Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Safety and effectiveness of controlled-release paroxetine in routine clinical practice: results of a postmarketing surveillance study of patients with depression
Authors Kato M ![]() , Kimura T
, Kimura T ![]() , Kimura T, Hara T
, Kimura T, Hara T
Received 14 November 2014
Accepted for publication 6 January 2015
Published 20 February 2015 Volume 2015:11 Pages 435—452
DOI https://doi.org/10.2147/NDT.S77542
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Masaki Kato,1 Toshifumi Kimura,2 Takeshi Kimura,3 Terufumi Hara3
1Department of Neuropsychiatry, Kansai Medical University, Moriguchi-shi, Osaka, Japan; 2Medical Affairs, 3Clinical Safety and PMS, Development and Medical Affairs Division, GlaxoSmithKline K.K., Shibuya-ku, Tokyo, Japan
Objective: Selective serotonin reuptake inhibitors are commonly used in the pharmacotherapy of depression. However, adverse events can lead to their early discontinuation. This study evaluated the safety and effectiveness of paroxetine controlled-release (CR) tablets in Japanese patients with depression/depressive state (hereafter referred to as depression) in routine clinical practice in Japan.
Patients and methods: This was an open-label, noninterventional, prospective, postmarketing surveillance study. A total of 3,213 patients aged 12–92 years with depression were prescribed paroxetine CR for 8 weeks at the physician’s discretion. Safety was evaluated on the basis of the reporting of adverse drug reactions. Effectiveness was evaluated on the basis of the physician’s assessment using the Clinical Global Impression-Global Improvement (CGI-GI) and the Clinical Global Impression-Severity of Illness (CGI-SI) scales, as well as on the basis of the patients’ self-reported satisfaction. The primary effectiveness outcome was the improvement rate based on the physician’s assessment using the CGI-GI.
Results: The incidence of adverse drug reactions was 11.2% (359/3,213; 95% confidence interval [CI]: 10.1%–12.3%). The common adverse drug reactions that accounted for 1.0% or more of the incidence were nausea (3.5%) and somnolence (2.7%). The proportion of patients who continued paroxetine CR at week 8 was 80.2% (2,577/3,213; 95% CI: 78.8%–81.6%). The improvement rate at week 8 (last observation carried forward) was 72.8% (2,132/2,927; 95% CI: 71.2%–74.4%). The proportion of patients with CGI-SI scores of moderately or severely ill decreased from 63.6% at baseline to 17.9% at week 8. The proportion of patients who were satisfied with paroxetine CR treatment was 69.8% (2,040/2,921; 95% CI: 68.1%–71.5%).
Conclusion: The results of this study suggest that paroxetine CR is a well-tolerated and efficacious treatment for depression in routine clinical practice.
Keywords: paroxetine, controlled-release, postmarketing surveillance, depression
Introduction
The recent mainstream pharmacotherapy for depression is the use of second-generation antidepressants, such as selective serotonin reuptake inhibitors (SSRIs) and serotonin and noradrenaline reuptake inhibitors (SNRIs). An analysis of the US Claims Database revealed that the first choice of treatment was SSRIs in 69.6% and SNRIs in 9.6% (in total, 79.2%) of depressive patients.1 According to the analysis of the prescription patterns of antidepressants in East Asia, SSRIs and other newer antidepressants were also frequently prescribed.2 Different guidelines that have been developed to treat depressive disorder recommend SSRIs among the first drugs of choice.3–6
However, one of the important limitations of SSRIs is their tolerability. Adverse drug reactions (ADRs) frequently reported in the early stage of treatment, such as gastrointestinal symptoms, often lead to the early discontinuation of SSRIs.7,8 Early discontinuation is associated with delayed remission of depression,9 as well as with the increased risk of relapse or recurrence.10 Therefore, reducing the risk of ADRs is essential to provide a more successful treatment option for depression.
Paroxetine hydrochloride hydrate controlled-release tablets (GlaxoSmithKline K.K., Tokyo, Japan; hereafter, referred to as paroxetine CR) are a controlled-release formulation of paroxetine, an SSRI antidepressant.11 This enteric, film-coated, controlled-release formulation allows for the slow dissolution and gradual release of paroxetine only after the tablets have passed the stomach.12,13 Because of these characteristics, the drug can avoid the excessive exposure of paroxetine in the upper gastrointestinal tract, which is related to gastrointestinal symptoms such as nausea.12 Moreover, the gradual absorption in the gastrointestinal tract leads to slow increases and smaller fluctuations in the blood concentration of paroxetine over time, as compared with the immediate-release tablets (paroxetine IR).12,13 These properties of paroxetine CR are expected to be associated with a reduced risk of ADRs and improved tolerability when compared with paroxetine IR because dose reduction and slower titration are clinical strategies that are commonly recommended to manage the ADRs of antidepressants.6
The safety and efficacy information regarding paroxetine CR, mainly derived from clinical studies, suggests that paroxetine CR features improved tolerability, though it has antidepressant activity that is comparable to that of paroxetine IR. Golden et al14 reported that the incidence of nausea in the early stage of treatment was significantly lower for paroxetine CR (14%) than for paroxetine IR (23%; P≤0.05) and that the rate of dropouts due to adverse events (AEs) was comparable between paroxetine CR and placebo (10% and 6%, respectively; P=0.14), but this rate was significantly higher for paroxetine IR (16%) than for placebo (P=0.0008).
However, there is a limitation to the applicability of the results from clinical studies to routine clinical practice, as subjects of clinical studies are typically selected on the basis of rigorous inclusion and exclusion criteria. The sample size in most clinical studies is too small to detect very rare ADRs. So far, no large-scale studies have been conducted to assess ADRs and the effectiveness of paroxetine CR in a real-world scenario. Thus, we conducted this postmarketing surveillance study to evaluate the safety and effectiveness of paroxetine CR in patients with depression in routine clinical practice. Regarding effectiveness outcomes, the patients’ satisfaction with treatment with paroxetine CR, as well as their treatment preferences, were evaluated with the objective measures frequently used in clinical studies, as those are known to be positively associated with improved adherence or persistence.15–17 The interim analysis of safety and effectiveness in this study has already been reported in patients who completed the study by the end of January 2013 (safety population, n=1,150).18
In routine clinical practice, some patients fail to continue antidepressant treatment because of an inadequate response or intolerability. One of the recommended strategies for addressing inadequate response or intolerability is switching to another antidepressant.4,6 In a survey of clinicians who treated depressive patients with a lack of response to adequate SSRI treatment, switching to another antidepressant was the most frequently chosen option.19 Nevertheless, to the authors’ knowledge, there are no reports of a comprehensive evaluation of the safety and effectiveness of paroxetine CR in Japanese and Asian patients with depression when the treatment has been switched from antidepressants of other classes or from those with other active ingredients. Thus, in this study, stratified analyses were performed on the basis of class and ingredients of the prior antidepressant when the patients switched from another antidepressant.
This study was completed in June 2013, and the target sample size of 3,000 patients for the safety population was achieved. This report will provide new findings regarding the safety and effectiveness outcomes in the final analysis.
Materials and methods
Subjects
This surveillance study enrolled patients who were diagnosed with depression/depressive state (hereafter referred to as depression) by the investigator on the basis of information obtained from a medical interview, as well as on the basis of the patients’ course of treatment, and the patients were prescribed paroxetine CR for the first time. Depression/depressive state was defined as both a diagnosis of depression and having a clinically significant depressive episode. Patients who were taking any antidepressants (defined as drugs used to treat depression, including paroxetine IR) were also enrolled. The target sample size of 3,000 patients for the safety population was determined to give a 95% probability of detecting any unknown ADR with an incidence of 0.1%.
Study method
Patients were registered for the study through a central registration system. Investigators documented the relevant information about the individual patients prescribed paroxetine CR in the case report form (CRF) from the start of treatment to the end of the observation period. The institutional ethical committee’s approval and the patients’ informed consent were not necessarily required because this was a postmarketing surveillance study of standard medical practice. Treatment details were determined by the investigators. This study was conducted in compliance with the Japanese Good Post-Marketing Surveillance Practice (Ordinance of the Ministry of Health, Labour and Welfare No 171, dated December 20, 2004), which specifies how manufacturers should carry out appropriate postmarketing surveillance to evaluate the safety and effectiveness of drug products after marketing.
Observation period and measures
The patients were observed for 8 weeks from the start of treatment with paroxetine CR (or until discontinuation or termination of treatment, if applicable), because at least 8 weeks of treatment were required to assess the patients’ responses.3,4
The data collected during the observation period included patient demographics and other characteristics, treatment adherence, concomitant medications, safety data, and effectiveness data (the Clinical Global Impression-Global Improvement [CGI-GI],20 the Clinical Global Impression-Severity of Illness [CGI-SI],20 and the patients’ satisfaction and treatment preferences). For data analysis by the class of prior antidepressants, the drugs used to treat depression just before switching to paroxetine CR were classified into one of the following six categories: immediate-release SSRIs (paroxetine IR, fluvoxamine, sertraline, and escitalopram); SNRIs (milnacipran and duloxetine); noradrenergic and specific serotonergic antidepressants (NaSSA; mirtazapine); tricyclic antidepressants (imipramine, clomipramine, amitriptyline, nortriptyline, amoxapine, trimipramine, lofepramine, and dosulepin); tetracyclic antidepressants (mianserin, maprotiline, and setiptiline); and others (sulpiride and trazodone). Fluoxetine (an SSRI) and venlafaxine (an SNRI) were not included because they were unapproved antidepressants in Japan.
Criteria for the safety and effectiveness evaluation
For the safety evaluation, all data pertaining to AEs (eg, diseases, symptoms, and laboratory abnormalities) that occurred after the start of treatment with paroxetine CR were collected regardless of the causal relationship. An AE that had a relationship with paroxetine CR that could not be ruled out by the investigator was regarded as an ADR. A serious AE (SAE) was defined as an AE that was considered by the investigator to result in death or to be life-threatening, to require hospitalization or the prolongation of existing hospitalization, to result in a disability that interferes with daily activities, as well as to lead to important medical events that were as serious as the other conditions listed here, or to the development of a congenital anomaly/birth defect in the patient’s offspring. If a patient had received branded paroxetine IR tablets (GlaxoSmithKline K.K.) before starting treatment with paroxetine CR and he or she had switched to paroxetine CR because of an AE with the branded paroxetine IR tablets, the AE was investigated and recorded in detail.
Effectiveness was evaluated using the CGI-GI and CGI-SI. The CGI-GI rating was performed at weeks 2, 4, and 8, or at the discontinuation/termination of treatment with paroxetine CR. Specifically, the investigator assessed each patient’s total improvement on the basis of changes in symptoms according to the following scale: very much improved; much improved; minimally improved; no change; minimally worse; much worse; very much worse; and not assessed. The improvement rate, defined as the proportion of patients who were considered minimally, much, or very much improved, was calculated because even a minimal improvement is considered to be clinically significant in patients pretreated with antidepressants who are often seen in routine clinical practice. For the analysis of improvement rate at week 8 (or at discontinuation/termination, if applicable), missing values at week 8 were inputted using the last observation carried forward approach.
The CGI-SI rating was performed at the start of treatment with paroxetine CR (baseline) and at week 8 (or at the discontinuation/termination of treatment). Specifically, the investigator assessed the severity of depression for each patient in accordance with the following scale: 0= not assessed; 1= normal (not at all ill); 2= borderline mentally ill; 3= mildly ill; 4= moderately ill; 5= markedly ill; 6= severely ill; and 7= among the most extremely ill. This severity score was used to assess the severity of illness over time.
The investigator interviewed individual patients to determine their level of satisfaction with their treatment with paroxetine CR, as well as their treatment preferences. Patient satisfaction was assessed at week 8 or at the discontinuation/termination of treatment. The satisfaction level was rated according to the following 7-point scale: very satisfied; moderately satisfied; slightly satisfied; neutral; slightly dissatisfied; moderately dissatisfied; and very dissatisfied. The satisfaction rate, defined as the proportion of patients who were very, moderately, or slightly satisfied, was calculated. In addition, patients were asked to select every applicable reason for treatment satisfaction/dissatisfaction from the following options: realization of efficacy/no realization of efficacy; absence of AEs or fewer AEs/experience of AEs; and other. Patients who switched to paroxetine CR from another antidepressant were asked at week 8 of treatment to assess their preference for paroxetine CR, as compared with the prior antidepressant, according to the following 7-point scale: much better; moderately better; slightly better; similar; slightly worse; moderately worse; and much worse. The preference rate, defined as the proportion of patients who assessed paroxetine CR as much better, moderately better, or slightly better, was calculated. In addition, patients were asked to select every applicable reason for their better/worse evaluation of paroxetine CR from the following options: realization of efficacy/no realization of efficacy; absence of AEs or fewer AEs/experience of AE(s); and other. The calculation of satisfaction rate and preference rate included the categories of slightly satisfied and slightly better, respectively, because these ratings were also considered to be clinically significant for patients pretreated with antidepressants who were often seen in routine clinical practice.
Statistical analysis
Chi-square or Fisher’s exact tests were used to compare the incidence of ADRs or the improvement rates between patients in the absence and presence of each main patient characteristic. The Wilcoxon signed-rank test was used to compare the CGI-SI scores before and after the start of treatment with paroxetine CR. All of these analyses were performed using SAS version 9.2 with a two-sided significance level of 0.05.
For data from patients who were switched from another antidepressant, stratified analyses were performed on the basis of the class and ingredient of the prior antidepressant. Patients who had received no antidepressant within 4 weeks before the start of treatment with paroxetine CR were regarded as patients without prior pharmacological treatment for depression.
Results
Patient disposition and characteristics
This study was conducted between June 2012 and June 2013. A total of 3,431 patients were enrolled at 594 medical institutions, and CRFs for 3,423 patients were collected. The safety population was composed of 3,213 patients (Figure 1). The effectiveness population was composed of all patients in the safety population, except those who met the exclusion criteria for the effectiveness population. A total of 2,927 patients were included in the effectiveness population.
 | Figure 1 Disposition of the patients. |
Table 1 shows the characteristics of patients in the safety population. At baseline, 1,047 patients (32.6%) were mildly ill, 1,644 (51.2%) were moderately ill, and 340 (10.6%) were markedly, severely, or among the most extremely ill. The disease duration before the start of treatment was ≤6 months for 1,772 patients (55.2%), >6 months and ≤2 years for 605 patients (18.8%), >2 and ≤5 years for 378 patients (11.8%), and >5 years for 249 patients (7.7%).
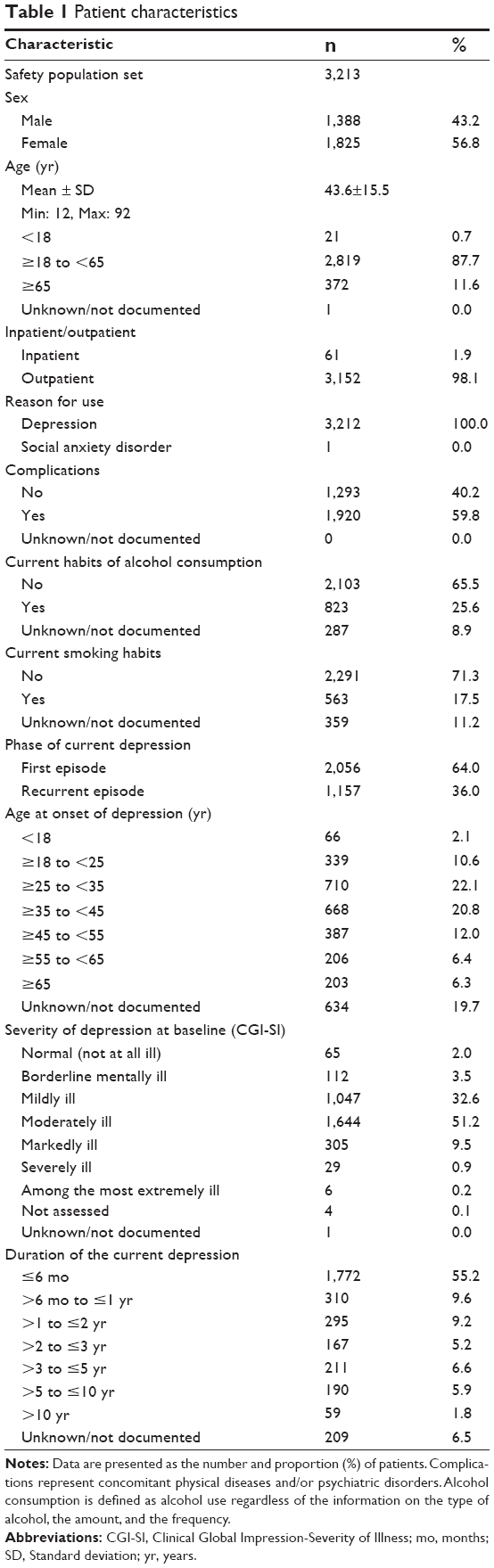 | Table 1 Patient characteristics |
In the safety population, 1,533 patients (47.7%) had no prior pharmacological treatment for depression, whereas 1,680 patients (52.3%) did, and of these patients, 1,476 (45.9% of the total) were switched to paroxetine CR from another antidepressant (these patients were referred to as “switched patients”). Among the 1,476 switched patients, the duration of treatment with the maintenance dose or the maximum dose of the prior antidepressant (just prior to beginning treatment with paroxetine CR) was ≤1 month for 244 patients (16.5%), >1 and ≤6 months for 378 patients (25.6%), >6 months and ≤2 years for 420 patients (28.5%), and >2 years for 381 patients (25.8%). During treatment with paroxetine CR, 1,074 patients (33.4%) also received other antidepressants. The most commonly used concomitant antidepressant was sulpiride (n=263). In addition, 606 patients (18.9%) received concomitant nondrug therapy for depression, such as cognitive behavior therapy. In the safety population, 1,920 patients (59.8%) had complications; 2,444 patients (76.1%) received concomitant medications for the treatment of diseases other than depression; and 1,544 patients (48.1%) received benzodiazepine (BZD) anxiolytics. The mean dose (± standard deviation [SD]) of paroxetine CR was 18.1±10.2 mg/day (d) at baseline and 26.0±13.2 mg/d at week 8.
Among the 3,213 patients in the safety population, 2,577 patients remained under treatment with paroxetine CR at week 8 (continuation rate: 80.2%; 95% confidence interval [CI]: 78.8%–81.6%), whereas 635 patients (19.8%) discontinued/terminated the treatment (one patient’s data pertaining to his/her continuation status at week 8 were missing). The most frequent reason for discontinuation/termination was failure to visit in 232 patients (7.2%), followed by AEs in 199 patients (6.2%), inadequate responses in 81 patients (2.5%), and improvement of symptoms in 56 patients (1.7%).
Safety
Adverse drug reactions
There were 458 cases of ADRs (defined as AEs that had a causal relationship with paroxetine CR that could not be ruled out by the investigator) for 359 (11.2%) of the 3,213 patients in the safety population. The most common ADRs (with an incidence of ≥1%) were nausea (113 cases) and somnolence (87 cases) (Table 2).
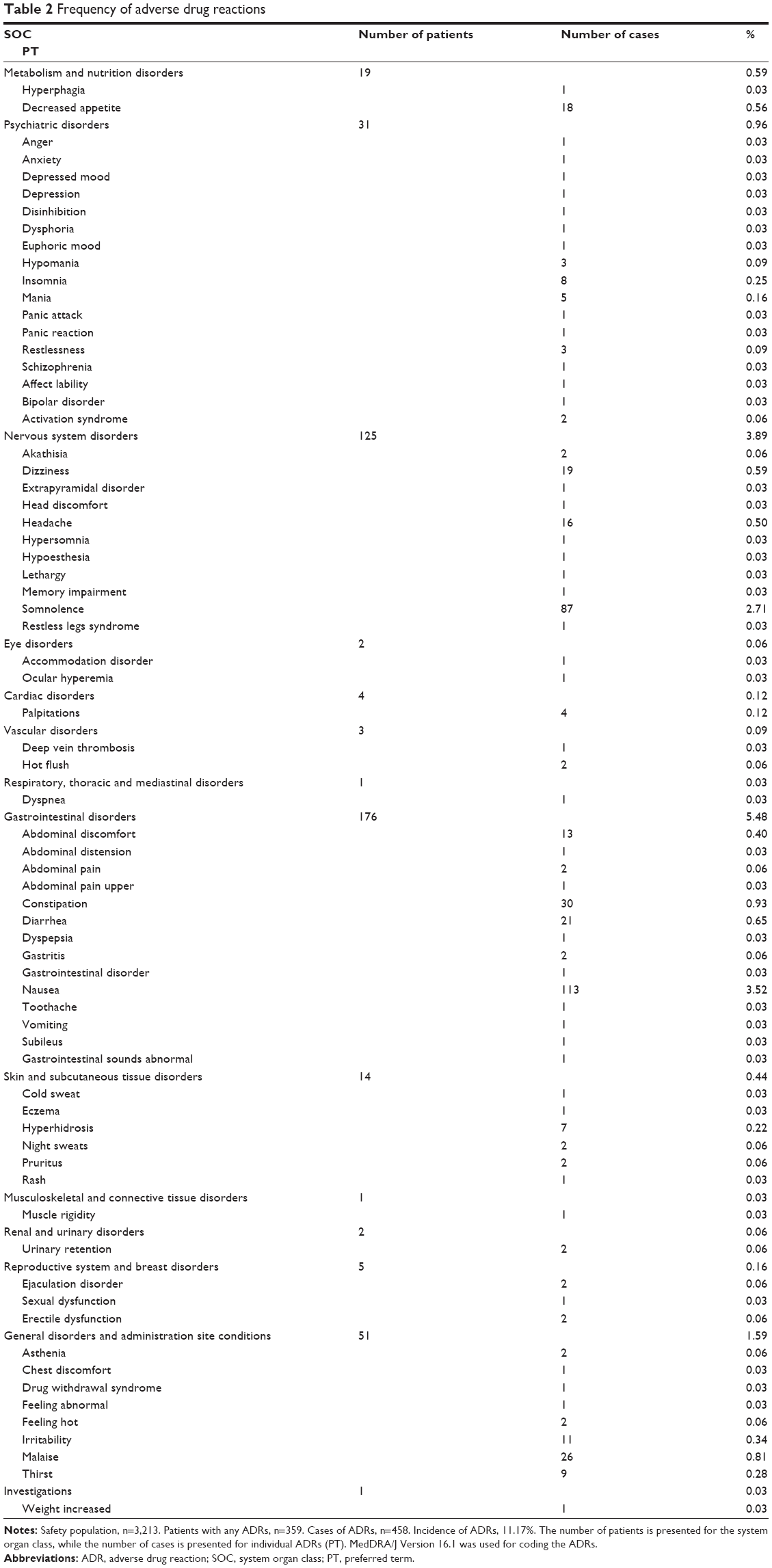 | Table 2 Frequency of adverse drug reactions |
There were 22 cases of SAEs in 18 patients. They included two fatal cases (suicide and myocardial infarction), a case of self-injurious behavior, and a case of suicidal ideation. In addition, long QT syndrome was reported in one patient. These SAEs were not considered to be related to paroxetine CR. There were 12 treatment-related SAEs in eight patients: dizziness (two cases); urinary retention (two cases); and nausea, abdominal discomfort, anxiety, irritability, schizophrenia, bipolar disorder, deep vein thrombosis, and mania (one case each). All of the aforementioned treatment-related SAEs, except for bipolar disorder, resolved or were resolving during treatment continuation (one case of urinary retention), or at or after treatment discontinuation. The patient who experienced bipolar disorder received paroxetine CR monotherapy (without mood stabilizers or antipsychotics) for the treatment of bipolar depression. This patient discontinued treatment with paroxetine CR after aggravation of symptoms and did not recover from this event.
There were two cases of so-called activation symptoms or activation syndrome that were reported in two patients. Both cases were nonserious and had resolved. There is currently no established definition of so-called activation symptoms. Thus, the symptoms listed in the warning by the US Food and Drug Administration in 2004, as related to activation syndrome21 and other related symptoms, were defined as activation syndrome-related symptoms to take a conservative approach to evaluating the incidence of so-called activation symptoms. As a result, 37 cases of such symptoms in 32 patients (including the aforementioned two cases in two patients) were found, with an incidence of 1.0% (32/3,213).
The onset time of ADRs was investigated in patients without prior pharmacological treatment for depression. Of the 292 ADRs reported in this subset of patients, 209 ADRs (71.6%) occurred in the first 2 weeks of treatment with paroxetine CR. The most common ADRs reported in the first 2 weeks were gastrointestinal disorders (100 cases), including 71 cases of nausea and 13 of diarrhea, as well as nervous system disorders (59 cases), including 40 cases of somnolence.
The effects of dose increases on the incidence of ADRs were assessed in patients without prior pharmacological treatment for depression. In this subset of patients from the safety population, 90.7% (1,390/1,533) started treatment with paroxetine CR at 12.5 mg/d, which is the approved initial dose in Japan. The incidence of ADRs was analyzed on the basis of the mean daily dose of the drug in patients who started treatment with paroxetine CR at the initial dose of 12.5 mg/d (excluding one patient with ADRs whose mean daily dose was <12.5 mg/d). The incidence was 15.9% (189/1,188) for ≥12.5 and <25 mg/d, 12.9% (20/155) for ≥25 and <37.5 mg/d, and 6.5% (3/46) for ≥37.5 and <50 mg/d, indicating that no increase in the incidence of ADRs was associated with the increase in the mean daily dose (Figure 2).
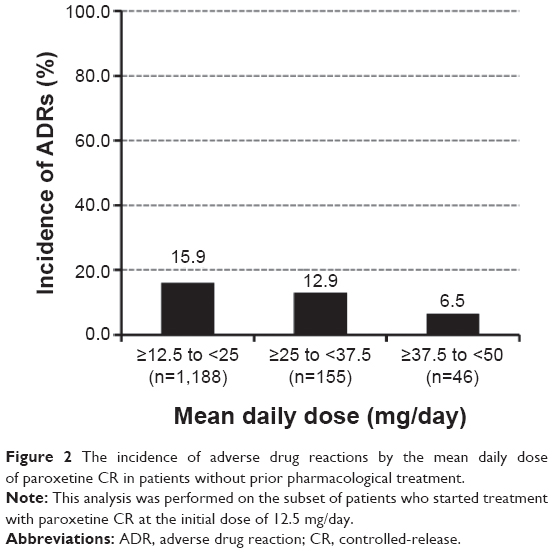 | Figure 2 The incidence of adverse drug reactions by the mean daily dose of paroxetine CR in patients without prior pharmacological treatment. |
Adverse drug reactions by patient characteristics
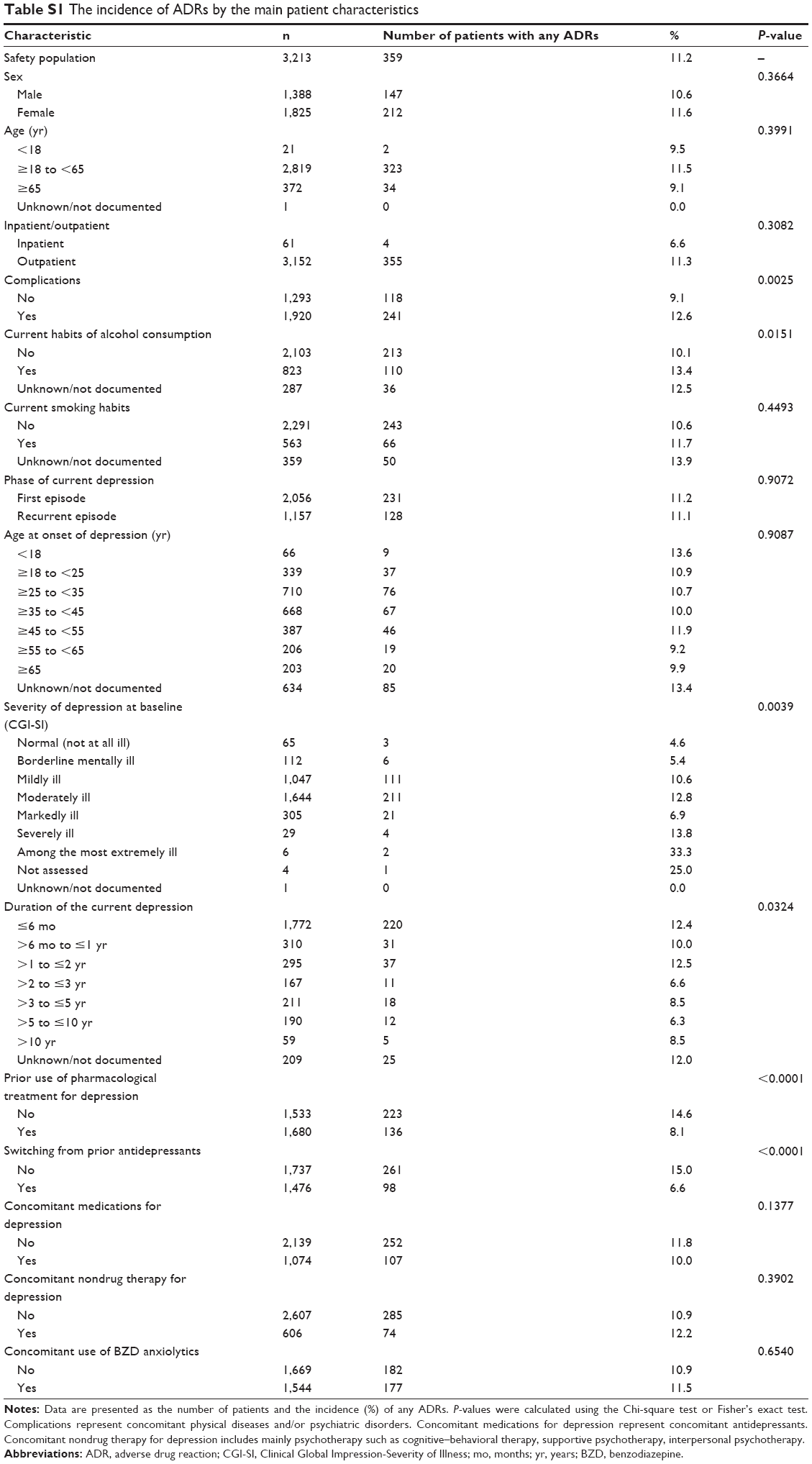
We compared the incidence of ADRs between patients in the absence and presence of each main patient characteristic (Table S1). Significant differences in the incidence of ADRs were demonstrated in relation to the following features: complications; drinking habits; the severity of depression at baseline; the duration of depression; prior use of pharmacological treatment for depression; and switching from a prior antidepressant.
Effectiveness
Improvement rate and severity over time
The improvement rate increased in relation to the duration of treatment: 52.1% (1,431/2,748; 95% CI: 50.2%–54.0%) at week 2; 68.9% (1,879/2,727; 95% CI: 67.1%–70.6%) at week 4; and 77.3% (1,982/2,565; 95% CI: 75.6%–78.9%) at week 8. The improvement rate at week 8 (or at discontinuation/termination, if applicable) was 72.8% (2,132/2,927; 95% CI: 71.2%–74.4%) (Figure 3).
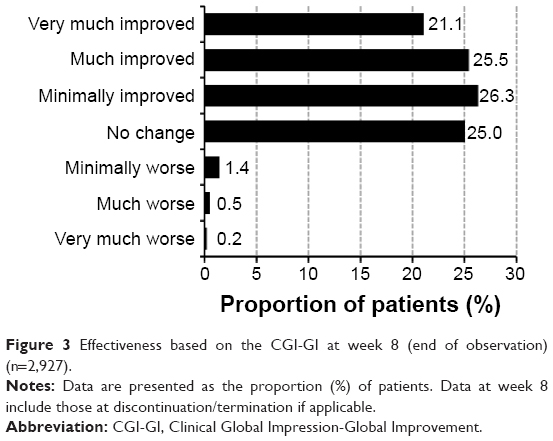 | Figure 3 Effectiveness based on the CGI-GI at week 8 (end of observation) (n=2,927). |
The change in severity over time from baseline to week 8 was assessed in 2,910 patients in the effectiveness population for whom the CGI-SI data both at baseline and at week 8 (or at discontinuation/termination, if applicable) were available. The proportion of patients rated as moderately or more severely ill decreased from 63.6% at baseline to 17.9% at week 8 (Figure 4). Moreover, the severity score at week 8 (mean ± SD; 2.74±0.92) was significantly lower than that at baseline (3.72±0.74; P<0.0001).
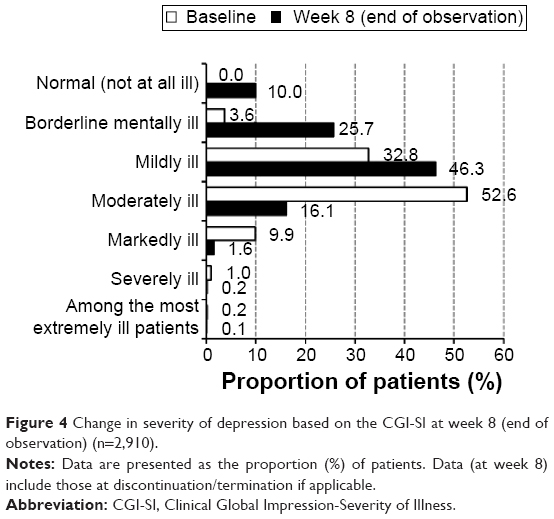 | Figure 4 Change in severity of depression based on the CGI-SI at week 8 (end of observation) (n=2,910). |
Improvement rate by patient characteristics
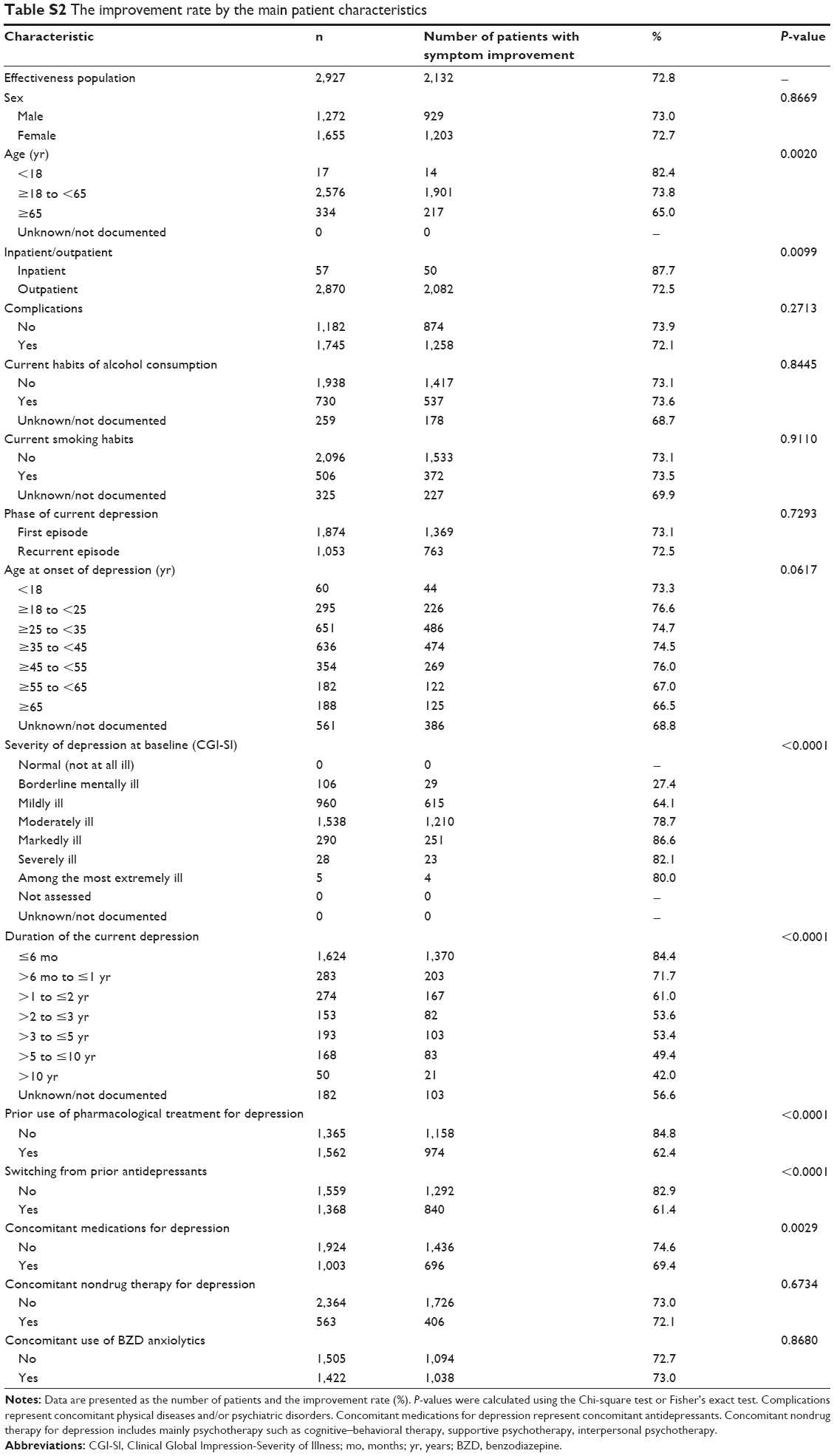
We compared the improvement rate between patients in the absence and presence of each main patient characteristic (Table S2). Significant differences in the improvement rate were demonstrated in relation to the following features: age; inpatient/outpatient; severity of depression at baseline; duration of depression; prior use of pharmacological treatment for depression; switching from a prior antidepressant; and use of concomitant medications for depression.
Satisfaction with and preference for paroxetine CR
Patient satisfaction with treatment using paroxetine CR was investigated at week 8 or at discontinuation/termination (Table 3). The satisfaction rate was 69.8% (2,040/2,921; 95% CI: 68.1%–71.5%). The most common reason for satisfaction (multiple answers allowed) was realization of efficacy (79.7%; 1,625/2,040), followed by the absence of AEs (33.7%; 688/2,040), fewer AEs (2.0%; 41/2,040), and other (1.4%; 28/2,040). The most common reason for dissatisfaction (multiple answers allowed) was the experience of AEs (53.3%; 123/231), followed by no realization of efficacy (45.5%; 105/231), and other (4.3%; 10/231).
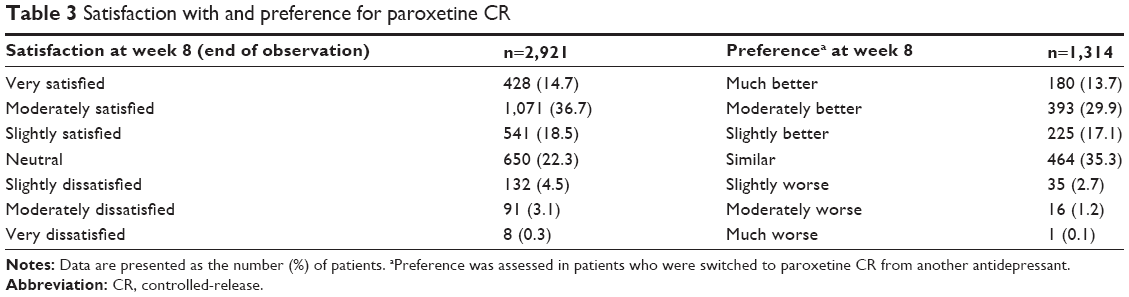 | Table 3 Satisfaction with and preference for paroxetine CR |
In switched patients, the patient preference for paroxetine CR, as compared with the prior antidepressant, was assessed at week 8 (Table 3). The preference rate was 60.7% (798/1,314; 95% CI: 58.0%–63.4%). The most common reason for an improved evaluation (multiple answers allowed) was the realization of efficacy (69.7%; 556/798), followed by the absence of AEs (39.4%; 314/798), other (3.1%; 25/798), and fewer AEs (0.8%; 6/798). The most common reason for a worse evaluation (multiple answers allowed) was the experience of AEs (55.8%; 29/52), followed by no realization of efficacy (34.6%; 18/52), and other (13.5%; 7/52).
The safety and effectiveness of paroxetine CR in patients switched from another antidepressant
In this study, approximately half of the patients in the safety population (52.3%; 1,680/3,213) had received prior pharmacological treatment for depression, and 87.9% of them (1,476/1,680) were switched to paroxetine CR. Among these 1,476 patients, the most commonly used class of prior antidepressants was immediate-release SSRIs for 1,282 patients (86.9%), followed by SNRIs for 62 (4.2%), NaSSA for 47 (3.2%), tricyclic/tetracyclic antidepressants for 30 (2.0%), multiple classes for 29 (2.0%), and others for 26 (1.8%) patients (Table 4). Among the 1,282 patients who used immediate-release SSRIs before switching, most were switched from paroxetine IR, and only 226 patients (17.6%) were switched from immediate-release SSRIs other than paroxetine. In the patients who switched from an antidepressant (not from multiple antidepressants), the most commonly used ingredient of the prior antidepressant was paroxetine IR in 1,054 patients (71.4%), followed by sertraline in 103, escitalopram in 82, duloxetine in 52, mirtazapine in 47, fluvoxamine in 41, and sulpiride in 21 patients. The reasons for switching included an inadequate response (54.5%; 805/1,476), the experience of AEs (6.8%; 114/1,476), and other (37.7%; 557/1,476). The proportions of patients who switched because of an inadequate response, the experience of AEs, or other reasons by the class of the prior antidepressant are demonstrated in Table 4. The prior antidepressant was switched to paroxetine CR because of inadequate response among most of the patients who had been treated with drugs other than paroxetine.
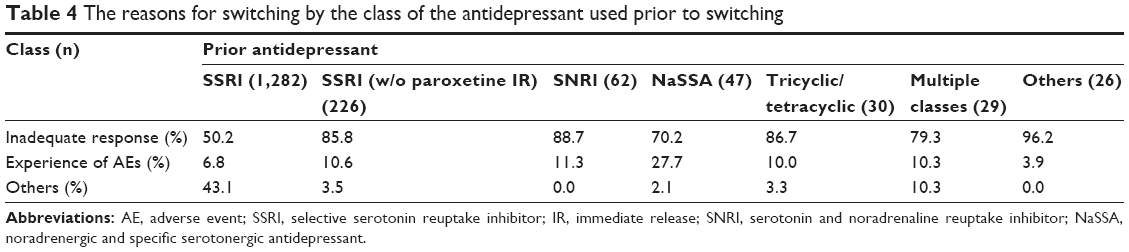 | Table 4 The reasons for switching by the class of the antidepressant used prior to switching |
The incidence of ADRs in switched patients by class and ingredient of the prior antidepressant is presented in Table 5. The analyses presented were performed only for antidepressant classes or ingredients used in at least ten patients. Patients who switched from multiple antidepressant classes or multiple ingredients to paroxetine CR were excluded from the analyses. The treatment continuation rate at week 8 was also presented by class and ingredient of the prior antidepressant (Table 5). Treatment was continued in approximately 80%–90% of the patients who were switched from another antidepressant, except for those patients who switched from sulpiride.
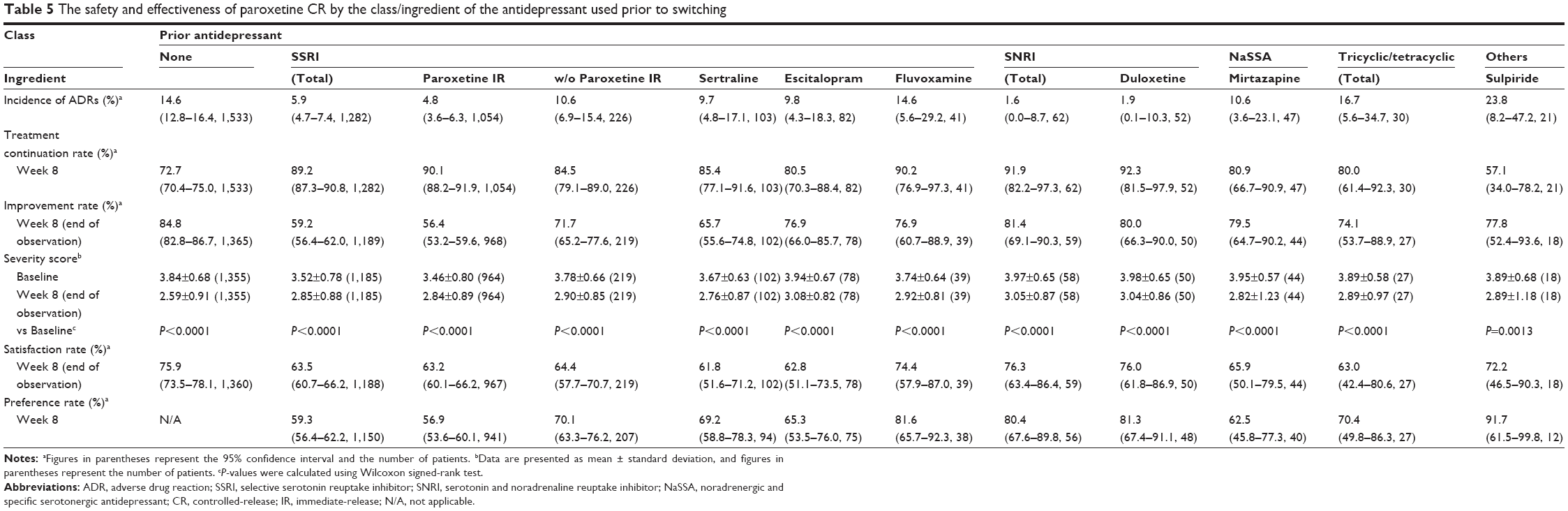 | Table 5 The safety and effectiveness of paroxetine CR by the class/ingredient of the antidepressant used prior to switching |
The effectiveness variables are also shown by class and ingredient of the prior antidepressant in Table 5. The point estimate of the improvement rate was generally over 70% in most of the switched patients, except those patients who switched from paroxetine IR. The physician-assessed severity score at week 8 significantly decreased from baseline regardless of the class or ingredient of the prior antidepressant.
The point estimates of patient satisfaction and preference rates by class or ingredient of the prior antidepressant ranged from 62% to 76% and from 57% to 92%, respectively.
We focused on patients who switched from paroxetine IR, which was the most common antidepressant used prior to switching in this study. The safety and effectiveness variables by the reasons for switching in this subset of patients are shown in Table 6, and they were compared with those of patients who switched from a nonparoxetine SSRI. The incidence of ADRs tended to be higher in patients who switched because of AEs than in those who switched owing to inadequate responses, whether they were switched from paroxetine IR or from a nonparoxetine SSRI. Moreover, patients who were switched from paroxetine IR to paroxetine CR at a dose ratio of 1:1.25 owing to AEs, and who were treated with the constant or increased dose of paroxetine CR after switching, were specifically followed to determine the outcome of ADRs commencing upon prior paroxetine IR treatment during the treatment with paroxetine CR. A total of 27 ADRs of paroxetine IR were followed up: ten cases of somnolence; three cases each of nausea and dizziness; two cases of constipation; and one case each of libido decrease, nightmare, gastrointestinal disorder, malaise, bruxism, decreased appetite, palpitations, dysuria, and mania. One case each of libido decrease, nightmare, somnolence, and dizziness did not resolve, while 85.2% (23/27 cases) of these reactions resolved or were in the process of resolving during treatment with paroxetine CR (specifically, 18 cases resolved, and two cases of somnolence and one case each of palpitations, dysuria, and constipation were resolving).
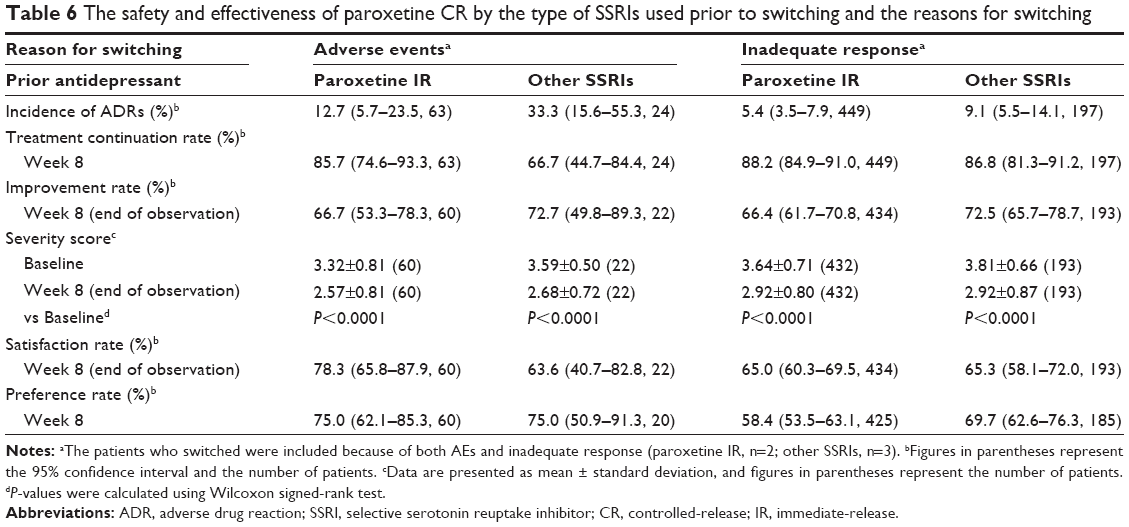 | Table 6 The safety and effectiveness of paroxetine CR by the type of SSRIs used prior to switching and the reasons for switching |
The improvement rate based on the physician-assessed effectiveness endpoint was similar between patients who switched because of AEs and those who switched because of inadequate responses, whether they were switched from paroxetine IR or from a nonparoxetine SSRI. On the other hand, the satisfaction and preference rates were similar in patients switched from a nonparoxetine SSRI, whether because of AEs or inadequate responses, but these rates were higher in patients who were switched owing to AEs, as compared with those who were switched owing to inadequate responses when switched from paroxetine IR.
Discussion
This postmarketing surveillance study suggested the good tolerability and high effectiveness profile of paroxetine CR in patients with depression in routine clinical practice.
The incidence of ADRs was 11.2%. The common ADRs of nausea and somnolence found in this study were generally consistent with those reported by a previous clinical study.11 Most of the reported ADRs were not serious, and all serious ADRs, except for bipolar disorder, had resolved or were resolving. Although further accumulation of safety data is required for rare events, the overall data revealed no new issues/concerns. The patient who experienced bipolar disorder as a serious ADR received paroxetine CR monotherapy (without mood stabilizers or antipsychotics) for the treatment of bipolar depression. It should be noted that the management of bipolar depression with antidepressant monotherapy is not recommended because of the potential risk of a manic switch and no definitive conclusion regarding efficacy.22 In addition, the incidence rate of the activation syndrome-related symptoms in this study, which was conducted with more than 3,000 patients (1.0%), was lower than that reported in a systematic review of all evidence pertaining to activation syndrome by Sinclair et al23 (4%–65%), although the difficulty in discriminating these symptoms from those caused by the worsening of depression should be taken into account.
The onset time of ADRs was analyzed only in patients without prior pharmacological treatment to avoid the possible effects of prior antidepressants. The majority of ADRs (71.6%) occurred in the first 2 weeks of treatment with paroxetine CR. The most common ADRs reported in the first 2 weeks were gastrointestinal disorders such as nausea, and nervous system disorders such as somnolence. This ADR profile was consistent with that reported by a clinical study of paroxetine CR11 and the postmarketing surveillance of paroxetine IR in patients with depression and panic disorder.24
The effects of dose increases from the approved initial dose in Japan of 12.5 mg/d on the incidence of ADRs were assessed in patients without prior pharmacological treatment for depression. Dose increases were not associated with any increases in the incidence of ADRs, which supports the findings that the majority of ADRs occurred in the first 2 weeks of treatment with paroxetine CR.
The improvement rate at week 8 or at discontinuation/termination (72.8%) was similar to that reported in the postmarketing surveillance study of paroxetine IR (75.9%),24 although there are some differences in the observation period and methods of assessment regarding the rate of improvements between the two studies. The calculation of the improvement rate in our study included the categories of not only very much or much improved, but also minimally improved. The previous randomized, double-blind, placebo-controlled study conducted in Japan and Korea demonstrated that the response rates, defined as CGI-GI ratings of very much or much improved, at week 8 were 71% for paroxetine CR and 53% for placebo.11 In this clinical study, paroxetine CR was started after a washout period if enrolled patients were pretreated with any antidepressants. For comparison, the response rates of our study (defined in a similar way to the aforementioned clinical study) at week 8 were calculated in patients with no prior pharmacological treatment for depression. The response rate in our study was 61%, which was between the values observed for paroxetine CR (71%) and placebo (53%), indicating that the antidepressive effect of paroxetine CR could be detected. The high effectiveness of paroxetine CR was also supported by the significant decrease in the severity score from baseline to week 8.
The effectiveness assessments of this study included not only the physician’s assessments, but also the patients’ assessments. The patient satisfaction rate at week 8 (or at discontinuation/termination, if applicable) was 69.8%. It has been reported that patient satisfaction with antidepressants was positively correlated with adherence to antidepressants.15 In the future, further investigations will be needed to determine whether patient satisfaction with treatment using paroxetine CR has a beneficial effect on adherence and long-term treatment outcomes.
Exploratory analyses were conducted to determine the incidence of ADRs and the improvement rate by each patient’s characteristic features. The observed influence of the prior use of pharmacological treatment for depression on the incidence of ADRs suggested that patients with prior exposure to antidepressants may be less likely to experience new ADRs. In fact, 87.9% of the patients with prior pharmacological treatment received one or more antidepressants just before switching to paroxetine CR, and it should be noted that 70% of them received paroxetine IR and thus had already been exposed to paroxetine. The observed influence of the prior use of pharmacological treatment on the improvement rate of depression may possibly be explained by the fact that depression in some patients may have been partially improved by prior antidepressant use before treatment with paroxetine CR, and that the severity of depressive symptoms at baseline may have been lower in switched patients, who accounted for the majority of patients with prior pharmacological treatment compared with patients without prior pharmacological treatment. There is currently no clear explanation for the observed influence of the other patient features on the incidence of ADRs, or on the improvement rate. It is necessary to perform further analyses that are adjusted for potential confounding factors – for example, severity of depression at baseline, which was found to affect the incidence of ADRs and the improvement rate – to identify patient features that are associated with the occurrence of ADRs and the response to paroxetine CR.
In the safety population, 45.9% of the patients were switched from other antidepressants to paroxetine CR, which indicated that paroxetine CR is used not only in patients without prior pharmacological treatment, but also frequently as a treatment when switching from other antidepressants. In patients who were switched from antidepressants with other active ingredients except for duloxetine (an SNRI), the incidence of ADRs (point estimate) was more than twofold higher than that in patients switched from paroxetine IR who had already been exposed to paroxetine. The incidence of ADRs was relatively low in patients switched from SNRIs; however, the reason remains unclear.
The treatment continuation rate of paroxetine CR at week 8 in patients switched from antidepressants other than sulpiride ranged from approximately 80% to 90%, which was higher than the rate observed in patients without prior pharmacological treatment. It has been reported that many patients discontinue treatment in the first month.25,26 In the present study, 79.9% of the switched patients received treatment with the prior antidepressant for more than 1 month. This means that many patients with good adherence among the switched patients may have influenced the observed continuation rate. Although the number of patients who switched from sulpiride was small, the incidence of ADRs and the continuation rate in such patients tended to be higher and lower than those in patients switched from other antidepressants, respectively. On the other hand, the preference rate was more than 90% among the patients who completed the 8-week treatment with paroxetine CR. Sulpiride is an antipsychotic drug that has dopamine D2 receptor antagonistic action; it does not act directly to regulate serotonin (as is observed in other antidepressants), and it exerts its antidepressant effect at relatively low doses.27 The distinctly different features on the safety and effectiveness of paroxetine CR observed in patients switched from sulpiride when compared with the other antidepressants may be due to switching to an antidepressant with an absolutely different mechanism of action. To our knowledge, there has been no report of switching from sulpiride to SSRIs, but it was reported that sulpiride, which was approved in Japan for the treatment of depression, is quite frequently prescribed there, unlike the situation with overseas populations.2 Further investigations of switching from sulpiride will be needed to promote proper use.
In switched patients, the lower the severity of depression at baseline, the lower the improvement rate tended to be. Although a comparison among different classes or ingredients of prior antidepressants has some limitations – especially due to the lack of adjustment for patient characteristics among subgroups (as based on class or ingredient of the prior antidepressants) – the improvement, patient satisfaction, and preference rates were generally over 60% in the switched patients regardless of the class or ingredient of the prior antidepressants, which suggested that paroxetine CR is also effective in the setting of treatment switching. This feature was supported by the results of the stratified analyses for SSRIs by the reason for switching (AEs or inadequate responses).
In patients who switched from paroxetine IR to paroxetine CR at a dose ratio of 1:1.25 owing to AEs and who were treated with a constant or increased dose of paroxetine CR after switching, the majority (85.2%) of ADRs that occurred during treatment with paroxetine IR resolved or was in the process of resolving after switching to paroxetine CR. Since ADRs of SSRIs tend to occur during the early stage of treatment, longer exposure to paroxetine may result in fewer events. The open nature of the study in which patients are aware of change in treatment is a potential limitation. It is also likely that the more gradual increase and smaller fluctuation in blood concentration after switching from paroxetine IR to paroxetine CR may lead to a reduced risk of ADRs.
There are some limitations to this study. The diagnosis of depression was made on the basis of the physician’s clinical findings because there are currently no established diagnostic criteria for the depression/depressive state, which is the approved indication for paroxetine CR administration in Japan. The patients were not registered consecutively, which could lead to the risk of sample selection bias. In order to reduce the selection bias, a registration period was set during the course of 7 days after the start of treatment with paroxetine CR. Moreover, the lack of a control group and blinding made it difficult to eliminate the possible influence of a placebo effect, an antidepressive effect of routine clinical management by psychiatrists, or spontaneous improvement on the effectiveness outcomes. However, we believe that the large sample size of this study (>3,000 patients) provided safety and effectiveness data reflecting the actual reality of treatment with paroxetine CR in routine clinical practice in Japan, and that these findings are directly applicable to real-life clinical settings in Japan. In the current study, paroxetine CR appeared to be an effective treatment option for depressive patients in the presence or absence of pretreatment in real-world clinical settings.
Conclusion
The results of this study suggest that paroxetine CR is a well-tolerated and efficacious treatment for depression in routine clinical practice.
Acknowledgments
The authors wish to thank the investigators at the 594 medical institutions across the country for their cooperation with the study and for the provision of valuable data.
Disclosure
Masaki Kato has received grant funding from the Grant-in-Aid for Scientific Research (C) (grant number: 23591684) from the Japan Society for the Promotion of Science (JSPS), and speaker’s honoraria from Dainippon-Sumitomo Pharma, Otsuka, Meiji-Seika Pharma, Eli Lilly, MSD K.K., GlaxoSmithKline K.K., Pfizer, Shionogi, and Ono Pharmaceutical within the past 3 years. Toshifumi Kimura, Takeshi Kimura, and Terufumi Hara are full-time employees of GlaxoSmithKline K.K.; Terufumi Hara is also a stockholder of GlaxoSmithKline K.K. GlaxoSmithKline K.K. funded this study (study number 116488). The authors report no other conflicts of interest in this work.
References
Milea D, Guelfucci F, Bent-Ennakhil N, Toumi M, Auray JP. Antidepressant monotherapy: a claims database analysis of treatment changes and treatment duration. Clin Ther. 2010;32(12):2057–2072. | ||
Uchida N, Chong MY, Tan CH, et al. International study on antidepressant prescription pattern at 20 teaching hospitals and major psychiatric institutions in East Asia: analysis of 1898 cases from China, Japan, Korea, Singapore, and Taiwan. Psychiatry Clin Neurosci. 2007;61(5):522–528. | ||
Work Group on Major Depressive Disorder, American Psychiatric Association. Practice Guideline for the Treatment of Patients with Major Depressive Disorder. 3rd ed. October 2010. Available from: http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed November 14, 2014. | ||
Bauer M, Bschor T, Pfennig A, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders in primary care. World J Biol Psychiatry. 2007;8(2):67–104. | ||
Japanese Society of Mood Disorders. Treatment Guideline II: Major Depressive Disorder, 2013 Ver. 1.1. Available from: http://www.secretariat.ne.jp/jsmd/mood_disorder/img/130924.pdf. Japanese. Accessed November 14, 2014. | ||
Anderson IM, Ferrier IN, Baldwin RC, et al. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2008;22(4):343–396. | ||
Mackay FJ, Dunn NR, Wilton LV, Pearce GL, Freemantle SN, Mann RD. A comparison of fluvoxamine, fluoxetine, sertraline and paroxetine examined by observational cohort studies. Pharmacoepidemiol Drug Saf. 1997;6(4):235–246. | ||
Bull SA, Hunkeler EM, Lee JY, et al. Discontinuing or switching selective serotonin-reuptake inhibitors. Ann Pharmacother. 2002;36(4):578–584. | ||
Melartin TK, Rytsälä HJ, Leskelä US, Lestelä-Mielonen PS, Sokero TP, Isometsä ET. Continuity is the main challenge in treating major depressive disorder in psychiatric care. J Clin Psychiatry. 2005;66(2):220–227. | ||
Melfi CA, Chawla AJ, Croghan TW, Hanna MP, Kennedy S, Sredl K. The effects of adherence to antidepressant treatment guidelines on relapse and recurrence of depression. Arch Gen Psychiatry. 1998;55(12):1128–1132. | ||
Higuchi T, Hong JP, Jung HY, Watanabe Y, Kunitomi T, Kamijima K. Paroxetine controlled-release formulation in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled study in Japan and Korea. Psychiatry Clin Neurosci. 2011;65(7):655–663. | ||
Golden RN. Efficacy and tolerability of controlled-release paroxetine. Psychopharmacol Bull. 2003;37(Suppl 1):176–186. | ||
DeVane CL. Pharmacokinetics, drug interactions, and tolerability of paroxetine and paroxetine CR. Psychopharmacol Bull. 2003;37(Suppl 1):29–41. | ||
Golden RN, Nemeroff CB, McSorley P, Pitts CD, Dubé EM. Efficacy and tolerability of controlled-release and immediate-release paroxetine in the treatment of depression. J Clin Psychiatry. 2002;63(7):577–584. | ||
Aljumah K, Hassali AA, AlQhatani S. Examining the relationship between adherence and satisfaction with antidepressant treatment. Neuropsychiatr Dis Treat. 2014;10:1433–1438. | ||
Barbosa CD, Balp MM, Kulich K, Germain N, Rofail D. A literature review to explore the link between treatment satisfaction and adherence, compliance, and persistence. Patient Prefer Adherence. 2012;6:39–48. | ||
Hunot VM, Horne R, Leese MN, Churchill RC. A cohort study of adherence to antidepressants in primary care: the influence of antidepressant concerns and treatment preferences. Prim Care Companion J Clin Psychiatry. 2007;9(2):91–99. | ||
Kato M, Kimura T, Kimura T, Hara T. [Safety and efficacy of paroxetine controlled-release in patients in depressive state/depression – interim report of post-marketing surveillance for patients in depressive state/depression]. Jpn Pharmacol Ther. 2013;41(8):749–764. Japanese. | ||
Fredman SJ, Fava M, Kienke AS, White CN, Nierenberg AA, Rosenbaum JF. Partial response, nonresponse, and relapse with selective serotonin reuptake inhibitors in major depression: a survey of current “next-step” practices. J Clin Psychiatry. 2000;61(6):403–408. | ||
Guy W. ECDEU assessment manual for psychopharmacology. (ADM) 76-338. Rockville, MD: US Department of Health, Education, and Welfare Publication, National Institute of Mental Health; 1976:217–222. | ||
US Food and Drug Administration. Worsening Depression and Suicidality in Patients Being Treated With Antidepressants. Available from: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders /ucm161696.htm. Accessed November 14, 2014. | ||
Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2013. Bipolar Disord. 2013;15(1):1–44. | ||
Sinclair LI, Christmas DM, Hood SD, et al. Antidepressant-induced jitteriness/anxiety syndrome: systematic review. Br J Psychiatry. 2009;194(6):483–490. | ||
Kamijima K, Nakamura J, Tsuboi K, Higuchi T. [Review of the efficacy and safety of paroxetine-from the results of post-marketing surveillance]. Jpn J Clin Psychopharmacol. 2007;10(6):1045–1061. Japanese. | ||
Olfson M, Marcus SC, Tedeschi M, Wan GJ. Continuity of antidepressant treatment for adults with depression in the United States. Am J Psychiatry. 2006;163(1):101–108. | ||
Sawada N, Uchida H, Suzuki T, et al. Persistence and compliance to antidepressant treatment in patients with depression: a chart review. BMC Psychiatry. 2009;9:38. | ||
Rüther E, Degner D, Munzel U, et al. Antidepressant action of sulpiride. Results of a placebo-controlled double-blind trial. Pharmacopsychiatry. 1999;32(4):127–135. |
Supplementary materials
 | Table S1 The incidence of ADRs by the main patient characteristics |
 | Table S2 The improvement rate by the main patient characteristics |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.