Back to Journals » OncoTargets and Therapy » Volume 15
Role of the JNK Pathway in Bladder Cancer
Authors Lee EH, Kim HT, Chun SY, Chung JW ![]() , Choi SH, Lee JN, Kim BS, Yoo ES, Kwon TG, Kim TH, Ha YS
, Choi SH, Lee JN, Kim BS, Yoo ES, Kwon TG, Kim TH, Ha YS ![]()
Received 16 May 2022
Accepted for publication 29 July 2022
Published 5 September 2022 Volume 2022:15 Pages 963—971
DOI https://doi.org/10.2147/OTT.S374908
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Eun Hye Lee,1,* Hyun Tae Kim,2,* So Young Chun,3 Jae-Wook Chung,2 Seock Hwan Choi,2 Jun Nyung Lee,2 Bum Soo Kim,2 Eun Sang Yoo,2 Tae Gyun Kwon,2 Tae-Hwan Kim,2 Yun-Sok Ha2
1Joint Institution of Regenerative Medicine, Kyungpook National University, Daegu, Korea; 2Department of Urology, School of Medicine, Kyungpook National University, Daegu, Korea; 3BioMedical Research Institute, Kyungpook National University Hospital, Daegu, Korea
*These authors contributed equally to this work
Correspondence: Tae-Hwan Kim; Yun-Sok Ha, Department of Urology, School of Medicine, Kyungpook National University, Daegu, Korea, Email [email protected]; [email protected]
Abstract: Bladder cancer, one of the most frequently diagnosed cancers worldwide, is associated with high morbidity and mortality and a poor prognosis. The bladder cancer types include 1) non-muscle invasive bladder cancer (NMIBC) and 2) muscle invasive bladder cancer (MIBC). Metastases and chemoresistance in MIBC patients are the leading causes of the high death rate. c-Jun N-terminal kinase (JNK) is an important factor for the undifferentiated state of cancer cells. JNK belongs to the mitogen-activated protein kinases (MAPKs) family; it is activated by various extracellular stimuli, such as stress, radiation, and growth factors and mediates diverse cellular functions, such as apoptosis, autophagy, proliferation, invasion, and migration by mediating AKT (Ak strain transforming), ATG (Autophagy related), mTOR (Mammalian target of rapamycin), and caspases 3, 8, and 9. This review describes the JNK-related functions, mechanisms, and signaling in bladder cancer.
Keywords: bladder cancer, JNK, proliferation, apoptosis, metastasis, chemoresistance, autophagy
Introduction
Bladder cancer (BC) is the second most common genitourinary malignancy worldwide.1,2 Over 430,000 men and women in the world and 74,000 in the USA are diagnosed with BC every year—and the incidence is four times higher in men than women. Urinary BC ranks fourth in the number of estimated new cases in the USA.3 Most BCs are urothelial carcinomas developing in the urinary bladder and derived from the stratified epithelium (also known as urothelium). About 20% of BC patients are younger than 75. The occurrence of new BC cases increases steadily with age. Reported risk factors for BC include cigarette smoking, older age, male gender, white race, occupational exposure to certain chemicals, radiation to the pelvis, use of medications (such as cyclophosphamide), chronic bladder infection, and family history.4
BC cases can be grouped by morphology and basement membrane invasion. There are three morphology types: (1) papillary, (2) sessile, and (3) mixed; most BCs are urothelial carcinomas with basement membrane invasion. Moreover, BC cases are classified into two groups: (1) non-muscle invasive bladder cancer (NMIBC, mostly low-grade BC) and (2) muscle invasive bladder cancer (MIBC, high-grade BC).5–7 Most NMIBC cases (50–70%) recur and 10–15% progress to invasion; the five-year-survival rate of NMIBC cases is above 90%. Meanwㄴㅇhile, MIBC has a poor prognosis with a low five-year-survival rate and tends to progress to metastasis.8 NMIBC is not life-threatening, while MIBC is clinically aggressive and produces lymph node, liver, lung, bone, and brain metastases. Invasion and metastases have increased the death rate of BC every year.9 Resistance to chemotherapy, cell adhesion molecule production, protease secretion, and angiogenesis at the tumor sites have been studied as main reasons of metastatic BCs. Thus, the molecular mechanism of metastasis is insufficiently studied.10–12
Besides metastasis occurrence, early diagnosis is critical for treating BC. Asymptomatic patients are frequently diagnosed from a routine checkup, and patients with urinary tract malignancies often experience hematuria and irritative voiding symptoms.13 Regarding standardized BC diagnosis, the gold standards of diagnostic procedures are cystoscopy and urine cytology. Cystoscopy is invasive and expensive and can cause urinary tract infections. However, cystoscopy method might miss a small flat tumor site and involves the risk of urethral injury, urinary tract infection due to instrumentation. Painless hematuria (blood or blood clot in urine) is the most common BC symptom. Less typical symptoms include frequent urination and painful urination. Meanwhile, advanced BC can cause pelvic pain, bone pain, and back pain.14–16
JNK (c-Jun N-terminal kinase, also called stress-activated protein kinase or stress-activated MAP kinase, SAPK) plays pivotal roles in BC. JNK belongs to the mitogen-activated protein kinase (MAPK) family and participates in various cellular processes, including cell proliferation, apoptosis, angiogenesis, differentiation, metastasis, invasion, and inflammation.17–19 Numerous cell stimuli activate JNK through autophosphorylation.20 Such stimuli include cytokines, such as tumor necrosis factor (TNF) and interleukin-1 (IL-1), and environmental stress factors, such as redox stress, radiation, and osmotic stress. JNK can suppress or promote tumor development. Continued abnormal functioning of JNK leads to various diseases such as cancers.21 JNK is a highly complex multi-functional protein associated with various signaling proteins, such as p38 and nuclear factor kappa B (NF-κB); crosstalks between these proteins are critical for cancer programming. JNK, p38, and NF-κB share upstream kinases and may act as synergistic regulators for cancer cell survival.22 Besides, autophagy activates JNK, counteracting apoptosis signaling and promoting cancer cell survival.23 Additionally, JNK-mediated prosurvival autophagy promotes chemotherapy resistance in cancer cells.24 Moreover, the cell proliferation mechanism based on the regulation of JNK mediates cancer cell survival.25
Key cancer recurrence factors include signaling pathway regulating proteins, such as p38/MAPK, transforming growth factor-beta (TGF-β), TWIST (Twist-related protein 1), NF-κB, Src (Proto-oncogene tyrosine-protein kinase Src), AKT, and JNK.1,26–30 This review provides a comprehensive summary of the specific JNK signaling pathway function in BC (Table 1 and Figure 1). It aims to improve the understanding of the JNK signaling pathway’s role in cancer growth mechanisms, such as tumorigenesis, apoptosis, autophagy, chemotherapy resistance, and metastasis (invasion and migration).
 |
Table 1 Function of JNK in Bladder Cancer |
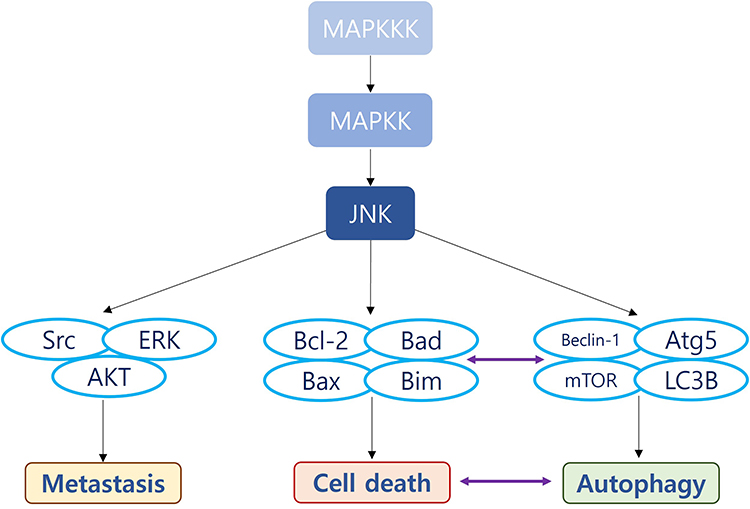 |
Figure 1 The functional categories of JNK in bladder cancer (BC) and the related biomolecules. JNK is downstream of Mitogen Activated Protein (MAP) kinase kinase kinase (MAPKKK) involved in tumorigenesis, cell proliferation, metastasis, chemoresistance, autophagy, metabolism, and immune escape. Abbreviations: MAPKKK, Mitogen-activated protein kinase kinase kinase; MAPKK, Mitogen-activated protein kinase kinase; AP-1, Activator protein 1; AKT, Ak strain transforming; JIP1, JNK-interacting protein 1; FAS, Fas Cell Surface Death Receptor; TNF, Tumor necrosis factor; JNK, c-Jun N-terminal kinases; p21WAF1, cyclin-dependent kinase inhibitor 1; TGF-β1, Transforming growth factor beta 1; SOD, Superoxide dismutase; Bcl-2, B-cell lymphoma 2; HMGB1, High mobility group box 1 protein; also known as high-mobility group protein; MEX3C, Mex-3 RNA Binding Family Member C; PD-L1, Programmed death-ligand 1. |
The Diverse Function of JNK Signaling Pathway in BC
Tumorigenesis
Tumorigenesis is the dynamic, complicated cancer formation process. It occurs in three stages: initiation, progression, and metastasis.31,32 Malignant cancer cells exhibit abnormal features such as uncontrolled cell proliferation, cell death signal resistance, metastasis formation, growth suppressor evasion, immune destruction evasion, genome instability, and angiogenesis (vascularization) stimulation.33 AKT-activated JNK participates in oncogenic transformation in urinary BC.34–36 In addition, JNK phosphorylates activator protein-1 (AP-1), which mediates in proliferation and apoptosis through cell cycle regulation. c-Jun and c-Fos constitute AP-1and JNK phosphorylates c-Jun to activate AP-1.37 Moreover, AKT binds to JNK interacting protein 1 (JIP1), preventing JNK activation and regulating apoptosis.38 In BPV (Bovine papillomavirus)-associated urothelial carcinomas, phosphorylated JNK (pJNK) and pJUN (a major JNK substrate) are overexpressed and PI3K (Phosphoinositide 3-kinases)-AKT pathway signaling is overactivated. Both pJUN and pJNK are associated with oncogenic transformation.35
Besides the activation of JNK by PI3K, the interaction of BPV-E5 (Bovine papillomavirus E5 protein) and pPDGF (platelet-derived growth factor)-βR plays a role in tumor formation by phosphorylating Jun and JNK.37 In 293T cells, JNK effectively triggers the ubiquitination and degradation of the tumor suppressor protein p53. A mass spectrometry analysis showed that p53 was activated by T81, the phosphorylation site of JNK.38 JNK2 down-regulation leads to p53 ubiquitination, resulting in p53 degradation and cell proliferation.39 Besides, in BC patients who underwent radical cystectomy, low JNK2 expression levels were strongly associated with poorer overall survival.40 These results indicate that JNK2 acts as a tumor suppressor in BC. Thus, JNK is a double-edged sword able to suppress or promote tumorigenesis through AP-1 and JIP1.
Cell Survival (Apoptosis and Proliferation)
Apoptosis, the programmed cell death, plays a pivotal role in living organisms. It is an organized process regulated by serial signaling transduction cascades and cellular proteins.41 The cell cycle consists of the G0, G1, S, and G2 phases. Diverse cyclins and cyclin-dependent kinases regulate mitosis. Normal tissue homeostasis disruption enables cancer cells to acquire invasion, metastasis, and angiogenesis capacities.42 Proliferation is essential for growth and proper tissue function and is maintained by cell cycle management in normal mammalian cells.43,44 Upon apoptosis, JNK, which can activate the pro-apoptotic factor caspase-3, moves to the membrane of mitochondria and releases cytochrome C. Apoptosis signal-regulating kinase 1 (ASK1) mediates TNF-α, and oxidative stress activates JNK/p38 and apoptosis. Besides, TNF-α regulates the apoptosis signaling pathway through the JNK/p38 pathway.44
Besides TNF-α, reactive oxygen species (ROS), such as H2O2, also activate JNK and p38.45 ROS can severely damage DNA, leading to apoptosis. In human BC cells, sanguinarine (a benzophenanthridine alkaloid isolated from plants belonging to the Papaveraceae family) activated JNK in a ROS-dependent manner. However, treating these cells with a JNK inhibitor did not reverse the sanguinarine-induced cell death. Similarly, ROS-induced JNK activation was reported in T24, EJ, and 5637 cells.46 In T24 cells, vitamin K2 induced mitochondria-related apoptosis via ROS-induced JNK/p38 MAPK signaling pathway activation. Vitamin K2 induced cytochrome C release from mitochondria to the cytosol and turned on the caspase cascade, resulting in mitochondria-related apoptosis.47 In addition, actein strongly activated p38 and JNK by phosphorylation and inactivated autophagy-related proteins, such as AKT/mTOR.2
Furthermore, JNK affects apoptosis by regulating TGF-β1 in urological, prostate cancer (PC3), and BC (T24) cells. TGF-β1 participates in the apoptotic process of various cell types. In PC3 and T24 cells, it activates JNK and other stress-activated kinases, and JNK acts as a direct tumor suppressor on invasive prostate cancer and BC cells by inducing apoptosis and inhibiting cell proliferation.48 Besides TGF-β1, AATBC (apoptosis-associated transcript in bladder cancer, also known as LOC284837) down-regulation modulated JNK activity and affected BC cell apoptosis. Additionally, the JNK inhibitor SP600125 attenuated the pro-apoptotic ability of AATBC down-regulation, clearly indicating that AATBC modulates BC cell death through JNK signaling.49 Cordycepin, an organic component found in garlic, activated JNK by phosphorylation, causing T24 cell apoptosis. It also inhibited cancer cell growth and p21WAF1-mediated G2/M phase cell cycle arrest.30 JNK inhibition suppressed cleaved caspase-3, poly-ADP ribose polymerase, caspase-8, and caspase-9 in actein treated BC cells. Actein triggered autophagy and apoptosis by cleaving caspase-3, PARP (Poly (ADP-ribose) polymerase), caspase-8, and caspase-9. N-acetyl cysteine (NAC) significantly reversed the expression of apoptosis-related proteins. Thus, JNK plays critical roles in the apoptosis process in cancer cells, acting both as an anti-and pro-apoptotic protein by mediating FAS, TNF, p21WAF1, and TGF-β1.
Metastasis (Migration and Invasion)
Metastases are responsible for a considerable part (nearly 90%) of the yearly worldwide cancer-associated deaths. Metastases come from cancer cells escaping the primary tumor mass and colonizing other body parts.50,51 Metastases occur when cancer cells acquire migratory and invasive capabilities52 because of basement membrane and extracellular matrix degradation. Extracellular matrix hydration through matrix metalloproteinase-2 (MMP-2) and MMP-9 secretion is critical in the metastasis process.53,54 Cell migration implies multiple processes: cell-cell adhesion loss, membrane protrusion in the direction of movement, de-adhesion from the extracellular matrix, forward re-adhesion, and actin cytoskeleton contraction to drive the cell body forward.
Less than 30% of all BC patients are diagnosed with MIBC, the most life-threatening BC type lacking effective treatment options. At the molecular level, MIBC is heterogenous, with genomic instability and a high genomic mutation rate.55 JNK participates in tumorigenesis, notably in the metastasis and invasion of multiple cancers.56–59 In gastric cancer cells, JNK regulates fascin1 expression through TGF- β1 and the ERK (extracellular-signal-regulated kinase) signaling pathway. Specific MAPK signaling pathway inhibitors PD98059 (ERK inhibitor) and SP600125 (JNK 1/2/3 inhibitor) suppressed fascin1 expression.56 In isoliquiritigenin-treated prostate cancer cells, JNK regulated cellular invasion and migration ability. SP600125 treatment suppressed uPA (Urokinase-Type Plasminogen Activator), VEGF (Vascular endothelial growth factor), MMP, and TIMP-1 (Tissue inhibitor matrix metalloproteinase-1) secretion and DNA binding activity.57 In colon cancer cells, JNK modulated cancer progression and metastasis via epithelial-mesenchymal transition. Chemokine ligand 7 (CCL7) interacts with CC chemokine receptor 3 (CCR3), promoting cellular proliferation, invasion, and migration via the ERK and JNK signaling pathways.58 In osteosarcoma cells, nobiletin exerted anti-migratory effects through the ERK and JNK signaling pathways. Cotreating U2OS and HOS cells with nobiletin and ERK and JNK inhibitors suppressed DNA the binding activity of NF-κB, cAMP response element-binding protein, and specificity protein 1 (SP-1). In addition, the co-treatment reduced the migration and invasion ability of U2OS cells.59
JNK also acts as a key migration and invasion regulator in BC. Metastatic cells express higher JNK levels than non-metastatic cells. Superoxide dismutase 2 (SOD2) activates JNK, and SOD2 inhibition promotes migration and invasion in T24 cells. SOD2 is a critical factor in BC cell migration regulation. It reduces CDC2 and Rac1 protein expression and decreases migration by inhibiting JNK activation in T24 cells.51 Programmed cell death 4 (PDCD4) and SRY (sex determining region Y)-box 2 (SOX2) also modulate JNK activation.60,61 PDCD 4 is a tumor suppressor localized in the nucleus and involved in tumor progression and prognosis in various human cancers. PDCD 4 inhibited human BC cells invasion and reversed the epithelial-mesenchymal transition by activating the JNK/c-Jun signaling pathway.60 Cheliensisin A, a styryl-lactone isolated from Goniothalamus cheliensis Hu, inhibited human BC cells by activating the MAPK/JNK/c-Jun cascade and increased microRNA 200c (miR-200c) levels via SOX2 protein translation inhibition.61 These results indicate that JNK promotes invasion and migration—and, therefore, metastasis—via miR-200c and SOD2.
Autophagy
Autophagy (meaning “self-eating” or “self-digestion”) is a process discovered more than 40 years ago. It consists in capturing and degrading intracellular components to maintain cellular homeostasis; autophagy defects can lead to various diseases, such as cancer, aging, or lysosomal disorders. Autophagy protects cells from endoplasmic reticulum stress, ontological development, microbial infection, and diseases involving protein aggregation. It starts with the formation of a double-membrane vesicle (known as an autophagosome) near the cargo macromolecules (organelles or regions of the cytosol) under cellular stress conditions, such as nutrient deprivation (caloric restriction), oxidative stress, hypoxia, protein aggregation, and molecular toxicity.62 First, stress inactivates mTOR, initiating ATG (autophagy-related gene) 1 (an Ulk1 and Ulk2 homolog) kinase activation. Next, Atg1/Ulk1–2 activation induces Atg13 and FIP200 phosphorylation and Ulk (Unc-51 like autophagy activating kinase) proteins autophosphorylation. The activated and phosphorylated Atg13-Ulk-FIP200 (focal adhesion kinase family-interacting protein of 200 kDa) complex then recruits other Atg proteins, forming an autophagosome. Autophagosomes then fuse with lysosomes to form autophagolysosomes, and proteases degrade their components. Autophagosomes can be named according to the targeted organelle. For instance, a phagosome targeting mitochondria is a mitophagosome, and one targeting the peroxisome is a pexophagosome. For lysosome degradation, selective autophagy (or chaperone-mediated autophagy) occurs. In this case, the lysosome fuses with targeted proteins within the cytoplasm.
The role of autophagy in cancer remains unclear and somewhat controversial.63,64 mTORC2 indirectly suppresses autophagy by activating mTORC1. Previous studies reported that mTORC1 inhibition increases the autophagic reaction.65 LC3B (1A/1B-light chain 3) and p62 are widely used autophagy markers. During autophagy, LC3B-I (the cytosolic form) is converted to LC3B-II (the lapidated form) and becomes an integral part of the autophagosome membrane.66 JNK is one of the regulators of autophagy inducers in diverse cancers.67,68
In ovarian cancer, neferine induced autophagy by forming autophagosomes and converting LC3B-I to LC3B-II. Moreover, neferine activated the p38 MAPK and JNK signaling pathways. In addition, JNK and p38 MAPK specific inhibitors attenuated neferine induced LC3B-II accumulation in A2780 and HO8910 cells.69 In BC cells, epirubicin, an anthracycline drug, induced JNK phosphorylation, leading to Bcl-2/Beclin-1 complex breakdown and autophagy initiation. Tea polyphenols suppressed epirubicin-induced JNK-mediated autophagy through the JNK/Bcl2 (B-cell lymphoma 2)/Beclin-1 signaling pathway.70 Furthermore, blocking JNK suppressed the autophagic reaction and increased apoptosis. In T24 and BIU-87 cells (human BC cell lines), HMGB1 (High mobility group box 1 protein) knockdown attenuated gemcitabine-induced JNK and ERK activation. Gemcitabine affects BC cell apoptosis and induces a cytoprotective autophagic reaction that involves HMGB1-mediated JNK and ERK signaling pathway activation.1 Besides, activated JNK inhibits gemcitabine-induced apoptosis via an autophagy suppression mechanism. A JNK inhibitor effectively blocked gemcitabine-induced autophagy by suppressing LC3B-II.24 Thus, JNK regulates autophagy through Bcl, Beclin-1, and HMGB1.
Regulation of Tumor Cell Metabolism and Immune Escape
Genome instability and mutations in oncogenes and tumor suppressor genes are enabling characteristics of cancer.71 Mex-3 proteins are associated with numerous cancers. For instance, MEX3C (Mex-3 RNA Binding Family Member C) is related to fatty acid metabolism in BC through the MAPK/JNK signaling pathway. MEX3C overexpression increases the accumulation of lipids and triglycerides in BC cells.72 As cancers grow in the host, immune escape occurs as the final phase of cancer immunoediting, facilitating death signal modulation and escape for cancer cells.73 JNK plays a crucial part in the immune escape in BC by modulating PD-L1 expression. Inhibiting JNK suppressed PD-L1 and enhanced the immune response of BC.74
Chemoresistance
Chemotherapy is the main treatment for most cancer patients, and BC is a chemosensitive malignant tumor. Several patients show positive responses at the beginning of treatment; however, repeating treatments may progressively become ineffective. This drug resistance is a significant obstacle to the successful cure of BC patients. Thus, identifying the chemoresistance mechanism is paramount to predict treatment outcome, develop effective chemotherapeutic agents, and cure BC patients. The poor prognosis and mortality of BC are mostly due to chemoresistance-related recurrence.61 Many cisplatin-resistant cancers seem unaffected by other chemotherapies. Therefore, chemotherapy resistance is a critical obstacle to BC treatment. Unfortunately, there is no consensus on how to manage cisplatin-resistant BC.75 Inhibiting JNK can increase the sensitivity of hepatocellular carcinoma cells to cisplatin. Cisplatin-resistant hepatocellular carcinoma cells overexpress the MDR1 (Multidrug resistance) gene, encoding for the drug efflux protein Pgp (P-glycoprotein 1). In addition, this acquired resistance involved JNK over-activation. These results indicate that the MDR1 gene is closely linked to cisplatin resistance via the JNK signaling pathway.76
In T24 cells, JNK down-regulation promoted mitomycin C chemoresistance by decreasing p53 stability. JNK2 protects p53 from MDM2 (Murine double minute 2 homolog)-mediated degradation by phosphorylating p53 on Thr81. Moreover, JNK2 down-regulation attenuated mitomycin C-induced BC cell death.39 Thus, the activity of JNK critically affects chemotherapy efficacy by regulating p53. However, the role of JNK in cancer chemoresistance remains controversial.
Conclusions
JNK signaling pathway research has made major advances in recent years. The current paper provides a comprehensive overview of the relationship between JNK and BC. BC is a global burden, and MIBC treatment remains a major challenge because of the migratory and invasive characteristics, deregulated cell proliferation, chemoresistance, and cell death evasion of cancer cells. This study reports the involvement of the JNK signaling pathway in BC. The JNK signaling pathway is deeply associated with many cancer growth factors. For instance, AKT-mediated JNK activation promotes tumorigenesis; however, JNK, as a double-edged sword, can also suppress tumorigenesis. As a tumor suppressor, activated JNK initiates apoptotic signaling, killing cancer cells.
In metastasis, JNK modulates diverse factors like PDCD4, micro RNAs, and SOX2. JNK also regulates the chemosensitivity to multiple anticancer drugs. Additionally, the JNK signaling pathway manages autophagy-related genes maintaining homeostasis. The mechanisms involving JNK in autophagy and cancer need deeper research. Furthermore, inhibiting JNK significantly increases cisplatin sensitivity. Based on these reported papers, JNK is one of important molecules in BC, since JNK is involved in many categories (tumorigenesis, cell proliferation, metastasis, autophagy, chemoresistance, metabolism, and immunity of cancer). The current body of knowledge shows that targeting the JNK pathway could lead to the discovery of new therapies and biomarkers; however, the present study highlights the necessity of further investigating the mechanisms linking the JNK signaling pathway and tumorigenesis, cell proliferation, metastasis, chemoresistance, and autophagy in BC.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2019R1A2C1004046) (2019R1H1A1079839) (2020R1A2B5B03002344) (2020R1I1A3071568) (2021R1G1A1092985) (2022R1I1A3069482), and Business for Cooperative R&D between Industry, Academy, and Research Institute funded Korea Small and Medium Business Administration (S3106172).
Disclosure
The authors declare no conflict of interest.
References
1. Babjuk M, Burger M, Comperat EM, et al. European association of urology guidelines on non-muscle-invasive bladder cancer (TaT1 and carcinoma in situ) - 2019 update. Eur Urol. 2019;76(5):639–657. doi:10.1016/j.eururo.2019.08.016
2. Powles T, Bellmunt J, Comperat E, et al. Bladder cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33(3):244–258. doi:10.1016/j.annonc.2021.11.012
3. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi:10.3322/caac.21654
4. Kirkali Z, Chan T, Manoharan M, et al. Bladder cancer: epidemiology, staging and grading, and diagnosis. Urology. 2005;66(6 Suppl 1):4–34. doi:10.1016/j.urology.2005.07.062
5. Griffiths TR. Current perspectives in bladder cancer management. Int J Clin Pract. 2013;67(5):435–448. doi:10.1111/ijcp.12075
6. Patel VG, Oh WK, Galsky MD. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J Clin. 2020;70(5):404–423. doi:10.3322/caac.21631
7. Ha Y-S. Developing biomarkers and new therapeutic targets in muscle invasive bladder cancer. Korean J Urol Oncol. 2020;18(1):1–10. doi:10.22465/kjuo.2020.18.1.1
8. Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15(1):25–41. doi:10.1038/nrc3817
9. He W, Zhong G, Jiang N, et al. Long noncoding RNA BLACAT2 promotes bladder cancer-associated lymphangiogenesis and lymphatic metastasis. J Clin Invest. 2018;128(2):861–875. doi:10.1172/JCI96218
10. Kim EY, Seo JM, Kim C, Lee JE, Lee KM, Kim JH. BLT2 promotes the invasion and metastasis of aggressive bladder cancer cells through a reactive oxygen species-linked pathway. Free Radic Biol Med. 2010;49(6):1072–1081. doi:10.1016/j.freeradbiomed.2010.06.023
11. Youssef RF, Mitra AP, Bartsch G, Jones PA, Skinner DG, Cote RJ. Molecular targets and targeted therapies in bladder cancer management. World J Urol. 2009;27(1):9–20. doi:10.1007/s00345-008-0357-x
12. Yun-Sok H, Kim T-H. Chemotherapy in advanced urothelial carcinoma. Korean J Urol Oncol. 2016;14(2):47–53. doi:10.22465/kjuo.2016.14.2.47
13. Grossman HB, Messing E, Soloway M, et al. Detection of bladder cancer using a point-of-care proteomic assay. JAMA. 2005;293(7):810–816. doi:10.1001/jama.293.7.810
14. Tran L, Xiao JF, Agarwal N, Duex JE, Theodorescu D. Advances in bladder cancer biology and therapy. Nat Rev Cancer. 2021;21(2):104–121. doi:10.1038/s41568-020-00313-1
15. Droller MJ; Urologic Oncology: Seminars and Original Investigations. A twenty-fifth anniversary history. Urol Oncol. 2021;39(9):506–513. doi:10.1016/j.urolonc.2021.01.024
16. Pashos CL, Botteman MF, Laskin BL, et al. Bladder cancer: epidemiology, diagnosis, and management. Cancer Pract. 2002;10(6):311–322. doi:10.1046/j.1523-5394.2002.106011.x
17. Gkouveris I, Nikitakis NG. Role of JNK signaling in oral cancer: a mini review. Tumour Biol. 2017;39(6):1010428317711659. doi:10.1177/1010428317711659
18. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi:10.1038/sj.onc.1210421
19. Bubici C, Papa S. JNK signalling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol. 2014;171(1):24–37. doi:10.1111/bph.12432
20. Zhou YY, Li Y, Jiang WQ, Zhou LF. MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep. 2015;35(3). doi:10.1042/BSR20140141
21. Suzuki S, Okada M, Shibuya K, et al. JNK suppression of chemotherapeutic agents-induced ROS confers chemoresistance on pancreatic cancer stem cells. Oncotarget. 2015;6(1):458–470. doi:10.18632/oncotarget.2693
22. Ruan J, Qi Z, Shen L, et al. Crosstalk between JNK and NF-kappaB signaling pathways via HSP27 phosphorylation in HepG2 cells. Biochem Biophys Res Commun. 2015;456(1):122–128. doi:10.1016/j.bbrc.2014.11.045
23. Li J, Zhao L, Zhao X, Wang P, Liu Y, Ruan J. Foxo1 attenuates NaF-induced apoptosis of LS8 cells through the JNK and mitochondrial pathways. Biol Trace Elem Res. 2018;181(1):104–111. doi:10.1007/s12011-017-1015-1
24. Huang XL, Zhang H, Yang XY, et al. Activation of a c-Jun N-terminal kinase-mediated autophagy pathway attenuates the anti-cancer activity of gemcitabine in human bladder cancer cells. Anticancer Drugs. 2017;28(6):596–602. doi:10.1097/CAD.0000000000000499
25. Vucur M, Reisinger F, Gautheron J, et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep. 2013;4(4):776–790. doi:10.1016/j.celrep.2013.07.035
26. Kumar B, Koul S, Petersen J, et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010;70(2):832–841. doi:10.1158/0008-5472.CAN-09-2918
27. Fan Y, Shen B, Tan M, et al. TGF-beta-induced up-regulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin Cancer Res. 2014;20(6):1531–1541. doi:10.1158/1078-0432.CCR-13-1455
28. Wu D, Zhang J, Qian T, et al. IFN-gamma regulates the expression of MICA in human corneal epithelium through miRNA4448 and NFkappaB. Front Immunol. 2018;9:1530. doi:10.3389/fimmu.2018.01530
29. Chung JW, Kim SW, Kang HW, et al. Efficacy of modified radical prostatectomy technique for recovery of urinary incontinence in high-grade prostate cancer. Minerva Urol Nefrol. 2020;72(5):605–614. doi:10.23736/S0393-2249.20.03633-4
30. Lee SJ, Kim SK, Choi WS, Kim WJ, Moon SK. Cordycepin causes p21WAF1-mediated G2/M cell-cycle arrest by regulating c-Jun N-terminal kinase activation in human bladder cancer cells. Arch Biochem Biophys. 2009;490(2):103–109. doi:10.1016/j.abb.2009.09.001
31. Wang M, Zhao J, Zhang L, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8(5):761–773. doi:10.7150/jca.17648
32. Kalea AZ, See F, Harja E, Arriero M, Schmidt AM, Hudson BI. Alternatively spliced RAGEv1 inhibits tumorigenesis through suppression of JNK signaling. Cancer Res. 2010;70(13):5628–5638. doi:10.1158/0008-5472.CAN-10-0595
33. Balani S, Nguyen LV, Eaves CJ. Modeling the process of human tumorigenesis. Nat Commun. 2017;8:15422. doi:10.1038/ncomms15422
34. Contreras-Paredes A, De la Cruz-Hernandez E, Martinez-Ramirez I, Duen-as-Gonzalez A, Lizano M. E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (akt/PI3K) signaling pathway. Virology. 2009;383(1):78–85. doi:10.1016/j.virol.2008.09.040
35. Corteggio A, Urraro C, Roperto S, Roperto F, Borzacchiello G. Phosphatidyl-inositol-3-kinase-AKT pathway, phospho-JUN and phospho-JNK expression in spontaneously arising bovine urinary bladder tumours. J Comp Pathol. 2010;143(2–3):173–178. doi:10.1016/j.jcpa.2010.03.001
36. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi:10.1016/S0092-8674(00)00116-1
37. Papachristou DJ, Batistatou A, Sykiotis GP, Varakis I, Papavassiliou AG. Activation of the JNK-AP-1 signal transduction pathway is associated with pathogenesis and progression of human osteosarcomas. Bone. 2003;32(4):364–371. doi:10.1016/S8756-3282(03)00026-7
38. Kim AH, Yano H, Cho H, et al. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. 2002;35(4):679–709.
39. Petti LM, Ricciardi EC, Page HJ, Porter KA. Transforming signals resulting from sustained activation of the PDGFbeta receptor in mortal human fibroblasts. J Cell Sci. 2008;121(Pt 8):1172–1182. doi:10.1242/jcs.018713
40. Pan, CW, Liu H, Zhao Y, et al. JNK2 downregulation promotes tumorigenesis and chemoresistance by decreasing p53 stability in bladder cancer. Oncotarget. 2016;7(23):35119–35131. doi:10.18632/oncotarget.9046
41. Buschmann T, Potapova O, Bar-Shira A, et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol. 2001;21(8):2743–2754. doi:10.1128/MCB.21.8.2743-2754.2001
42. Abotaleb M, Samuel SM, Varghese E, et al. Flavonoids in cancer and apoptosis. Cancers. 2018;11(1):28. doi:10.3390/cancers11010028
43. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337
44. Fiandalo MV, Kyprianou N. Caspase control: protagonists of cancer cell apoptosis. Exp Oncol. 2012;34(3):165–175.
45. Ouyang L, Shi Z, Zhao S, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45(6):487–498. doi:10.1111/j.1365-2184.2012.00845.x
46. Yang SH, Chien CM, Lu CM, Chen YL, Chang LS, Lin SR. Involvement of c-Jun N-terminal kinase in G2/M arrest and FasL-mediated apoptosis induced by a novel indoloquinoline derivative, IQDMA, in K562 cells. Leuk Res. 2007;31(10):1413–1420. doi:10.1016/j.leukres.2007.02.014
47. Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2(3):222–228. doi:10.1093/embo-reports/kve046
48. Han MH, Park C, Jin CY, et al. Apoptosis induction of human bladder cancer cells by sanguinarine through reactive oxygen species-mediated up-regulation of early growth response gene-1. PLoS One. 2013;8(5):e63425. doi:10.1371/journal.pone.0063425
49. Duan F, Yu Y, Guan R, Xu Z, Liang H, Hong L. Vitamin K2 Induces mitochondria related apoptosis in human bladder cancer cells via ROS and JNK/p38 MAPK signal pathways. PLoS One. 2016;11(8):e0161886. doi:10.1371/journal.pone.0161886
50. Al-Azayzih A, Gao F, Goc A, Somanath PR. TGFbeta1 induces apoptosis in invasive prostate cancer and bladder cancer cells via Akt-independent, p38 MAPK and JNK/SAPK-mediated activation of caspases. Biochem Biophys Res Commun. 2012;427(1):165–170. doi:10.1016/j.bbrc.2012.09.035
51. Zhao F, Lin T, He W, et al. Knockdown of a novel lincRNA AATBC suppresses proliferation and induces apoptosis in bladder cancer. Oncotarget. 2015;6(2):1064–1078. doi:10.18632/oncotarget.2833
52. Wittekind C, Neid M. Cancer invasion and metastasis. Oncology. 2005;69(Suppl 1):14–16. doi:10.1159/000086626
53. Jin H, Yu Y, Hu Y, et al. Divergent behaviors and underlying mechanisms of cell migration and invasion in non-metastatic T24 and its metastatic derivative T24T bladder cancer cell lines. Oncotarget. 2015;6(1):522–536. doi:10.18632/oncotarget.2680
54. Said AH, Raufman JP, Xie G. The role of matrix metalloproteinases in colorectal cancer. Cancers. 2014;6(1):366–375. doi:10.3390/cancers6010366
55. Kenny HA, Lengyel E. MMP-2 functions as an early response protein in ovarian cancer metastasis. Cell Cycle. 2009;8(5):683–688. doi:10.4161/cc.8.5.7703
56. Li H, Qiu Z, Li F, Wang C. The relationship between MMP-2 and MMP-9 expression levels with breast cancer incidence and prognosis. Oncol Lett. 2017;14(5):5865–5870. doi:10.3892/ol.2017.6924
57. Bai Y, Yang H, Zhang G, et al. Inhibitory effects of resveratrol on the adhesion, migration and invasion of human bladder cancer cells. Mol Med Rep. 2017;15(2):885–889. doi:10.3892/mmr.2016.6051
58. Fu H, Hu Z, Wen J, Wang K, Liu Y. TGF-beta promotes invasion and metastasis of gastric cancer cells by increasing fascin1 expression via ERK and JNK signal pathways. Acta Biochim Biophys Sin. 2009;41(8):648–656. doi:10.1093/abbs/gmp053
59. Kwon GT, Cho HJ, Chung WY, Park KK, Moon A, Park JH. Isoliquiritigenin inhibits migration and invasion of prostate cancer cells: possible mediation by decreased JNK/AP-1 signaling. J Nutr Biochem. 2009;20(9):663–676. doi:10.1016/j.jnutbio.2008.06.005
60. Lee YS, Kim SY, Song SJ, et al. Crosstalk between CCL7 and CCR3 promotes metastasis of colon cancer cells via ERK-JNK signaling pathways. Oncotarget. 2016;7(24):36842–36853. doi:10.18632/oncotarget.9209
61. White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. doi:10.1172/JCI73941
62. Massari F, Santoni M, Ciccarese C, et al. Emerging concepts on drug resistance in bladder cancer: implications for future strategies. Crit Rev Oncol Hematol. 2015;96(1):81–90. doi:10.1016/j.critrevonc.2015.05.005
63. Liu XY, Liu SP, Jiang J, Zhang X, Zhang T. Inhibition of the JNK signaling pathway increases sensitivity of hepatocellular carcinoma cells to cisplatin by down-regulating expression of P-glycoprotein. Eur Rev Med Pharmacol Sci. 2016;20(6):1098–1108.
64. Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25(19):1999–2010. doi:10.1101/gad.17558811
65. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–542. doi:10.1038/nrc.2017.53
66. Follo C, Vidoni C, Morani F, Ferraresi A, Seca C, Isidoro C. Amino acid response by halofuginone in cancer cells triggers autophagy through proteasome degradation of mTOR. Cell Commun Signal. 2019;17(1):39. doi:10.1186/s12964-019-0354-2
67. Wang K, Du B, Xu B, Lv X. JMJD6-STAT3(Y705ph) axis promotes autophagy in osteosarcoma cancer cells by regulating ATG. Biofactors. 2020;46(5):839–848. doi:10.1002/biof.1614
68. Xu L, Zhang X, Li Y, et al. Neferine induces autophagy of human ovarian cancer cells via p38 MAPK/ JNK activation. Tumour Biol. 2016;37(7):8721–8729. doi:10.1007/s13277-015-4737-8
69. Gu W, Lin Y, Gou X, He W. Tea Polyphenol inhibits autophagy to sensitize Epirubicin-induced apoptosis in human bladder cancer cells. Neoplasma. 2017;64(5):674–680. doi:10.4149/neo_2017_504
70. Romero-Garcia, S, Lopez-Gonzalez JS, Báez-Viveros JL, Aguilar-Cazares D, Prado-Garcia H. Tumor cell metabolism: an integral view. Cancer Biol. 2011;12(11):939–948. doi:10.4161/cbt.12.11.18140
71. Chao H, Deng, L;, Xu F, et al. MEX3C regulates lipid metabolism to promote bladder tumorigenesis through JNK pathway. Onco Targets Ther. 2019;1(12):3285–3294.
72. Bhatia, A, Kumar Y. Cellular and molecular mechanisms in cancer immune escape: a comprehensive review. Expert Rev Clin Immnunol. 2014;10(1):41–62. doi:10.1586/1744666X.2014.865519
73. Ni, Z, Sun P, Zheng J, et al. JNK signaling promotes bladder cancer immune escape by regulating METTL3-mediated m6A modification of PD-L1 mRNA. Cancer Res. 2022;82(9):1789–1802. doi:10.1158/0008-5472.CAN-21-1323
74. Cheng HL, Hsieh MJ, Yang JS, et al. Nobiletin inhibits human osteosarcoma cells metastasis by blocking ERK and JNK-mediated MMPs expression. Oncotarget. 2016;7(23):35208–35223. doi:10.18632/oncotarget.9106
75. Amling CL. Diagnosis and management of superficial bladder cancer. Curr Probl Cancer. 2001;25(4):219–278. doi:10.1067/mcn.2001.117539
76. Fleshner N, Garland J, Moadel A, et al. Influence of smoking status on the disease-related outcomes of patients with tobacco-associated superficial transitional cell carcinoma of the bladder. Cancer. 1999;86(11):2337–2345. doi:10.1002/(SICI)1097-0142(19991201)86:11<2337::AID-CNCR23>3.0.CO;2-6
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Metformin Inhibits HaCaT Cell Proliferation Under Hyperlipidemia Through Reducing Reactive Oxygen Species via FOXO3 Activation
Zhang L, Liu X, Huang M, Wang R, Zhu W, Li Y, Shen L, Li C
Clinical, Cosmetic and Investigational Dermatology 2022, 15:1403-1413
Published Date: 22 July 2022
A 5`-tRNA Derived Fragment NamedtiRNA-Val-CAC-001 Works as a Suppressor in Gastric Cancer
Zheng J, Li C, Zhu Z, Yang F, Wang X, Jiang P, Yan F
Cancer Management and Research 2022, 14:2323-2337
Published Date: 4 August 2022
Eriochloa villosa Alleviates Progression of Benign Prostatic Hyperplasia in vitro and in vivo
Baek EB, Hwang YH, Park S, Hong EJ, Won YS, Kwun HJ
Research and Reports in Urology 2022, 14:313-326
Published Date: 24 September 2022
Antitumor Activity of Berberine by Activating Autophagy and Apoptosis in CAL-62 and BHT-101 Anaplastic Thyroid Carcinoma Cell Lines
Shi XZ, Zhao S, Wang Y, Wang MY, Su SW, Wu YZ, Xiong C
Drug Design, Development and Therapy 2023, 17:1889-1906
Published Date: 26 June 2023
The Progress of Platelets in Breast Cancer
Wang L, Zhang K, Feng J, Wang D, Liu J
Cancer Management and Research 2023, 15:811-821
Published Date: 11 August 2023