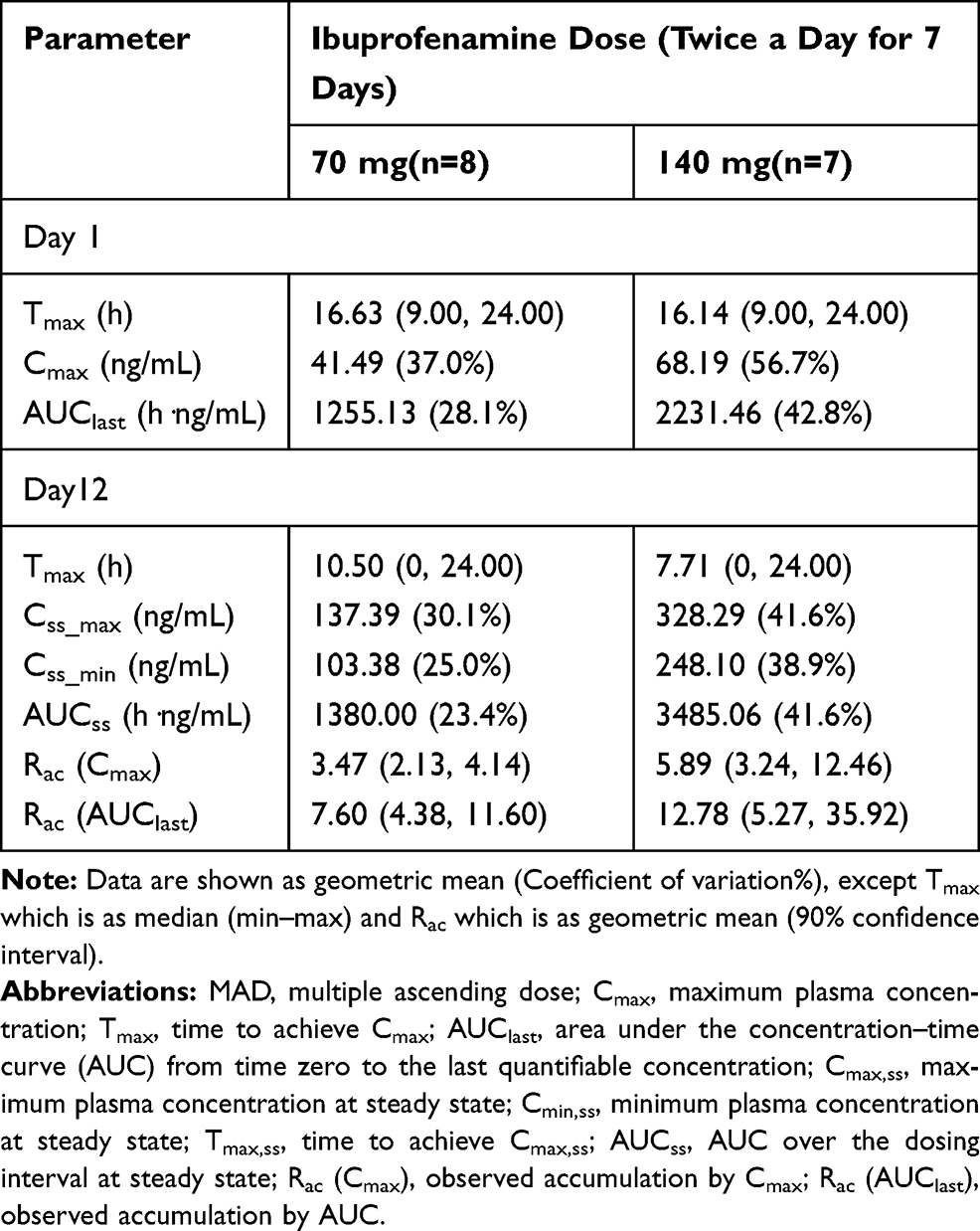 |
Table 7 Pharmacokinetic Parameters of Ibuprofen in the MAD Study |
Discussion
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used for relieving pain and inflammation in patients with rheumatoid arthritis and osteoarthritis. One of the main concerns regarding the use of conventional NSAIDs is the gastrointestinal toxicity, which is dependent on dose and duration of use.5,6 Transdermal products are one of the most common and important classes of drug products due to their unique advantages relative to other dosage forms and routes of administration. They can provide sustained drug delivery for several days (to improve patient compliance), avoid first pass metabolism of a drug by the liver. Transdermal drug products are administered to the skin, through which the drugs permeate into the systemic circulation and are delivered throughout the body to the site of action, avoid gastrointestinal adverse effect.7–9
Ibuprofenamine hydrochloride spray is new transdermal NSAIDs, is the prodrug of ibuprofen, which will be converted into therapeutic ibuprofen soon after absorption. Oral and topical administration of ibuprofen has been described with similar efficacy, this drug is poorly soluble in aqueous media and thus the rate of dissolution from the currently available solid dosage forms is limited.10–13 This leads to poor bioavailability at high doses after oral administration, thereby increasing the risk of unwanted adverse effects.14 The bioavailability of ibuprofenamine hydrochloride is 75%, reaching the level of many oral drugs, and administered to the skin, can avoid gastrointestinal adverse effect.
This is the first study to evaluate the safety, tolerability and pharmacokinetics of different doses of ibuprofenamine hydrochloride in healthy Chinese subjects. In the SAD study, for ibuprofenamine, only few samples with quantifiable concentration were observed rendering it impossible to calculate PK parameters such as λz, t1/2, AUCinf and CL/F, reflecting the rapid conversion to the Ibuprofen. For ibuprofen, the terminal phase t1/2 was 7.59 hours for the lower dose (35 mg) and increased to 14.37–22.68 hours for the higher doses. The dose-dependent nature of t1/2 may be the result of unmasking the true t1/2 values as the concentrations in the terminal phase increased with higher doses. In the MAD study, both ibuprofenamine and ibuprofen steady-state exposure increased obviously more than dose-proportional manner across the evaluated dose range (twice a day for 7 days) with the maximum ibuprofenamine concentrations after administration. On the other hand, obvious accumulation of ibuprofenamine and Ibuprofen were found probably related to ibuprofenamine hydrochloride have high tissue affinity and the tissue half time would be much longer than in blood, thus causing an accumulation tendency in multiple dosing when ibuprofenamine was released from the tissue and entered into the blood again.15–18 The results of the multiple dosing pharmacokinetics indicated that the dosing interval of ibuprofenamine hydrochloride should be extended in the following clinical phase study.
As expected, more AEs were observed in the MAD study because treatment was administered twice a day for 7 days, mainly focused on skin reactions. Treatment with ibuprofenamine hydrochloride did not raise any safety findings in terms of clinically significant changes in vital signs, ECG, laboratory values. In general, the administration of ibuprofenamine hydrochloride was safe and well tolerated and resulted in no SAEs or deaths. The skin irritation assessment of the ibuprofenamine hydrochloride was evaluated after administration according to the skin irritation assessment system, and shown to be non-irritation.
Conclusion
Ibuprofenamine hydrochloride spray, new transdermal NSAIDs, exhibited favorable pharmacokinetic and safety profiles in healthy Chinese subjects. In this Phase I study, single and multiple doses of ibuprofenamine hydrochloride spray were adequately tolerated in single dose up to 280 mg, and multiple doses up to 140 mg twice daily. These results support dose selection and further clinical evaluation of ibuprofenamine hydrochloride spray for a Phase II study.
Data Sharing Statement
The raw data of this study will not be shared because of confidentiality.
Acknowledgments
The study was funded by Xin Chen Taifei Biochemical Technology Co. Ltd, Tianjin, China. The authors thank all the volunteers who participated in this study, as well as all the investigators, site staff, and operations staff who participated in the study. Additionally, the authors thank Rundong pharmaceutical R & D (Shanghai) Co. Ltd of medical writing for providing medical writing assistance, which was funded by Xin Chen Taifei Biochemical Technology Co. Ltd, in accordance with Good Publication Practice guidelines.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Yeomans ND, Graham DY, Husni ME, et al. Randomised clinical trial: gastrointestinal events in arthritis patients treated with celecoxib, ibuprofen or naproxen in the PRECISION trial. Aliment Pharmacol Ther. 2018;47:1453–1463. doi:10.1111/apt.14610
2. García-Rayado G, Navarro M, Lanas A. NSAID induced gastrointestinal damage and designing GI-sparing NSAIDs. Expert Rev Clin Phar. 2018;11:1031–1043. doi:10.1080/17512433.2018.1516143
3. N’Da DD. Prodrug strategies for enhancing the percutaneous absorption of drugs. Molecules. 2014;19:20780–20807. doi:10.3390/molecules191220780
4. Mikulak SA, Vangsness CT, Nimni ME, et al. Transdermal delivery and accumulation of indomethacin in subcutaneous tissues in rats. J Pharm Pharmacol. 1998;50:153–158. doi:10.1111/j.2042-7158.1998.tb06170.x
5. Bjarnason I, Sancak O, Crossley A, et al. Differing disintegration and dissolution rates, pharmacokinetic profiles and gastrointestinal tolerability of over the counter ibuprofen formulations. J Pharm Pharmacol. 2018;70:223–233. doi:10.1111/jphp.12827
6. Bjarnason I, Scarpignato C, Takeuchi K, et al. Determinants of the short-term gastric damage caused by NSAIDs in man. Aliment Pharm Ther. 2007;26:95–106. doi:10.1111/j.1365-2036.2007.03348.x
7. Prausnitz MR, Langer R. Transdermal drug delivery. Nat Biotechnol. 2008;26:1261–1268. doi:10.1038/nbt.1504
8. Al Hanbali A, Khan H, Sarfraz M, et al. Transdermal patches: design and current approaches to painless drug delivery. Acta Pharm. 2019;69(2):197–215. doi:10.2478/acph-2019-0016
9. Lewis F, Connolly MP, Bhatt A, et al. A pharmacokinetic study of an ibuprofen topical patch in healthy male and female adult volunteers. Clin Pharm Drug Dev. 2018;7:684–691. doi:10.1002/cpdd.423
10. Underwood M, Ashby D, Cross P, et al. Advice to use topical or oral ibuprofen for chronic knee pain in older people: randomised controlled trial and patient preference study. BMJ. 2008;336:138–142. doi:10.1136/bmj.39399.656331.25
11. Campbell J, Dunn T. Evaluation of topical ibuprofen cream in the treatment of acute ankle sprains. Emerg Med J. 1994;11(3):178–182. doi:10.1136/emj.11.3.178
12. Tiso RL, Tong-Ngork S, Fredlund KL. Oral versus topical ibuprofen for chronic knee pain: a prospective randomized pilot study. Pain Physician. 2010;13:457–467.
13. Derry S, Conaghan P, Da Silva JA, et al. Topical NSAIDs for chronic musculoskeletal pain in adults. Cochrane Database Syst Rev. 2008;4:CD007400.
14. Irvine J, Afrose A, Islam N. Formulation and delivery strategies of ibuprofen: challenges and opportunities. Drug Dev Ind Pharm. 2018;44:173–183. doi:10.1080/03639045.2017.1391838
15. Gu R, He Y, Han S, et al. Pharmacokinetics and bioavailability of tuftsin-derived T peptide, a promising antitumor agent, in beagles. Drug Metab Pharmacok. 2016;31:51–56. doi:10.1016/j.dmpk.2015.08.005
16. Kivisto KT, Kallio A, Neuvonen PJ. Pharmacokinetics and pharmacodynamics of transdermal dexmedetomidine. Eur J Clin Pharmacol. 1994;46:345–349. doi:10.1007/BF00194403
17. Liu W, Teng L, Yu K, et al. Design of hydrogels of 5-hydroxymethyl tolterodine and their studies on pharmacokinetics, pharmacodynamics and transdermal mechanism. Eur J Pharm Sci. 2017;96:530–541. doi:10.1016/j.ejps.2016.10.024
18. Park I, Kim D, Song J, et al. BupredermTM, a new transdermal delivery system of buprenorphine: pharmacokinetic, efficacy and skin irritancy studies. Pharm Res. 2008;25:1052–1062. doi:10.1007/s11095-007-9470-6
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.