Back to Journals » Journal of Inflammation Research » Volume 14
Rosuvastatin Enhances Alveolar Fluid Clearance in Lipopolysaccharide-Induced Acute Lung Injury by Activating the Expression of Sodium Channel and Na,K-ATPase via the PI3K/AKT/Nedd4-2 Pathway
Authors Xu HR, Yang Q, Xiang SY, Zhang PH, Ye Y, Chen Y, Xu KW, Ren XY, Mei HX, Shen CX, Ma HY, Smith FG, Jin SW ![]() , Wang Q
, Wang Q ![]()
Received 10 January 2021
Accepted for publication 18 March 2021
Published 16 April 2021 Volume 2021:14 Pages 1537—1549
DOI https://doi.org/10.2147/JIR.S299267
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Hao-Ran Xu,1 Qian Yang,1 Shu-Yang Xiang,1 Pu-Hong Zhang,1 Yang Ye,1 Yan Chen,1 Ke-Wen Xu,2 Xi-Ya Ren,2 Hong-Xia Mei,1 Chen-Xi Shen,1 Hong-Yu Ma,1 Fang-Gao Smith,1,3 Sheng-Wei Jin,1 Qian Wang1
1Department of Anesthesia and Critical Care, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, 325027, People’s Republic of China; 2Wenzhou Medical University, Wenzhou, People’s Republic of China; 3Institute of Inflammation and Aging, College of Medical and Dental Sciences, University of Birmingham, Birmingham, UK
Correspondence: Sheng-Wei Jin; Qian Wang
Department of Anesthesia and Critical Care, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, 109 Xueyuan Road, Wenzhou, Zhejiang Province, 325027, People’s Republic of China
Tel +86 577-88002806
Fax +86 577-88832693
Email [email protected]; [email protected]
Background: Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are devastating clinical conditions characterized by pulmonary epithelial damage and protein-rich fluid accumulation in the alveolar spaces. Statins are a class of HMG-CoA reductase inhibitors, which exert cholesterol-lowering and anti-inflammatory effects.
Methods: Rosuvastatin (1 mg/kg) was injected intravenously in rats 12 h before lipopolysaccharide (LPS, 10 mg/kg) administration. Eight hours later after LPS challenge, alveolar fluid clearance (AFC) was detected in rats (n = 6– 8). Rosuvastatin (0.3 μmol/mL) and LPS were cultured with primary rat alveolar type II epithelial cells for 8 h.
Results: Rosuvastatin obviously improved AFC and attenuated lung-tissue damage in ALI model. Moreover, it enhanced AFC by increasing sodium channel and Na,K-ATPase protein expression. It also up-regulated P-Akt via reducing Nedd4-2 in vivo and in vitro. Furthermore, LY294002 blocked the increase in AFC in response to rosuvastatin. Rosuvastatin-induced AFC was found to be partly rely on sodium channel and Na,K-ATPase expression via the PI3K/AKT/Nedd4-2 pathway.
Conclusion: In summary, the findings of our study revealed the potential role of rosuvastatin in the management of ALI/ARDS.
Keywords: acute respiratory distress syndrome, rosuvastatin, alveolar fluid clearance, sodium channel, Na,K-ATPases
Background
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are potentially dangerous clinical conditions characterized by pulmonary epithelial damage and the accumulation of protein-rich fluid in the alveolar spaces.1,2 Excessive alveolar edema fluid causes refractory hypoxemia and pulmonary hypertension.3 Therefore, the timely and effective removal of excess pulmonary edema is important for the recovery from ALI/ARDS.4
The clearance of alveolar fluid dependent on the Na+ transport. Active Na+ transport across the alveolar epithelium via sodium channels (ENaC) and Na,K-ATPases produce an osmotic gradient that drives fluid from the alveolar spaces into the interstitium.5–7 We have previously reported that ENaC and Na,K-ATPases subunits play important roles in the resolution of edema.8
Statins belong to the class of selective HMG-CoA reductase inhibitors, which are commonly prescribed to regulate lipid parameters including triglycerides and high-density lipoprotein cholesterol (HDL-C).9,10 In addition to their well-established lipid-lowering properties, statins also exert significant anti-inflammatory effects by reducing the extent of infiltration of the pulmonary neutrophils and decreasing cytokine levels.11,12 Rosuvastatin is a hydrophilic statin documented to provide end-organ protection in stroke-prone rats owing to its anti-inflammatory propensity.13,14 This finding suggests that rosuvastatin could potentially be used as an anti-inflammatory drug in humans. However, there are no studies that have addressed the effects of rosuvastatin in pulmonary edema.
The present study was designed to investigate whether rosuvastatin played a therapeutic role in lipopolysaccharide (LPS)-induced lung injury. We investigated the effect of rosuvastatin on ENaC and Na,K-ATPase expression. The PI3K inhibitor, LY294002, was used in in vivo and in vitro studies to investigate how the PI3K signaling pathway regulated ENaC, Na,K-ATPase, and AFC and to elucidate its underlying mechanisms.
Methods
Materials
Rosuvastatin, LY294002 was purchased from MedChemExpress (MJ, USA), and LPS was obtained from Sigma (MO, USA). Tumor necrosis factor (TNF)-α, myeloperoxidase (MPO), interleukin (IL)-1β and IL-10 enzyme-linked immunosorbent assay (ELISA) kits were procured from R&D Systems (MN, USA). MK-2206 was purchased from Target Molecule Corp (BSN, USA). Anti-ENaC α, β, and γ antibodies were procured from Biorbyt (CS, UK). Anti-Na,K-ATPase α1 and β1 antibodies were from Abcam (MA, USA). Anti-total Akt (T-Akt), anti-phosphorylated Akt (P-Akt), and anti-Nedd4-2 antibodies were from CellSignalling Technology (MA, USA).
Animal Studies
The male Sprague-Dawley rats (200–250 g) were obtained from Zhejiang Vital River Laboratory Animal Technology Co. and the experimental protocols were approved by Wenzhou Medical University. Chinese guidelines for the welfare of the laboratory animals (GB/T 35823-2018) were followed in the present study.
In the CTR and LPS group, rats were induced by an administration of saline or LPS (10 mg/kg) via the caudal vein. In the LPS+Rosuvastatin, LPS+Rosuvastatin+LY294002 and LPS+Rosuvastatin+MK-2206 groups, rats were pretreated with Rosuvastatin (pre-treatment group) in the presence or absence of 3 mg/kg LY294002 or 300 μg/kg MK-2206 via the caudal vein 12 h prior to LPS (10 mg/kg) stimulation. The AFC was measured for 8 h and the survival rate was detected for 48 h (Figure 1A).
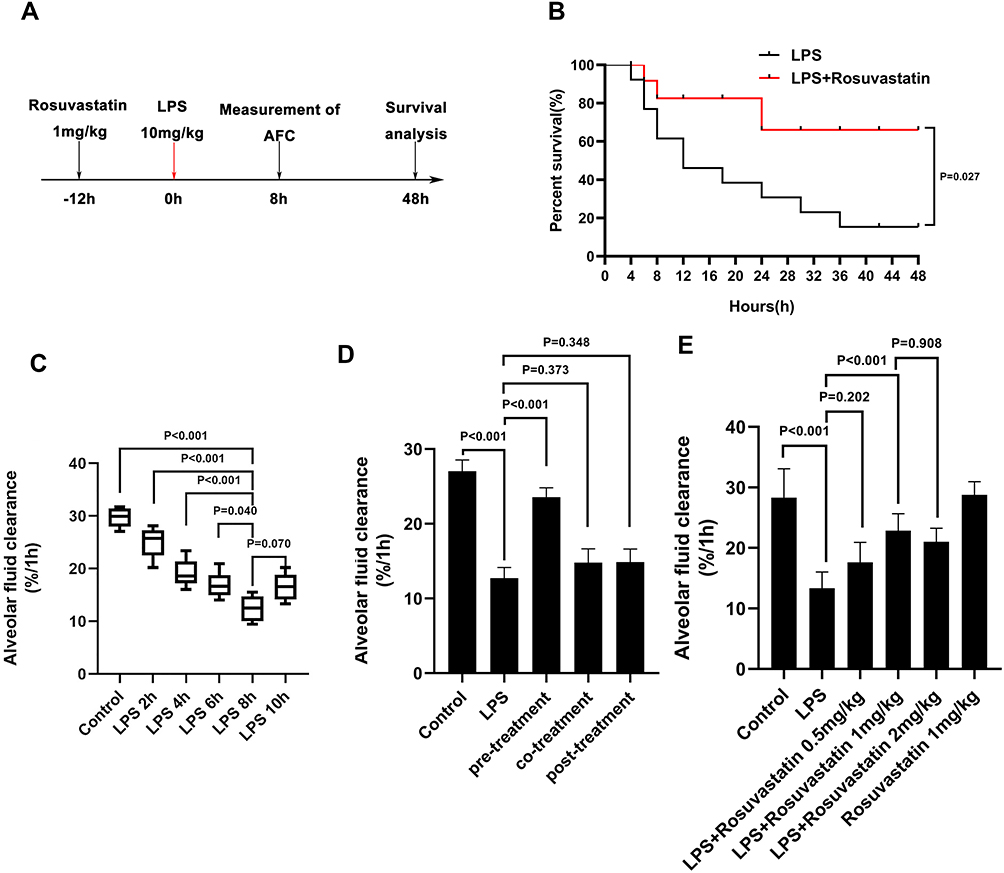 |
Figure 1 Effect of rosuvastatin on survival rate and alveolar fluid clearance in LPS-induced ALI/ARDS in vivo. Rosuvastatin (1mg/kg, tail vein injection) was administered to rat 12 h before LPS (10mg/kg, tail vein injection) stimulation, the AFC was measured for 8 h and the survival rate was detected for 48 h (A). Survival rate per group was evaluate by Kaplan–Meier survival curves and compare by Log rank test (B). The evans blue solution (5mL/kg) containing albumin was instilled into the left lung of the rat through a thin tube. After 1 hour of mechanical ventilation, the absorbance of the lavage fluid was measured and the alveolar fluid clearance rate was calculated. Rats AFC was detected under different action time of LPS (2, 4, 6, 8 and 10h) (C). After intravenous injection of different concentrations of rosuvastatin (0.5 mg/kg, 1 mg/kg and 2 mg/kg) 12 h, LPS was administered for 8 h, and the AFC was detected (D). For the pre-treatment group, rosuvastatin (1 mg/kg) was intravenously injected 12 h before LPS administration; For the co-treatment group, rosuvastatin was intravenously injected with the LPS; For the post-treatment group, rosuvastatin was intravenously injected 8 h after LPS administration (E). The data are presented as the mean ± SD, n =10. |
Rats in the co-treatment group received LPS and rosuvastatin for 8 h and the AFC was determined. Rats in the post-treatment group received LPS 8 h before rosuvastatin administration, and AFC was measured one hour later.
Survival Analysis
Rats were pretreated with rosuvastatin (1mg/kg) via a caudal vein injection 12 h before LPS (10 mg/kg) administration. The survival rate was recorded every 4 h and monitored for 48 h.
AFC
AFC was measured as described in a previous study.15 The rate of AFC was calculated based on the following equation: AFC = (1 −C0/C1), where C0 is the initial concentration of the perfusion solution and C1 is the final concentration of the alveolar lavage fluid, which was detected using spectrophotometry at a wavelength of 621 nm. The original perfusion solution was 5% albumin with Evans blue (0.15 mg/mL). This solution was infused into the left lung after the rats were anesthetized, and collected after 1 h using mechanical ventilation.
Histopathological Studies
Lung tissues were fixed with 4% paraformaldehyde and stained with hematoxylin and eosin (H&E). Lung-injury scores were determined based on the following parameters proposed by the American Thoracic Society (ATS): thickening of the alveolar septum; paratracheal or perivascular neutrophil infiltration; infiltration of the protein edema fluid in the alveolar cavity; hyaline membrane formation.16
ELISA
The lung-tissue homogenate TNF-α, MPO, IL-1β and IL-10 concentrations were determined using an ELISA kit according to the manufacturer’s protocol.
Western Blot
Lung tissue and the ATII cell proteins were extracted using a previously published method.17 BCA kit was used to measure the protein concentration. After the primary and secondary antibody culture, the PVDF membranes were photographed using an Odyssey CLx imager and the bands were analyzed using ImageJ.
Primary ATII Cells
Lung tissues were separated and digested using elastase to extract the primary ATII cells. Cells were cultured in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum, 0.1 mg/mL streptomycin, 100 U/mL penicillin, and 2 mmol/L L-glutamine. Primary ATII cells were incubated with LPS (1 µg/mL) with or without rosuvastatin (0.3 µmol/mL) and/or LY294002 (10 µM) for 24 h.18
Immunofluorescence
Cells were cultured overnight with the primary antibody at 4°C, followed by incubation with the secondary antibody at 37°C for 1 h. Laser confocal microscopy was used for observation and the images were further analyzed using ImageJ.
Statistical Analysis
Data are presented as the mean ± SD. All data were analyzed using one-way ANOVA followed by Tukey’s post-hoc test for multiple comparisons. Significance was determined at the p<0.05 level. Statistical analyses were performed using Prism 7.0 (GraphPad Software, San Diego, CA)
Results
Pre-Treatment with Rosuvastatin Up-Regulated the Survival Rate and AFC in LPS-Induced Lung Injury in vivo
We determined the effect of rosuvastatin (Figure 1A) on the survival rate after LPS-induced lung injury. Compared to that in the LPS group, the survival rate was obviously increased in the LPS+rosuvastatin group (Figure 1B). Moreover, we found that LPS treatment (2, 4, 6, 8, and 10 h) improved AFC in a time-dependent manner and the optimal effect was observed at 8 h (Figure 1C). Therefore, an LPS intervention of 8 h was used in our study.
In the present study, co-treatment or post-treatment with rosuvastatin did not change the AFC compared to that in the LPS group (Figure 1D). However, compared to that in the LPS group, pre-treatment with 0.5 mg/kg rosuvastatin did not increase AFC; however, treatment with 1 and 2 mg/kg of rosuvastatin improved LPS-induced AFC. The effects of rosuvastatin were similar at doses of 1 and 2 mg/kg; therefore, 1 mg/kg rosuvastatin was chosen for our experiments (Figure 1E). There was no significant difference of AFC between the control and rosuvastatin group (Figure 1E).
Rosuvastatin Protected Lung Tissues in the ALI Model Induced by LPS in vivo
Next, the effect of rosuvastatin on LPS-induced lung injury was determined using H&E staining. As shown in Figure 2A, tissues in the LPS group were found to be severely damaged; however, pre-treatment with rosuvastatin reduced infiltration of the inflammatory cells and thickening of the alveolar walls. The acute lung-injury scores correlated well with the morphologic changes (Figure 2B). The concentrations of TNF-α, MPO, IL-1β and IL-10 in the lung tissue homogenates were increased in the LPS group compared to those in the control group and significantly decreased in the rosuvastatin treatment group compared to those in the LPS group (Figure 2C–F). There were no significant changes between the control and rosuvastatin groups (Figure 2A–F).
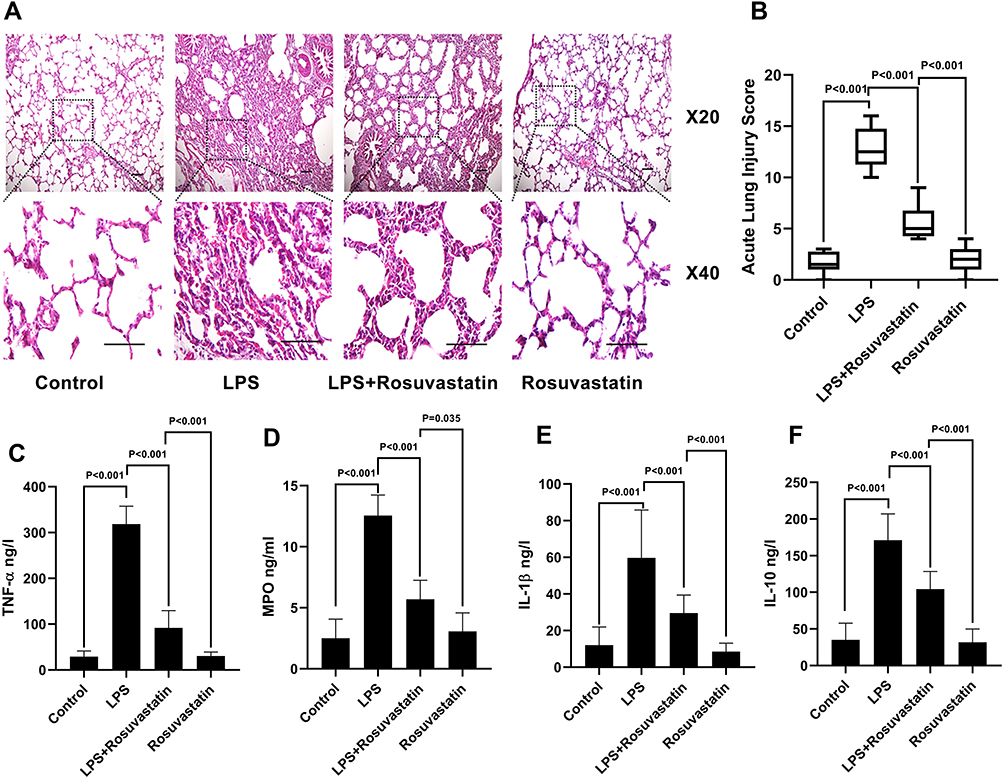 |
Figure 2 Effect of rosuvastatin on lung injury and inflammatory factors. After intravenous injection of different concentrations of 1 mg/kg rosuvastatin was intravenous injected 12 h before 10 mg/kg LPS administration. Eight hours later, the lung tissues was harvest. The protective effect of rosuvastatin on lung injury was assayed by H&E (A), and the acute lung injury score was calculated (B). The concentrations of inflammatory factors in lung tissue homogenate, such as TNF‐α (C), MPO (D), IL‐1β (E) and IL‐10 (F) were measured by ELISA. The data are presented as the mean ± SD, n = 9. Abbreviations: H&E, hematoxylin and eosin; TNF, α-tumor necrosis factor‐α; IL, interleukin; MPO, myeloperoxidase. |
Rosuvastatin Increased Sodium Channel and Na,K-ATPase Expression in LPS-Induced Lung Injury in PI3k/AKT/Nedd4-2 Signaling Pathway
We evaluated the mechanism of rosuvastatin on the enhancement of AFC in vivo. The lung-tissue homogenate was collected to determine the sodium channel and Na,K-ATPase level using Western blotting (Figure 3A). Compared to that in the control group, LPS treatment significantly downregulated the protein expression of ENaC α, β, and γ subunits (Figure 3B–D) and the Na,K-ATPase α1 and β1 subunits (Figure 3E and F), whereas pre-treatment with rosuvastatin upregulated the protein levels compared to those in the LPS group. However, the efficacy of rosuvastatin was abolished after treatment with the PI3K inhibitor, LY294002 (Figure 3B–F). Besides, the effect of rosuvastatin on the protein expression of ENaC α was relieved by AKT inhibitor, MK-2206 (Figure 4C).
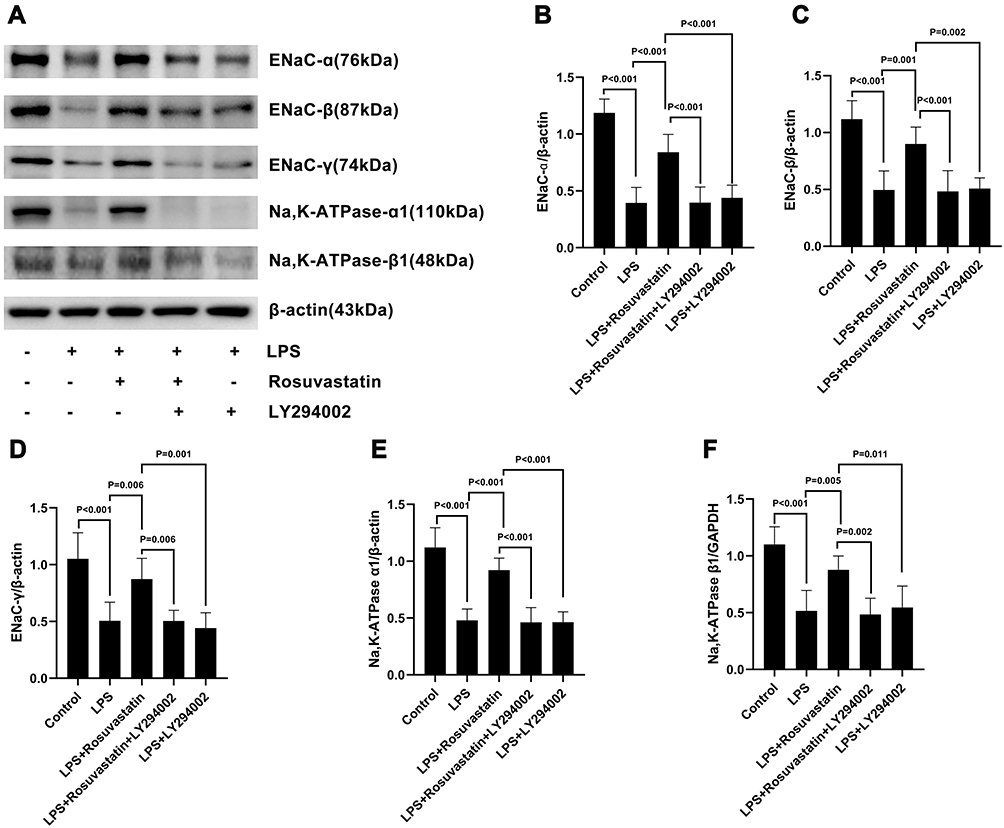 |
Figure 3 Effect of rosuvastatin on ENaC α, β, and γ subunits and Na,K-ATPase α1 and β1 subunits expression in LPS-induced lung injury. The rats were treated with LY294002 (PI3K inhibitor, 3 mg/kg) 1 hour before injection of rosuvastatin (1 mg/kg), twelve hours later, LPS was administered, and the lung tissues of each group were collected 8 h after LPS administration. Lung tissue homogenate was harvest to measure the ENaC α, β, and γ subunits and Na,K‐ATPase α1 and β1 subunits protein level (A–F). The data are presented as the mean ± SD, n = 6. |
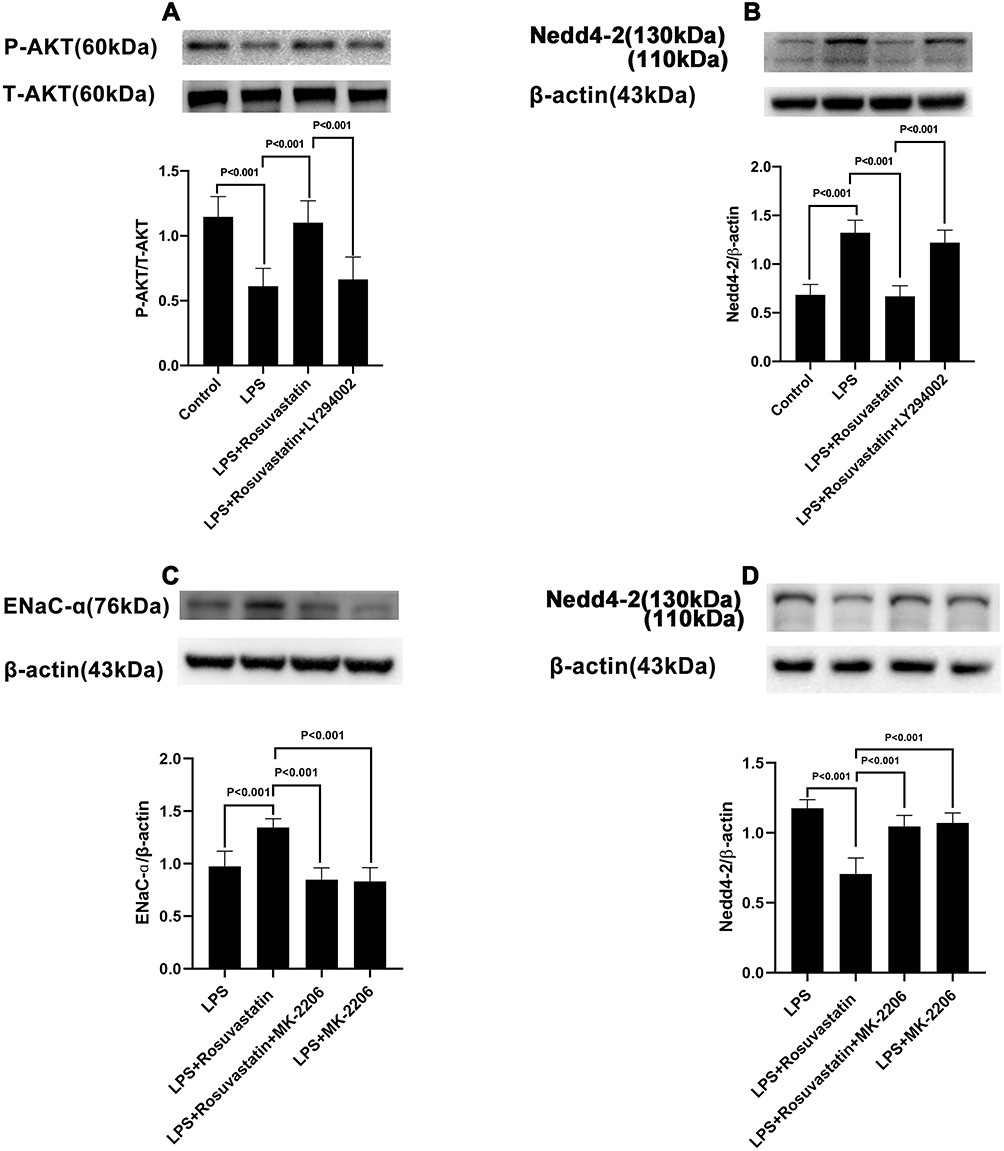 |
Figure 4 Effect of rosuvastatin on AKT and Nedd4-2 expression. The rats were treated with LY294002 (PI3K inhibitor, 3 mg/kg) or MK-2206 (AKT inhibitor, 300 μg/kg) 1 hour before injection of rosuvastatin (1 mg/kg), twelve hours later, LPS was administered, and the lung tissues of each group were collected 8 h after LPS administration. The level of P-Akt (A), ENaC α (C) and Nedd4-2 (B and D) protein expression was measured by Western blot. The data are presented as the mean ± SD, n = 6. |
Compared to that in the LPS group, p-AKT expression was increased in the LPS+rosuvastatin group, whereas the improvement in p-AKT level was partly abolished by treatment with LY294002 (Figure 4A). Additionally, Nedd4-2 expression was reduced in the LPS+rosuvastatin group compared to that in the LPS group, and the decreased Nedd4-2 level was partly abrogated by LY294002 and MK-2206 (Figure 4B and D).
Rosuvastatin Improved AFC in LPS-Induced ALI via the PI3k Signaling Pathway
To determine if rosuvastatin could enhance AFC through the PI3K pathway, rats were co-administered LY294002 and rosuvastatin via the caudal vein. The AFC in the LPS+rosuvastatin+LY294002 group was lower compared to that in the LPS+rosuvastatin group, but there was no significant difference in AFC between the LPS and LPS+LY294002 groups (Figure 5).
 |
Figure 5 Effect of rosuvastatin on alveolar fluid clearance in LPS-induced lung injury is blocked by LY294002 (PI3K inhibitor). The rats were treated with LY294002 (PI3K inhibitor, 3 mg/kg) 1 hour before injection of rosuvastatin (1 mg/kg), twelve hours later, LPS was administered, and the lung tissues of each group were collected 8 h after LPS administration. After 1 hour of mechanical ventilation, the absorbance of the lavage fluid was measured and the alveolar fluid clearance rate was calculated. The data are presented as the mean ± SD, n = 6. |
Rosuvastatin Enhanced the Expression of Na,K-ATPase in LPS-Stimulated Primary at II Cells in a Time- and Dose-Dependent Manner in vitro
Primary AT II cells were cultured with 0.1, 0.3, and 30 μmol/mL of rosuvastatin and then stimulated with LPS (1 µg/mL) for 24 h. We found that the Na,K-ATPase α1 subunit protein level was significantly downregulated by LPS and upregulated by rosuvastatin in a dose‐dependent manner; the maximum effect was achieved at 0.3 μmol/mL of rosuvastatin (Figure 6A). Next, primary ATII cells were cultured with 0.3 μmol/mL rosuvastatin for 4, 8, and 12 h and then interrupted with LPS (1 µg/mL) for 24 h. We found that the expression of the Na,K‐ATPase α1 subunit protein improved significantly at 8 h (Figure 6B).
 |
Figure 6 Effect of LPS on Na, K-ATPase α1 expression on primary ATII cells was ameliorated by rosuvastatin with dose and time dependency. Primary ATII cells was cultured with LPS (1 µg/mL) in the presence or absence of different concentration of rosuvastatin (0.1, 0.3, 3µM) for 12h, then the Na,K‐ATPase α1 expression was measured by Western blot (A). Next, Primary ATII cells were cultured with LPS (1 µg/mL) in the presence or absence of 0.3 µM rosuvastatin for 4h,8h and 12h to measure the Na,K‐ATPase α1 protein level (B). The data are presented as the mean ± SD, n = 6. |
Rosuvastatin Promoted ENaC and Na,K-ATPase Protein Level via the PI3K/Akt/Nedd4-2 Signaling Pathway in vitro
The protein expression of ENaC α and Na,K‐ATPase α1 subunits in LPS-stimulated primary AT II cells was detected using immunofluorescence (Figure 7A) and was found to be reduced in the LPS group compared to that in the control group. On the other hand, pre-treatment with rosuvastatin could upregulate the protein expression of the ENaC α and Na,K‐ATPase α1 subunits (Figure 7A). ENaC α, β, γ and Na,K‐ATPase α1, β1 subunits protein level were detected using Western blot (Figure 7B). The expression of ENaC α, β, γ; and Na,K‐ATPase α1, β1 subunits were markedly reduced by LPS, whereas rosuvastatin increased the expression of ENaC α, β, γ; and Na,K‐ATPase α1, β1 subunits. However, the effect of rosuvastatin on the protein expression of ENaC α, β, γ; and Na,K‐ATPase α1, β1 subunits was abrogated after treatment with LY294002 (Figure 7B–G).
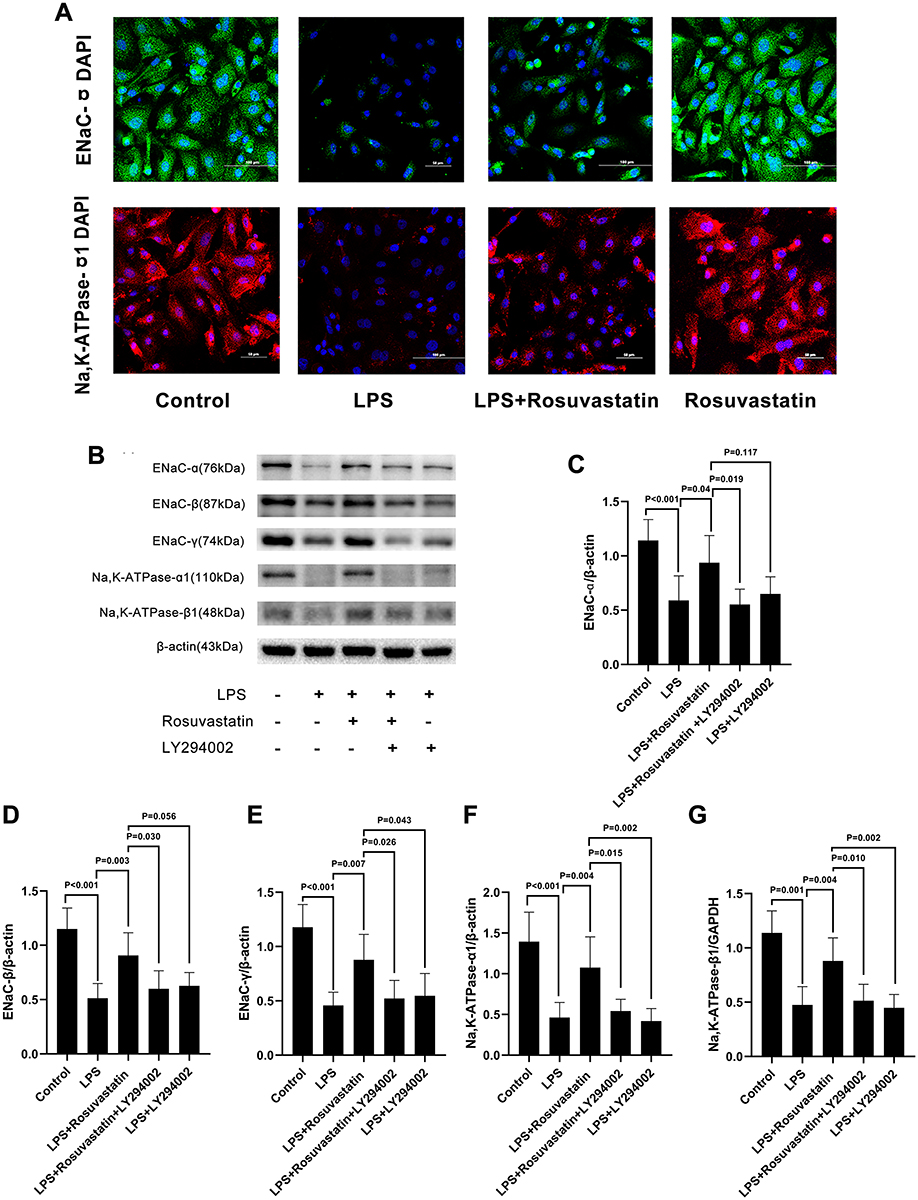 |
Figure 7 Effect of LPS on ENaC α, β, and γ subunits and Na,K-ATPase α1 and β1 subunits expression in primary ATII was ameliorated by rosuvastatin partly through activation PI3K/Akt/Nedd4-2 pathway. Primary ATII cells were incubated with LPS (1 µg/mL) with or without rosuvastatin (0.3 µM) and/or LY294002 (10 mM) for 12 h. Na,K‐ATPase α1 level was measured by immunofluorescence (A). ENaC α, β, and γ subunits and Na,K‐ATPase α1 and β1 subunits expression (B–G) was analysis by Western blot. The data are presented as the mean ± SD, n = 6. |
Compared to that in the LPS group, pre-treatment with rosuvastatin could upregulate the p-AKT level. However, the enhanced effect of rosuvastatin on P-AKT could be partially blocked by LY294002 (Figure 8A). Moreover, LPS enhanced Nedd4-2 expression, whereas pre-treatment with rosuvastatin could reduce Nedd4-2 levels. The decreased role of rosuvastatin on Nedd4-2 could be partially blocked by LY294002 (Figure 8B).
 |
Figure 8 Effect of rosuvastatin on p-Akt and Nedd4-2 expression in LPS stimulated primary ATII cells. Primary ATII cells were incubated with LPS (1 µg/mL) with or without rosuvastatin (0.3 µM) and/or LY294002 (10 mM) for 12 h. P‐Akt (A) and Nedd4-2 (B) protein expression was measured by Western blot. The data are presented as the mean ± SD, n = 6. |
Discussion
In this study, we provided evidence that pre-treatment with rosuvastatin significantly upregulated AFC, ameliorated pulmonary damage, reduced the levels of the inflammatory mediators, and improved survival rate in the LPS-induced murine lung injury model. However, co-treatment or post-treatment with rosuvastatin had no effect on AFC in the LPS-induced murine model of ALI. Additionally, pre-treatment with rosuvastatin enhanced the protein expression of sodium channels and Na,K-ATPase both in vivo and in vitro. The PI3K inhibitor, LY294002, abrogated the effect of rosuvastatin on AFC and the protein expression of ENaCs and Na,K-ATPases. AKT inhibitor, MK-2206, abolished the effect of rosuvastatin on the protein expression of ENaC α and Nedd4-2. Furthermore, rosuvastatin promoted Akt phosphorylation and decreased the expression of Nedd4-2 in vivo and in vitro. These results indicated that pre-treatment with rosuvastatin increased the expression of sodium channel and Na,K-ATPase to promote AFC via the PI3K/Akt/Nedd4-2 signaling pathway (Figure 9).
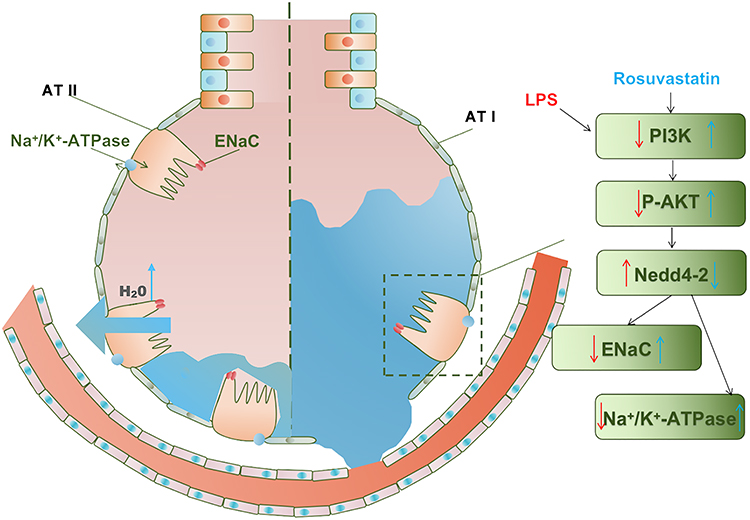 |
Figure 9 Effect of rosuvastatin on alveolar fluid clearance by up-regulating the expression of ENaC and Na, K-ATPase in vivo and in vitro. Rosuvastatin activates the PI3K/AKT/Nedd4-2 pathway to up-regulate the expression of ENaC protein, which locates at the top of alveolar type II epithelial cells, and promotes the active transport of sodium ions to the alveolar type II epithelial cells; at the same time, Rosuvastatin up-regulated the expression of Na, K‐ATPase protein in the basal layer of alveolar type II epithelial cells, actively transporting sodium ions to the interstitium of lung tissue, and the sodium ion concentration gradient formed in this process drives the transfer of water molecules from the alveolar cavity to the lung interstitium. Blue arrows indicate the effects of rosuvastatin. Red arrows indicate the effects of LPS. ATI-type I alveolar epithelial cell; ATII-type II alveolar epithelial cell. |
ALI/ARDS is a serious condition characterized by pulmonary epithelial damage, which results in impaired AFC. Therefore, the removal of excess alveolar edematous fluid is crucial for the recovery and survival of patients with lung injury.2 Previous studies show that statins exert both anti-inflammatory and immunomodulatory effects.19 Several clinical trials indicate that statins significantly reduce mortality in patients with sepsis and bacterial pneumonia and improve the prognosis of patients with ARDS.20–23
Rosuvastatin is one of the most commonly prescribed HMG-CoA reductase inhibitors.
Neukamm A reported that rosuvastatin treatment improves pulmonary function and attenuates systemic inflammation in patients with chronic obstructive pulmonary disease.24 Previous study showed that continuous use of statins in patients with sepsis who have previously received statin therapy and developed severe ARDS has an independent beneficial effect on the 28-day survival rate.23 Another study showed that rosuvastatin improved the survival rate of patients with hyperinflammatory subphenotype of ARDS.25 Drug prophylaxis can prevent the occurrence and development of diseases and their complications.26,27 Hence, in this study, pre-treatment with rosuvastatin, but not co-treatment or post-treatment, improved AFC and survival rate. Moreover, pre-treatment with rosuvastatin ameliorated pulmonary damage, inflammatory cell infiltration, and the levels of the inflammatory factors. Therefore, these findings revealed the new value of rosuvastatin in the precise treatment of ALI/ARDS.
Na+ transport plays an important role in the alleviation of pulmonary edema. The cellular and molecular mechanisms responsible for the vectorial transport of Na+ from the alveoli to the interstitium relies on the sodium channels in the ATI and ATII. The intracellular Na+ is expelled by basolateral Na,K‐ATPase and results in the production of an osmotic gradient that constitutes the primary driving force for the reabsorption of the pulmonary fluid across the alveolar epithelium.28 We found that rosuvastatin not only upregulated the LPS-induced reduction of the protein expression of the sodium channel α, β, and γ subunits, but also increased the protein expression of Na,K‐ATPase α1 and β1 both in vivo and in vitro, indicating that this drug regulated Na+ transport by promoting the expression of ENaC and Na,K‐ATPase. Consistent with our findings, previous studies have shown that pre-treatment with rosuvastatin could increase the myocardial Na,K‐ATPase levels in rats with doxorubicin-induced cardiotoxicity.29 Thus, these findings suggested that rosuvastatin promoted AFC by upregulating the sodium channel and the expression of Na,K‐ATPase.
PI3K is known to regulate ENaC-mediated AFC by insulin and Specialized Pro-resolving Mediator (SPM).15,30,31 We found that LY294002 markedly attenuated rosuvastatin-induced ENaC expression and Na,K-ATPase level in vivo and in vitro, indicating that the effect of rosuvastatin in the lungs was PI3K-dependent. AKT is a crucial mediator of PI3K signals that has profound effects on several physiological events.32,33 Our study suggested that rosuvastatin treatment caused the LPS-induced reduction in Akt phosphorylation in vivo and in vitro, which was partially reversed by LY294002. The effect of rosuvastatin on the protein expression of ENaC α and Nedd4-2 was also abolished by AKT inhibitor, MK-2206. This finding indicated that rosuvastatin could activate the PI3K/Akt signaling pathway.
Nedd4-2, an E3 ubiquitin-protein ligase, participates in negatively regulating sodium channel expression by ubiquitination and targeting for degradation.34,35 Moreover, Akt phosphorylation competes with Nedd4‐2 for the binding sites of ENaC to increase ENaC expression.36,37 Consistently, rosuvastatin reduced the LPS-induced increase in Nedd4-2 protein levels, and that the beneficial effects of rosuvastatin in inhibiting Nedd4-2 protein expression were abrogated by LY294002, indicating that rosuvastatin improved ENaC and Na,K-ATPase expression through activation of the PI3K/Akt/Nedd4-2 pathway. Additionally, rosuvastatin-enhanced AFC was found to be abolished by LY294002 in vivo. Collectively, these results indicated that rosuvastatin could increase ENaC and Na,K‐ATPase level to enhance AFC via the PI3K/Akt/Nedd4-2 pathway.
Conclusion
To summarize, the results of our study indicated that pretreatment with rosuvastatin caused a reduction in mortality in LPS-induced rats. Rosuvastatin partially upregulated the expression of ENaC and Na,K-ATPase by activating the PI3K/Akt/Nedd4-2 pathway in a murine model of LPS-induced lung injury to promote AFC. Thus, rosuvastatin could be used for reabsorption of pulmonary fluid in an edematous condition after ALI, indicating its potential use in the management and treatment of ALI/ARDS.
Acknowledgments
This work was sponsored by grants from the National Natural Science Foundation of China (no. 81571862 and no. 81870065) and by the Natural Science Foundation of Zhejiang Province (LY18H010005), Provincial Medical and health science and technology project (2021455293) and Research Fund for Lin He’s Academician Workstation of New Medicine and Clinical Translation (19331102).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Pham T, Rubenfeld GD. Fifty years of research in ARDS. The epidemiology of acute respiratory distress syndrome. a 50th birthday review. Am J Respir Crit Care Med. 2017;195(7):860–870. doi:10.1164/rccm.201609-1773CP
2. Thompson BT, Chambers RC, Liu KD, Drazen JM. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):1904–1905. doi:10.1056/NEJMra1608077
3. Morty RE, Eickelberg O, Seeger W. Alveolar fluid clearance in acute lung injury: what have we learned from animal models and clinical studies? Intensive Care Med. 2007;33(7):1229–1240. doi:10.1007/s00134-007-0662-7
4. Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18.
5. Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163(6):1376–1383. doi:10.1164/ajrccm.163.6.2004035
6. Dumasius V, Sznajder JI, Azzam ZS, et al. β2 -adrenergic receptor overexpression increases alveolar fluid clearance and responsiveness to endogenous catecholamines in rats. Circ Res. 2001;89(10):907–914. doi:10.1161/hh2201.100204
7. Zhou G, Dada LA, Sznajder JI. Regulation of alveolar epithelial function by hypoxia. Eur Respir J. 2008;31(5):1107–1113. doi:10.1183/09031936.00155507
8. Zhang PH, Han J, Cao F, et al. PCTR1 improves pulmonary edema fluid clearance through activating the sodium channel and lymphatic drainage in lipopolysaccharide-induced ARDS. J Cell Physiol. 2020;235(12):9510–9523. doi:10.1002/jcp.29758
9. Ferreira TS, Lanzetti M, Barroso MV, et al. Oxidative stress and inflammation are differentially affected by atorvastatin, pravastatin, rosuvastatin, and simvastatin on lungs from mice exposed to cigarette smoke. Inflammation. 2014;37(5):1355–1365. doi:10.1007/s10753-014-9860-y
10. Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation. 2000;101(2):207–213. doi:10.1161/01.CIR.101.2.207
11. Morikawa S, Takabe W, Mataki C, et al. The effect of statins on mRNA levels of genes related to inflammation, coagulation, and vascular constriction in HUVEC. Human umbilical vein endothelial cells. J Atheroscler Thromb. 2002;9(4):178–183. doi:10.5551/jat.9.178
12. Jacobson JR, Barnard JW, Grigoryev DN, Ma SF, Tuder RM, Garcia JG. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;288(6):L1026–1032. doi:10.1152/ajplung.00354.2004
13. Sironi L, Gianazza E, Gelosa P, et al. Rosuvastatin, but not simvastatin, provides end-organ protection in stroke-prone rats by antiinflammatory effects. Arterioscler Thromb Vasc Biol. 2005;25(3):598–603. doi:10.1161/01.ATV.0000157145.98200.55
14. Chang CC, Wang SS, Hsieh HG, et al. Rosuvastatin improves hepatopulmonary syndrome through inhibition of inflammatory angiogenesis of lung. Clin Sci (Lond). 2015;129(6):449–460. doi:10.1042/CS20140622
15. Wang Q, Lian QQ, Li R, et al. Lipoxin A4 activates alveolar epithelial sodium channel, Na,K-ATPase, and increases alveolar fluid clearance. Am J Respir Cell Mol Biol. 2013;48(5):610–618. doi:10.1165/rcmb.2012-0274OC
16. Feldman AT, Wolfe D. Tissue processing and hematoxylin and eosin staining. Methods Mol Biol. 2014;1180:31–43.
17. Hnasko TS, Hnasko RM. The western blot. Methods Mol Biol. 2015;1318:87–96.
18. Dobbs LG, Williams MC, Gonzalez R. Monoclonal antibodies specific to apical surfaces of rat alveolar type I cells bind to surfaces of cultured, but not freshly isolated, type II cells. Biochim Biophys Acta. 1988;970(2):146–156. doi:10.1016/0167-4889(88)90173-5
19. Arnaud C, Mach F. Potential antiinflammatory and immunomodulatory effects of statins in rheumatologic therapy. Arthritis Rheum. 2006;54(2):390–392. doi:10.1002/art.21757
20. Hackam DG, Mamdani M, Li P, Redelmeier DA. Statins and sepsis in patients with cardiovascular disease: a population-based cohort analysis. Lancet. 2006;367(9508):413–418. doi:10.1016/S0140-6736(06)68041-0
21. Martin CP, Talbert RL, Burgess DS, Peters JI. Effectiveness of statins in reducing the rate of severe sepsis: a retrospective evaluation. Pharmacotherapy. 2007;27(1):20–26. doi:10.1592/phco.27.1.20
22. Mortensen EM, Restrepo MI, Copeland LA, et al. Impact of previous statin and angiotensin II receptor blocker use on mortality in patients hospitalized with sepsis. Pharmacotherapy. 2007;27(12):1619–1626. doi:10.1592/phco.27.12.1619
23. Mansur A, Steinau M, Popov AF, et al. Impact of statin therapy on mortality in patients with sepsis-associated acute respiratory distress syndrome (ARDS) depends on ARDS severity: a prospective observational cohort study. BMC Med. 2015;13(1):128. doi:10.1186/s12916-015-0368-6
24. Neukamm A, Hoiseth AD, Einvik G, et al. Rosuvastatin treatment in stable chronic obstructive pulmonary disease (RODEO): a randomized controlled trial. J Intern Med. 2015;278(1):59–67. doi:10.1111/joim.12337
25. Calfee CS, Delucchi KL, Sinha P, et al.; Irish Critical Care Trials Group. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med. 2018;6(9):691–698. doi:10.1016/S2213-2600(18)30177-2
26. Almog Y, Shefer A, Novack V, et al. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation. 2004;110(7):880–885. doi:10.1161/01.CIR.0000138932.17956.F1
27. Chalmers JD, Singanayagam A, Murray MP, Hill AT. Prior statin use is associated with improved outcomes in community-acquired pneumonia. Am J Med. 2008;121(11):1002–1007e1001. doi:10.1016/j.amjmed.2008.06.030
28. Matthay MA, Folkesson HG, Verkman AS. Salt and water transport across alveolar and distal airway epithelia in the adult lung. Am J Physiol. 1996;270(4 Pt 1):L487–503. doi:10.1152/ajplung.1996.270.4.L487
29. Sharma H, Pathan RA, Kumar V, Javed S, Bhandari U. Anti-apoptotic potential of rosuvastatin pretreatment in murine model of cardiomyopathy. Int J Cardiol. 2011;150(2):193–200. doi:10.1016/j.ijcard.2010.04.008
30. Soundararajan R, Melters D, Shih IC, Wang J, Pearce D. Epithelial sodium channel regulated by differential composition of a signaling complex. Proc Natl Acad Sci U S A. 2009;106(19):7804–7809. doi:10.1073/pnas.0809892106
31. Deng W, Li CY, Tong J, Zhang W, Wang DX. Regulation of ENaC-mediated alveolar fluid clearance by insulin via PI3K/Akt pathway in LPS-induced acute lung injury. Respir Res. 2012;13(1):29. doi:10.1186/1465-9921-13-29
32. Lee IH, Dinudom A, Sanchez-Perez A, Kumar S, Cook DI. Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J Biol Chem. 2007;282(41):29866–29873. doi:10.1074/jbc.M701923200
33. Mattes C, Laube M, Thome UH. Rapid elevation of sodium transport through insulin is mediated by AKT in alveolar cells. Physiol Rep. 2014;2(3):e00269. doi:10.1002/phy2.269
34. Rotin D, Staub O. Nedd4-2 and the regulation of epithelial sodium transport. Front Physiol. 2012;3:212. doi:10.3389/fphys.2012.00212
35. Duerr J, Leitz DHW, Szczygiel M, et al. Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat Commun. 2020;11(1):2012. doi:10.1038/s41467-020-15743-6
36. Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J. 2001;15(1):204–214. doi:10.1096/fj.00-0191com
37. Zhou R, Patel SV, Snyder PM. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J Biol Chem. 2007;282(28):20207–20212. doi:10.1074/jbc.M611329200
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.