")
Back to Journals » OncoTargets and Therapy » Volume 11
Role of ubenimex as an anticancer drug and its synergistic effect with Akt inhibitor in human A375 and A2058 cells
Authors Wang X, Liu Y, Wu R, Guo F, Zhang L, Cui M, Wu X, Zhang Y, Liu W
Received 19 November 2017
Accepted for publication 12 January 2018
Published 22 February 2018 Volume 2018:11 Pages 943—953
DOI https://doi.org/10.2147/OTT.S157480
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Xiaoqing Wang,1 Yang Liu,1 Rongde Wu,1 Feng Guo,1 Lijuan Zhang,1 Mingyu Cui,1 Xiangyu Wu,1 Yongfei Zhang,2 Wei Liu1
1Department of Pediatric Surgery, Shandong Provincial Hospital affiliated to Shandong University, Jinan, People’s Republic of China; 2Department of Dermatology, Shandong Provincial Qianfoshan Hospital affiliated to Shandong University, Jinan, People’s Republic of China
Background: Malignant melanoma (MM) is a malignant tumor produced by changes in melanocytes in the skin or other organs. In the classification of skin tumor mortality, skin melanoma ranks the highest. Ubenimex, an Aminopeptidase N (APN) inhibitor, is now widely used for cancer as an adjunct therapy, conferring antitumor effects. Apoptosis and the induction of autophagy have both been found to be closely associated with tumor cell death.
Methods: In this study, the A375 and A2058 cell lines were treated with ubenimex. Cell viability was measured using the Cell Counting Kit 8 assay. Apoptosis and autophagic cell death were assessed using flow cytometry and acridine orange/ethidium bromide staining. Protein expression was assessed by Western blot analyses and immunofluorescence. Matrigel invasion and migration assays were used to examine the metastatic ability of melanoma cells.
Results: The results revealed that ubenimex inhibited the expression of APN in melanoma cells, which may be connected with the inhibition of metastasis. In addition, it increased melanoma cell death by inducing apoptosis and autophagic cell death. This effect was accompanied by increased levels of p-JNK. Moreover, treatment with ubenimex induced protective Akt activation, and combined use of an Akt inhibitor with ubenimex provided a better effect for inducing tumor cell death.
Conclusion: As an effective anti-tumor drug in vitro, ubenimex might be an excellent adjunctive therapy for the treatment of melanoma, with greater effects when combined with the use of an Akt inhibitor.
Keywords: melanoma, ubenimex, jnk, Akt, mixed cell death, metastasis
Introduction
Malignant melanoma (MM) is a malignant tumor produced by changes in melanocytes in the skin or other organs. In the classification of skin tumor mortality, skin melanoma ranks the highest.1 Skin melanoma manifests a significant change in pigmented lesions within months or years. In recent years, the incidence of malignant melanoma has posed a huge threat to human health.2 The disease is characterized by a high rate of metastasis in the early stage, poor sensitivity to chemotherapy, and poor prognosis.3 Therefore, understanding the reasons for chemotherapy treatment resistance and exploring possible adjuvant medications is the final “life-saving straw” for melanoma patients, especially those with advanced cases.
Ubenimex, also known as bestatin, has been used as an adjunct therapy for many tumors, enhancing the function of immunocompetent cells and conferring antitumor effects,4 and the effect is also Aminopeptidase N (APN) related. APN, also called CD13, is involved in various cellular processes, and, in particular, it has been revealed to correlate with the invasion/metastasis of various malignancies.4 In malignant melanoma, the inhibition of APN always induces the inhibition of metastasis.5 However, few studies have examined the functions of ubenimex in melanoma cells in vitro.
Cell death is divided into programmed cell death and non-programmed death. Programmed cell death, which is often called apoptosis, is caspase-dependent cell death, whereas autophagic cell death is caspase independent.6 In many cases, autophagy is unequivocally the mode of tumor cell death.7 The JNK pathway plays an important role as a classical signaling pathway in the regulation of tumor cell apoptosis and autophagic death.8 The function of JNK as a regulator in apoptosis and autophagic cell death has been demonstrated in many tumor cells, such as bladder tumor, osteosarcoma, breast cancer, and hepatocellular carcinoma.8–11 In melanoma, JNK also plays a vital role in proliferation and cell death.12 The Akt pathway is often regulated in response to DNA-damaging chemotherapeutics, and participates in regulating tumor cell death.13 One of our previous papers also revealed its functions in tumor cell death and radiotherapy resistance following treatment with ubenimex.14
Referring to the drug ubenimex, our previous study proved its efficacy in renal cell carcinoma, prostate cancer, and glioma cells;14–16 although all experiments indicated that ubenimex could work as an anti-cancer drug, the mechanisms differ. Therefore, this study aimed to investigate whether ubenimex could still work as an anti-tumor drug in malignant melanoma cells and to determine the underlying potential mechanisms.
Materials and methods
Tumor cell lines
Malignant melanoma cell lines A375 and A2058 were purchased from the Chinese Academy of Sciences (Beijing, People’s Republic of China) Cell Bank. Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM), a high-glucose medium (Macgene, Beijing, People’s Republic of China), supplemented with 1% penicillin–streptomycin and 10% fetal bovine serum (Biological Industries, Kibbutz Beit Haemek, Israel). The cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
CCK-8 cell proliferation assay
A375 and A2058 cells in an exponential phase of growth were harvested and seeded into 96-well plates at a density of 6,000 cells/well in DMEM high-glucose medium supplemented with different concentrations of ubenimex. After the cells underwent specific treatments for the indicated time periods, 10 μL of Cell Counting Kit-8 (CCK-8) solution (CCK-8 cell proliferation and cytotoxicity assay kit-8; Dojindo, Kumamoto, Japan) was added to each well. The plates were then incubated for an additional 2 h at 37°C, and the absorbance was determined using a microplate reader (EL340; Bio-Tek Instruments, MA, USA) at 450 nm.
Acridine orange/ethidium bromide double staining
Cells were cultured in six-well plates for 24 h and then treated with different doses of ubenimex for 48 h. After the indicated treatment times, the acridine orange/ethidium (AO/EB) (Leagene, Beijing, China) operating fluid was mixed with reagents A (AO solution), B (EB solution), and C (AO/EB Dilution Buffer) at a ratio of 1:1:8. To each sample, 2 μL of AO/EB operating fluid was added, and the supernatant was discarded after low-speed centrifugation at 500 g/min. The cells were then resuspended in AO/EB dilution buffer at a concentration of 5×106 cells/mL, and 1 μL of AO/EB operating fluid was added to 25 μL of cell suspension. Next, the cells were observed under a fluorescence microscope (Nikon Inc., Tokyo, Japan).
Western blot analysis
To determine APN, LC3B, JNK/P-JNK, Akt/P-Akt, P62, and cleaved-caspase-3/pro-caspase-3 levels, proteins were extracted from the cells by suspension in radioimmunoprecipitation assay buffer. Samples were centrifuged at 12,000 rpm at 4°C for 30 min, and the supernatants were recovered for analysis. The protein concentrations were determined using the Bradford protein method and the bicinchoninic acid protein assay kit (Sigma, St Louis, MO, USA). Protein (40 μg) was electrophoresed on a pre-cast bis-Tris polyacrylamide gel (8%~12%) and then transferred onto a polyvinylidene difluoride membrane. Membranes were blotted with rabbit anti-APN (1:1,000), rabbit anti-LC3B (1:500), rabbit anti-JNK (1:1,000), rabbit anti-p-JNK (1:1,000), rabbit anti-Akt (1:1,000), rabbit anti-p-Akt (1:1,000), mouse anti-P62 (1:1,000), rabbit anti-Caspase-3 (1:1,000) (all from Abcam, San Francisco, CA, USA), and mouse anti-actin (1:1,000; Proteintech, NY, USA), followed by horseradish peroxidase-conjugated secondary antibodies (1:5,000; ZsBio, Beijing, China). Immunoblots were visualized using enhanced chemiluminescence (LAS-4000).
Annexin V–phycoerythrin (PE)/7-aminoactinomycin D staining
The apoptotic cells were quantified (%) using an Annexin V–PE/7-aminoactinomycin D (7AAD) kit (BD Biosciences, NJ, USA) and detected by flow cytometry. The A375 and A2058 cells were harvested after 48 h of treatment with ubenimex or combined with Akt inhibitor. Next, the cells were resuspended in a binding buffer (10 mmol/L 4-(2-hydroxyethyl)-1-pipera-zine-ethanesulfonic acid, 140 mmol/L NaCl, and 2.5 mmol/L CaCl2, pH 7.4) and incubated with Annexin V–PE/7AAD in the dark for 15 min. A total of 10,000 cells/sample were analyzed using a FACSCalibur or an EPICS XL flow cytometer (BD Biosciences).
Immunofluorescence
We performed cell concentration smears and fixed the cells with 4% paraformaldehyde for 20 min. We thoroughly rinsed the cells with 0.01 M PBS (5 min ×3) and incubated the cells in 0.3% Triton at room temperature for 20 min, followed by a wash in 0.01 M PBS (5 min ×3). Next, 30 μL/sample goat serum blocking solution was added at room temperature for 60 min, followed by a 1:100 dilution of primary antibody at 30 μL/sample (diluted in goat serum). The cells were placed in a wet box at 4°C overnight and then washed in 0.01 M PBS (5 min ×3). On the next day, 4′,6-diamidino-2-phenylindole was added for 10 min, and the cells were thoroughly rinsed with 0.01 M PBS (5 min ×3), followed by the addition of 1:100 fluorescent secondary antibody at 30 μL/sample (10% skim milk powder) at room temperature and in the dark for 60 min. Next, the cells were washed in 0.01 M PBS (5 min ×3) and mounted with an anti-fluorescence quencher.
Wound healing migration assays
The A375 and A2058 cells were plated in six-well culture plates and grown to 100% confluency before scratching with a sterile P200 pipette tip across the monolayer. The cell debris was removed by washing with PBS, and the cells were cultured in DMEM and 2% fetal bovine serum (FBS) supplemented with different concentrations of ubenimex. The area of the scratch was measured at different times (A375 for 0 h, 12 h, 24 h; A2058 for 0 h, 18 h, 36 h), and quantification was performed by measuring the area of cell migration at different time points compared to the scratch area at 0 h. Each experiment was repeated three times.
Matrigel invasion assay and matrigel migration assay
Migration assays were performed using transwell chambers. Control untreated cells or cells treated with ubenimex (0.5 or 1 mg/mL for 48 h) were trypsinized, and 1×105 cells were plated in the upper wells in serum-free medium, while medium with 10% FBS was added to the lower well as a stimulus. After 24 h of incubation, the cells on the matrigel side of the chambers were removed using a cotton swab. The inserts were fixed in methanol and stained using hematoxylin and eosin. The number of invading cells attached to the other side of the inserts was quantified under a light microscope using six random fields at a magnification of ×200. In addition, invasion assays were performed using transwell chambers that were pre-coated with 40 μL of 1 mg/mL matrigel matrix (BD Biosciences, Bedford, MA, USA). Each treatment group had a control containing 2.5×105 cells plated in the upper wells, with the remaining steps following the matrigel migration assay.
Statistical analysis
The data were statistically analyzed using Student’s t-test, the χ2 test, or Fisher’s exact test using SPSS version 19.0 software (IBM Corporation, Armonk, NY, USA). A P-value <0.05 was considered significant.
Results
Ubenimex could induce cell death and reduce the vitality of melanoma cells
CCK-8 was used to examine cell proliferation after treatment with different doses of ubenimex for different time periods in A375 and A2058 cells (Figure 1). The proliferation of tumor cells decreased over time. In addition, the proliferation of melanoma cells was significantly inhibited by the high dose of ubenimex after 48 h of exposure. Using CCK-8, we determined the appropriate concentration and time of drug use. Then, cell death was examined by AO/EB staining and flow cytometry (Figures 2 and 3). The cell death rate increased with ubenimex treatment, which showed a dose-dependent response (Figure 3). Therefore, we concluded that ubenimex could inhibit cell proliferation and induce cell death in both A375 and A2058 cells.
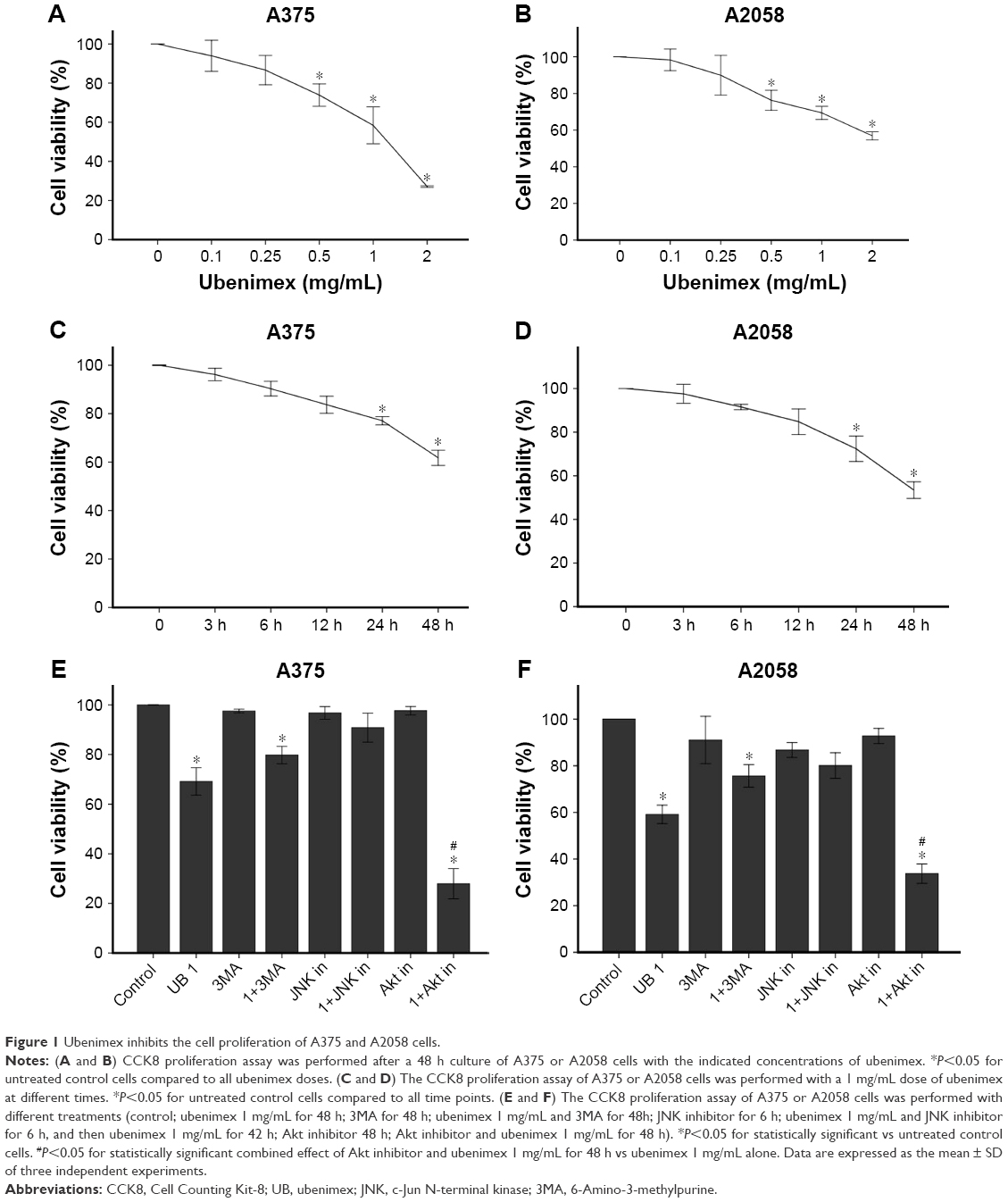 | Figure 1 Ubenimex inhibits the cell proliferation of A375 and A2058 cells. |
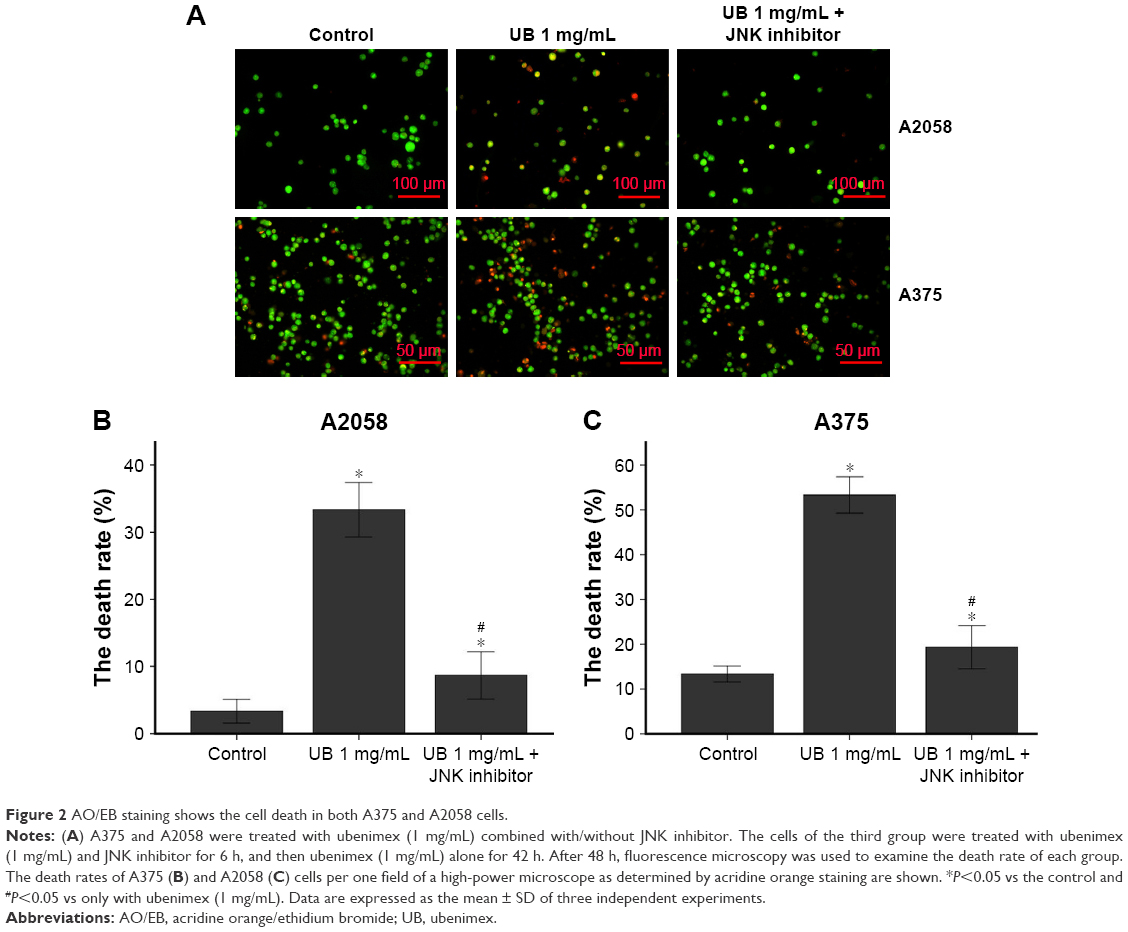 | Figure 2 AO/EB staining shows the cell death in both A375 and A2058 cells. |
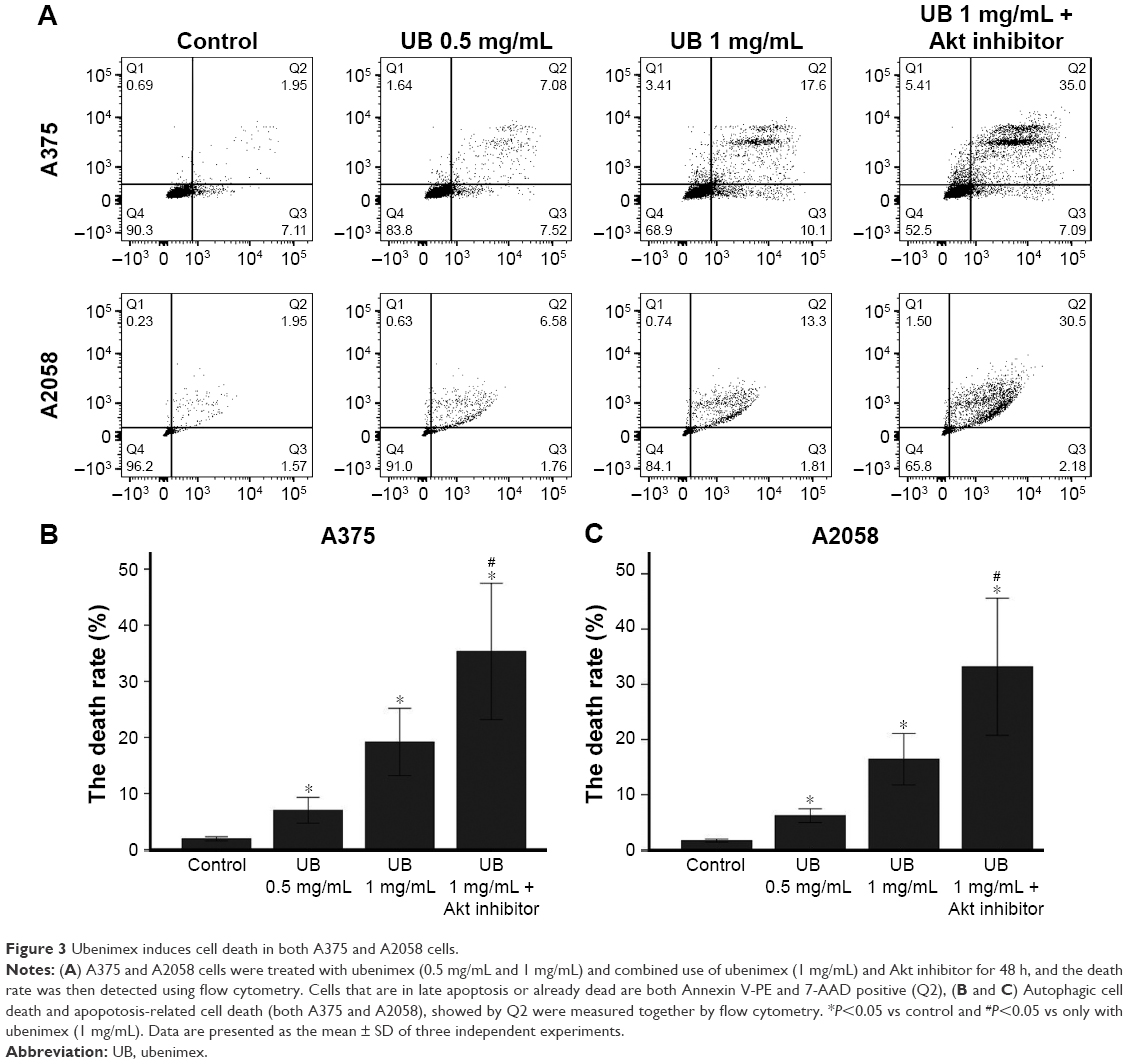 | Figure 3 Ubenimex induces cell death in both A375 and A2058 cells. |
Through transwell invasion and migration assays, we concluded that ubenimex could reduce invasion mobility in A375 cells
Wound healing assay and transwell assays were performed to determine whether ubenimex could affect the migration and invasion capacity of A375 cells (Figures 4 and 5). The migration capacity of A375 cells was significantly suppressed by ubenimex in a concentration-dependent manner after 48 h of exposure. Moreover, the dose of ubenimex used was also an important factor affecting A375 cells. We then further examined the effect of ubenimex on the invasive activity of A375 cells using matrigel invasion assays. Pretreatment with ubenimex markedly inhibited the invasive capacity of the A375 cells. Taken together, these results suggest that ubenimex inhibited the migration and invasion of A375 cells.
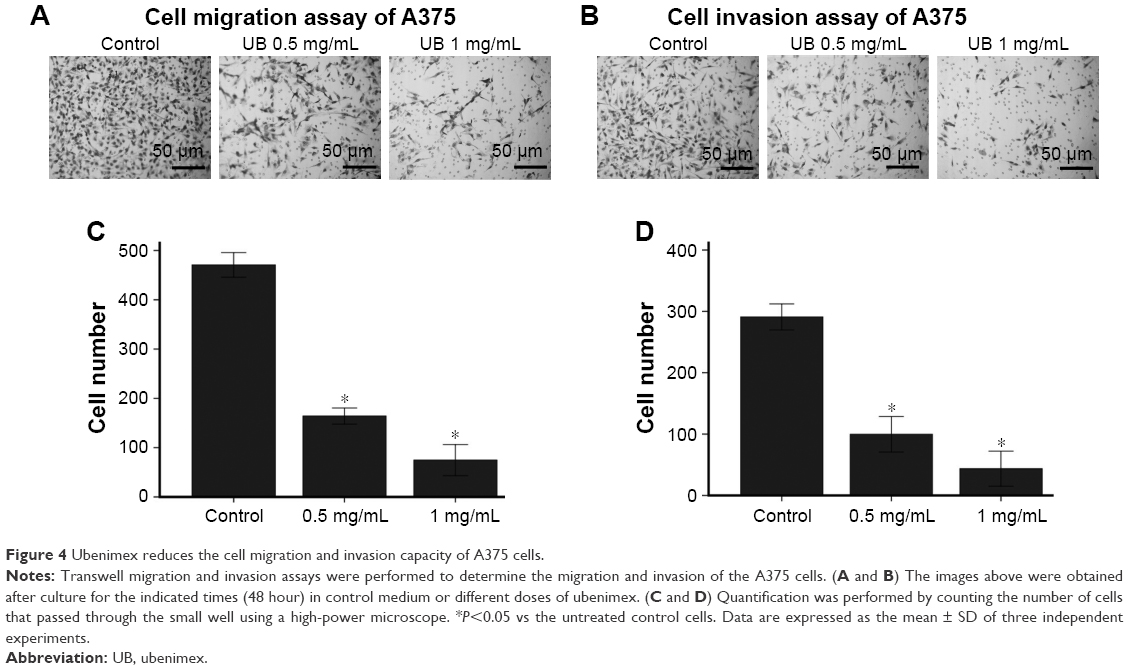 | Figure 4 Ubenimex reduces the cell migration and invasion capacity of A375 cells. |
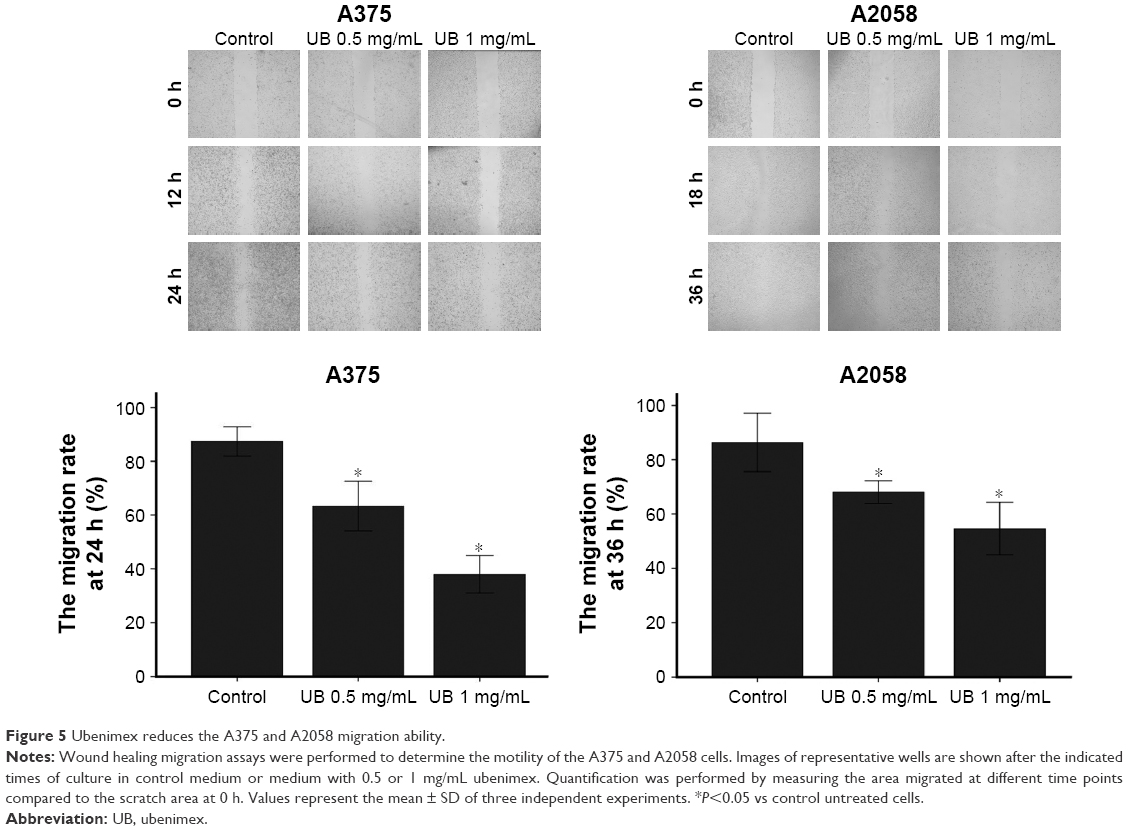 | Figure 5 Ubenimex reduces the A375 and A2058 migration ability. |
Ubenimex could function as an APN inhibitor in A375 and A2058 cells
Western blotting was used to test the expression of APN after ubenimex treatment (Figure 6). Additionally, A375 and A2058 cells were treated with ubenimex (1 mg/mL) for 48 h. The expression of APN was significantly inhibited by the increased dose of ubenimex.
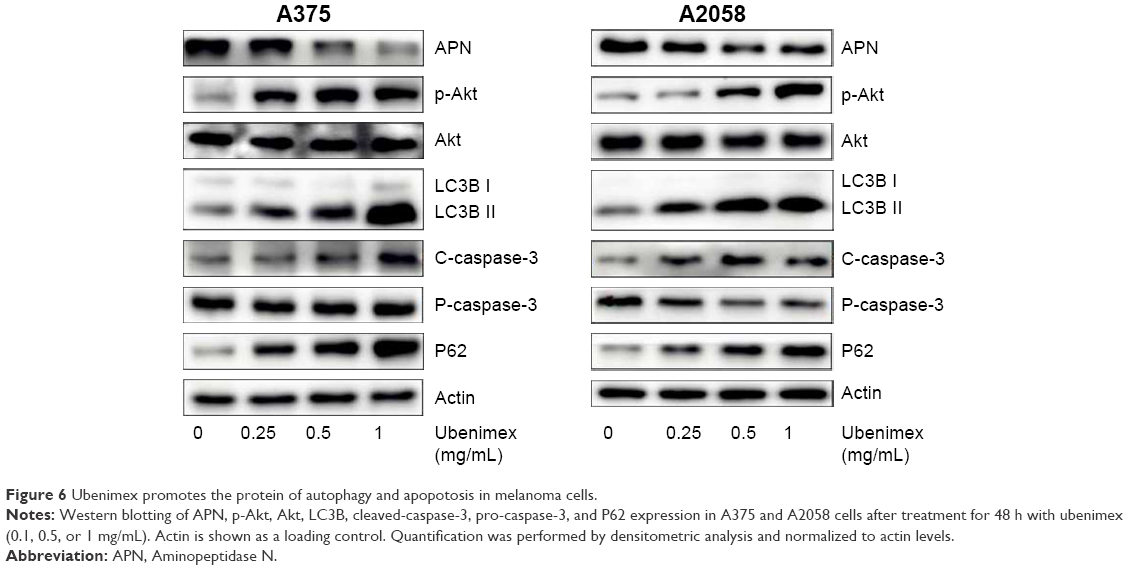 | Figure 6 Ubenimex promotes the protein of autophagy and apopotosis in melanoma cells. |
Treatment with ubenimex induced apoptosis in melanoma cells
Flow cytometry is a well-known experimental method for detecting cell apoptosis. As shown in Figure 3, the death rates of A375 and A2058 cells increased after ubenimex treatment and proved to be dose dependent. In addition, AO/EB staining also showed increased death rate of cells after treatment with ubenimex (Figure 2). In addition to autophagic cell death, apoptosis increased after the treatment. The CCK-8 assay provided additional proof: when we added 3MA to inhibit autophagy, the cell death induced by ubenimex persisted. Programmed cell death, which is often called apoptosis, is caspase-dependent cell death. Therefore, to further examine the increase in apoptosis, Western blotting was used to examine the level of caspase-3 (Figure 6). Treatment with ubenimex increased the level of cleaved-caspase-3, which is also evidence of increased cell apoptosis.
The cell death induced by ubenimex included not only apoptosis, but also autophagic cell death
The cell death induced by ubenimex is a combination of effects. In addition to apoptosis, autophagic cell death also occurs in melanoma cells. As mentioned above, 3MA was used to distinguish apoptosis from autophagic cell death. After the combined use of 3MA and ubenimex, the death rate was significantly decreased (Figure 1). We can easily conclude that ubenimex induced autophagic cell death in melanoma cells. Additionally, we used Western blotting and immunofluorescence to detect the expression of LC3B, an iconic protein in autophagy (Figures 6 and 7). As the dose of ubenimex increased, the expression of LC3B and autophagy-related P62 also increased.
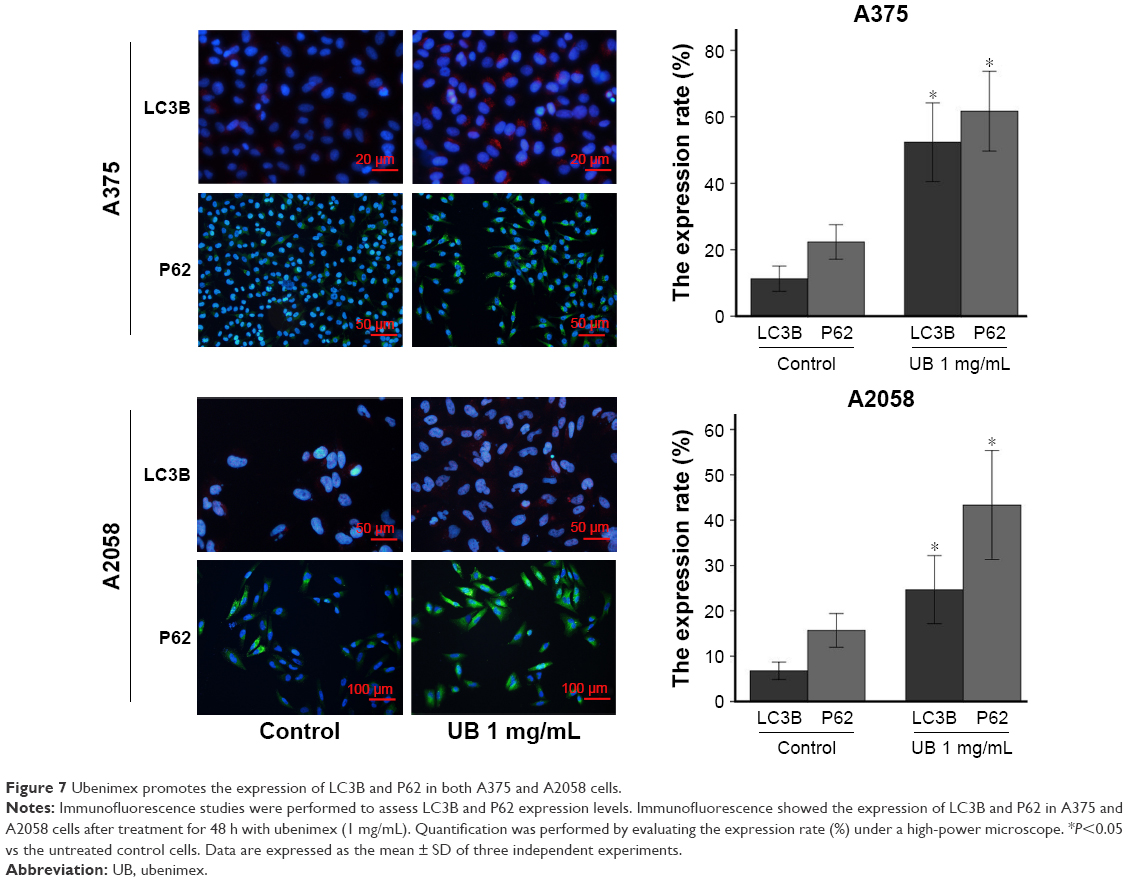 | Figure 7 Ubenimex promotes the expression of LC3B and P62 in both A375 and A2058 cells. |
The induced cell death may be regulated by the variation of p-JNK expression
We used Western blotting to examine the level of p-JNK (Figure 8). After treatment with ubenimex (1 mg/mL) for different lengths of time (0, 3 h, 6 h, 12 h, 24 h, 48 h), we examined the variation in p-JNK1 and p-JNK2. Despite slight differences, the expressions of these two proteins both increased after treatment with ubenimex at early time points and then decreased to a level lower than at 0 h. To further illustrate this point, we chose the 6-h time point for Western blot verification (Figure 9). We obtained the same result: the expression of p-JNK1 and p-JNK2 increased following treatment with ubenimex for 6 h. Moreover, CCK-8 was used to examine the role of the JNK pathway in melanoma cell death (Figure 1). We chose to use a JNK inhibitor (SP600125, Selleck, Houston, TX, USA) in combination with ubenimex for 6 h, and then used ubenimex alone for 42 h. The results showed decreased cell death in A375 and A2058 cells.
 | Figure 8 Ubenimex influences the expression of p-JNK and p-Akt in melanoma cells. |
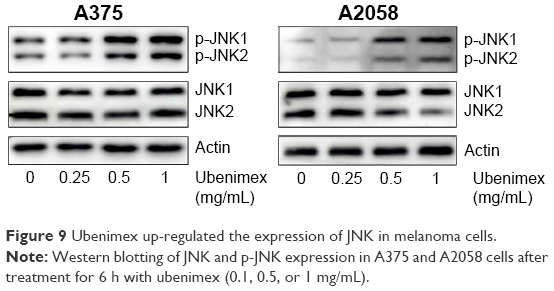 | Figure 9 Ubenimex up-regulated the expression of JNK in melanoma cells. |
Treatment with ubenimex could induce the Akt pathway, which shows a protective effect for melanoma cells
After treatment with different doses of ubenimex for 48 h, p-Akt was significantly increased in a dose-dependent manner (Figure 6). Then, we detected the expression at different time points (0, 3 h, 6 h, 12 h, 24 h, 48 h) after treatment with ubenimex (1 mg/mL) (Figure 8). p-Akt was also significantly increased. Then, we added an Akt inhibitor (PF-04691502, Selleck) to the ubenimex treatment. When combined with the Akt inhibitor, the effect of CCK-8 on cell proliferation was significantly inhibited, and cell death increased.
Discussion
Ubenimex continued to function as an APN inhibitor in A375 and A2058 cells. The inhibition of APN seems to be related to tumor metastasis, and reports have shown its function in many cancers, such as lung, pancreas, and colon cancers.17–19 In malignant melanoma cells, cell-surface APN expression could correlate to an invasive phenotype, and, through the inhibition of APN and ICAM, cell metastasis has been shown to significantly weaken.5 In particular, a previous study demonstrated that ubenimex, as an APN inhibitor, could inhibit tumor angiogenesis and may influence tumor growth and metastasis in B16-BL6 melanoma cells in vivo.20 However, there are few reports on the function of ubenimex in melanoma cells in vivo. Therefore, through Western blot analysis, when we added ubenimex, APN expression in A375 and A2058 cells was suppressed. These results are very consistent with previous findings in prostate cancer cells.15 Furthermore, matrigel invasion and migration assays were used to determine the migratory and invasion capacity of A375 cells. The capacity of A375 cells decreased with the use of ubenimex. Thus, we concluded that, in malignant melanoma cells, ubenimex could inhibit the ability to metastasize through the inhibition of APN.
Ubenimex could cause apoptosis and autophagic cell death in A375 and A2058 cells. Cell death is divided into programmed cell death and non-programmed death. Programmed cell death, which is often called apoptosis, is caspase dependent, whereas autophagic cell death is caspase independent.6 Both can cause tumor cell death. Although autophagy-associated cell death is different from apoptosis, there is sometimes a link between autophagy and apoptosis.21 In addition, the link is affected by cellular processes and determines whether the cells die or survive. In our previous study, ubenimex always served as an anti-tumor drug and induced cell death in prostate and renal cell carcinoma.14,15 Here, in malignant melanoma cells, we used 3MA to determine the presence of induced autophagic cell death. In addition, we found that ubenimex also induced caspase-related apoptosis, thus showing its anti-tumor capabilities. In our study, the apoptosis of malignant melanoma cells was assessed by Annexin V–PE/7AAD staining and Western blotting. Flow cytometry analysis showed that A375 and A2058 cell death increased with ubenimex treatment. Western blotting also demonstrated that caspase-3 was increased in this context. Thus, we detected a mixed phenotype of apoptosis and autophagic cell death in response to ubenimex treatment in malignant melanoma cells. Therefore, we concluded that ubenimex induced mixed programmed cell death in malignant melanoma cells.
Among mammals, there are three JNK genes: JNK1, JNK2, and JNK3.22 JNK1 and JNK2 are commonly expressed, while JNK3 is mainly manifested in the brain, heart, and testis.22 JNK, also known as stress-activated protein kinase, is closely related to apoptosis and autophagy.23–25 Moreover, the JNK signaling pathway is often stimulated by increased pressures from the external environment, including UV irradiation, oxidative stress, and drug treatment, which can induce apoptosis and autophagic cell death.23 Activation of the JNK pathway appears to lead to tumor cell death, including induced apoptosis and autophagy-related cell death.26 Here, we used Western blotting to analyze the JNK expression regulated by ubenimex. We found that JNK expression went up shortly before the drug was added, but, as time progressed, JNK expression decreased. We performed Western blotting at 6 h after treatment with ubenimex, and p-JNK expression was up-regulated. To determine the mechanism of cell death induced by the JNK pathway, we used a JNK inhibitor. In reference to the Western blot results, the JNK inhibitor was used to inhibit the increase in JNK in the first 6 h. The inhibition of JNK decreased tumor cell death and weakened the inhibition of proliferation. Thus, we can conclude that the JNK pathway is involved in response to treatment with ubenimex.
The JNK pathway can induce not only caspase-related apoptosis, but also P62-related autophagy.27 P62 protein (sequestosome 1) is a selective substrate that plays a key role in forming cytoplasmic inclusion bodies.27 In particular, the stress tumor cells face induces the activation of JNK and the expression of P62, and then autophagic cell death occurs.28 In our experiment, the alteration of LC3B expression was the same as that of P62. Therefore, activation of JNK may induce P62-related autophagic cell death in A375 and A2058 cells.
Phosphoinositide 3-kinase (PI3K)/Akt is mainly involved in cell death, and is considered to deliver survival signals to protect cells from cell death.29 Thus, many researchers have demonstrated that the inhibition of Akt could be a good method for cancer treatment.30 In melanoma, inhibition of the Akt pathway also suggested an anti-tumor effect, and the activation of Akt seemed to have a protective effect.31,32 Additionally, previous studies have revealed that stress activates the Akt signal transduction pathway in tumor cells, resulting in a protective effect.13 Through our experiments, the stress of ubenimex treatment also led to the activation of the Akt signaling pathway in melanoma cells. Over time, the activation effect became increasingly obvious. After we used an Akt inhibitor, we found a very significant induction of death and the inhibition of proliferation in melanoma cells. Therefore, the application of ubenimex could lead to the activation of the protective Akt pathway, which can be reversed by its specific inhibitors.
Conclusion
The results of this study demonstrated that ubenimex could inhibit cell proliferation, migration, and invasion, and induce autophagic cell death and apoptosis in A375 and A2058 cells, and these efforts might be associated with the down-regulation of APN and up-regulation of the JNK pathway. In addition, when treated with ubenimex, the melanoma cells up-regulated protective Akt activation, and the combined use of an Akt inhibitor with ubenimex provided a good effect for inducing tumor cell death. Therefore, this study suggests that ubenimex might be an excellent adjunctive therapy in the treatment of melanoma, with improved results when combined with an Akt inhibitor.
Acknowledgments
Thanks are given for the funding provided by the National Natural Science Foundation (81400575), the Science and Technology Development Plan Project of Shandong Province, China (2014GSF118144), and the Shandong Provincial Natural Science Foundation (ZR2017MH091).
Disclosure
The authors report no conflicts of interest in this work.
References
Savoia P, Fava P, Nardò T, Osella-Abate S, Quaglino P, Bernengo MG. Skin metastases of malignant melanoma: a clinical and prognostic survey. Melanoma Res. 2009;19(5):321–326. | ||
Gladfelter P, Darwish NHE, Mousa SA. Current status and future direction in the management of malignant melanoma. Melanoma Res. 2017;27(5):403–410. | ||
Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–284. | ||
Liu S, Xie F, Wang H, et al. Ubenimex inhibits cell proliferation, migration and invasion in renal cell carcinoma: The effect is autophagy-associated. Oncol Rep. 2015;33(3):1372–1380. | ||
Laube F. Immunoluminescent detection of intercellular adhesion molecule-1 and aminopeptidase N on human melanoma cells. Anticancer Res. 2007;27(4A):2047–2052. | ||
Xu JW, Li CG, Huang XE, Li Y, Huo JG. Ubenimex capsule improves general performance and chemotherapy related toxicity in advanced gastric cancer cases. Asian Pac J Cancer Prev. 2011;12(4):985–987. | ||
Riaz Ahmed KB, Kanduluru AK, Feng L, Fuchs PL, Huang P. Antitumor agent 25-epi Ritterostatin GN1N induces endoplasmic reticulum stress and autophagy mediated cell death in melanoma cells. Int J Oncol. 2017;50(5):1482–1490. | ||
Huang XL, Zhang H, Yang XY, et al. Activation of a c-Jun N-terminal kinase-mediated autophagy pathway attenuates the anticancer activity of gemcitabine in human bladder cancer cells. Anticancer Drugs. 2017;28(6):596–602. | ||
Wang G, Zhang T, Sun W, et al. Arsenic sulfide induces apoptosis and autophagy through the activation of ROS/JNK and suppression of Akt/mTOR signaling pathways in osteosarcoma. Free Radic Biol Med. 2017;106:24–37. | ||
Sun ZL, Dong JL, Wu J. Juglanin induces apoptosis and autophagy in human breast cancer progression via ROS/JNK promotion. Biomed Pharmacother. 2017;85:303–312. | ||
He JD, Wang Z, Li SP, et al. Vitexin suppresses autophagy to induce apoptosis in hepatocellular carcinoma via activation of the JNK signaling pathway. Oncotarget. 2016;7(51):84520–84532. | ||
Wang Y, Zhang G, Jin J, Degan S, Tameze Y, Zhang JY. MALT1 promotes melanoma progression through JNK/c-Jun signaling. Oncogenesis. 2017;6(7):e365. | ||
Wu J, Hu G, Dong Y, et al. Matrine induces Akt/mTOR signalling inhibition-mediated autophagy and apoptosis in acute myeloid leukaemia cells. J Cell Mol Med. 2017;21(6):1171–1181. | ||
Liu S, Wang X, Lu J, et al. Ubenimex enhances the radiosensitivity of renal cell carcinoma cells by inducing autophagic cell death. Oncol Lett. 2016;12(5):3403–3410. | ||
Wang X, Niu Z, Jia Y, et al. Ubenimex inhibits cell proliferation, migration and invasion by inhibiting the expression of APN and inducing autophagic cell death in prostate cancer cells. Oncol Rep. 2016;35(4):2121–2130. | ||
Han L, Zhao Q, Liang X, et al. Ubenimex enhances Brd4 inhibition by suppressing HEXIM1 autophagic degradation and suppressing the Akt pathway in glioma cells. Oncotarget. 2017;8(28):45643–45655. | ||
Ikeda N, Nakajima Y, Tokuhara T, et al. Clinical significance of aminopeptidase N/CD13 expression in human pancreatic carcinoma. Clin Cancer Res. 2003;9(4):1503–1508. | ||
Tokuhara T, Hattori N, Ishida H, et al. Clinical significance of aminopeptidase N in non-small cell lung cancer. Clin Cancer Res. 2006;12(13):3971–3978. | ||
Hashida H, Takabayashi A, Kanai M, et al. Aminopeptidase N is involved in cell motility and angiogenesis: Its clinical significance in human colon cancer. Gastroenterology. 2002;122(2):376–386. | ||
Aozuka Y, Koizumi K, Saitoh Y, Ueda Y, Sakurai H, Saiki I. Anti-tumor angiogenesis effect of aminopeptidase inhibitor bestatin against B16-BL6 melanoma cells orthotopically implanted into syngeneic mice. Cancer Lett. 2004;216(1):35–42. | ||
Xiang XY, Yang XC, Su J, et al. Inhibition of autophagic flux by ROS promotes apoptosis during DTT-induced ER/oxidative stress in HeLa cells. Oncol Rep. 2016;35(6):3471–3479. | ||
Kops GJ, Dansen TB, Polderman PE, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904);316–321. | ||
Deng R, Li W, Guan, Z et al. Acetylcholinesterase expression mediated by c-Jun-NH2-terminal kinase pathway during anticancer drug-induced apoptosis. Oncogene. 2006;25(53):7070–7077. | ||
Mizukami Y, Yoshioka K, Morimoto S, Yoshida Ki. A novel mechanism of JNK1 activation. Nuclear translocation and activation of JNK1 during ischemia and reperfusion. J Biol Chem. 1997;272(26):16657–16662. | ||
Zhang P, Miller BS, Rosenzweig SA, Bhat NR. Activation of C-jun N-terminal kinase/stress-activated protein kinase in primary glial cultures. J Neurosci Res. 1996;46(1):114–121. | ||
He JD, Wang Z, Li SP, et al. Vitexin suppresses autophagy to induce apoptosis in hepatocellular carcinoma via activation of the JNK signaling pathway. Oncotarget. 2016;7(51):84520–84532. | ||
De Luca A, Carpanese D, Rapanotti MC, et al. The nitrobenzoxadiazole derivative MC3181 blocks melanoma invasion and metastasis. Oncotarget. 2017;8(9):15520–15538. | ||
Go DH, Lee YG, Lee DH, et al. 3-Decylcatechol induces autophagy-mediated cell death through the IRE1α/JNK/p62 in hepatocellular carcinoma cells. Oncotarget. 2017;8(35):58790–58800. | ||
Krajarng A, Chulasiri M, Watanapokasin R. Etlingera elatior Extract promotes cell death in B16 melanoma cells via down-regulation of ERK and Akt signaling pathways. BMC Complement Altern Med. 2017;17(1):415. | ||
Díaz M, González R, Plano D, Palop JA, Sanmartín C, Encío I. A diphenyldiselenide derivative induces autophagy via JNK in HTB-54 lung cancer cells. J Cell Mol Med. 2018;22(1):289–301. | ||
Pappalardo F, Russo G, Candido S, et al. Computational modeling of PI3K/AKT and MAPK signaling pathways in melanoma cancer. PLoS One. 2016;11(3):e0152104. | ||
Tang R, Xu X, Yang W, et al. MED27 promotes melanoma growth by targeting AKT/MAPK and NF-κB/iNOS signaling pathways. Cancer Lett. 2016;373(1):77–87. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.