")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Rosiglitazone attenuates the metalloprotease/anti-metalloprotease imbalance in emphysema induced by cigarette smoke: involvement of extracellular signal-regulated kinase and NFκB signaling
Authors Hou G, Yin Y, Han D, Wang Q, Kang J
Received 14 November 2014
Accepted for publication 21 January 2015
Published 7 April 2015 Volume 2015:10(1) Pages 715—724
DOI https://doi.org/10.2147/COPD.S77514
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Gang Hou, Yan Yin, Dan Han, Qiu-yue Wang, Jian Kang
Department of Respiratory Medicine, the First Hospital of China Medical University, Shenyang, People’s Republic of China
Objective: We investigated how rosiglitazone attenuated cigarette smoke (CS)-induced emphysema in a rat model. In particular, we focused on its possible effects on the imbalance between metalloprotease (MMP) and anti-MMP activity, mitogen-activated protein kinase (MAPK) phosphorylation, and nuclear factor kappa-light-chain-enhancer of activated B cell (NFκB) signaling pathway over-activation.
Methods: A total of 36 Wistar rats were divided into three groups (n=12 each): animals were exposed to CS for 12 weeks in the absence (the CS group) or presence of 30 mg/kg rosiglitazone (the rosiglitazone-CS [RCS] group); a control group was treated with the rosiglitazone vehicle only, without any CS exposure. Histopathology of lung tissue in all groups was evaluated to grade severity of the disease. Expression levels of peroxisome proliferator-activated receptor γ (PPARγ), MMP2, and MMP9 in lung tissue were determined and compared using Western blotting and immunohistochemistry. Activation of MAPKs, NFκB, and the nuclear factor of kappa light polypeptide gene enhancer in B-cell inhibitor, alpha (IκBα) phosphorylation in lung tissue was examined by Western blotting.
Results: Emphysema-related pathology, based on inter-alveolar wall distance and alveolar density, was less severe in the RCS group than in the CS group. Compared with the CS group, levels of PPARγ were higher in the RCS group, and levels of MMP2 and MMP9 proteins were lower in the RCS rats. Levels of activated MAPKs and NFκB were also lower, while the IκBαphosphorylation was increased in the lung tissue of RCS rats.
Conclusion: Our findings suggest that oral administration of rosiglitazone attenuates the metalloprotease activity induced by CS, and the underlying mechanism might involve the activation of signaling pathways dependent on MAPKs or NFκB. Our results further suggest that PPARγ contributes to the pathogenesis of emphysema as well as airway inflammation induced by CS.
Keywords: emphysema, chronic obstructive pulmonary disease, matrix metalloprotease9, matrix metalloprotease2, PPAR, NFκB
Introduction
Chronic obstructive pulmonary disease (COPD) of the lungs involves irreversible airway obstruction and abnormal, chronic inflammation of the airway.1 Smoking is a key risk factor of COPD: it strongly induces the release of inflammatory mediators, including oxidants and proteases, which ultimately trigger chronic airway inflammation and emphysema.2 Emphysema is a complication of COPD that arises when chronic inflammation in the lung destroys the normal balance between the activity of metalloproteases like matrix metalloproteinases (MMPs) and the activity of anti-metalloproteases like tissue inhibitor of metalloproteinase (TIMPs), resulting in abnormally enhanced MMP activity.3,4
Smoking cessation may partially restore pulmonary function and lung density in patients with stable COPD,5,6 whereas co-administration of anti-inflammatory drugs is a must in order to control chronic airway inflammation and progression of emphysema. Not all patients with COPD respond equally well to the medical treatment; for example, patients without emphysema respond much better than those with severe emphysema do.7
Efforts have been made to identify more universally effective anti-inflammatory drugs to treat COPD, and researchers’ interest has been gradually focused on peroxisome proliferator-activated receptors (PPARs). These ligand-activated transcription factors belong to a nuclear hormone receptor family8 and appear to regulate immune and inflammatory responses by inhibiting the expression of proinflammatory genes.9,10 The most extensively studied PPAR subtype is PPARγ, which is involved in a wide spectrum of lung diseases, including pulmonary vascular disease, acute lung injury, asthma, and lung cancer.11–14 PPARγ agonists have immunomodulating effects that lead to reduced secretion of inflammatory cytokines in articular cell culture or experimental models of arthropathy.15,16 They also repress the synthesis of extracellular matrix components in various cell types, such as collagen synthesis by thiazolidinediones in mesangial cells from diabetic rats17 and Wy14643 synthesis in hepatic stellate cells from rats with cholestatic liver fibrosis.18 Recently, PPARγ agonists have begun to be used as an anti-inflammatory therapy for COPD.10,15
One unresolved question is whether PPARγ agonists help reverse the metalloprotease/anti-metalloprotease imbalance associated with emphysema. In this study, we used a rat model of smoking-induced emphysema to examine how rosiglitazone (Sigma-Aldrich Co., St Louis, MO, USA), one of the PPARγ agonists, affected COPD pathology. We examined whether rosiglitazone altered the metalloprotease/anti-metalloprotease imbalance, and explored the molecular mechanism underlying the imbalance.
Materials and methods
Animal model
All experiments were conducted in accordance with the guidelines of the Research Ethics Committee of China Medical University. Male Wistar rat weanlings (weighing 170–210 g) were obtained from the Laboratory Animal Center of China Medical University and randomly assigned into three groups with 12 animals in each group. The cigarette smoke (CS) group was exposed to smoke from 16 commercial cigarettes (Altria Group Inc, Richmond, VA, USA); each of these cigarettes contained 0.9 mg nicotine, 14 mg CO, and 12 mg tar oil, according to the manufacturer’s specifications. Animals were exposed to smoke (scope, 8% v/v) with the cigarette filters removed for 30 minutes, twice a day, 6 days per week for 12 weeks. The rosiglitazone-CS (RCS) group was exposed to CS in the same way as the CS group but in the presence of rosiglitazone, intragastrically administered once daily (30 mg/kg, dissolved in normal saline). The control group and CS group received a daily orally administered injection of saline vehicle without any smoke.
At the end of the 12th week, rats were sacrificed by exsanguination and lungs were excised. The right upper lobes were removed and stored at −80°C, and the right median lobes were embedded in paraffin.
Lung tissue histology and morphometric analysis
Paraffin-embedded sections of tissues were stained with hematoxylin-eosin and examined by light microscopy at a total magnification of 100×. Interalveolar wall distance was measured as mean linear intercept (MLI), defined as the total length of the test line divided by the number of alveolar walls intersecting it.19 Alveolar density was measured as mean alveolar number (MAN), defined as the number of alveoli in each field.19 MLI and MAN were calculated for each sample based on ten random fields.
Levels of MMP2 and MMP9 proteins in lung tissue
Lung tissues were homogenized and lysed. Nuclear and cytoplasmic fractions were prepared as previously described.20 The cell lysates (20 μg for each lane) were subjected to 10% polyacrylamide gel electrophoresis (PAGE), and the separated protein was subsequently transferred onto a nitrocellulose membrane by electroblotting. The membrane was blocked for 1 hour at room temperature with blocking solution. The blot was then incubated overnight at 4°C with mouse anti-MMP2 antibody (1:500; Abcam PLC, Cambridge, UK) or anti-MMP9 antibody (1:500; Abcam PLC). The primary antibody was removed by washing the blot three times, and the membrane was next incubated with secondary antibody (1:2,000 dilution) for 2 hours at room temperature. The signals were detected using enhanced chemiluminescence (GE Healthcare UK Ltd, Little Chalfont, UK), and the protein bands were desitometrically analyzed with β-actin as a loading control (Metamorph/Evolution MP 5.0/B X51, Olympus, US).
To complement these Western blotting studies, we also examined protein signal intensity in situ using immunohistochemistry of lung tissue sections. Sections were cleared of paraffin, endogenous peroxidases were blocked using 3% H2O2, and sections were washed and incubated with rabbit serum for 10 minutes at room temperature. Sections were then incubated overnight at 4°C with mouse antibody against MMP2 (1:50; Abcam) or MMP9 (1:50; Abcam). Finally, sections were incubated with biotinylated rabbit anti-mouse or goat anti-rabbit immunoglobulin G (IgG) (1:2,000; Santa Cruz Biotechnology Inc, Dallas, TX, USA). Sections were stained with diaminobenzidine (Sinopharm, Shanghai, People’s Republic of China) according to the manufacturer’s instructions. Five sections from each animal were analyzed, and four images were acquired from one randomly selected location per section. Digital photos were analyzed using Image-Pro Plus 6.0; the analyst was blind to group allocation of the tissue sections.
To verify signal specificity for MMP2 and MMP9, sections were incubated with only primary or secondary antibody in parallel with the experimental samples. Positive staining was not observed in any case.
Determination of MMP2 and MMP9 activity in lung tissues
The resected lung tissues of each group were homogenized and the activity of MMP2 and MMP9 was quantitatively determined using MMP2 and MMP9 Biotrak™ activity assay kit (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA).
Apoptosis analysis
The alveolar cell type I was isolated as previously described,21 the isolated cells were resuspended in phosphate-buffered saline (PBS), and then fixed in ethanol under room temperature overnight. The cells were washed with PBS and resuspended in staining solution (50 mg/mL propidium iodide, 1 mg/mL RNase A, and 0.1% Triton X-100 in PBS, all purchased from Thermo Fisher Scientific, Waltham, MA, USA). The stained cells were then analyzed for apoptosis with a BD flow cytometer (BD Biosciences, San Jose, CA, USA).
Levels of PPARγ, phospho-MAPK and activated NFκB proteins in lung tissue
Cytoplasmic fractions containing 50 μg of total protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. The membrane was blocked for 1 hour at room temperature with a blocking solution. The blot was then incubated overnight at 4°C with mouse anti-di-phosphorylated extracellular-signal-regulated kinases1 and 2 (ERK1/2) (1:500; Abcam), anti-phospho-JNK (c-Jun N-terminal kinase) (1:500; Abcam), or anti-phospho-p38 (1:500; Abcam). After three washing steps, the membrane was incubated with secondary antibody (1:2,000 dilution) for 2 hours at room temperature. The signals were detected using enhanced chemiluminescence (GE Healthcare UK Ltd), and the protein bands were densitometrically analyzed with β-actin as a loading control (Metamorph/Evolution MP 5.0/B X51).
To determine the level of NFκB activation in lung tissue, the p65 fraction of NFκB and IκBα (1:500; Abcam) in nuclear extracts was analyzed by Western blotting. Nuclear extracts containing 50 μg of total protein were resolved by 10% SDS-PAGE, and NFκB was detected using rabbit polyclonal antibody against NFκB p65 (1:500; Abcam) or anti-IκBα (1:500, Abcam). As a loading control, actin levels were determined using a mouse antibody against actin (1:1,000; Abcam). Blots were incubated with secondary antibody, processed for chemiluminescence, and analyzed as described in this section. Band intensities were quantified using ImageJ software 1.42q and normalized to levels in the control group, as described in this section. Experiments were performed three times for each experimental condition.
Statistical analysis
Statistical analyses were performed using SPSS version 11.5 (SPSS Inc, Chicago, IL, USA). All data were expressed as means ± standard deviation (SD). Differences among more than two groups were compared using one-way analysis of variance (ANOVA); differences between two groups were compared using Fisher’s least significant difference test. Significance of correlation between two variables was assessed using the Pearson correlation coefficient. The threshold for significance was set at P<0.05.
Results
PPARγ upregulation by rosiglitazone partially protects against emphysema induced by CS
In this study, we analyzed the possible effect of PPARγ activation by administration of rosiglitazone on lung morphology when exposed to cigarette smoking. MLI and MAN were measured to compare CS-induced lung emphysema and rosiglitazone-treated emphysema to evaluate the protective effect of PPARγ. Cigarette smoke exposure caused significantly decreased expression of PPARγ (Figure 1) and increases in MLI and MAN in the CS group when compared to the control group (Figure 2). When simultaneously treated with rosiglitazone and CS exposure, the changes of emphysema were partially prevented with increased MLI and MAN and the increased expression of PPARγ compared with the CS group (Figures 1 and 2; Table 1).
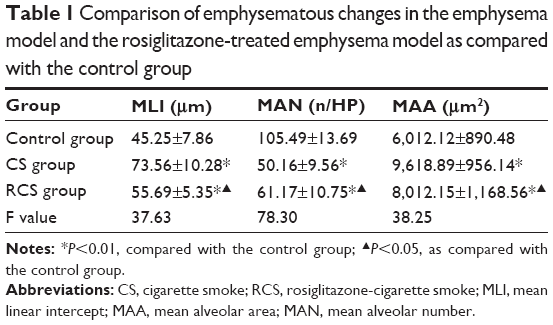 | Table 1 Comparison of emphysematous changes in the emphysema model and the rosiglitazone-treated emphysema model as compared with the control group |
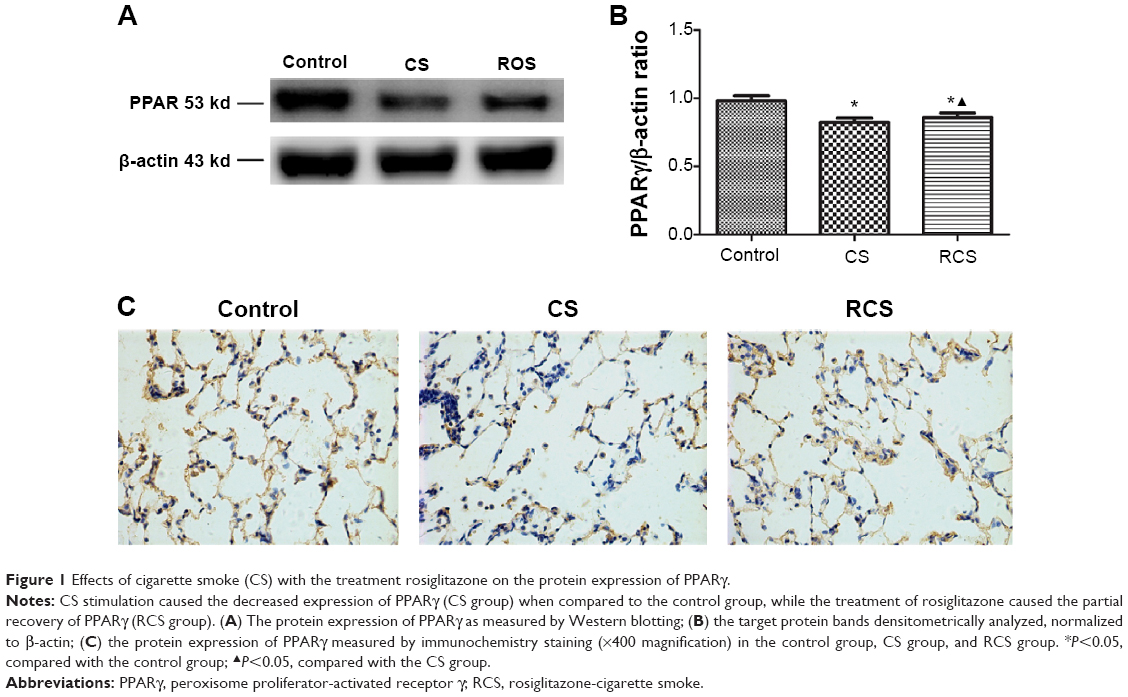 | Figure 1 Effects of cigarette smoke (CS) with the treatment rosiglitazone on the protein expression of PPARγ. |
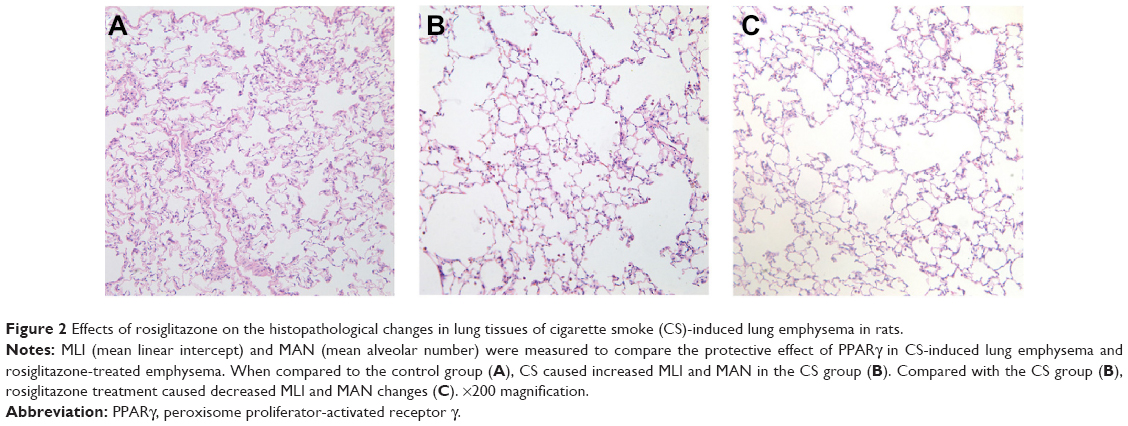 | Figure 2 Effects of rosiglitazone on the histopathological changes in lung tissues of cigarette smoke (CS)-induced lung emphysema in rats. |
We compared the lung tissue damage caused by CS in the presence or absence of the PPARγ agonist rosiglitazone. The CS group showed significantly downregulated expression of PPARγ protein (Figure 1) as well as decreases in MLI and MAN (Figure 2), as compared with the control group. The RCS group showed higher levels of PPARγ protein than that of the CS group and lower than that of the control group; both the Western blotting and immunochemistry revealed similar results (Figure 1). Meanwhile, the values of MLI and MAN were higher than those of the CS group and lower than those of the control group (Figure 2).
Rosiglitazone suppressed CS-induced increases in MMP2 and MMP9 protein levels
In order to assess the inhibitory effect of rosiglitazone on the abnormally enhanced MMP activity associated with emphysema, which thereby helps to restore the metalloprotease/anti-metalloprotease balance, we measured protein levels and activities of MMP2 and MMP9 in rats exposed to CS with or without treatment of rosiglitazone. The CS group showed significantly higher levels of both protein expression levels (Western blot, Figure 3A, B; immunohistochemistry, Figure 3E), and MMP2 and MMP9 activities (Figure 3C, D), than the other two groups. The protein expression levels and activities of MMP2 and MMP9 in the RCS group were all higher than in the control group (Figure 3).
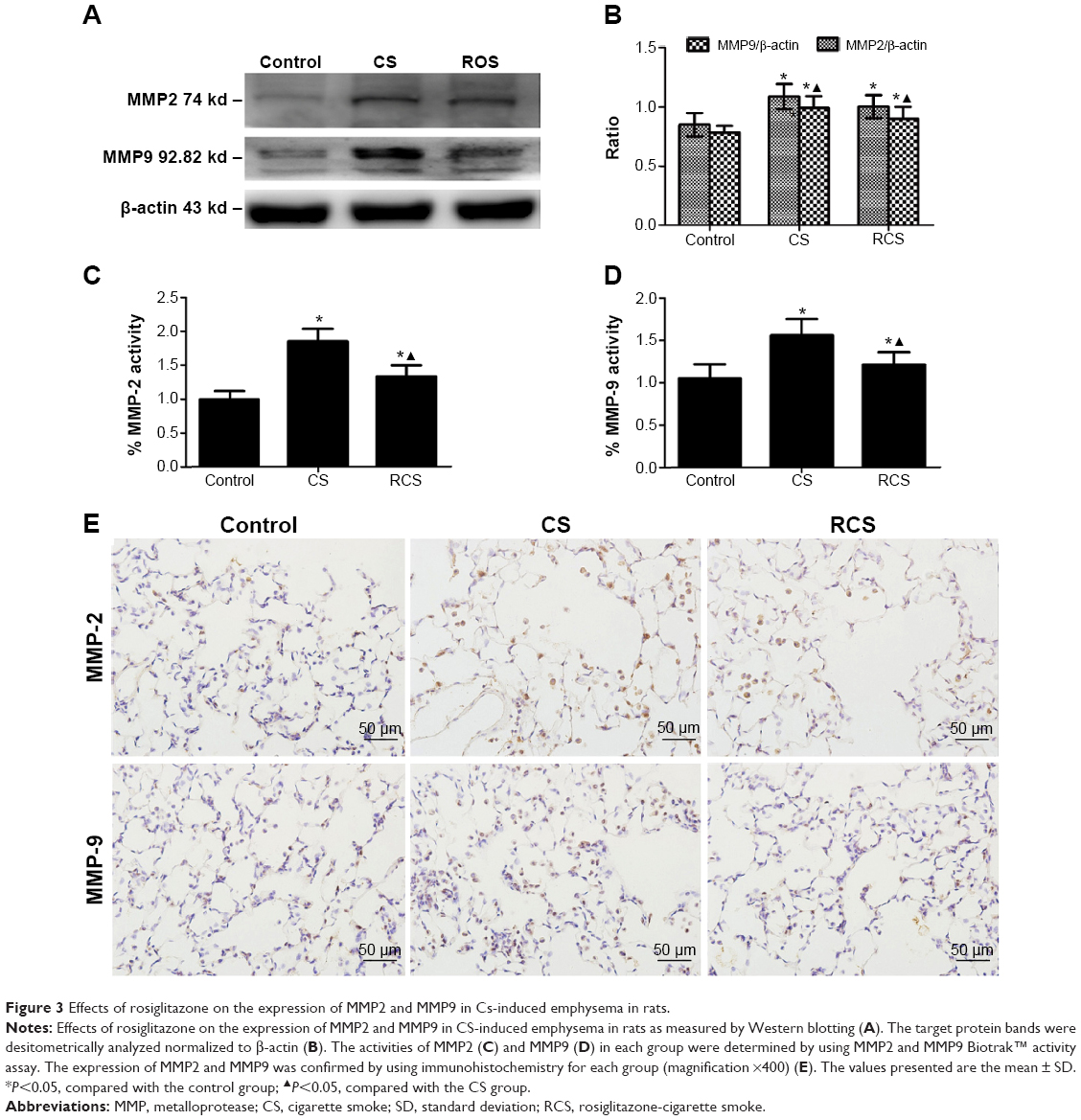 | Figure 3 Effects of rosiglitazone on the expression of MMP2 and MMP9 in Cs-induced emphysema in rats. |
Rosiglitazone inhibits CS-induced activation of NFκB
The CS group showed higher levels of NFκB p65 protein than the other two groups; levels in the RCS group were higher than the control group, as determined by Western blot (Figure 4) and immunohistochemistry (Figure 5). In addition to examining levels of NFκB p65, we measured phosphorylation levels of IκBα, which inhibited nuclear NFκB. Levels of IκBα phosphorylation were lower in the CS group than in either of the other two groups; the level in the RCS group was lower than levels in the control group (Figure 4).
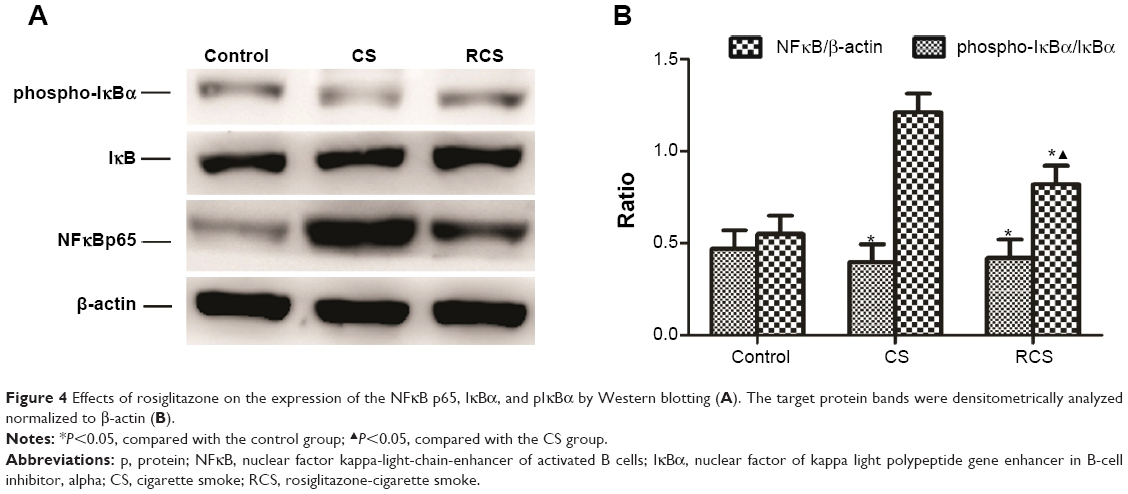 | Figure 4 Effects of rosiglitazone on the expression of the NFκB p65, IκBα, and pIκBα by Western blotting (A). The target protein bands were densitometrically analyzed normalized to β-actin (B). |
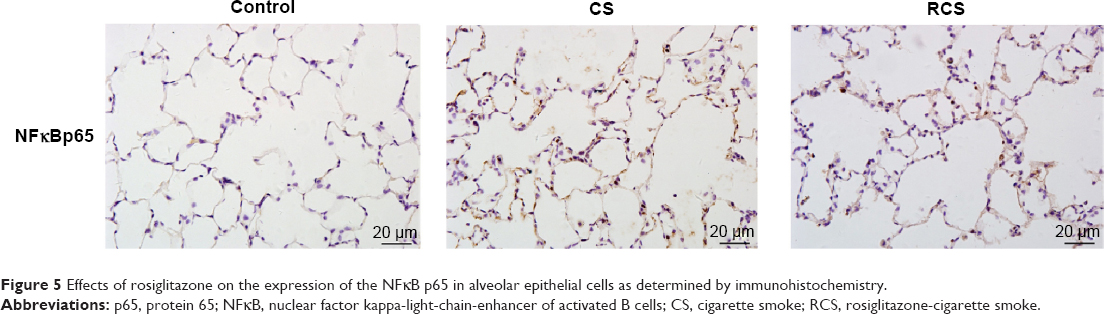 | Figure 5 Effects of rosiglitazone on the expression of the NFκB p65 in alveolar epithelial cells as determined by immunohistochemistry. |
Inhibition of NFκB activation by rosiglitazone involves MAPK signaling
We wanted to identify which one of several pathways known to regulate NFκB expression also functions as a mediator for the CS-induced activation of transcription factors, as well as for the rosiglitazone-mediated inhibition of this activation. Western blotting showed that levels of phosphorylated p38 and ERK1/2 were higher in the CS group than in the other two groups; these levels in the RCS group were higher than those in the control group (Figure 6). In contract, the levels of phosphorylated JNK in the three groups presented no significant differences between groups.
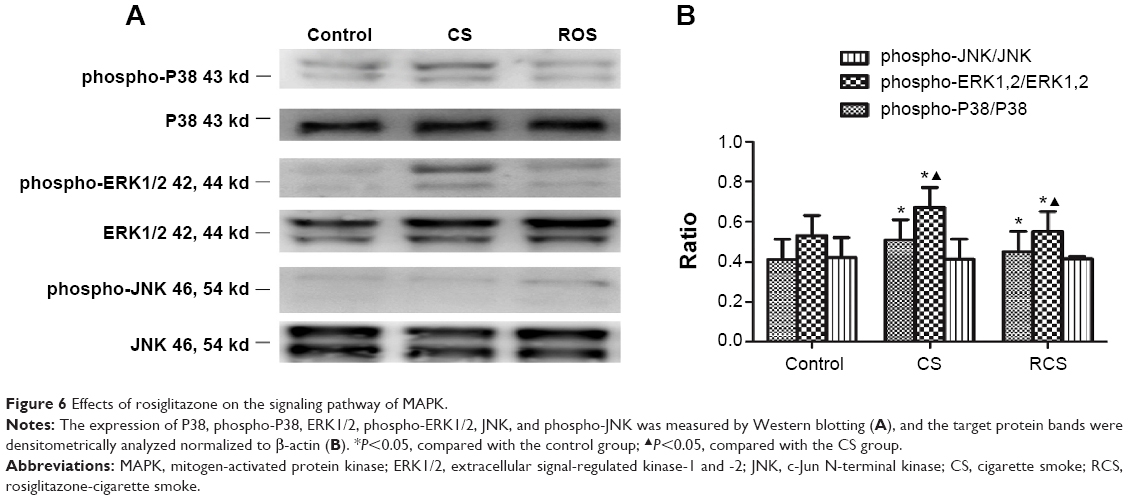 | Figure 6 Effects of rosiglitazone on the signaling pathway of MAPK. |
Effect of rosiglitazone on the apoptosis status of alveolar cells harvested from rats treated with CS
To quantitatively determine the number of apoptotic cells, we isolated alveolar cells (type I) following the protocol as described previously,21 and carried out flow cytometry analysis to determine the ratio of apoptotic cells. As shown in Figure 7, apoptosis indexes in control, CS, and RCS groups were 5.1%, 28.8%, and 14.4%, respectively.
 | Figure 7 Effect of rosiglitazone on apoptosis status of alveolar cells (type I) isolated from control group (A), CS group (B), and RCS group (C). |
Discussion
In this rat emphysema model, CS exposure upregulated expression of MMP2 and MMP9, creating an imbalance in the metalloprotease/anti-metalloprotease equilibrium, which is associated with histopathological changes in alveoli. These effects were partially but significantly reversed by administering the PPARγ agonist rosiglitazone, together with the CS. Rosiglitazone appears to work by activating NFκB via a MAPK-dependent signaling pathway.
Our observation that exposure to CS induced over-expression of MMP2 and MMP9 is consistent with the widely accepted notions that proteases make a critical contribution to all the pathologic processes detected in the lungs of COPD patients;22–24 with more specific findings that expression of MMPs 1–3 and 7–9 are higher in COPD patients than in healthy people; and that MMP9 levels correlated with Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage of pulmonary function test.24
Our observation that PPARγ activation reduced the expression of both MMP2 and MMP9, as well as the lung tissue histopathology characteristic of emphysema, is consistent with the idea that PPARγ protects against CS-induced emphysema. Indeed, we have already shown that exposure to CS decreases PPARγ expression and increases inflammatory cytokine expression in bronchial epithelial cells in vivo and in vitro.25
Our study adds emphysema to the types of tissue damage and disease against which PPARγ protects by inhibiting production of the extracellular matrix-degrading MMP9. This has already been shown in human bronchial epithelial cells treated with tumor necrosis factor alpha (TNFα) and phorbol myristate acetate (PMA) in a concentration-dependent manner,26 as well as in type 2 diabetic patients treated with rosiglitazone.27 Similarly, the PPARγ agonist pioglitazone inhibits the increase in MMP2 and MMP9 expression induced by renal ischemia/reperfusion injury, reducing the severity of renal injury.27 Atorvastatin inhibits expression of both MMP9 messenger RNA (mRNA) and protein, while upregulating the expression of all PPAR isoforms; pretreatment with PPAR inhibitors attenuates this inhibition of MMP9 expression.28 Our study is the first to extend the possibility of PPARγ protection to emphysema and the imbalance of MMPs in emphysema.
We found that while CS increased NFκB expression, PPARγ activation by rosiglitazone partially inhibited this NFκB upregulation. We observed the inverse trend with expression of IκBα, a negative regulator of NFκB. These results are consistent with the idea that NFκB is a key promoter of inflammation, whose activation in the lower airways of patients with COPD will lead to pulmonary emphysema,29,30 which is negatively regulated by PPARγ. Once activated, NFκB translocates to the nucleus, where it induces the transcription of proinflammatory genes. Based on experiments in adipose tissue, the RelA subunit of NFκB binds to PPARγ and prevents dimers of PPARγ and retinoid X receptor from binding to target genes.31 This binding event also leads to nuclear export of NFκB, thereby reducing NFκB-mediated inflammatory gene expression.32 While signal transduction “cross-talk” between PPARγ and NFκB has been documented in several types of tissue damage and disease, this is the first report suggesting that such cross-talk also occurs in CS-induced emphysema.
Evidence of such cross-talk is consistent with previous work from our laboratory in macrophages harvested from rats exposed to CS for 3 months in vivo, which showed that rosiglitazone counteracted CS-induced leukotriene B4 (LTB4) and interleukin 8 (IL8) release and PPARγ downregulation, markedly lowering the expression of toll-like receptor 4 (TLR4) and TLR2,25 and consistent with a study in a mouse model of polymicrobial sepsis due to cecal ligation and puncture.33 In the latter mouse model, sepsis upregulated NFκB expression and downregulated IκBα expression; both changes were partially inhibited by pioglitazone.
In order to assess what signaling pathways lead to NFκB activation in emphysema, we analyzed levels of p38 MAPK, ERK1/2, and JNK after exposure to CS in the presence or absence of rosiglitazone. We found that smoke exposure increased phosphorylation of ERK1/2 and p38 and decreased PPARγ expression, with no obvious effect on JNK phosphorylation. Exposure to smoke in the presence of rosiglitazone led to smaller increases in phosphorylation of ERK1/2 and p38. These results provide the first evidence that CS activates PPARγ by triggering the phosphorylation of ERK1/2 and p38 MAPK. These findings extend the results of a previous study indicating that MAPK kinase signaling mediates some of the effects of CS on cytokine production.34 How rosiglitazone inhibits the smoke-induced activation of ERK1/2 and p38 requires further study.
The mechanism of PPARγ regulation observed in the current study in CS-induced emphysema appears to be analogous to that described for other disease models.35,36 In a study of apoptosis regulation of human neuroblastoma cells, the PPARγ agonist 15-deoxy-delta 12,14-prostaglandin J2 induced PPARγ expression and activated ERK2, while both these changes were partially inhibited by the PPARγ antagonist GW9662.37 In a study exploring the mechanism of the rosiglitazone inhibition of TNFα and gamma interferon (IFNγ) inflammatory effects in human endothelial cells, rosiglitazone attenuated the inflammatory response by inhibiting ERK1/2 phosphorylation.38 Rosiglitazone also rapidly inhibited the homocysteine-induced phosphorylation of ERK1/2 and p38 MAPK in endothelial cells and vascular smooth muscle cells.39
This mechanism of PPARγ regulation may not be universal. In a mouse model of polymicrobial sepsis, an increase in nuclear levels of phosphorylated ERK1/2 correlated with a decrease in PPARγ phosphorylation in peripheral blood mononuclear cells; in lung tissue, however, the increase in phosphorylated ERK1/2 was associated with an increase in PPARγ phosphorylation.40 In MCF7 breast cancer cells, α-eleostearic acid simultaneously inhibited phosphorylation of ERK1/2 and PPARγ in a time-dependent manner.41 The PPARγ agonist troglitazone upregulated PPARγ expression in the human lung cancer cell lines NCI-H23 and CRL-2066, as well as to a lesser extent in the normal human lung cell line CRL-202.42 However, in contrast to the results observed in the present study, these troglitazone-induced increases in PPARγ expression were associated with increases in the levels of phosphorylated ERK1/2 and p38 and with decreases in stress-activated protein kinase (SAPK)/JNK activity. The difference between these results and those in our study and related work highlights the multiple signaling pathways that regulate PPARγ activity and the likelihood that the precise pathway involved depends on the physiological process (eg, inflammation, aging, proliferation) and the stimulus (eg, cigarette smoking, inflammatory cytokines).
Conclusion
In summary, our animal model suggests that CS works via signaling pathways dependent on MAPK and NFκB pathways to activate PPARγ, leading in turn to excess MMP2 and MMP9 activity. This metalloprotease/anti-metalloprotease imbalance plays a large part in the emphysematous changes induced by CS. These mechanistic insights may help lead to new therapies for COPD and emphysema.
Acknowledgment
This research was supported by project grant 2012BAI05B01 from the Ministry of Science and Technology of the People’s Republic of China.
Author contributions
GH helped design the study, establish the animal model, collect and analyze samples, and write the manuscript. YY and DH helped establish the animal model, performed Western blotting, immunohistochemistry analyses. YY and DH also helped analyse data and revise the manuscript. QYW and JK contributed to study planning, analyse and interpret data, review and revise the manuscript. All authors have approved the final version of this article to be published and agreed to be accountable for all aspects of the work in ensuring the questions related to the accuracy or integrity of any part of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Angelis N, Porpodis K, Zarogoulidis P, et al. Airway inflammation in chronic obstructive pulmonary disease. J Thorac Dis. 2014;6(Suppl 1):S167–S172. | ||
Mocchegiani E, Giacconi R, Costarelli L. Metalloproteases/anti-metalloproteases imbalance in chronic obstructive pulmonary disease: genetic factors and treatment implications. Curr Opin Pulm Med. 2011;17(Suppl 1):S11–S19. | ||
Churg A, Zhou S, Wright JL. Series “matrix metalloproteinases in lung health and disease”: matrix metalloproteinases in COPD. Eur Respir J. 2012;39(1):197–209. | ||
Dhariwal J, Tennant RC, Hansell DM, et al. Smoking cessation in COPD causes a transient improvement in spirometry and decreases micronodules on high-resolution CT imaging. Chest. 2014;145(5):1006–1015. | ||
Coxson HO, Dirksen A, Edwards LD, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. The presence and progression of emphysema in COPD as determined by CT scanning and biomarker expression: a prospective analysis from the ECLIPSE study. Lancet Respir Med. 2013;1(2):129–136. | ||
Carolan BJ, Sutherland ER. Clinical phenotypes of chronic obstructive pulmonary disease and asthma: Recent advances. J Allergy Clin Immunol. 2013;131(3):627–634. | ||
Ahmadian M, Suh JM, Hah N, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557–566. | ||
Kostadinova R, Wahli W, Michalik L. PPARs in diseases: control mechanisms of inflammation. Curr Med Chem. 2005;12(25):2995–3009. | ||
Belvisi MG, Hele DJ, Birrell MA. Peroxisome proliferator-activated receptor gamma agonists as therapy for chronic airway inflammation. Eur J Pharmacol. 2006;533(1–3):101–109. | ||
Yoo SH, Abdelmegeed MA, Song BJ. Activation of PPARalpha by Wy-14643 ameliorates systemic lipopolysaccharide-induced acute lung injury. Biochem Biophys Res Commun. 2013;436(3):366–371. | ||
Lu X, Bijli KM, Ramirez A, Murphy TC, Kleinhenz J, Hart CM. Hypoxia downregulates PPARgamma via an ERK1/2-NF-kappaB-Nox4-dependent mechanism in human pulmonary artery smooth muscle cells. Free Radic Biol Med. 2013;63:151–160. | ||
Wechsler ME, Laviolette M, Rubin AS, et al; Asthma Intervention Research 2 Trial Study Group. Bronchial thermoplasty: long-term safety and effectiveness in patients with severe persistent asthma. J Allergy Clin Immunol. 2013;132(6):1295–1302. | ||
Kim SH, Hong JH, Lee YC. Ursolic acid, a potential PPARgamma agonist, suppresses ovalbumin-induced airway inflammation and Penh by down-regulating IL-5, IL-13, and IL-17 in a mouse model of allergic asthma. Eur J Pharmacol. 2013;701(1–3):131–143. | ||
Lea S, Plumb J, Metcalfe H, et al. The effect of peroxisome proliferator-activated receptor-gamma ligands on in vitro and in vivo models of COPD. Eur Respir J. 2014;43(2):409–420. | ||
Yamasaki S, Nakashima T, Kawakami A, et al. Functional changes in rheumatoid fibroblast-like synovial cells through activation of peroxisome proliferator-activated receptor gamma-mediated signalling pathway. Clin Exp Immunol. 2002;129(2):379–384. | ||
Ko GJ, Kang YS, Han SY, et al. Pioglitazone attenuates diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. Nephrol Dial Transplant. 2008;23(9):2750–2760. | ||
Miyahara T, Schrum L, Rippe R, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275(46):35715–35722. | ||
Xu L, Cai BQ, Zhu YJ. Pathogenesis of cigarette smoke-induced chronic obstructive pulmonary disease and therapeutic effects of glucocorticoids and N-acetylcysteine in rats. Chin Med J (Engl). 2004;117(11):1611–1619. | ||
Qi B, Chen HL, Shang D, Dong Y, Zhang GX, Yu L. Effects of hypoxia-inducible factor-1alpha and matrix metalloproteinase-9 on alveolar-capillary barrier disruption and lung edema in rat models of severe acute pancreatitis-associated lung injury. Exp Ther Med. 2014;8:899–906. | ||
Gonzalez RF, Dobbs LG. Isolation and culture of alveolar epithelial Type I and Type II cells from rat lungs. Methods Mol Biol. 2013;945:145–159. | ||
Mocchegiani E, Giacconi R, Costarelli L. Metalloproteases/anti-metalloproteases imbalance in chronic obstructive pulmonary disease: genetic factors and treatment implications. Curr Opin Pulm Med. 2011;17(Suppl 1):S11–S19. | ||
Antunes MA, Rocco PR. Elastase-induced pulmonary emphysema: insights from experimental models. An Acad Bras Cienc. 2011;83(4):1385–1396. | ||
Navratilova Z, Zatloukal J, Kriegova E, Kolek V, Petrek M. Simultaneous up-regulation of matrix metalloproteinases 1, 2, 3, 7, 8, 9 and tissue inhibitors of metalloproteinases 1, 4 in serum of patients with chronic obstructive pulmonary disease. Respirology. 2012;17(6):1006–1012. | ||
Yin Y, Hou G, Li ER, Wang QY, Kang J. Regulation of cigarette smoke-induced toll-like receptor 4 expression by peroxisome proliferator-activated receptor-gamma agonists in bronchial epithelial cells. Respirology. 2013;18(Suppl 3):30–39. | ||
Hetzel M, Walcher D, Grüb M, Bach H, Hombach V, Marx N. Inhibition of MMP-9 expression by PPARgamma activators in human bronchial epithelial cells. Thorax. 2003;58(9):778–783. | ||
Marx N, Froehlich J, Siam L, et al. Antidiabetic PPAR gamma-activator rosiglitazone reduces MMP-9 serum levels in type 2 diabetic patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2003;23(2):283–288. | ||
Han L, Li MG, Liu YX, Han CG, Ye P. Atorvastatin may delay cardiac aging by upregulating peroxisome proliferator-activated receptors in rats. Pharmacology. 2012;89(1–2):74–82. | ||
Kobayashi Y, Wada H, Rossios C, et al. A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-kappaB inhibition. J Pharmacol Exp Ther. 2013;345(1):76–84. | ||
Brown V, Elborn JS, Bradley J, Ennis M. Dysregulated apoptosis and NFkappaB expression in COPD subjects. Respir Res. 2009;10:24. | ||
Suzawa M, Takada I, Yanagisawa J, et al. Cytokines suppress adipogenesis and PPAR-function through the TAK1/TAB1/NIK cascade. Nat Cell Biol. 2003;5(3):224–230. | ||
Kelly D, Campbell JI, King TP, et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol. 2004;5(1):104–112. | ||
Kaplan J, Nowell M, Chima R, Zingarelli B. Pioglitazone reduces inflammation through inhibition of NF-kappa B in polymicrobial sepsis. Innate Immun. 2014;20(5):519–528. | ||
Mao J, Liu J, Pang X, et al. Nicotine induces the expression of C-reactive protein via MAPK-dependent signal pathway in U937 macrophages. Mol Cells. 2012;34(5):457–461. | ||
Burgermeister E, Seger R. MAPK kinases as nucleo-cytoplasmic shuttles for PPAR gamma. Cell Cycle. 2007;6(13):1539–1548. | ||
Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771(8):952–960. | ||
Kim EJ, Park KS, Chung SY, et al. Peroxisome proliferator-activated receptor-gamma activator 15-deoxy-Delta(12,14)-prostaglandin J(2) inhibits neuroblastoma cell growth through induction of apoptosis: Association with extracellular signal-regulated kinase signal pathway. J Pharmacol Exp Ther. 2003;307(2):505–517. | ||
Lombardi A, Cantini G, Piscitelli E, et al. A new mechanism involving ERK contributes to rosiglitazone inhibition of tumor necrosis factor-alpha and interferon-gamma inflammatory effects in human endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28(4):718–724. | ||
Bai YP, Liu YH, Chen J, et al. Rosiglitazone attenuates NF-kappaB-dependent ICAM-1 and TNF-alpha production caused by homocysteine via inhibiting ERK1/2/p38MAPK activation. Biochem Biophys Res Commun. 2007;360(1):20–26. | ||
Kaplan JM, Hake PW, Denenberg A, Nowell M, Piraino G, Zingarelli B. Phosphorylation of extracellular signal-regulated kinase (ERK)-1/2 Is associated with the downregulation of peroxisome proliferator-activated receptor (PPAR)-γ during polymicrobial sepsis. Mol Med. 2010;16(11–12):491–497. | ||
Moon HS, Guo DD, Lee HG, et al. Alpha-eleostearic acid suppresses proliferation of MCF-7 breast cancer cells via activation of PPARgamma and inhibition of ERK 1/2. Cancer Sci. 2010;101(2):396–402. | ||
Li M, Lee TW, Mok TS, Warner TD, Yim AP, Chen GG. Activation of peroxisome proliferator-activated receptor-gamma by troglitazone (TGZ) inhibits human lung cell growth. J Cell Biochem. 2005;96(4):760–774. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.