")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Safety and Efficacy of Centanafadine Sustained-Release in Adults With Attention-Deficit Hyperactivity Disorder: Results of Phase 2 Studies
Authors Wigal SB, Wigal T, Hobart M, Madera JJ, Baker RA , Kohegyi E , McKinney A , Wilens TE
Received 12 December 2019
Accepted for publication 12 May 2020
Published 8 June 2020 Volume 2020:16 Pages 1411—1426
DOI https://doi.org/10.2147/NDT.S242084
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Sharon B Wigal,1 Tim Wigal,1 Mary Hobart,2 Jessica J Madera,3 Ross A Baker,3 Eva Kohegyi,3 Anthony McKinney,4 Timothy E Wilens5
1AVIDA Inc., Newport Beach, California, USA; 2Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, Maryland, USA; 3Otsuka Pharmaceutical Development & Commercialization, Inc., Princeton, New Jersey, USA; 4Ethismos Research, Inc, Cambridge, Massachusetts, USA; 5Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA
Correspondence: Timothy E Wilens
Division of Child and Adolescent Psychiatry, Center for Addiction Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA
Tel +1 617-643-3481
Fax +1 617-724-3742
Email [email protected]
Purpose: Two phase 2 studies evaluated the efficacy and tolerability of centanafadine sustained-release (SR) for adults with attention-deficit/hyperactivity disorder (ADHD).
Patients and Methods: In a phase 2a, flexible-dose, single-blind study, 41 male patients (aged 18‒55 years) with a diagnosis of ADHD (based on Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition) were titrated with centanafadine-SR 200‒300, 400, or 500 mg/d for 2 weeks, and then were treated with the titrated dose for 2 weeks. In a phase 2b, randomized, double-blind, placebo-controlled, crossover study, 85 male and female patients (aged 18‒60 years) with a diagnosis of ADHD (Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition) were titrated to target doses of centanafadine-SR 400, 500, 600, or 800 mg/d over the course of 1 week, and then received their titrated dose for 3 weeks. The primary outcome in both studies was mean total ADHD Rating Scale-IV (ADHD-RS-IV) score.
Results: In the phase 2a study, mean ADHD-RS-IV total score decreased by 21.41 (standard deviation 10.74) from the start of active centanafadine-SR treatment to the end of week 4 (P< 0.001). In the phase 2b study, centanafadine-SR treatment resulted in a statistically significant improvement in ADHD-RS-IV from baseline to week 3 compared with placebo (least-squares mean − 16.5 vs − 8.4; P< 0.001; effect size 0.66), with significant efficacy demonstrated as early as week 1. Centanafadine-SR was generally well tolerated at doses ≤ 400 mg. Most treatment-emergent adverse events (TEAEs) were mild or moderate; decreased appetite, headache, and nausea were the most frequently reported. In the 2 studies, 13 of 120 patients discontinued centanafadine-SR due to TEAEs; however, only 1 patient who received ≤ 400 mg discontinued due to a TEAE. No serious TEAEs were reported at any dose.
Conclusion: These results support the continued development of centanafadine-SR at doses up to 400 mg/d.
Keywords: ADHD Rating Scale-IV, efficacy, norepinephrine-dopamine-serotonin reuptake inhibitor, tolerability
Introduction
Attention-deficit hyperactivity disorder (ADHD) is a neurobehavioral disorder characterized by 3 core symptoms: hyperactivity, inattentiveness, and impulsivity.1 Based on the efficacy of available ADHD pharmacotherapies currently used in pediatric and adult patients, the pathophysiology of ADHD is believed to involve abnormalities of dopaminergic and noradrenergic tone in the prefrontal cortex (PFC). Preclinical research has shown that administration of methylphenidate at low doses that improve cognition in rats preferentially increases norepinephrine and dopamine efflux in the PFC relative to subcortical structures, further supporting the hypothesis that dopaminergic and noradrenergic activation in the PFC region is a primary mediator of the therapeutic effects of ADHD medications.2 Other neurotransmitter systems, including glutamate via α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, are thought to be involved.3
Pharmacotherapies available for the treatment of ADHD comprise 2 major classes of drugs: stimulants such as methylphenidate and amphetamines, which are generally first-line treatments, and nonstimulants such as the norepinephrine reuptake inhibitor atomoxetine, and the α-adrenergic agonists guanfacine and clonidine. In general, both stimulants and nonstimulants are more effective than placebo for the short-term treatment of ADHD; however, they may be less effective and not as well tolerated in adults as they are in children and adolescents.4
Amphetamines act primarily through dopamine and norepinephrine release.5 Methylphenidate is believed to act through dopamine and norepinephrine reuptake inhibition, as well as dopamine transporter inverse agonism.6 These individual agents have a rapid onset of efficacy, address all 3 core symptom domains, and have each been associated in clinical trials with response rates of ~70%.7 Their use is, however, limited by concerns about misuse, which is highly prevalent,8 and adverse reactions, such as sleep disturbance.7,9
Nonstimulant agents may be an option for patients who have difficulty tolerating stimulants or for whom misuse is a particular concern. Approved nonstimulants are, however, generally less effective than stimulants and are limited by adverse effects of their own.4 Atomoxetine is associated with liver toxicity, and increases in blood pressure and heart rate,10 and has a boxed warning in its label regarding the risk of suicidal ideation in children and adolescents,11 although this has not been shown to be a concern in adults.12 The α-adrenergic agonists clonidine and guanfacine are even less effective than atomoxetine,13 and guanfacine has been associated with high discontinuation rates.14
Centanafadine is a triple monoamine inhibitor of norepinephrine (half-maximal inhibitory concentration [IC50] 6 nM), dopamine (IC50 38 nM), and serotonin (IC50 83 nM) transporter reuptake. Thus, centanafadine demonstrates the highest activity for norepinephrine reuptake inhibition, 6 times less for dopamine reuptake inhibition, and 14 times less for serotonin reuptake inhibition.15 Extracellular norepinephrine and dopamine levels are increased in the PFC, with the greatest increase in extracellular norepinephrine. Dopamine levels also are increased in the striatum. Preferential affinity for norepinephrine transporters relative to dopamine transporters suggests that centanafadine-sustained-release (SR) may effectively address core features of ADHD with mixed neurotransmitter effects that are comparable to those achieved with stimulants, but without other unwanted stimulant effects. Activity at the serotonin receptor may address associated ADHD symptoms such as anxiety and depression and mitigate adverse effects of treatment such as sleep disturbances and changes in appetite. Finally, the nonstimulant profile of centanafadine-SR appears to be favorable compared with the stimulant drugs because it is less likely to cause sleep issues that are a problematic side effect of stimulants.
Centanafadine was evaluated in the National Institute on Drug Abuse drug discrimination model as a potential treatment for cocaine abuse in rats and rhesus monkeys. This study was designed to ascertain if the test drug substituted for the discriminative stimulus of cocaine. In rats, centanafadine substituted fully for the discriminative stimulus effects of cocaine; however, centanafadine substituted only at a dose that reduced response rates, possibly indicating it was aversive at higher doses. In monkeys, centanafadine dose-dependently substituted for cocaine. As demonstrated in a Phase 1 exploratory human abuse liability study using an immediate-release formulation of centanafadine (ClinicalTrials.gov NCT02144415), centanafadine may have less abuse potential than stimulants commonly prescribed for ADHD. In the human abuse liability study, the immediate-release (IR) formulation of centanafadine, like other triple reuptake inhibitors (eg, tesofensine,16 NS-235917) and bupropion,18 was initially aversive and believed to be unlikely to be abused by known stimulant users.
The phase 1 trial program for centanafadine-SR demonstrated that it was safe and well tolerated in healthy subjects when administered as a bid dose (up to 500 mg/d) for 10 days (unpublished data). Centanafadine-SR has a half-life of just over 4.5 hours, enabling bid administration, while centanafadine-IR has a plasma half-life of less than 1.5 hours. Thus, the SR formulation has the advantage of requiring no greater than bid administration, which is likely to improve patient compliance and/or satisfaction for chronic oral administration. Results from a pilot phase 2a study that demonstrated the safety and the pharmacokinetic parameters of centanafadine-SR versus the IR formulation indicate that centanafadine-SR may provide a level of efficacy similar to stimulants with a rapid onset of action.
This article presents results from two phase 2 trials conducted in adults with ADHD: the phase 2a study conducted to provide initial signals of efficacy at centanafadine doses up to 500 mg/d (ClinicalTrials.gov NCT01939353) and a phase 2b study conducted to further evaluate the efficacy and tolerability of centanafadine-SR at the target dose identified as best in the phase 2a study (NCT02547428).
Patients and Methods
Patient Population
The phase 2a study enrolled male patients aged of 18 and 55 years who met Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-4)19 criteria for ADHD (combined, predominantly hyperactive-impulsive types) on the Mini-International Neuropsychiatric Interview-Plus, Version 6.0.0 (MINI-Plus). Eligible patients had a baseline score ≥28 on the ADHD Rating Scale-IV (ADHD-RS-IV) and a Clinical Impression of Severity (CGI-S) score ≥4, and were deemed by investigators to be functioning at an age-appropriate intellectual level so that they could be included for rating on psychiatric scales required by the protocol.
The phase 2b study enrolled male and female patients aged 18‒60 years, with a DSM-520 primary diagnosis of ADHD and ≥5 of the 9 DSM-5 subtype criteria met, as determined using the Conners’ Adult ADHD Diagnostic Interview for DSM-IV™. As in the phase 2a study, eligible patients had a baseline score ≥28 on the ADHD-RS-IV and a CGI-S score ≥4, and were considered by investigators to be functioning at an age-appropriate intellectual level.
Centanafadine was evaluated initially in adults because relatively few drugs were indicated for adult ADHD, compared to childhood ADHD, at the time of initial Investigational New Drug Application filing. Given the increasing rate of ADHD diagnosis in adults, and the fact that centanafadine is a new chemical entity, it was thought an initial program in adults offered the highest benefit/risk ratio. There are slight differences in inclusion criteria between the phase 2a and phase 2b studies to expand the upper range in age and BMI, for instance, to enable enhanced recruitment. In addition, only males were enrolled into the phase 2a study because the results from the reproductive toxicology studies enabling inclusion of females were not yet available at the time the phase 2a study was designed.
The key exclusion criteria in both studies were major psychiatric comorbidity, presence or history of suicidality, current or recent history of substance abuse, and significant medical comorbidities, including history of seizures, cardiovascular disease, elevated blood pressure, bleeding disorders, cancer, and any chronic or current acute illness or disability that might confound safety findings or increase the risk of adverse events during treatment with centanafadine-SR. Patients were also excluded if they had a body mass index <18 or >35 kg/m2 (phase 2b), or <18.5 or ≥40 kg/m2 (phase 2b). Patients would be withdrawn from their study prior to receiving centanafadine-SR if they showed improvement ≥30% in ADHD symptoms or a score <28 on the ADHD-RS-IV after the 1-week, blinded placebo lead-in in the phase 2a study or prior to randomization in the phase 2b study.
Study Design
Phase 2a Study
The phase 2a study was a flexible-dose, single-blind, exploratory trial conducted at 3 US sites between October 2013 and February 2014. The study included a screening phase, a 1-week placebo lead-in phase, and a 4-week centanafadine-SR treatment phase. During screening, investigators determined whether patients met eligibility criteria. Eligible patients tapered and discontinued disallowed medications, which included methylene blue, drugs metabolized by cytochrome P450 2D6 or 1A2, and any prescription or over-the-counter medication with central nervous system effects.
Starting on day 1 of the placebo lead-in (baseline-1), eligible patients received 1 placebo tablet in the morning, with or without food, for 1 week. At the start of active treatment (baseline-2), patients received a single centanafadine-SR 100-mg tablet in the morning on days 1 and 2, then took 1 tablet in the morning and another 5 hours later on days 3 and 4, and 2 tablets in the morning and 1 tablet 5 hours later on days 5 through 7. During week 2 of treatment, the dose was twice increased by 100 mg to a total daily dose (TDD) of 500 mg, after which treatment continued at the TDD for 2 weeks subject to dose reduction due to adverse events. Dosage groups were defined based on the final dose taken before study conclusion.
Phase 2b Study
The phase 2b study was a randomized, double-blind, 2-period, 2-treatment, crossover trial conducted at 4 US sites between August 2015 and June 2016. The study included a screening period (up to 6 weeks), a baseline visit, and a 7-week, double-blind, crossover treatment period (Figure 1). During screening, patients meeting eligibility criteria underwent a 1-week washout of any sedating antihistamine or stimulant therapies for ADHD. Therapies prohibited during the phase 2a study were allowed during the phase 2b study; therefore, the population in the phase 2b study was treated under conditions more consistent with those likely to be encountered in clinical practice.
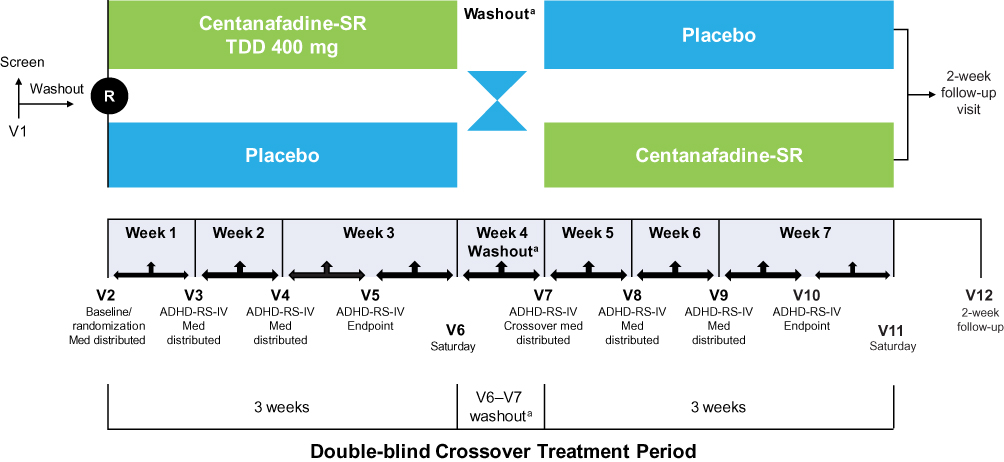 |
Figure 1 Phase 2b study design. aWashout length was variable for each patient depending on the day their weekly clinic visit was scheduled. Abbreviations: ADHD-RS-IV, Attention-Deficit Hyperactivity Disorder Rating Scale-IV; Med, investigational product; R, randomization; SR, sustained-release; TDD, total daily dose; V, visit. |
During the double-blind treatment period, patients received 3 weeks of centanafadine-SR or placebo, after which they underwent a 1-week washout before crossing over to the opposite treatment (centanafadine-SR or placebo) for an additional 3 weeks of treatment. The original TDD was centanafadine-SR 500 mg/d, which was amended due to emerging safety and tolerability data during the study to a TDD of 400 mg/d. The final amended study protocol called for centanafadine-SR or matching placebo to be initiated at a dose of 100 mg in the morning on days 1 and 2, after which patients received 100 mg in the morning (~7
Ethics
The protocols for both studies were approved by the institutional review board/independent ethics committee for each study site, and all patients provided written informed consent prior to participation. Both studies were conducted in full compliance with the International Conference on Harmonization guidelines, including Good Clinical Practice, and all other applicable local laws and regulations.
Assessments
Efficacy in both studies was based primarily on results of the ADHD-RS-IV, an 18-item scale based on DSM-IV criteria for ADHD. The first 9 items assess inattentiveness and the second 9 assess hyperactivity/impulsivity. Scoring is based on a 4-point Likert-type severity scale: 0=none, 1=mild, 2=moderate, and 3=severe. The potential range for ADHD-RS-IV total scores is 0‒54 based on the sum of the individual scores on the 18 items.
In the phase 2a study, the primary efficacy assessment was change from baseline-2 (start of centanafadine-SR treatment) to the end of week 4 of treatment in ADHD symptoms, as assessed by the ADHD-RS-IV. Individual scores on the ADHD-RS-IV inattentiveness and hyperactivity/impulsivity subscales, as well as results by dosage group, were also assessed.
In the phase 2b study, assessments were made at baseline and the end of each of two consecutive double-blind treatment periods. The primary efficacy outcome was ADHD-RS-IV total score at the end of 3 weeks of double-blind treatment, both overall and in the subgroup of adults with ADHD treated with a target centanafadine-SR dose of 400 mg/d. The ADHD-RS-IV total scores after 1 and 2 weeks of treatment, and scores on the ADHD-RS-IV inattentiveness and hyperactivity/impulsivity subscales were also reported.
In the phase 2b study, patients were also assessed using the Clinical Global Impression of Improvement (CGI-I) and the Permanent Product Measure of Performance (PERMP). The CGI-I is a 7-point investigator-rated scale with scores of 1 (very much improved), 2 (much improved), 3 (minimally improved), 4 (no change from baseline), 5 (minimally worse), 6 (much worse), and 7 (very much worse). The CGI-I was administered after 1, 2, and 3 weeks of each treatment, and 2 weeks after the completion of each treatment period. The PERMP is a 10-minute, individual performance-adjusted math test that provides an objective measure of performance over the course of a single day.21–23 The PERMP was administered on the last day (Saturday) of each double-blind treatment sequence in a highly controlled setting approximating an adult workplace environment (AWE). The AWE day is considered a suitable environment for capturing cognitive changes under conditions that simulate a standard workday in the real world. For this assessment, groups of patients were given 5 pages with 80 math problems on each page, and were instructed to work at their assigned table seats and complete as many problems as possible in 10 minutes. The PERMP score is the composite of the number of problems attempted and the number completed correctly. The PERMP pretest was administered at baseline to establish each patient’s pretreatment ability level. PERMP testing to evaluate treatment effects was performed at the end of each 3-week treatment period ~0.5 hours predose and ~1, 3, 5, 7, 9, 11, and 13 hours postdose. Between the pretest and end-of-treatment test, additional practice math tests were given at each clinic visit to ensure that the final PERMP test results reflected treatment effects rather than learning or practice effects during the test days in the AWE setting.
Safety and tolerability assessment in both studies was based on the occurrence of treatment-emergent adverse events (TEAEs). Relationship of AEs to study drug and intensity of AEs were recorded during the course of the event, including the start and stop dates for each change in intensity: mild (easily tolerated and does not interfere with usual activity), moderate (interferes with usual activity, but patient is still able to function), and severe (incapacitating, and patient is unable to work or complete usual activity). An AE was considered “serious” if, in the view of either the investigator or sponsor, it was life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, or resulted in a persistent or significant incapacity or disability).
Statistical Analysis
In the phase 2a study, it was expected that 10% of patients would drop out during the placebo period due to an early response and 20% would drop out overall. Approximately 40 adult patients would, therefore, be required to enter the treatment period, from which the expected sample of 32 completers would be sufficient to determine statistical significance (P<0.05) on t-test comparisons of baseline and endpoint values with 90% power. Additional patients were enrolled for evaluation of dose-response and tolerability to inform future, larger, placebo-controlled trials.
In the phase 2b study, it was assumed that for the primary outcome measure, the between-patient standard deviation (SD) of ADHD-RS-IV measurements would be ~12.5 points, and that a within-patient minimum difference between centanafadine-SR and placebo of 5 points would be clinically relevant (equivalent to a minimum detectable effect size of 0.4). Approximately 60 patients were to be randomized to the double-blind crossover phase of the study (30 patients/treatment sequence) to provide a minimum 85% power to detect an effect size of 0.4 at a 2-sided significance level of 5% using a paired t-test.
In the phase 2a study, efficacy analyses were performed in the per-protocol population, which included any patient who met inclusion/exclusion criteria and completed the trial (4 weeks of centanafadine-SR) without major protocol violations. Safety was evaluated in any patient who received ≥1 dose of centanafadine-SR. Inferential statistical tests were performed on efficacy parameters, which were considered appropriate given the exploratory design of the study.
In the phase 2b study, efficacy was assessed in the full analysis set, which consisted of all patients who were randomized, received ≥1 dose of centanafadine-SR or placebo, and had ≥1 postdose efficacy assessment. Safety was assessed in the safety population, which included all patients who received ≥1 dose of centanafadine-SR or placebo.
The primary efficacy comparison for the phase 2b study was performed in patients who completed ADHD-RS-IV assessments after 3 weeks of double-blind treatment in both treatment periods. The primary efficacy analysis compared the ADHD-RS-IV total score changes from baseline (visits 2 and 7) between centanafadine-SR and placebo at the end of 3 weeks of double-blind treatment (visits 5 and 10). A mixed-effects analysis of covariance model was used to compare the differences in ADHD-RS-IV total score changes from baseline between centanafadine-SR and placebo. The model included terms for period, sequence, and treatment as fixed effects, baseline (visit 2) score as a covariate, and patient-within-sequence as a random effect. The least-squares (LS) mean within-patient difference, the associated 95% confidence interval (CI) for the difference, the effect size, and P-value were calculated. Data captured at the early termination visit were used in the efficacy analyses; for example, if a patient attended visit 2 then discontinued, the efficacy data collected at early termination were used for the next scheduled visit (visit 3). Incomplete post-dose data within each period were handled using the last-observation-carried-forward approach. For example, if a patient had data at visit 3 but not visit 4 or 5, then the visit 3 value was carried forward to visits 4 and 5. Data from the first period were not carried forward to the second period. In addition, observations from baseline (visit 2) and visit 7 (pre-dose) were not carried forward into the double-blind treatment phase time points for either period.
Effect size was calculated as the difference in least-squares (LS) means between treatments and its estimate of the SD using the following formula in patients providing values for both treatment periods in this crossover study:
Effect size = LS mean difference between treatments/SD estimate
The CGI-I scores were dichotomized into 2 categories, with “very much improved” and “much improved” (scores 1–2) combined into a single “improved” category, and the remaining scores (3–7) combined into a single “not improved” category. This dichotomized comparison was performed using McNemar’s test at the same time points.
The PERMP scores were compared between centanafadine-SR and placebo after a minimum of 3 weeks of double-blind treatment. At each time point, a mixed-effects analysis of variance model was used to compare the difference in scores between centanafadine-SR and placebo. The model included terms for period, sequence, and treatment as fixed effects, and patient-within-sequence as a random effect. The difference in LS means, the associated 95% CI for the difference, and P-value were calculated.
All comparisons of centanafadine-SR with placebo were made at a nominal α level of 0.05. Per the statistical analysis plan, all efficacy analyses were repeated for the subgroup of patients treated with a centanafadine-SR TDD of 400 mg/d and compared with results for the overall population.
Results
Phase 2a Study
Patient Disposition and Characteristics
Forty-five patients were enrolled at baseline-1 and received placebo. Of these patients, 4 discontinued treatment (3 withdrew consent and 1 was lost to follow-up), leaving 41 enrolled at baseline-2 who received ≥1 dose of centanafadine-SR and were evaluable for safety. Four patients discontinued treatment for the following reasons: TEAE (allergic contact dermatitis; n=1), serious TEAE (cerebrovascular accident; n=1), being lost to follow-up (n=1), and withdrawn consent (n=1). The patient who discontinued the study due to a serious TEAE received the last dose of centanafadine-SR and was, therefore, included in the per-protocol population. Further, another patient was missing week 4 data and was, therefore, excluded from the per-protocol population. No patients were withdrawn from the study due to achieving improvement ≥30% in ADHD symptoms or a score <28 on the ADHD-RS-IV. In all, 37 patients completed 4 weeks of drug treatment with evaluable data after week 4, had no major protocol violations, and were included in the per-protocol efficacy analysis.
Thirty-three of the 37 per-protocol patients achieved the intended maximum dose of 500 mg at least once during the trial period. Dose groups for data analyses were 200‒300 mg (n=4), 400 mg (n=8), and 500 mg (n=25).
The mean age of the 41 patients who received centanafadine-SR at baseline-2 was 38 years (range 19‒55) and most were white (n=37). Mean ADHD-RS-IV and CGI-S scores at baseline-2 were 38.7 (SD 6.19) and 4.68 (SD 0.47), respectively.
Efficacy Outcomes
Centanafadine-SR therapy was associated with significant improvements in ADHD symptoms (Table 1). Mean (SD) ADHD-RS-IV total score decreased by 21.41 (10.74) from baseline-2 to week 4 (P<0.001). For inattention symptoms and hyperactive/impulsive symptoms, mean (SD) scores decreased by 12.41 (6.66; P<0.001) and 9.00 (5.24; P<0.001), respectively.
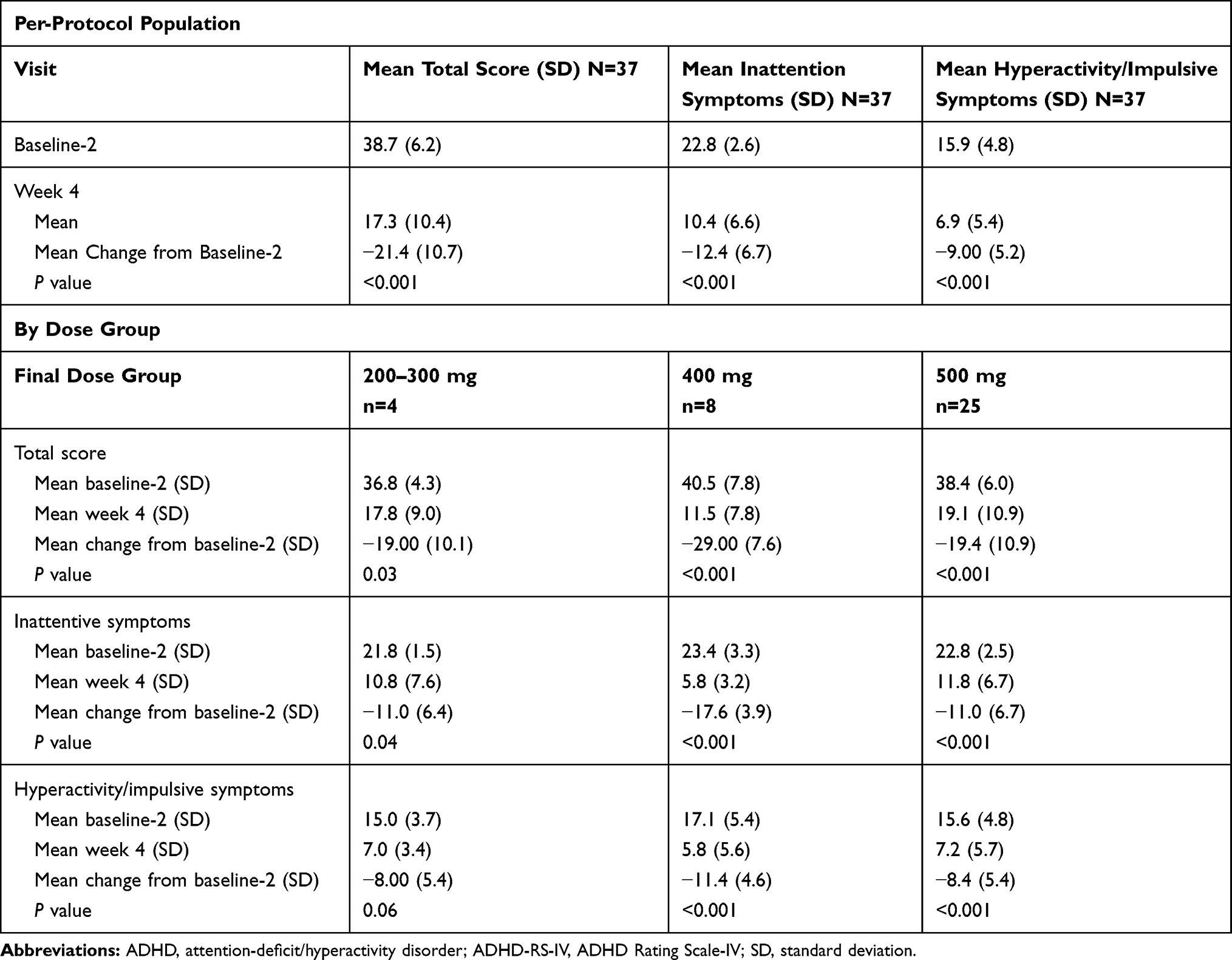 |
Table 1 Phase 2a Study: Change in ADHD Symptoms from Baseline-2 to Treatment Week 4 as Assessed by Mean (SD) ADHD-RS-IV Scores (Per-Protocol Population and Final Dose Group) |
When analyzed by final dose group, mean (SD) decreases in ADHD-RS-IV score from baseline-2 to week 4 were greater for the centanafadine-SR 400-mg final dose group (29.00 [7.60]; n=8) than for the 200‒300-mg (19.00 [10.13]; n=4) and 500-mg (19.36 [10.89; n=25]) groups. Similar results were observed for the inattentive symptoms and hyperactivity/impulsive symptoms subscale scores.
Safety
Thirty-four of 41 patients (83%) who received ≥1 dose of centanafadine-SR had ≥1 TEAE. More TEAEs occurred in higher doses of centanafadine-SR, including 23 occurring in patients on 500 mg at time of onset. Thirty-one patients (76%) had a total of 69 TEAEs considered as related to treatment, all of which were mild-moderate. The most commonly reported TEAEs were diarrhea (n=12 [29%]), headache (n=7 [17%]), decreased appetite (6 [15%]), and dry mouth (n=6 [15%]), none of which led to patient discontinuation.
No deaths were reported. There were no severe TEAEs and 1 serious TEAE (cerebrovascular accident in a patient with hypertension and history of headache) not related to treatment.
Phase 2b Study
Patient Disposition and Characteristics
In all, 117 patients were screened, and 85 were eligible for enrollment (Figure 2). No patient experienced an improvement ≥30% in ADHD symptoms or a score <28 on the ADHD-RS-IV prior to randomization; hence, all 85 patients were eligible for randomization to 1 of the 2 treatment sequences. Forty-two patients were randomized to a centanafadine-SR/placebo sequence and 43 to a placebo/centanafadine-SR sequence.
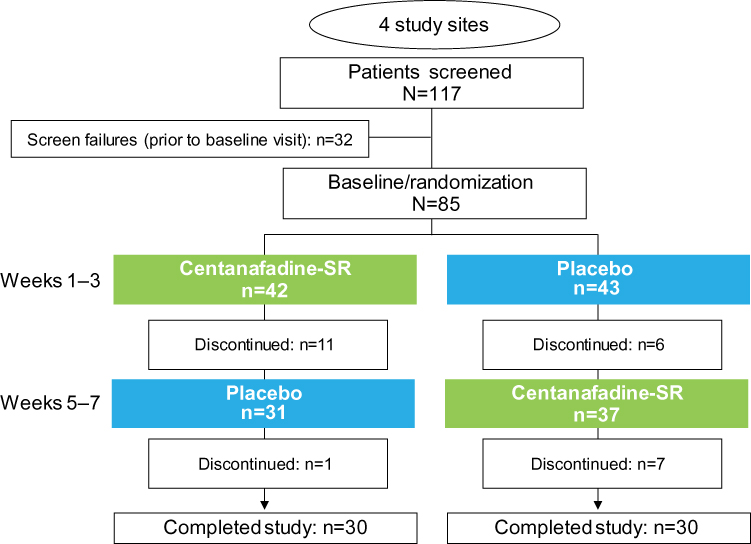 |
Figure 2 Phase 2b study: patient disposition. Abbreviation: SR, sustained-release. |
The best total daily dose for each patient was determined by the applicable protocol version at the time of enrollment. Forty-seven patients (55%) were assigned a TDD of 400 mg/d, and 20 (24%) and 18 (21%) patients were assigned TDDs of 600 and 800 mg/d, respectively.
Sixty patients completed the study and 25 discontinued, including 18 who discontinued while receiving centanafadine-SR and 7 while on placebo (Figure 2 and Table 2). Completion rates by dose were 94% (43/47) at the 400-mg/d TDD, 75% (15/20) at the 600-mg/d TDD, and 44% at the 800-mg/d TDD.
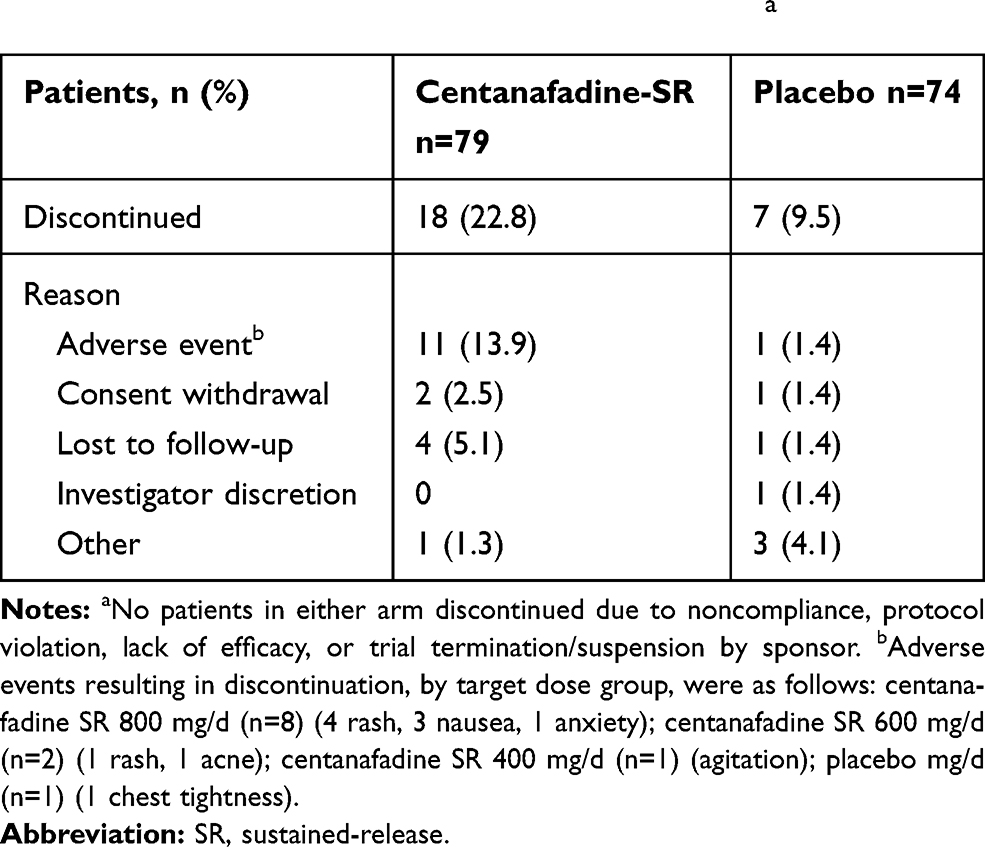 |
Table 2 Phase 2b Study: Patient Discontinuationsa |
The 2 treatment groups were similar with respect to age, gender, racial and ethnic makeup, and body composition, both overall and within dosage groups, with the exception that there were more male patients in the 400-mg/d group (68%) and more female patients in the 800-mg/d group (72%; Table 3).
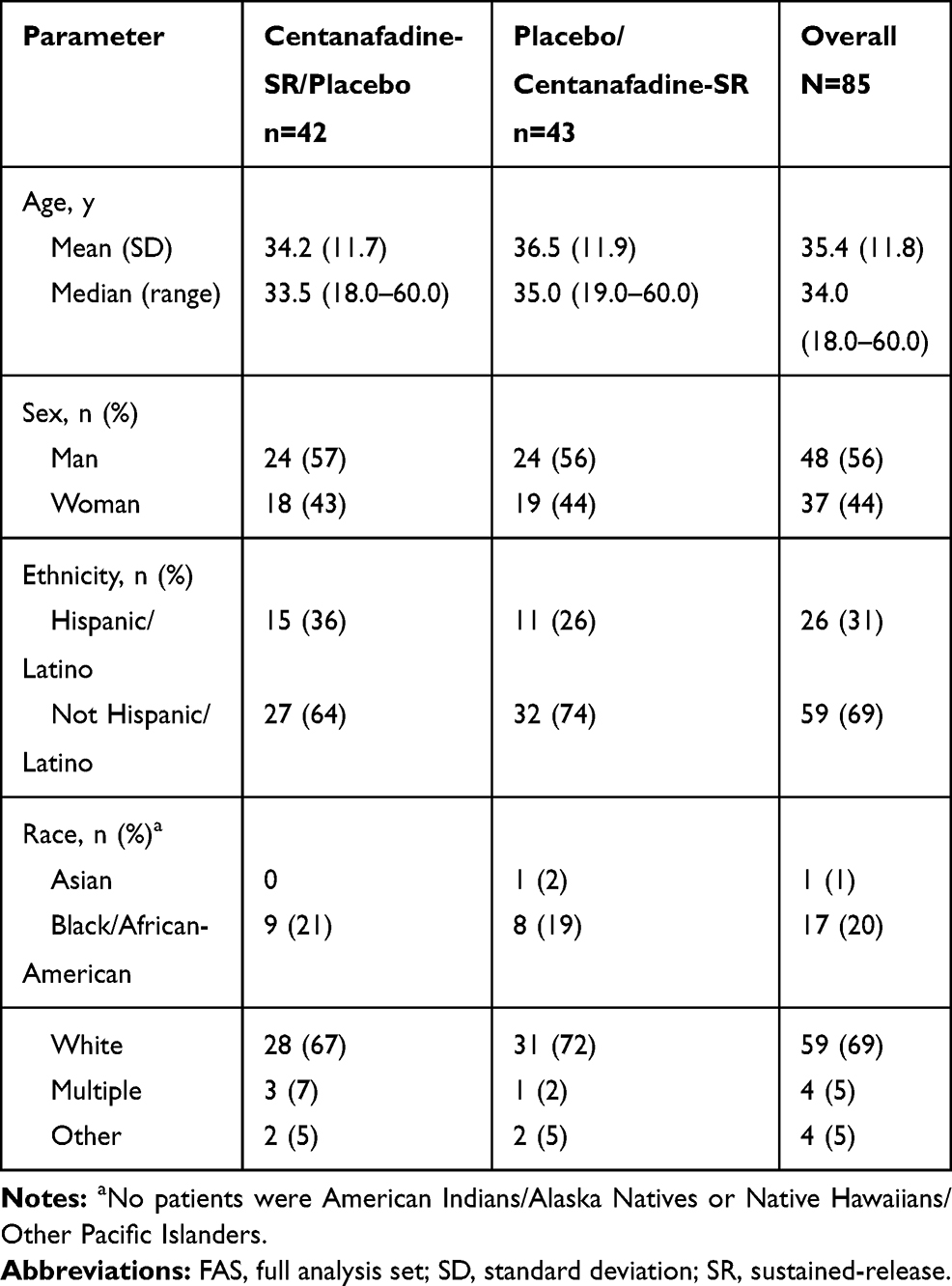 |
Table 3 Phase 2b Study: Patient Demographics (Safety/FAS Population) |
Efficacy Outcomes
Mean baseline ADHD-RS-IV total score for the entire 85-patient study population was 37.6 (range 28‒54). Mean ADHD-RS-IV total scores at week 3 (combined visits 5 and 10) were 21.5 (range 0‒47) for the centanafadine-SR treatment group (n=79) and 29.7 (range 2‒49) for the placebo group (n=74). In patients who completed 3-week assessments in both treatment periods (primary efficacy population [n=68]), centanafadine-SR met its primary efficacy endpoint of a significant decrease in ADHD-RS-IV total score from baseline after 3 weeks of treatment compared with adults who received placebo (Figure 3). A statistically significant improvement (P<0.001) in ADHD-RS-IV total score from baseline to week 3 was observed for centanafadine-SR (LS mean −16.5) compared with placebo (LS mean −8.4), resulting in an LS mean difference of −8.1 (standard error [SE] 1.48; 95% CI −11.0, −5.1). The effect size for centanafadine-SR in the entire population was 0.66.
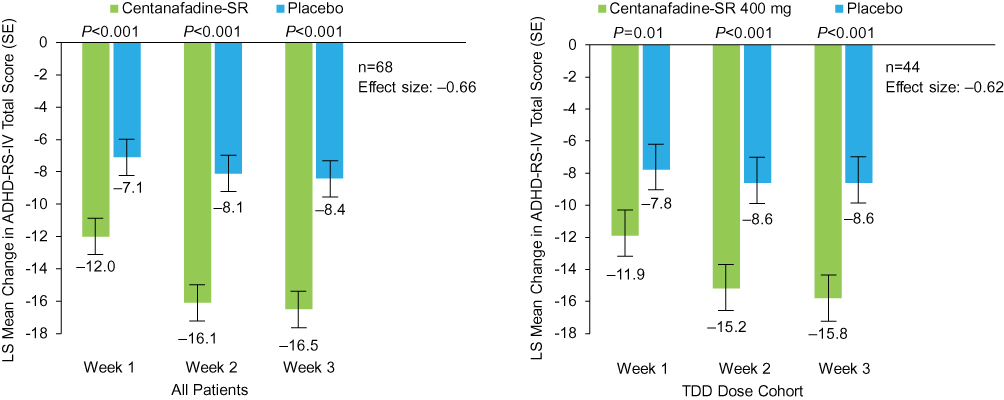 |
Figure 3 Phase 2b study: mean Attention-Deficit Hyperactivity Disorder Rating Scale-IV (ADHD-RS-IV) total score changes from baseline (last observation carried forward) after 1, 2, and 3 weeks of double-blind treatment (full analysis set population). Abbreviations: LS, least-squares; TDD, total daily dose; SE, standard error; SR, sustained-release. |
Similar results were reported for the ADHD-RS-IV inattention and hyperactivity/impulsivity subscales. A statistically significant improvement (P<0.001) in the inattention subscale score from baseline to week 3 was observed for centanafadine-SR (LS mean −9.5) compared with placebo (LS mean −4.5), for a LS mean difference of −5.0 (SE 0.94; 95% CI −6.8, +3.1). The LS mean improvements in the hyperactivity/impulsivity subscale score from baseline to week 3 in patients receiving the 400-mg TDD were −7.0 and −3.9 for the centanafadine-SR and placebo groups, respectively, which represents a significant (P=0.001) LS mean difference of −3.1 between treatments (SE 0.74; 95% CI −4.4, −0.8).
Mean baseline ADHD-RS-IV total score for the 47 patients treated with the centanafadine-SR 400-mg/d TDD was 36.6 (range 28‒51). Mean ADHD-RS-IV total scores at week 3 (combined visits 5 and 10) were 20.1 (range 0‒47) for the centanafadine-SR treatment group (n=45) and 28.0 (range 11‒48) for the placebo group (n=46). In the primary efficacy subpopulation (n=44), a statistically significant difference (P<0.001) in the improvement of ADHD symptom severity from baseline was observed with the centanafadine-SR 400-mg/d TDD (LS mean −15.8; n=44) compared with placebo (LS mean −8.6; n=44), resulting in an LS mean difference of −7.1 (standard error 1.7; 95% CI −10.7, −3.6; effect size 0.62; Figure 3). Statistically significant LS mean differences between centanafadine-SR and placebo in the Inattention (P<0.001) and hyperactivity/impulsivity (P=0.01) subscale scores were −4.5 (SE 1.05; 95% CI −6.7, −2.4) and −2.6 (SE 0.89; 95% CI −4.4, −0.8), respectively. A post hoc sensitivity analysis was conducted in all 85 subjects and the 47 subjects who received a target dose of 400 mg/day. The mean ADHD-RS-IV total score at Weeks 1, 2, and 3 and the improvement in severity of ADHD symptoms from baseline of CTN SR compared to placebo were similar to the primary efficacy results. There were no carryover effects seen with the sensitivity analysis results in assessing difference between treatments in ADHD-RS-IV total score.
For the centanafadine-SR 600-mg/d TDD and centanafadine-SR 800-mg/d TDD groups, LS mean differences in mean ADHD-RS-IV total score at week 3 were −5.1 (SE 3.4; 95% CI −12.4, 2.3; effect size 0.39) and −16.0 (SE 2.9; 95% CI −22.8, −9.2; effect size −1.85), respectively. No efficacy differences in dose response were observed among the 400 mg, 600 mg, and 800 mg target dose groups.
Centanafadine-SR demonstrated a relatively rapid onset of action for a nonstimulant. Specifically, it demonstrated efficacy 1 week after treatment initiation. As assessed by ADHD-RS-IV total score, by the end of week 1, centanafadine-SR demonstrated significant improvement vs placebo whether grouped by all doses (P<0.001) or by the 400 mg dose (P=0.01). Effect sizes for centanafadine-SR at weeks 1 and 2 were 0.49 and 0.70, respectively. Significant improvements with centanafadine-SR vs placebo were also apparent by week 1 for the inattention (P=0.047) and hyperactivity/impulsivity (P=0.01) subscales.
Clinician ratings also favored centanafadine-SR over placebo, with an early onset of action. Dichotomized CGI-I analysis results showed that significantly more patients treated with centanafadine-SR vs placebo had improved (CGI-I score 1–2) ADHD symptoms from baseline at week 1 (23 [34%] vs 8 [12%] patients; P=0.002), week 2 (33 [49%] vs 11 [16%]; P<0.001) and week 3 (34 [50%] vs 10 [15%]; P<0.001; Figure 4). Similar CGI-I results were reported for the subgroup of patients who received the 400 mg/d TDD. In the 400 mg/d TDD subgroup, more patients had improvement over baseline with centanafadine-SR vs placebo at week 2 (21 [48%] vs 7 [16%] patients; P<0.003) and week 3 (22 [50%] vs 6 [14%]; P=0.001), but not at week 1 (13 [30%] vs 6 [14]; P=0.052).
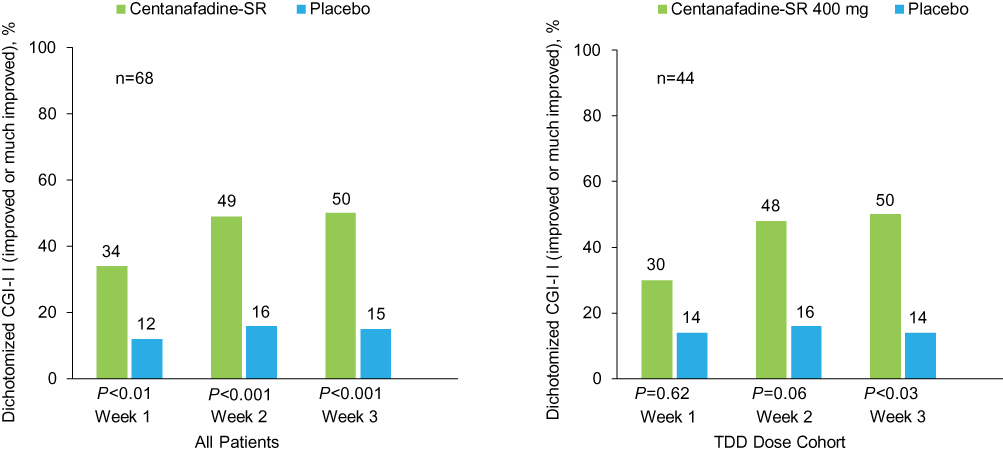 |
Figure 4 Phase 2b study: Clinical Global Impression of Improvement (CGI-I) ratings after 1, 2, and 3 weeks of double-blind treatment (full analysis set population). Abbreviations: SR, sustained-release; TDD, total daily dose. |
In contrast, over the entire study population, there were no statistically significant differences observed between the centanafadine-SR and placebo groups in the PERMP, which was used as an assessment of the duration of cognitive effect after each administration (Figure 5). Similar nonsignificant results were obtained in patients who received the centanafadine-SR 400-mg/d TDD as compared with placebo.
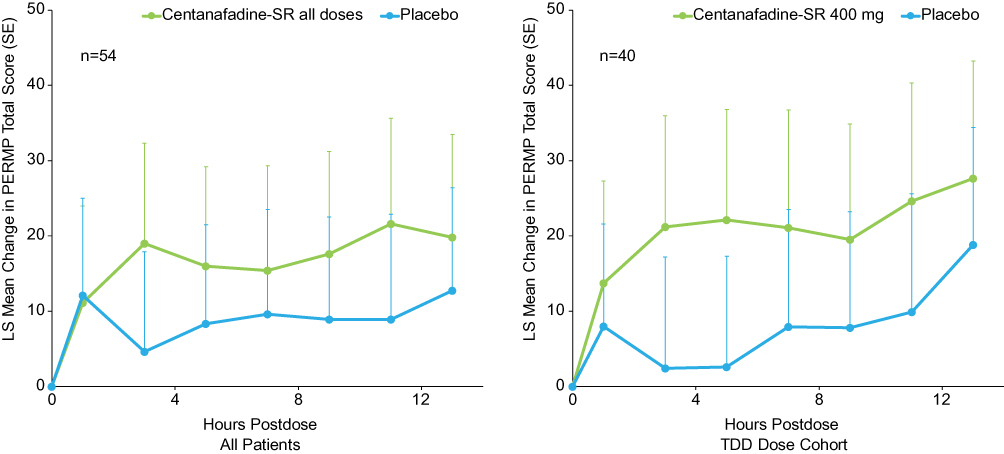 |
Figure 5 Phase 2b study: Permanent Product Measure of Performance (PERMP) total score change from baseline through 13 hours postdose. Abbreviations: LS, least-squares; SE, standard error; SR, sustained-release; TDD, total daily dose. |
Safety
Sixty-three of the 79 patients (80%) who received centanafadine-SR experienced ≥1 TEAE during treatment, and of these, 55 (70%) experienced a TEAE that was considered possibly, probably, or definitely related to treatment (Table 4). The most frequently experienced TEAEs that were considered possibly, probably, or definitely related to centanafadine-SR treatment were decreased appetite (24%), headache (18%), and nausea (20%). In general, the centanafadine-SR 400-mg TDD was well tolerated, with TEAE rates that were lower than at higher doses. The most frequently experienced, treatment-related TEAEs were decreased appetite (16%), and nausea and headache (each 13%). Of the 19 patients with centanafadine-SR 600-mg/d TDD, the most frequently experienced treatment-related TEAEs were diarrhea (26%) and decreased appetite, dry mouth, dizziness, and headache (each 15.9%). Of the 15 patients with centanafadine-SR 800-mg/d TDD, nausea and decreased appetite (each 60%) and fatigue, dry mouth, and headache (each 33.3%) were the most frequently experienced TEAEs.
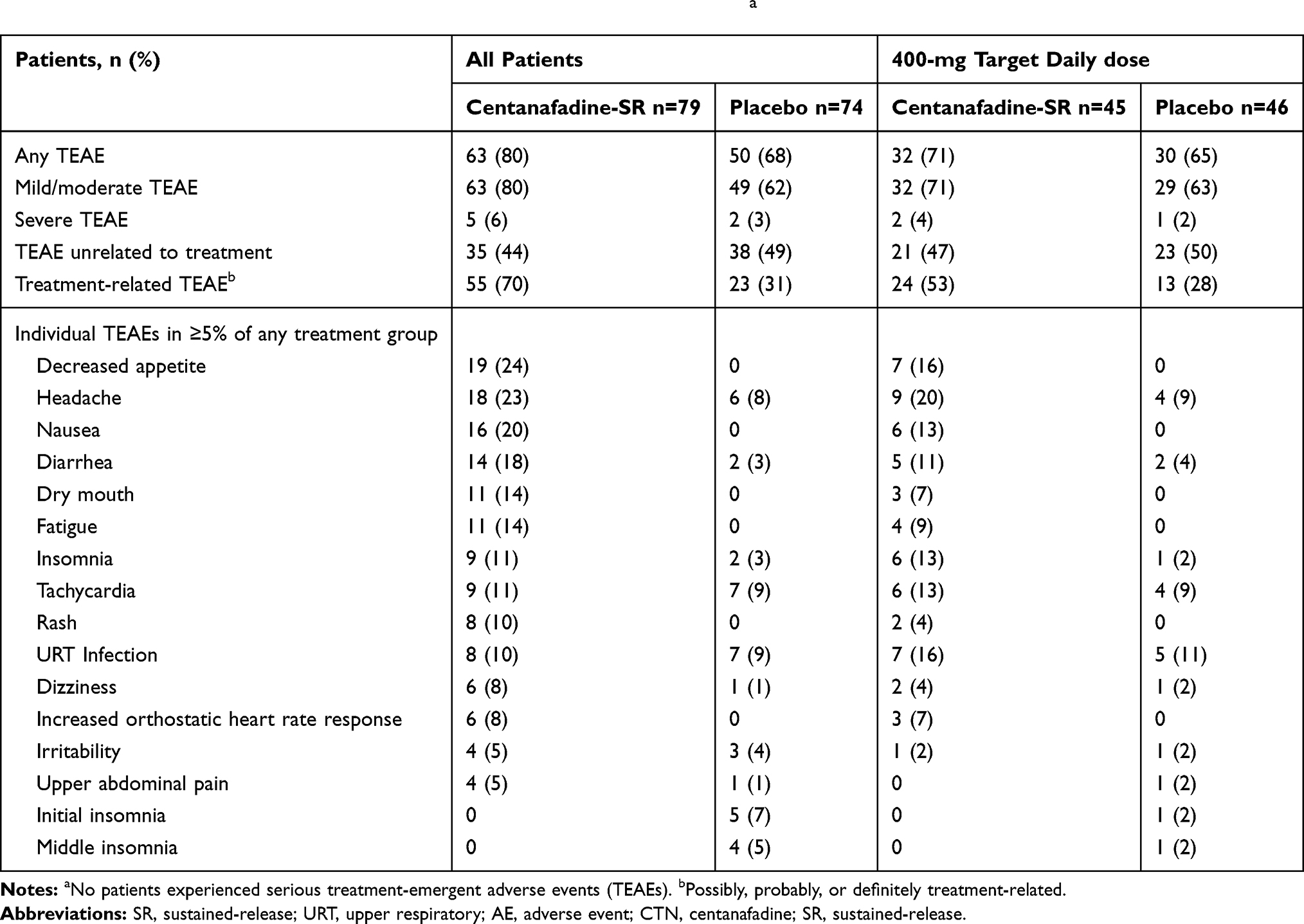 |
Table 4 Phase 2b Study: Patients with Treatment-Emergent Adverse Eventsa |
Most TEAEs were mild or moderate in severity (Table 4). Five patients experienced a total of 6 severe TEAEs (dry mouth, headache, insomnia, pollakiuria, and 2 rashes). Only the rashes led to treatment discontinuation. All TEAEs resolved within 2 weeks with continued treatment, dose reduction, or discontinuation, with the exceptions of 1 patient who experienced a severe nonserious rash and 1 who experienced acne. Both of these TEAEs were ongoing at the end of the study. At the 400-mg dose, 1 patient experienced severe insomnia and 1 experienced severe pollakiuria while on centanafadine-SR 400 mg; both TEAEs were considered treatment related. One patient experienced severe cholelithiasis while taking placebo, which was not considered treatment related. None of the severe TEAEs in patients receiving centanafadine-SR 400 mg necessitated treatment discontinuation.
Twelve patients discontinued treatment due to TEAEs (11, centanafadine-SR; 1, placebo), all of which were considered possibly, probably, or definitely related to treatment. Of the 11 patients who discontinued from centanafadine-SR treatment, 10 were assigned and received TDD ≥600 mg within 1 day of onset of the AE. One patient receiving the centanafadine-SR 400 mg TDD discontinued treatment due to a TEAE— moderate agitation—which was considered probably treatment related. None of the TEAEs were, however, considered serious and all but one (acne) resolved. No deaths or serious TEAEs occurred during the study.
Potentially abuse-related TEAEs were examined as per US Food and Drug Administration guidance.24 Events of this kind included 7 occurrences of dizziness, 2 of somnolence, and 1 of feeling abnormal. All but 1 occurrence of dizziness and 1 of somnolence, and the single case of feeling abnormal were reported during treatment with centanafadine-SR. Among the 8 potentially abuse-related TEAEs reported during centanafadine-SR, all but 1 occurrence of dizziness and the single occurrence of somnolence was considered probably, possibly, or potentially treatment related. On review, however, none of these TEAEs were considered indicative of misuse. There were no events of drug abuse, dependence, diversion, or euphoria observed during the study.
Insomnia was reported in 11% of all patients who received centanafadine-SR (vs 3% with placebo) and in 13% who received the 400-mg/d TDD (vs 2% with placebo). Poor-quality sleep was reported in 2 patients during centanafadine-SR treatment, including 1 in the 400-mg/d TDD group.
Eight patients experienced rash, and of these, 5 were discontinued from the study. Four of the rashes were in the centanafadine-SR 600-mg/d TDD group, and two each were in the 400- and 800-mg/d TDD groups. No patient in the 400-mg/d TDD group discontinued due to rash. A review by dermatologists concluded that none of the rashes exhibited a profile consistent with a rash that may progress to a serious or otherwise life-threatening adverse event. The dermatologist review also determined that the 2 rashes in patients assigned to the 400-mg/d TDD were not representative of rashes observed at higher doses, but were rather related to underlying dermatologic conditions that were aggravated by treatment: viral exanthema in 1 patient and an unidentified external factor in the other.
Discussion
Two phase 2 trials provided initial efficacy data and additional safety data to inform the Phase 3 trial program for centanafadine-SR. In the phase 2a study, centanafadine-SE administered in split doses of 200‒300, 400, and 500 mg/d resulted in statistically significant decreases in ADHD-RS-IV scores; however, mean changes were greatest in the 400-mg group and with fewer TEAEs observed relative to the 500-mg group. In the phase 2b study, patients administered centanafadine 400, 600, and 800 mg/d experienced significant improvements in ADHD symptoms when on active treatment, as measured by the ADHD-RS-IV, compared with placebo. Significant improvements with centanafadine-SR vs placebo were also observed for the individual ADHD-RS-IV inattention and impulsivity/hyperactivity subscales. There was no difference in efficacy between the centanafadine-SR 400-mg/d dose and higher doses. Similar results with ADHD-RS-IV total and subscale scores were observed at weeks 1 and 2 for patients in all dose groups, indicating a rapid onset of action. The effect sizes for centanafadine-SR in the phase 2b study during 3 weeks of treatment were 0.66 overall and 0.62 for the 400-mg/d dose, which compare favorably with other nonstimulants. For example, clinical trials of atomoxetine have generally shown effect sizes of ~0.50.25
Dichotomized CGI-I analysis results showed that significantly more patients treated with centanafadine-SR had improvement in their baseline ADHD symptoms at weeks 1 (P=0.003), 2 (P<0.001), and 3 (P<0.001) than with placebo. Significantly greater improvements with centanafadine-SR vs placebo were also observed at the 400 mg/d TDD at weeks 2 and 3, but were not significant at week 1 (P=0.052).
Although results on the PERMP as a pilot secondary outcome measure did not significantly favor centanafadine-SR over placebo, the study was not powered for a statistical comparison. The PERMP has been used to assess time course effects of the efficacy of stimulants in children and adults; its usefulness as a measure in adults requires sufficient preparatory math tests in individual subjects at all study sites to reduce or eliminate the practice effect.22,23 In the phase 2b study, some sites did not administer all of the practice PERMPs required by the protocol, which may have reduced the sensitivity of this measure and contributed to substantial variability at the 4 study sites that reduced the likelihood of a statistically significant drug effect. Effects of centanafadine-SR on cognitive outcomes remain to be determined in phase 3 trials designed to evaluate efficacy under tightly controlled conditions over a longer duration of time.
As in the phase 2a study, the centanafadine 400-mg dose in the phase 2b study was substantially better tolerated than were higher doses. Three of 47 patients assigned to the 400-mg TDD discontinued treatment and in only 1 patient was the discontinuation due to a TEAE. The most frequent TEAEs were decreased appetite, headache, and nausea, each of which was less frequent at the 400-mg/d dose than at higher doses. There were no treatment-emergent changes in laboratory parameters, vital signs, or physical examination findings associated with centanafadine-SR.
Rash was the TEAE most frequently associated with discontinuation; however, no patient discontinued the 400-mg/d dose and no rash occurring at any dose was considered serious or likely to progress to a serious condition. The rashes that occurred in the 2 subjects who received 400 mg TDD were not considered representative of the drug-induced rash eruptions seen at higher doses. Rash, therefore, appears unlikely to be a particular concern at the 400-mg dose selected for evaluation in phase 3 trials.
Based on these results, centanafadine-SR may offer several advantages compared with other available therapies. Currently available stimulants are known to be effective, but are associated with abuse potential and sleep disturbance. Sleep disturbance is prevalent in 43–80% of patients with ADHD and in clinical trials of stimulants, insomnia was reported as a TEAE in 10–45% of patients.26 Although sleep quality was not formally assessed in the present phase 2 studies, the modified Pittsburgh Sleep Quality Index was used to assess the impact of treatment on sleep as an exploratory endpoint. The results showed no differences in sleep duration or the frequency of sleep disturbance between centanafadine-SR and placebo.27 Further, insomnia was reported as a TEAE in 11% of patients treated with centanafadine-SR (vs 3% with placebo), which is at the bottom of the range for this event in clinical trials of other agents.26
An estimated 30% of individuals prescribed stimulants misuse them.8 In the phase 2b study, there were 8 TEAEs that occurred during centanafadine treatment that the US Food and Drug Administration considers potential warning signs for misuse.24 On review, however, none of these TEAEs were considered to be related to misuse, and there were no instances of abuse, dependence, diversion, or euphoria. These findings are consistent with results from an abuse liability study suggesting that centanafadine-SR at doses as high as 800 mg/d may have less abuse liability than lisdexamphetamine or d-amphetamine (ClinicalTrials.gov NCT02144415). In the phase 2b study, however, the short treatment period precludes drawing conclusions about the likelihood of abuse in clinical practice.
Centanafadine is a triple reuptake inhibitor with in vitro activity at the norepinephrine transporter (IC50, 6 nM), dopamine transporter (IC50 38 nM), and serotonin transporter (IC50 83 nM). Most importantly, the increases in dopamine efflux evoked by centanafadine in microdialysis are relatively slow in onset, reaching a maximum ~60 minutes after intraperitoneal dosing.15 In contrast, dopamine effluxes associated with amphetamine and methylphenidate result in sharper peaks that are reached within 15–30 minutes6 and are thought to result in states in striatum that are highly rewarding.28 The low and slow increases in dopamine would be expected to make centanafadine less rewarding, particularly since norepinephrine is considerably higher in potency than dopamine. Given this mechanism of action, aversion would more likely be encountered than euphoria.
Results of this pair of phase 2 trials provide important information about the dosage to be employed in phase 3 trials. Because the results for the 400-mg group in the phase 2a study were in a small number of patients (n=8), the initial signals of efficacy and safety obtained during treatment did not allow for definitive conclusions about the best centanafadine-SR dose to evaluate in future trials. The phase 2b study focused, therefore, on a 400-mg/d TDD, but with additional patients assigned to receive 600 and 800 mg/d. The study enrolled a larger patient population than the phase 2a study, employed a placebo control, and had a crossover design so that centanafadine-SR and placebo could be compared within each individual patient. Results from the phase 2b study confirmed that titration to a centanafadine-SR dose >400 mg/d offers no clear advantage with respect to efficacy and would be associated with unnecessary additional TEAEs. As a result, a 400-mg/d centanafadine-SR TDD has been selected for evaluation during phase 3 of development.
Conclusion
Two phase 2 studies examined the efficacy and tolerability of centanafadine-SR in adult patients with ADHD. Both studies demonstrated that centanafadine-SR 400 mg/d was effective, as assessed using the ADHD-RS-IV and its individual subscale scores and was well tolerated. Treatment with centanafadine-SR 400 mg did not result in sleep disturbances and there were no signs of abuse or diversion. These results support the use of a centanafadine-SR 400-mg/d TDD in phase 3 trials, which are currently underway.
Abbreviations
ADHD, attention-deficit hyperactivity disorder; ADHD-RS-IV, Attention-Deficit Hyperactivity Disorder Rating Scale Version IV; AWE, adult workplace environment; CGI-I, Clinical Global Impression of Improvement; CGI-S, Clinical Global Impression of Severity; CI, confidence interval; DSM-IV, Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; DSM-V, Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition; LS, least squares; PERMP, Permanent Product Measure of Performance; PFC, prefrontal cortex; SR, sustained-release; TEAE, treatment-emergent adverse events; TDD, total daily dose.
Data Sharing Statement
 |
|
Acknowledgments
Editorial support for this project was provided by Bioscience Communications, New York, NY, and funded by Otsuka Pharmaceutical Development & Commercialization, Inc.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
These studies were funded by Otsuka Pharmaceutical Development & Commercialization, Inc.
Disclosure
Dr Sharon B. Wigal reports grants from Neurovance and Otsuka during the conduct of the studies; consultant, advisory board, and/or speakers fees from Cingulate Therapeutics, Ironshore, Neurovance, NLS, Otsuka, Pfizer, Purdue, Rho, Rhodes, Sunovion and Supernus outside the submitted work. Dr Tim Wigal received consultant, advisory board, and/or research fees from Neurovance and Otsuka during the conduct of the studies; and from Cingulate Therapeutics, Ironshore, Neurovance, NLS, Otsuka, Pfizer, Purdue, Rhodes, Sunovion, and Supernus outside the submitted work. Drs Mary Hobart, Jessica J Madera, Ross A Baker, and Eva Kohegyi are full-time employees for Otsuka Pharmaceuticals Development & Commercialization, Inc. Mr Anthony McKinney was the President and CEO of Neurovance, and inventor of relevant patents. He received salary, bonus, and stock from Neurovanceand has patents 9856217, 9839627, and 9708261 issued. Dr Timothy E Wilens consulted for Otsuka during the conduct of the studies; Massachusetts General Hospital received consulting fees from Otsuka, Ironshore, KemPharm, and Vallon on behalf of work conducted by Dr Wilens; Dr Wilens received grants from NIH (NIDA), is co-owner of a copyrighted diagnostic questionnaire—Before School Functioning Questionnaire—with Ironshore, received royalties for the published book “Straight Talk About Psychiatric Medications for Kids” from Guilford Press and for co-editing the textbook “ADHD in Adults and Children” from Cambridge University Press, and received personal fees from Gavin Foundation, Bay Cove Human Services, US National Football League (ERM Associates), and US Minor/Major League Baseball outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Sharma A, Couture J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann Pharmacother. 2014;48(2):209–225. doi:10.1177/1060028013510699
2. Berridge CW, Devilbiss DM, Andrzejewski ME, et al. Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry. 2006;60(10):1111–1120. doi:10.1016/j.biopsych.2006.04.022
3. Levy F. Dopamine vs noradrenaline: inverted-U effects and ADHD theories. Aust N Z J Psychiatry. 2009;43(2):101–108. doi:10.1080/00048670802607238
4. Cortese S, Adamo N, Mohr-Jensen C, et al. Comparative efficacy and tolerability of medications for attention deficit hyperactivity disorder in children, adolescents, and adults: a systematic review and network meta-analysis. Lancet Psych. 2018;5(9):727–738. doi:10.1016/S2215-0366(18)30269-4
5. Heal DJ, Smith SL, Gosden J, Nutt DJ. Amphetamine, past and present–a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479–496. doi:10.1177/0269881113482532
6. Heal DJ, Smith SL, Henningfield JE. CNS stimulants. Neuropharmacology. 2014;87:1–3. doi:10.1016/j.neuropharm.2014.09.025
7. Kolar D, Keller A, Golfinopoulos M, Cumyn L, Hechtman L, Hechtman L. Treatment of adults with attention-deficit/hyperactivity disorder. Neuropsychiatr Dis Treat. 2008;4(1):107–121. doi:10.2147/ndt.s1747
8. Substance Abuse and Mental Health Services Administration. Results from the 2017 national survey on drug use and health: detailed tables. Available at: https://www.samhsa.gov/data/sites/default/files/cbhsq-reports/NSDUHDetailedTabs2017/NSDUHDetailedTabs2017.htm#tab1-25D.
9. Bright GM. Abuse of medications employed for the treatment of ADHD: results from a large-scale community survey. Medscape J Med. 2008;10:111.
10. Upadhyaya H, Tanaka Y, Lipsius S, et al. Time-to-onset and -resolution in adult patients with ADHD. Postgrad Med. 2015;127(7):677–685. doi:10.1080/00325481.2015.1083394
11. Atomoxetine [Package Insert]. Indianapolis, IN: Eli Lilly & Co; 2017.
12. Bangs ME, Wietecha LA, Wang S, Buchanan AS, Kelsey DK. Meta-analysis of suicide-related behavior or ideation in child, adolescent, and adult patients treated with atomoxetine. J Child Adolesc Psychopharmacol. 2014;24(8):426–434. doi:10.1089/cap.2014.0005
13. Chan E, Fogler JM, Hammerness PG. Treatment of attention-deficit/hyperactivity disorder in adolescents: a systematic review. JAMA. 2016;315(18):1997–2008. doi:10.1001/jama.2016.5453
14. Sallee FR, Lyne A, Wigal T, McGough JJ. McGough. Long-term safety and efficacy of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2009;19(3):215–226. doi:10.1089/cap.2008.0080
15. Bymaster FP, Golembiowska K, Kowalska M, Choi YK, Tarazi FI. Pharmacologic characterization of the norepinephrine and dopamine reuptake inhibitor EB-1020: implications for treatment of attention-deficit Hypersensitivity disorder. Synapse. 2012;66(6):522–532. doi:10.1002/syn.21538
16. Schoedel KA, Meier D, Chakraborty B, Manniche PM, Sellers EM. Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users. Clin Pharmacol Ther. 2010;88(1):69–78. doi:10.1038/clpt.2010.67
17. Learned S, Graff O, Bye A, et al. A novel double blind, placebo-controlled, modified crossover study to assess the abuse potential of GSK372475 in comparison with d-amphetamine and pseudoephedrine in healthy adult, experienced stimulant drug users. Poster presented at: 2010. Ann Meeting Am Soc Clin Pharmacol Ther. 2010;87.
18. Griffith JD, Carranza J, Griffith C, Miller LL. Bupropion: clinical assay for amphetamine-like abuse potential. J Clin Psychiatry. 1983;44(5 Pt 2):206–208.
19. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. Washington, DC: American Psychiatric Publishing; 1994.
20. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders.
21. Swanson JM, Agler D, Fineberg E, et al. UCI laboratory school protocol for PK/PD studies. In: Greenhill LL, Osman BB, editors. Ritalin: Theory and Practice,
22. Wigal SB, Wigal TL. The laboratory school protocol: its origin, use, and new applications. J Atten Disord. 2006;10(1):92–111. doi:10.1177/1087054705286049
23. Wigal SB. Laboratory school protocol mini-review: use of direct observational and objective measures to assess ADHD treatment response across the lifespan. Front Psychol. 2019;10:1796. doi:10.3389/fpsyg.2019.01796
24. US Department of Health and Human Services, Food and Drug Administration, Centers for Drug Evaluation and Research (CDER). Assessment of abuse potential of drugs: guidance for industry. January 2017. Available at: https://www.fda.gov/downloads/drugs/guidances/ucm198650.pdf.
25. Faraone SV, Glatt SJ. A comparison of the efficacy of medications for adult attention-deficit/hyperactivity disorder using meta-analysis of effect sizes. J Clin Psychiatry. 2010;71(06):754–763. doi:10.4088/JCP.08m04902pur
26. Wynchank D, Bijlenga D, Beekman AT, Kooij JJS, Penninx BW. Adult attention-deficit/hyperactivity disorder (ADHD) and insomnia: an update of the literature. Curr Psychiatr Rep. 2017;30(12):98. doi:10.1007/s11920-017-0860-0
27. Wigal SB, Wigal T, Leoni M, et al. A Phase 2b, Randomized, Double-Blind, Multicenter, Placebo-Controlled, Crossover, Safety and Efficacy Study of Centanafadine Sustained-Release in Adults with Attention-Deficit Hyperactivity Disorder. Presented At: 2019 Annual Meeting of the American Professional Society of ADHD and Related Disorders. January 18–20, 2019. Washington, DC. poster S4.
28. Volkow ND, Swanson JM. Variables that affect the clinical use and abuse of methylphenidate in the treatment of ADHD. Am J Psychiatry. 2003;160(11):1909–1918. doi:10.1176/appi.ajp.160.11.1909
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.