")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Role of SIRT1-mediated mitochondrial and Akt pathways in glioblastoma cell death induced by Cotinus coggygria flavonoid nanoliposomes
Authors Wang G , Wang J, To T, Zhao HF, Wang J, Hong-Ming T
Received 5 February 2015
Accepted for publication 30 April 2015
Published 4 August 2015 Volume 2015:10(1) Pages 5005—5023
DOI https://doi.org/10.2147/IJN.S82282
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Gang Wang,1,2,* Jun Jie Wang,1,2,* Tony SS To,3 Hua Fu Zhao,3 Jing Wang3
1Department of Pharmaceutics, Shanghai Eighth People’s Hospital, Shanghai, People’s Republic of China; 2College of Pharmacy, Hubei University of Medicine, Shiyan, Hubei Province, People’s Republic of China; 3Department of Health Technology and Informatics, The Hong Kong Polytechnic University, Hong Kong SAR, People’s Republic of China
*These authors contributed equally to this work
Abstract: Flavonoids, the major polyphenol components in Cotinus coggygria (CC), have been found to show an anticancer effect in our previous study; however, the exact mechanisms of inducing human glioblastoma (GBM) cell death remain to be resolved. In this study, a novel polyvinylpyrrolidone K-30/sodium dodecyl sulfate and polyethyleneglycol-coated liposome loaded with CC flavonoids (CCFs) was developed to enhance solubility and the antibrain tumor effect, and the molecular mechanism regarding how CCF nanoliposomes (CCF-NLs) induce apoptotic cell death in vitro was investigated. DBTRG-05MG GBM cell lines treated with CCF-NLs showed potential antiproliferative effects. Regarding the underlying mechanisms of inducing apoptosis in DBTRG-05MG GBM cells, CCF-NLs were shown to downregulate the expression of antiapoptotic B-cell lymphoma/leukemia 2 (Bcl-2), an apoptosis-related protein family member, but the expression of proapoptotic Bcl-2-associated X protein was enhanced compared with that in controls. CCF-NLs also inhibited the activity of caspase-3 and -9, which is the initiator caspase of the extrinsic and intrinsic apoptotic pathways. Blockade of caspase activation consistently induced apoptosis and inhibited growth in CCF-NL-treated DBTRG-05MG cells. This study further investigated the role of the Akt pathway in the apoptotic cell death by CCF-NLs, showing that CCF-NLs deactivated Akt. Specifically, CCF-NLs downregulated the expression of p-Akt and SIRT1 as well as the level of phosphorylated p53. Together, these results indicated SIRT1/p53-mediated cell death was induced by CCF-NLs, but not by extracellular signal-regulated kinase, in DBTRG-05MG cells. Overall, this study suggested caspase-dependent activation of both the intrinsic and extrinsic signaling pathways, probably through blockade of the SIRT1/p53-mediated mitochondrial and Akt pathways to exert the proapoptotic effect of CCF-NLs in DBTRG-05MG GBM cells.
Keywords: Cotinus coggygria flavonoid nanoliposomes, cell death, SIRT1, mitochondrial, PI3K/Akt pathway
Introduction
Glioblastoma (GBM) is the most frequently occurring primary tumor of the central nervous system, and patients with malignant GBM have a very poor prognosis. Unfortunately, there is no balance between metabolism and cell growth in GBM cells. The recent discovery of sirtuins (SIRTs), which are capable of simultaneously regulating multiple downstream pathways, may play a key role in explaining the complex mechanisms leading to changes in metabolic and enzymatic pathways and/or modulating cell growth that may result in GBM. SIRTs, a family of nicotinamide adenine dinucleotide-dependent deacetylases, are implicated in energy metabolism and life span. Among SIRTs family members, SIRT1 is a conserved nicotinamide adenine dinucleotide-dependent protein deacetylase that regulates life span.1 It has been proven that mammalian SIRT1 is a key regulator of cell survival when exposed to cellular stresses that otherwise trigger apoptotic pathways via the activation of p53 and/or FoxO transcription factors.2,3 Importantly, SIRT1 is also involved in cancer cell growth, apoptosis, and tumorigenesis.4,5 It has been proposed that SIRT1 inactivates the Akt pathway in a SIRT1 deacetylase-dependent manner; thus, SIRT1 acts as a deacetylase for the tumor suppressor PTEN,6 a known negative regulator for the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, which is a key oncogenic pathway that promotes cell growth and survival. Overall, SIRT1 might be a promoting factor for the tumorigenesis of glioma through the PI3K/Akt signaling pathway.7
SIRTs appear to be the molecular/metabolic transducer for the adaptation to several external stimuli, representing a natural and quite obvious strategy for new therapeutic interventions. Thus, the modulation of their activity may be beneficial for the prevention, retardation and cure of tumors, metabolic diseases, and aging. In addition, SIRTs seem to play an important role in cancers that are associated with the mitochondrial and metabolic pathways, in addition to the implications of the SIRT function in glioma; the search for SIRT modulators is a hot topic because of their possible therapeutic uses in age-related diseases, metabolic diseases, and cancer. SIRT inhibitors, such as Sirtinol, have been shown to induce apoptosis and autophagic cell death in MCF-7 human breast cancer cells.8 Additionally, Sirtinol induced senescence-like growth arrest in human breast cancer MCF-7 and lung cancer H1299 cells9 and enhanced the chemosensitivity to camptothecin and cisplatin in PC3, DU145, and HeLa cells, resulting in a significant reduction of viable cells due to enhanced apoptotic cell death.10–12
Plant-derived natural flavonoids represent a wide variety of compounds that are enriched in fruits, vegetables, wine, tea, and other plant products. Accumulating evidence has suggested that there are beneficial components in Cotinus Coggygria (CC), such as flavonoids, when used both in herbal medicine and as a spice.13 It has been reported that CC is used as a natural drug in the treatment of acute icteric infectious hepatitis;14 moreover, pharmacological research has shown that CC reduces jaundice and enzyme and gallbladder cholagogic activities and strengthens immune function. CC has also been reported to play a role in the prevention and treatment of coronary heart disease, angina pectoris, or myocardial infarction, improving hypoxia in animals or dissolving thrombi.15 In human studies, CC has been shown to reduce blood pressure levels in hypertensive patients, improve antioxidant status, and decrease risk factors associated with cardiovascular diseases.16 However, current research on CC is lacking, and the pharmacological effects of CC flavonoids (CCFs) are mostly limited to anticoagulant, hemolytic, and antiliver chemical injuries, with only a few reports regarding anticancer effects.
In our previous study, anticancer effects were found in GBM cells by CCFs; however, the exact underlying mechanisms of induction of human GBM cell death remain largely unresolved. Conventional oral administration of flavonoids appears to be inefficient, based on various findings, such as low solubility and permeability, instability and extensive first pass metabolism, before reaching systemic circulation.17 By contrast, the development of novel drug delivery systems (DDSs), such as nanovehicles, has recently markedly improved the efficacy and safety of drugs with unfavorable physicochemical or pharmacokinetic parameters, including flavonoids, and has enhanced patient compliance. Thus, the present study was aimed to evaluate the design and development of particulate carrier systems, initially in general terms and subsequently in relation to the delivery of CCFs.
There are numerous nanobased DDSs, including dendrimers,18 micelles,19 nanoemulsion,20 self-emulsifying systems,21 cubic phase gels,22 inorganic nanoparticles,23 nanocrystals,24 nanotubes,25 and quantum dots.26 Liposomes are currently the nanocarrier system that is most widely studied for use as a vehicle for flavonoids. Multilamellar phosphatidylcholine liposomes, with the addition of surfactant agents, have been characterized for topical and intratumor administration of catechin, epicatechin (EC), and epigallocatechin-3-gallate (EGCG) in nude mice,27 and phosphatidylcholine liposomes have been used to deliver flavonoids or polyphenols to human cancer cells in vitro.28 Moreover, polyphenolic fractions in sodium dodecyl sulfate (SDS) nanoparticles were able to effectively protect α-tocopherol by reducing α-tocopheroxyl radicals29 as well as protect against oxidation phenomena.30 In this study, we will primarily concentrate on lipid-based liposomes and lipid nanoparticles,31 which are considered to be among the most suitable delivery systems for CCFs.
Although flavonoids have been reported to exert anticancer actions and their ability to inhibit the proliferation of various tumor cell types and to induce apoptosis may be one of the mechanisms of their anticancer effect through antioxidants and scavenging free radicals, it remains unclear whether such CCFs exert anticancer effects by modulating the functions of other prosurvival proteins, including SIRTs, B-cell lymphoma/leukemia 2 (Bcl-2), and Bcl-2-associated X protein (Bax), as well as intracellular signaling pathways mediated by mitochondria, PI3K, and mitogen-activated protein kinases.32 Furthermore, the anticancer effect of CCFs or CCF nanoliposomes (CCF-NLs) on human glioma and their role in SIRT1-mediated mitochondrial and PI3K/Akt or mitogen-activated protein kinase pathway-induced apoptotic death on GBM cells are not very clear. Herein, the effect of CCFs and CCF-NLs on the expression levels and the role of SIRT1 in human GBM cell lines were studied. Here, we investigated the effects of CCFs and CCF-NLs on the proliferation and apoptosis in DBTRG-05MG GBM cells. We also evaluated whether CCFs and CCF-NLs regulate transcription factor SIRT1 expression. Additionally, the antiapoptotic Bcl-2, proapoptotic Bax, Akt, extracellular signal-regulated kinase (ERK) and mitochondrial proteins were investigated.
Materials and methods
Reagents and antibodies
Polyethyleneglycol-4000 (PEG4000) was purchased from Japan Oil & Fat Co., Ltd (Tokyo, Japan); polysorbate 80, polyvinylpyrrolidone K-30 (PVP-K30), and SDS were purchased from BASF Aktiengesellschaft (Germany); cholesterol was purchased from Shanghai Youngsun Foods Co., Ltd. (Shanghai, People’s Republic of China); Rutin, fisetin, quercetin, and myricetin were obtained from National Institute for the Control of Pharmaceutical and Biological Products (NICPBP, Beijing, People’s Republic of China); Annexin V and propidium iodide (PI) were obtained from Sigma Chemical Co. (St Louis, MO, USA); caspase-3, -8, -9 activity ELISA kit was purchased from BestBio Biotechnologies (Shanghai, People’s Republic of China). Roswell Park Memorial Institute 1640 medium (RPMI 1640), penicillin–streptomycin, trypsin–EDTA, fetal bovine serum (FBS), and L-glutamine were obtained from GIBCO BRL (Invitrogen Corp, Carlsbad, CA, USA). Anti-p53, phosphorylated p53 antibody, and anti-Akt, phosphorylated Akt antibody, antitotal ERK antibody, antiphospho-ERK antibody, polyclonal anti-Bax antibody, anti-Bcl-2 antibody, and horseradish peroxidase-conjugated antigoat IgG secondary antibodies were purchased from Santa Cruz Biotechnology Inc., (Dallas, TX, USA). Anticytochrome c, anticaspase-3, anticaspase-8, and anticaspase-9 antibodies were purchased from Cell Signaling Technology (San Jose, CA, USA). Bicinchoninic acid (BCA) protein assay kit was purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Extraction of CCFs
The flavonoids were extracted and separated from CC by liquid–liquid extraction method. Briefly, crushed powder of the stem was subjected to reflux ethanol extraction. Then, the alcohol extract was decolorized by macroporous resin column and eluted with 95% ethanol, followed by purification using a polyamide column. Elution was dried under vacuum. Redissolved in 95% ethanol, CCFs was isolated. The identity and content of the flavonoid compounds were confirmed by DIONEX Utimate-3000 high-performance liquid chromatography (HPLC) spectrometry with a dual-λ absorbance detector, at 365 nm. A flow of the eluent was 1.0 mL/min, which was composed of 25% methanol/20% acetonitrile/55% (0.3%) phosphoric acid from 0 to 20 minutes.
Preparation of CCF-NLs
According to an established method, CCF-NLs were prepared by emulsion-evaporation and low temperature curing preparation. Aqueous phase that made of PEG4000, PVP-K30, and SDS (1:4:0.01, w/v) was dissolved in pure water and maintained at 45°C in a water bath, and oil phase was mainly made of cholesterol and polysorbate 80 to organic solvents (0.01:1, w/v). Cholesterol was melted in a warm water bath at 45°C. About 1% CCF (w/v) was dissolved in the ethanol–acetone mixed solvent (1:1, v/v) to obtain a weight ratio of CCF and cholesterol was 1:10, which was codissolved in the oil phase, and then the oil phase of the aforementioned mixed solvent was injected into aqueous phase by a plastic needle tubing (internal diameter 0.45 μm, administration rate 2.0 mL/min) under mechanical agitation at 1,000 rpm. After stirring for 2 hours, the liposomal solution was curing at low temperature of 0°C–4°C under mechanical agitation at 800 rpm. Then, the nanoparticles were filt1ugh a dialysis tubing to remove the nonincorporated drugs. To prepare the purified CCF-NLs, the nanoliposomes- loaded CCF were injected into the dialysis tubing, which then was swished in the fresh mannitol solution(mannitol maintains isotonic pressure for both sides of dialysis solution) for dialysising three times to remove free CCF at 0°C–4°C. Finally, the purified CCF-NLs were obtained and stored at 0°C–4°C. The control nanoliposomes were also prepared by the same method without adding CCFs at any stage of the preparation. Before applied in this experiment, the prepared CCF-NLs were well distributed in RPMI 1640 medium containing 10% (v/v) heating activated FBS by using ultrasound treatment in order to obtain a CCF-NLs suspension.
Characteristic of CCF-NLs
To observe the particle size and morphology of CCF-NLs, the sample was dispersed in deionized water and the morphology was measured by using a JEM-2100 transmission electron microscope (JEOL, Tokyo, Japan). The identity and content of the compounds were confirmed by comparing the elution time of pure controls and by HPLC spectrometry as described earlier. UV data were collected for the samples in 1 mL quartz cuvettes using a T90 UV-VIS spectrophotometer (Beijing Purkinje General Instrument Co., Ltd.). Samples were dissolved in deionized water. The CCF-NLs core concentration in each sample was determined by the absorbance at 365 nm using the experimentally determined extinction coefficient of 0.0101 L/mg.
Cell culture
DBTRG-05MG GBM cells were purchased from American Type Culture Collection (Manassas, VA, USA). Cells were cultured at 37°C with 5% CO2 in a humidified atmosphere in Dulbecco’s Modified Eagle’s Medium (DMEM, Thermo Fisher Scientific) supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 U/mL). DBTRG-05MG GBM cells were cultured in vitro and treated with the indicated amounts of CCF, CCF-NLs, or dimethyl sulfoxide (DMSO), for 24 hours or shorter times, taking blank nanoliposomes as control.
Morphological changes by electron microscopy
Different concentrations (25 μg/mL, 50 μg/mL, and 100 μg/mL) of CCFs or CCF-NLs were taken, and 0.1% DMSO as a solvent control was added to the cells. Cells were grown and incubated for indicating time periods. Electron microscopy and morphometric analysis were performed using a fluorescence microscope (Olympus, Tokyo, Japan).
Cell viability assay
Cell viability was evaluated by the MTT assay. DBTRG-05MG GBM cells were seeded into a 96-well microplate at a density of 1×105 cells/well. DBTRG-O5MG cells were exposed to CCFs or CCF-NLs at different concentrations of 25, 50, 100, 200 μg/mL, and control nanoliposomes were incubated in the same way before cytotoxicity. Kinetics of killing DBTRG-05MG cells were further detected at 12 hours, 24 hours, and 48 hours after treatment with 25 μg/mL, 50 μg/mL, 100 μg/mL, and 200 μg/mL of CCF or CCF-NLs. Finally, the plates were analyzed using an ELISA plate reader at a wavelength of 570 nm to measure optical density (OD) of each well, which is proportionate to the number of viable cells in each well. The percentage of viable cells relative to control was obtained by dividing the average OD for the treated wells by the OD for the control wells.
|
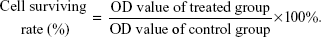
Flow cytometric analysis
Cell death rate was assessed by flow cytometric analysis of Annexin V/PI double staining. After DBTRG-05MG GBM cells were grown to 70%–80% confluence, they were treated with 25–100 μg/mL CCFs or CCF-NLs for 12 hours, 24 hours, and 48 hours. DBTRG-05MG cells were incubated with control nanoliposomes in the same way as the control group. Cells were then trypsinized and harvested by centrifugation before incubation with Annexin V and PI for 15 minutes at room temperature. Necrotic rates were examined and analyzed by flow cytometry with Annexin V-FITC/PI kit (BestBio Biotechnologies, Shanghai, People’s Republic of China). In this way, Annexin V binds to necrotic and apoptotic cells in which phosphatidylserine was exposed on the cell surface and the percentage of cell death was determined.
Effect of CCF or CCF-NLs on mitochondrial pathway
Based on our preliminary data, we determined the effects of CCF or CCF-NLs on the reactive oxygen species (ROS) level, mitochondrial release of cytochrome c, and the levels of caspase-3, -8, and -9 activity in DBTRG-05MG GBM cells.
Measurement of ROS level
DBTRG-05MG GBM cells were incubated with different concentrations (20 μg/mL, 50 μg/mL, and 100 μg/mL) of CCFs or CCF-NLs for 24 hours. Cells were harvested, washed twice with PBS, and then resuspended in 500 μL of DCFH-DA (10 μM) for the measurement of ROS levels, and the change in ROS production was assayed by flow cytometry, and referring to the study by Lu et al.33
Mitochondrial fraction isolation
A commercial isolation kit was used (Sigma) for isolation of mitochondria. The samples were prepared according to the manufacturer’s protocol. Briefly, after CCF or CCF-NLs treatment at the DBTRG-05MG GBM cells, whole-cell lysates were obtained as follows. In brief, pelleted cells were permeabilized for 1 minute in isotonic buffer containing 0.03% digitonin for 5 minutes on ice and then centrifuged at 15,000 rpm for 10 minutes. The supernatant (cytosolic fraction) and the pellet (mitochondrial fraction) were collected, and the pellet was further lysed as the final mitochondrial lysate for assays. The activity of caspase-3,-8,-9 was measured using commercially available kits, and western blot analyses for cytochrome c, cleaved caspase-3 and caspase-3.
Assays for caspase-3, -8, -9 activity
DBTRG-05MG GBM cells were treated with CCFs or CCF-NLs (50 μg/mL and 100 μg/mL) or DMSO alone for 12–24 hours. The caspase-3, -8, -9 activity and cell viability were determined and analyzed by using the Caspase-3, -8, -9 activity assay kit (10 μM), with referring to the study by Lee et al.34
Western blot assay for cytochrome c and caspase-3
For Western blotting, the protein concentration of the mitochondrial lysate was determined by using a BCA protein assay kit in accordance with the manufacturer’s instructions of Thermo Fisher. Total proteins were separated by denaturing 8%–12% SDS-polyacrylamide gel electrophoresis and were resolved over and electrotransferred by semi-dry blotting (Bio-Rad Laboratories Inc., Hercules, CA, USA) onto a nitrocellulose membrane. Membrane was incubated with antibodies to caspase-3 and β-actin (Santa Cruz Biotechnology Inc.). Protein signals were detected using an ECL detection system (Pierce Biotechnology, Rockford, IL, USA).
Western blot assay for the detection of Bcl-2 and Bax oligomers
In vitro crosslinking of associated Bax or Bcl-2 monomers was performed according to the method previously reported with some modifications. In brief, collected cells were lysed in crosslinking buffer (10 mM HEPES [pH 7.5], 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 250 mM sucrose, 2% CHAPS) on ice for 30 minutes, and then bismaleimidohexane was dissolved in DMSO and added to a final concentration of 10 mM. After incubation for 30 minutes at room temperature with rotation, the lysate was centrifuged at 15,000 rpm for 10 minutes. The supernatant was collected as crosslinked lysate, and after determination of the protein concentration using a BCA protein assay kit, the lysate was subjected to Western blotting analysis by using polyclonal anti-Bcl-2 and anti-Bax antibodies (Santa Cruz Biotechnology Inc.) and referring to the study by Tomiyama et al.35 Protein signals were detected using an ECL detection system (Pierce Biotechnology).
Western blot assay for SIRT1-mediated p53 pathways
Plasmid and transfection
The expression of plasmid was pcDNA-SIRT1 (presented from Dr SST TO, Hong Kong). Cells were subcultured and transfected using synthetic, HPLC-purified siRNAs (Thermo Fisher Scientific) formulated into liposomes (Oligofect AMINE, Thermo Fisher Scientific). The final volume of culture medium was 2 mL/well. Cells were monitored by microscopy and harvested for biochemical analyses at 24 hours, 48 hours, and 72 hours after transfection.
SIRT1 RNA silencing
The independent siRNA sequences were used to silence SIRT1 expression. Sequences: sense 5′-ACUU UGCUGUAACCCUGUA (dTdT)-3′ and antisense 5′-UACAGGGUUACAGCAAAGU (dTdT)-3′. Controls included liposomes formulated in the absence of siRNA. siRNA concentration was 0.39 μg per 1.0×105 cells.36
For transfection-based coimmunoprecipitation assays, cells were transfected with the indicated plasmids using Lipofectamine 2000 (Thermo Fisher Scientific), lysed in 0.5 mL lysis buffer, and immune-precipitated with Protein G Plus/Protein A Agarose Suspension beads (Calbiochem) for 3 hours at 4°C. After incubation for 6 hours, the media was replaced with DMEM containing 10% FBS. The beads were washed and eluted in SDS sample buffer. The eluted proteins were separated by SDS–PAGE, followed by Western blotting.
Western blot
Cells were harvested and lysed in RIPA buffer (EMD Millipore, Billerica, MA, USA) containing Complete Protease Inhibitor Cocktail (Hoffman-La Roche Ltd., Basel, Switzerland) plus 1 mM PMSF. Lysates were centrifuged at 12,000 rpm for 20 minutes at 4°C. After quantifications of the protein samples using the BCA Protein Assay Kit (Pierce Biotechonology), total protein was electrophoresed through a 10% SDS–polyacrylamide gel and then transferred to 0.45 lm nitrocellulose membranes (EMD Millipore, CA, USA) on a semi-dry electro transferring unit (Bio-Rad Laboratories Inc.). The following antibodies at indicated dilutions were used for Western blotting: SIRT 1 (1:500 dilution), β-actin antibody; Anti-p53, and phosphorylated p53 antibody (Santa Cruz Biotechnology Inc., 1:1,000 dilution). Protein signals were detected using an ECL detection system (Pierce Biotechonology).
Western blot assay for Akt and ERK pathways
For protein analysis, the cells were harvested at 12–24 hours as described earlier, washed with cold PBS, and then incubated in ice-cold RIPA buffer. Cell lysates were sonicated for 30 seconds on ice and then lysed at 4°C for 60 minutes. The cell lysates were centrifuged for 30 minutes at 4°C, 12,000 rpm. Protein concentrations in the supernatants were determined by the BCA reagent. Total proteins were separated by denaturing 8%–12% SDS-polyacrylamide gel electrophoresis, which were resolved over and electrotransferred by semi-dry blotting (Bio-Rad Laboratories Inc.) onto a nitrocellulose membrane. Membrane was incubated with antibodies to anti-Akt, phosphorylated-Akt antibody, antitotal ERK antibody, antiphospho-ERK antibody, and β-actin (Santa Cruz Biotechnology Inc.).
Statistics
Significance was determined using the one-way ANOVA test on the mean values of three different experiments. Significance was determined using the mean ± standard deviations (SDs) obtained by averaging values (95% CI) at three separate experiments (n=3), and analyzed by two-tailed Student’s t-tests using the Statistical Program for Social Sciences 13.0 software (SPSS Inc., Chicago, IL, USA). P<0.05 was used as the cutoff for statistically significant differences. Western blotting analysis used the corresponding strips gray value of the relative protein content to take a picture through Bio-Rad image analysis system With Image-Pro software analysis.
Results
Preparation and characterization of CCF-NLs
The size distribution (Figure 1A) of CCF-NLs, ranging from 60 nm to 400 nm, was observed using a laser particle analyzer, and the average diameter of the nanoparticles was 295.3±58.1 nm. Indeed, in transmission electron microscopy analysis, CCF-NLs formed spherical particles with a small diameter and a narrow size distribution (Figure 1B and C).
 | Figure 1 Characterization of CCF-NLs. |
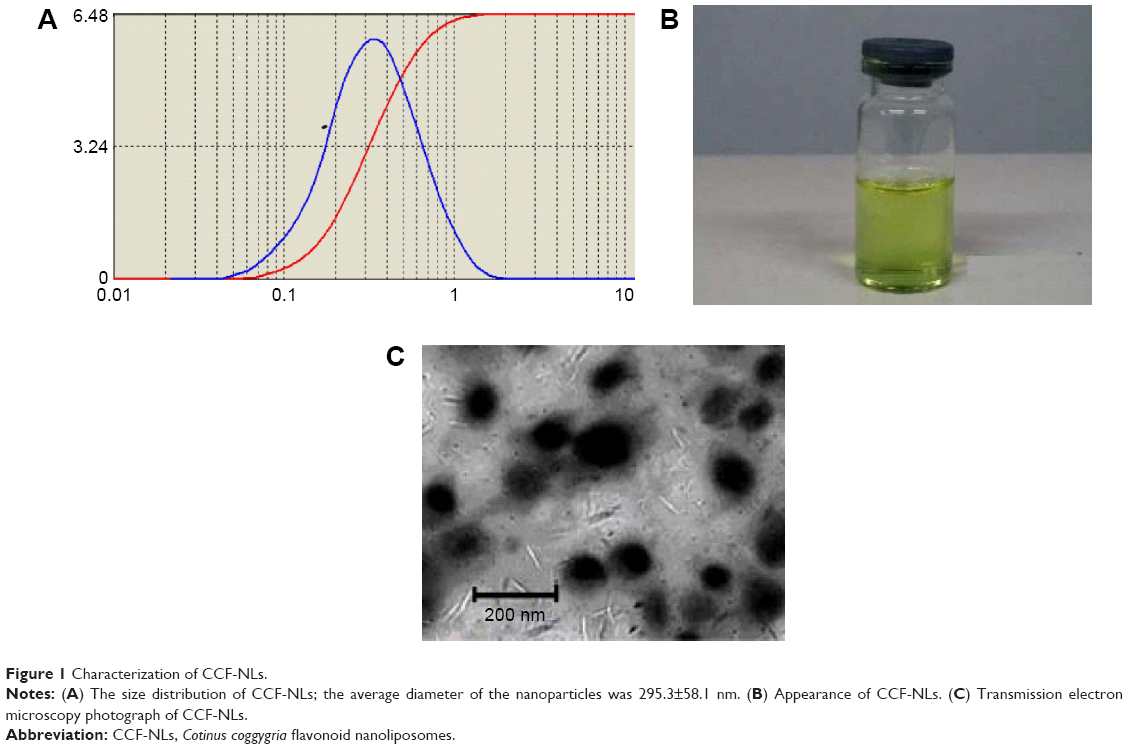
HPLC and UV-Vis spectroscopy for CCFs and CCF-NLs indicated that, as expected, the CCF-NLs core remained unchanged by the attachment of PEG4000 compared with the CCF or control nanoliposomes (Figure 2A, B, and D). HPLC also provided the simple technique for measuring the content of CCFs or CCF-NLs in samples. The CCF extract contained the following: 1.03% myricetin (with a molecular weight of 318), 1.41% CCF (with a molecular weight of 303), 1.40% rutin (with a molecular weight of 612), 0.09% quercetin (with a molecular weight of 338), and 0.08% of one type of flavonoid, the exact structure of which was not known. The percent of the total flavonoids of CC was quantitated as 4.01%. The dried CC was provided by Hubei University of Medicine (Shiyan City, Hubei Province, People’s Republic of China) and can also be prepared as described earlier.
 | Figure 2 Characterization of CCF-NLs and CCF. |
Cell death and morphological changes
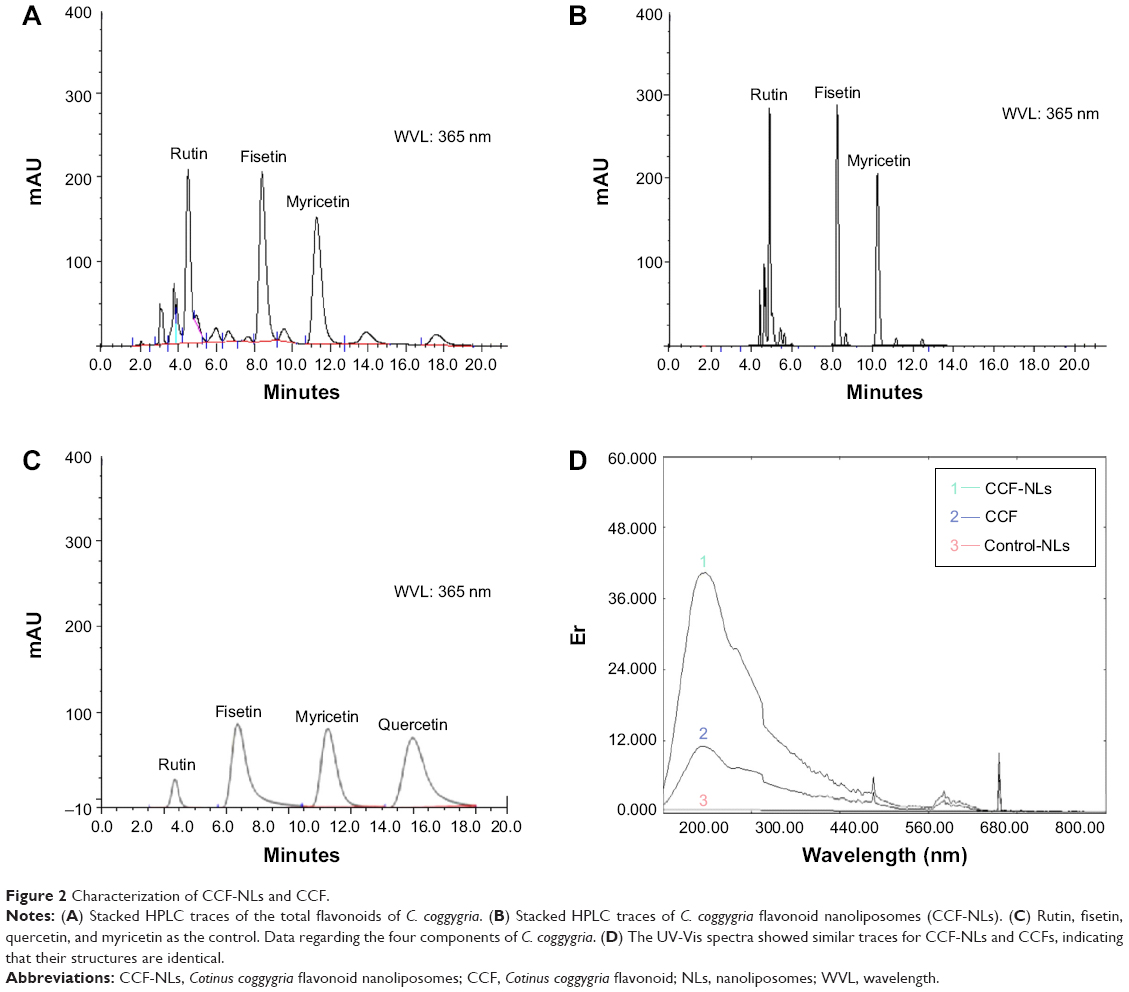
CCF-NL-induced morphological changes and cell death are shown in Figure 3A. The results demonstrated the percentage of viable cells with morphological changes caused by CCF-NLs, and the effects occurred in a dose- and time-dependent manner. Compared with CCF-NLs, the rate of cell death in DBTRG-05MG GBM cells was not remarkably enhanced (Figure 3B). Moreover, the control, 0.1% DMSO, had no significant effect on the viability of DBTRG-05MG cells (Figure 3C). Next, hematoxylin staining was used to detect the chromatin condensation of necrotic and apoptotic cells. In this study, the proportion of apoptotic cells was increased with concentrations of CCF-NLs at 50–100 μg/mL (Figure 3D) at 24 hours compared with treatment with CCF alone or the control (Figure 3E and F).
 | Figure 3 Morphology and viability of DBTRG cells. |
Cell viability assay
Regarding DBTRG-05MG cells, the cell-killing efficacy of CCF-NLs was assessed by the MTT assay, and the results demonstrated a significantly higher targeted killing efficacy in tumor cells at 12 hours, 24 hours, and 48 hours by CCF-NLs. Although free CCFs showed limited tumor cell killing, CCF-NLs provoked a significant cell death with the same CCF concentrations. Thus, CCF-NLs induced a 1.5-fold increase in cell death in GBM cells compared with the CCF concentration of 25–200 μM. A stronger time-dependent cell killing efficacy was also achieved in DBTRG-05MG GBM cells by CCF-NLs (Figure 4).
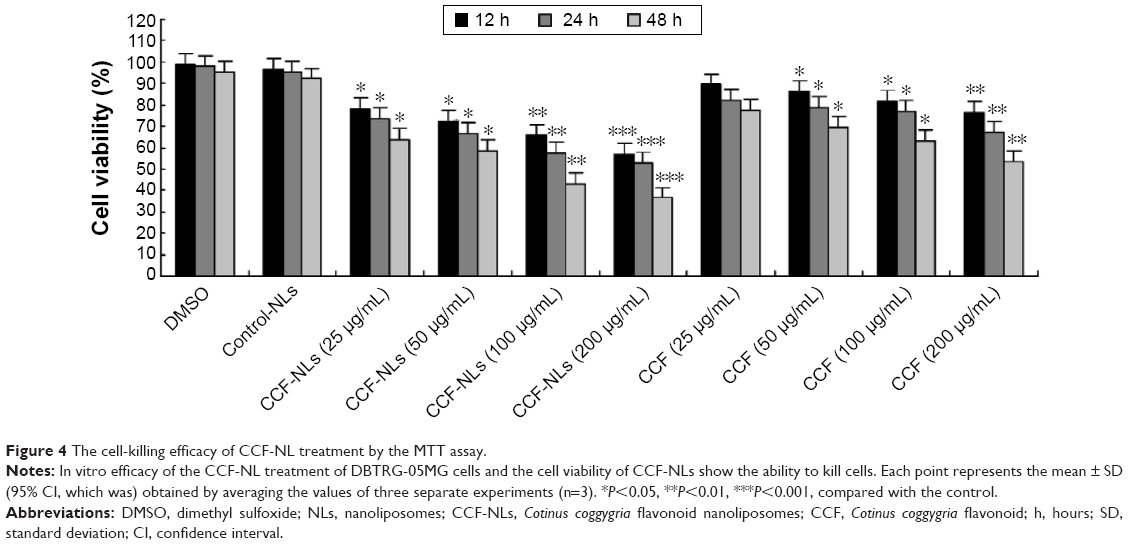 | Figure 4 The cell-killing efficacy of CCF-NL treatment by the MTT assay. |
Effect of CCF-NLs or CCFs on cell death
DBTRG-05MG cell apoptosis was significantly enhanced by CCF-NLs at concentrations of 25 μg/mL, 50 μg/mL, and 100 μg/mL for 24 hours (Figure 5A). However, when DBTRG-05MG cells were exposed to different concentrations (25–100 μg/mL) of CCF, necrotic cell death was displayed by the Annexin V/PI assay (Figure 5B), while 0.1% DMSO-treated and control cells indicated no significantly induced necrosis or apoptosis in GBM DBTRG-05MG cells (Figure 5C). These data were comparable with those in Figure 5D, showing that CCF-NLs enhanced the percentage of apoptotic cell death, while CCF-induced necrosis was the primary for of cell death found in DBTRG-05MG GBM cells (Figure 5E).
 | Figure 5 The percentage of apoptosis and necrosis in DBTRG-05MG glioma cells induced by CCF-NLs. |
ROS production of CCF-NLs or CCFs
To evaluate the function of ROS in cell death induced by CCF-NLs, DBTRG-05MG cells were treated with different concentrations (20 μg/mL, 50 μg/mL, and 100 μg/mL) of CCFs or CCF-NLs for 24 hours. The treatment efficiency was estimated by flow cytometer analysis. As shown in Figure 6A, the ROS level was markedly increased in DBTRG-05MG cells when the cells were treated with CCF-NLs alone, and the ROS levels at different concentrations (25 μg/mL, 50 μg/mL, 100 μg/mL) of CCF-NLs were 66.3%, 85.5%, and 95.6%, respectively, while the ROS level of control cells was ~4.7%–7.8%. By contrast, the levels of ROS were 30.8%, 43.4%, and 57.4%, respectively, according to the different concentrations of CCF (Figure 6B and C). These results indicated that there was a significant difference between CCFs and CCF-NLs in the ROS level and type in DBTRG-05MG cells, a finding that may be associated with endogenous ROS through different modes of cell death (Figure 6D).
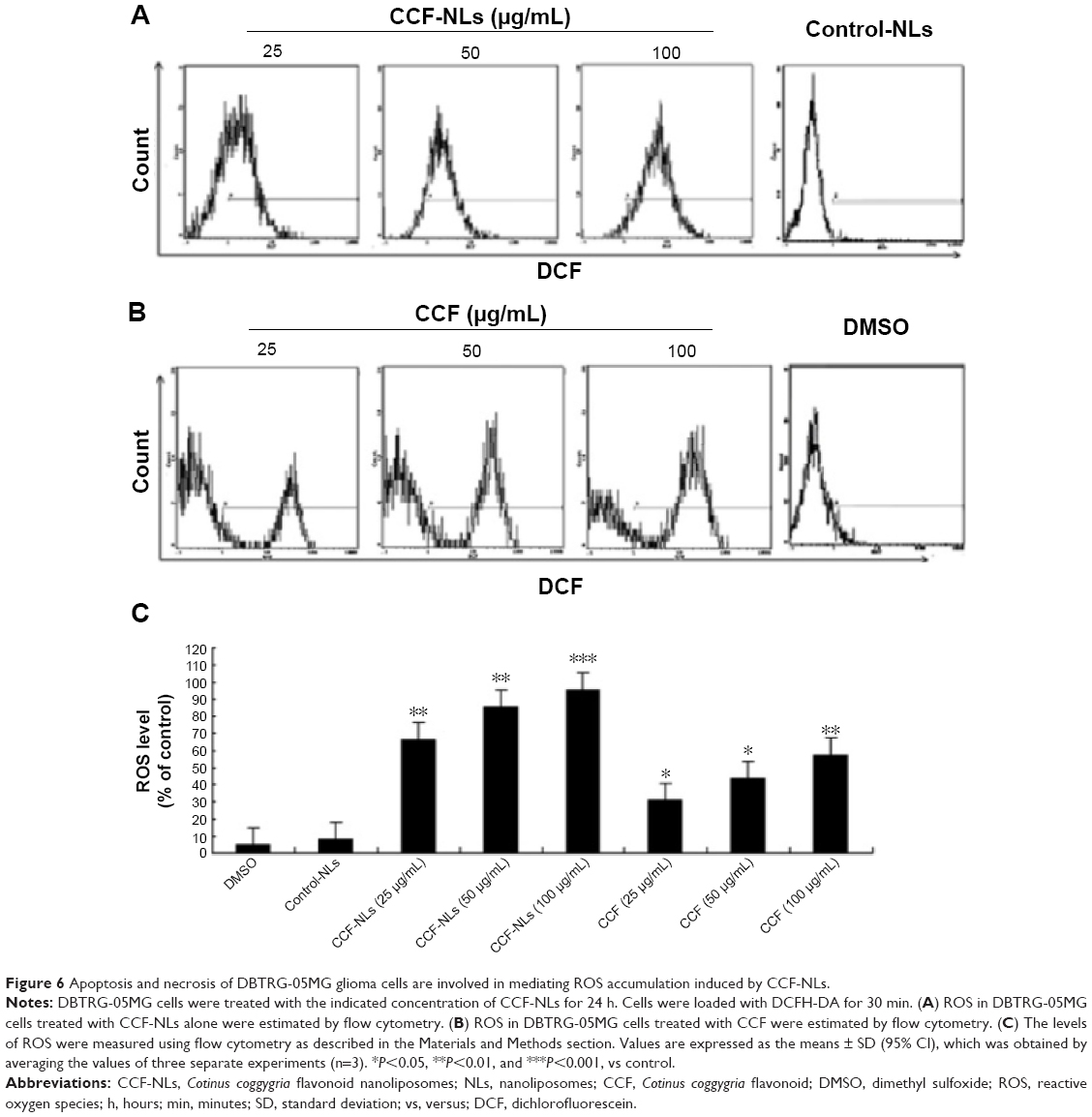 | Figure 6 Apoptosis and necrosis of DBTRG-05MG glioma cells are involved in mediating ROS accumulation induced by CCF-NLs. |
CCF-NL-induced cell death through the SIRT1-mediated p53 signaling pathway
Transfection for 48 hours with SIRT1 siRNA (transfection efficiency, 82.3%) caused a significant reduction in the expression of SIRT1 (Figure 7A and B). While transfecting with pCDNA-SIRT1 caused a slight decrease in the expression of SIRT1 in DBTRG-05MG cells, the results were similar to those of the control cells from SIRT1 pCDNA transfection and siRNA control transfection in DBTRG-05MG cells. Altogether, these results suggested that SIRT1 siRNA selectively silences SIRT1 protein expression. Significantly, there was downregulation of SIRT1 by CCF-NLs (P<0.05) in DBTRG-05MG cells after 24 hours (Figure 7C and D).
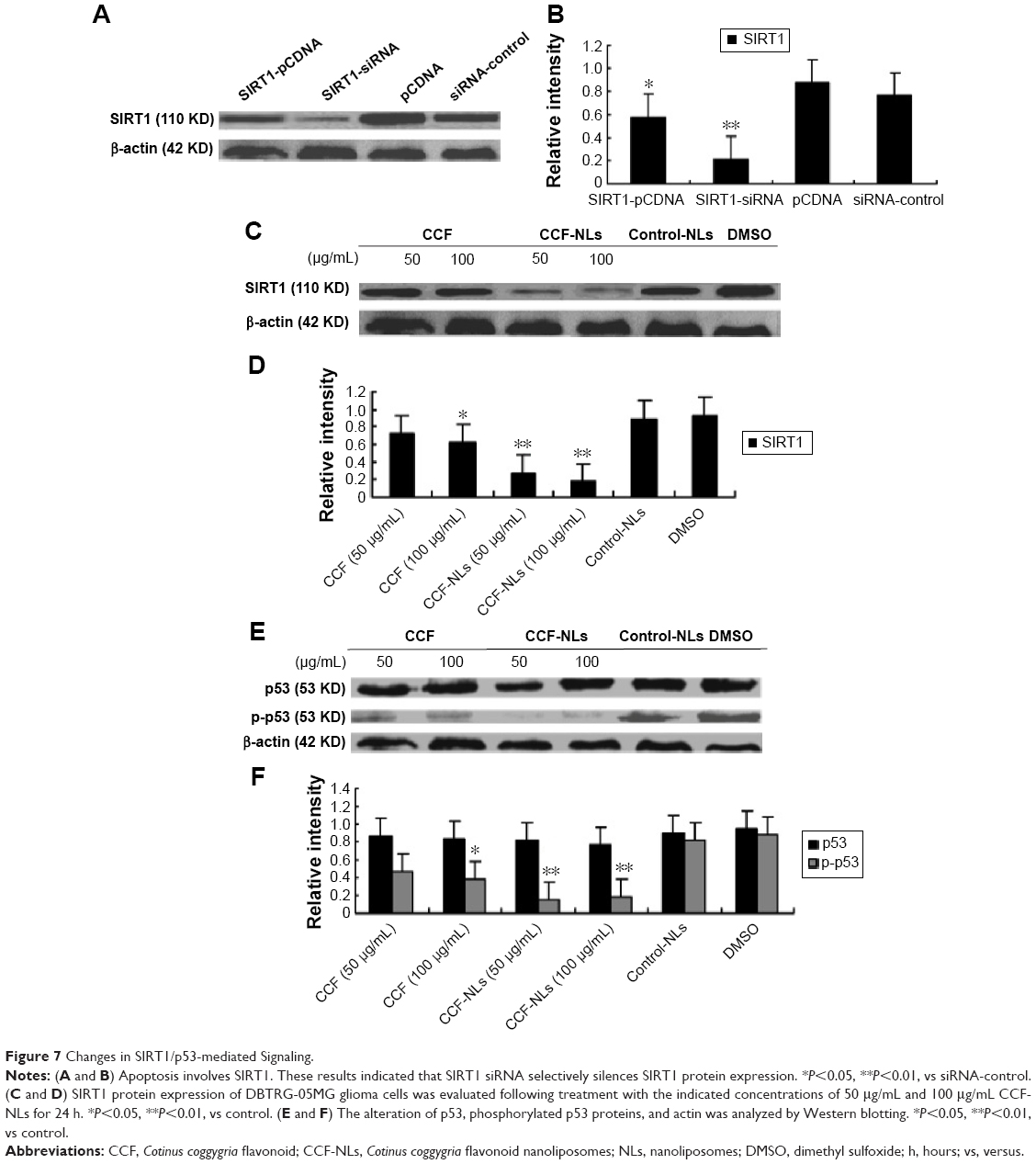 | Figure 7 Changes in SIRT1/p53-mediated Signaling. |
To identify the possible signaling pathways involved in SIRT1-mediated cell death in DBTRG-05MG GBM cells, we measured the expression of p53 and phosphorylated p53 after CCF-NL treatment by Western blot analysis.37 As shown in Figure 7E, phosphorylated p53 expression was downregulated by CCF-NLs (50–100 μg/mL), while there was no significant change in p53 expression in CCF-NL- or CCF-treated cells compared with control cells (Figure 7F). These results suggested that CCF-NL-induced cell death might have occurred through the SIRT1/p53-mediated pathway, primarily achieved by CCF-NL at a concentration of 50–100 μg/mL.
CCF-NLs induced cell death via the SIRT1-mediated mitochondrial pathway
CCF-NL-induced cell death involves caspase activation and cytochrome c release
Considering the mechanism of caspase activation, the mitochondrion is critical for relaying caspase cascade-activating signals. Therefore, we evaluated the caspase protein levels and activation with the involvement of the mitochondrial pathway. Downstream executioners of cell death induced by CCF-NLs were identified as caspase-3 and cytochrome c; moreover, the expression of cleaved caspase-3 protein was increased by CCF-NLs (Figure 8A). The caspase-3, cleaved caspase-3 activity, and cytochrome c protein levels were changed when DBTRG-05MG GBM cells were exposed to CCF-NLs (50–100 μg/mL) (Figure 8B). Particularly, the activities of caspase-3 and -9 were significantly changed in apoptotic cells by CCF-NLs, while CCFs (50 μg/mL) had no effect on the activities of caspase-3 and -9 in necrotic cells; these results were in agreement with those in other reports.38 CCF-NLs also decreased the protein expression of cytochrome c and the release of cytochrome c from the mitochondria to cytosol in DBTRG-05MG GBM cells.
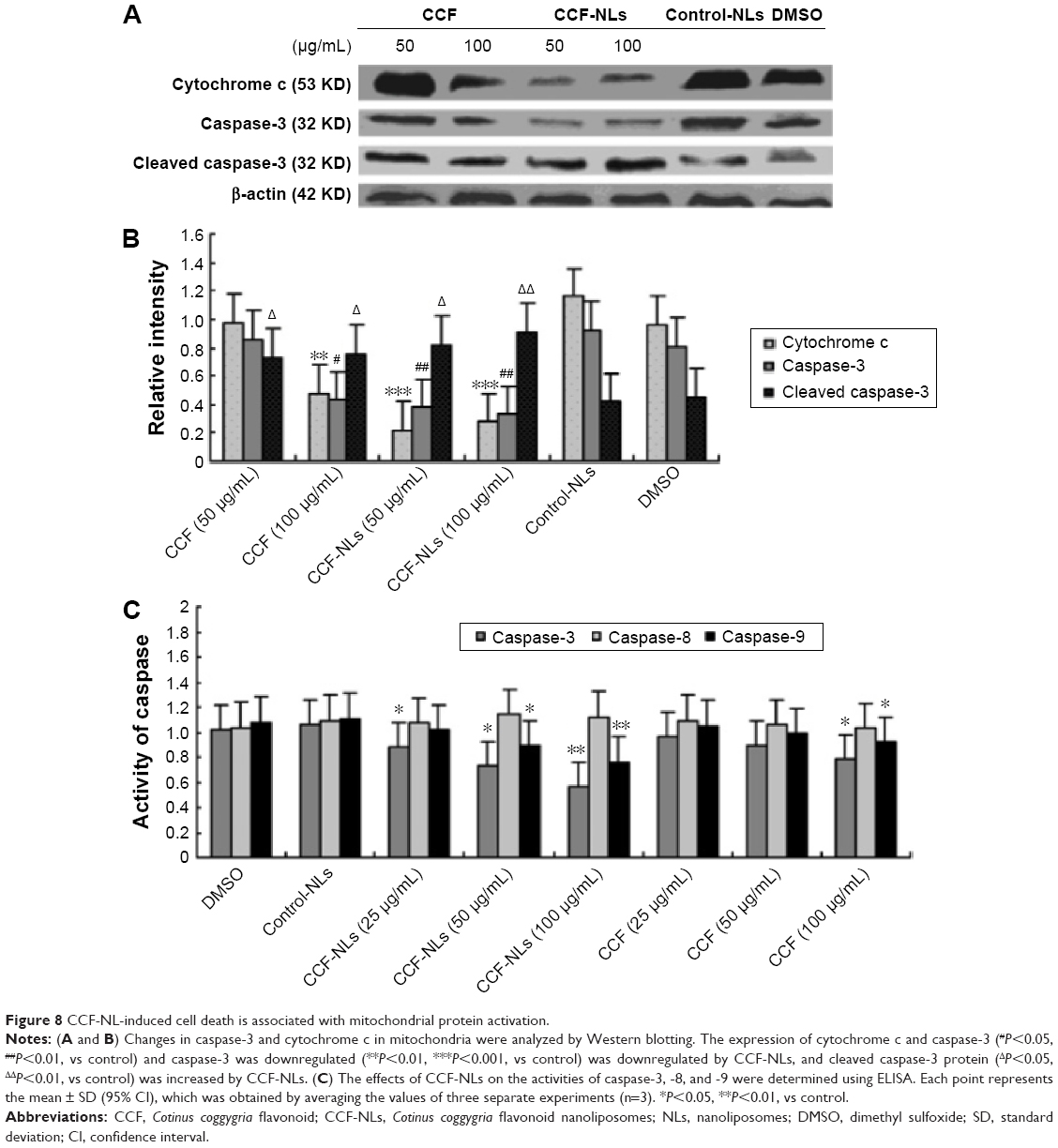 | Figure 8 CCF-NL-induced cell death is associated with mitochondrial protein activation. |
These results also demonstrated that CCF-NL-induced cell death was independent of caspase-8, whereas apoptotic cell death was dependent on caspase-3 and -9 when CCF-NLs were administered (Figure 8C). Notably, the cytochrome c level and cytochrome c release from mitochondria were both decreased by CCF-NLs in DBTRG-05MG GBM cells. Thus, cytochrome c is essential for CCF-NL-induced glioma cell death, and caspase-3 and -9, but not caspase-8, were activated downstream of the mitochondrial apoptotic pathway.
CCF-NL-induced cell death involved the Bcl-2 family
Proapoptotic Bcl-2 family proteins, particularly multidomain-type proapoptotic Bcl-2 family proteins, such as Bax and Bak, play an essential role in mitochondrial outer membrane permeabilization and the transduction of programed cell death by diverse cell death stimuli through the mitochondrial pathway.39,40 Considering the mechanism of caspase activation, the mitochondrion is the key intracellular organelle that relays caspase cascade-activating signals. Therefore, we monitored Bcl-2 activation and Bax in the mitochondria, and self-oligomerization of Bcl-2 and Bax was confirmed (Figure 9A and B). Because proapoptotic Bcl-2 family proteins, particularly multidomain type proapoptotic Bcl-2 family proteins, including Bax, play an essential role in mitochondrial outer membrane permeabilization and transduction by diverse cell death stimuli through the mitochondrial pathway,17,18 we monitored Bax activation because Bax formed a self-oligomer in the mitochondrial outer membrane.41
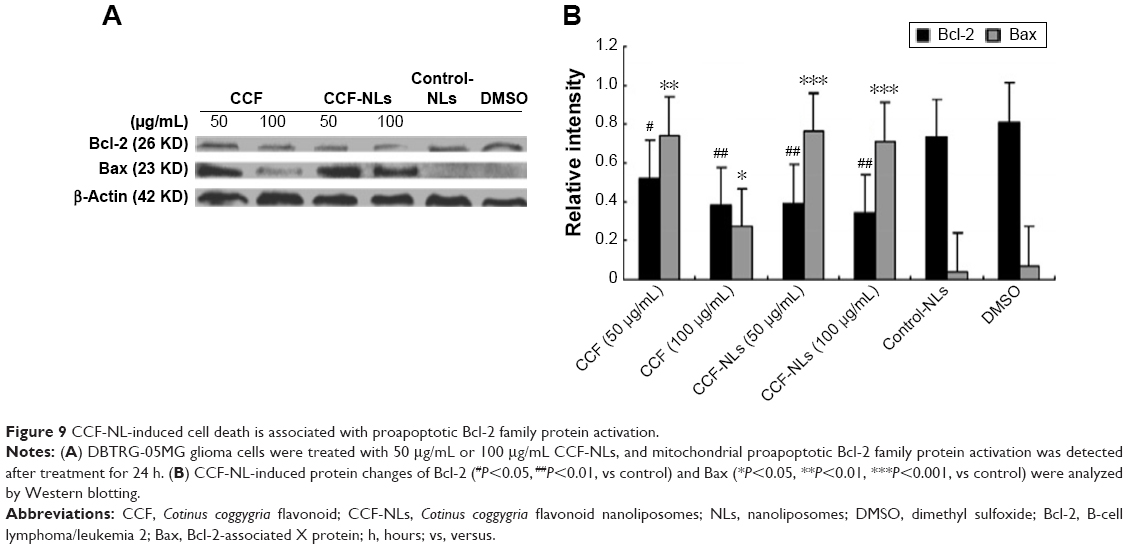 | Figure 9 CCF-NL-induced cell death is associated with proapoptotic Bcl-2 family protein activation. |
Consequently, in response to CCF-NLs, Bax was detected in the mitochondria, and self-oligomerization of Bcl-2 and Bax was also confirmed. Additionally, the caspase-3 activity and cytochrome c protein level were regulated by CCF-NLs. Overall, these results demonstrated that both Bcl-2 and Bax were essential for CCF-NL-induced DBTRG-05MG GBM cell death, and caspase-3 was activated downstream of mitochondrial proapoptotic Bcl-2 family protein activation.
SIRT1-mediated Akt signaling pathway by CCF-NLs
Akt phosphorylation (Ser 473) was significantly downregulated by CCF-NLs in DBTRG-05MG GBM cells. Additionally, there was downregulation of SIRT1 and Akt when SIRT1 was inhibited by CCF-NLs (Figures 10A and B). These data were comparable, as shown in Figure 7C and E, which shows that CCF-NLs suppressed SIRT1 activation constitutively by inducing apoptosis in DBTRG-05MG cells via downregulation of Akt and phosphorylation of Akt; thus, phosphorylated p53 expression was downregulated by CCF-NLs. Compared with the control, CCF-NLs had no effect on ERK and phosphor-ERK levels with the downregulation of SIRT1 levels (Figures 10C and D).
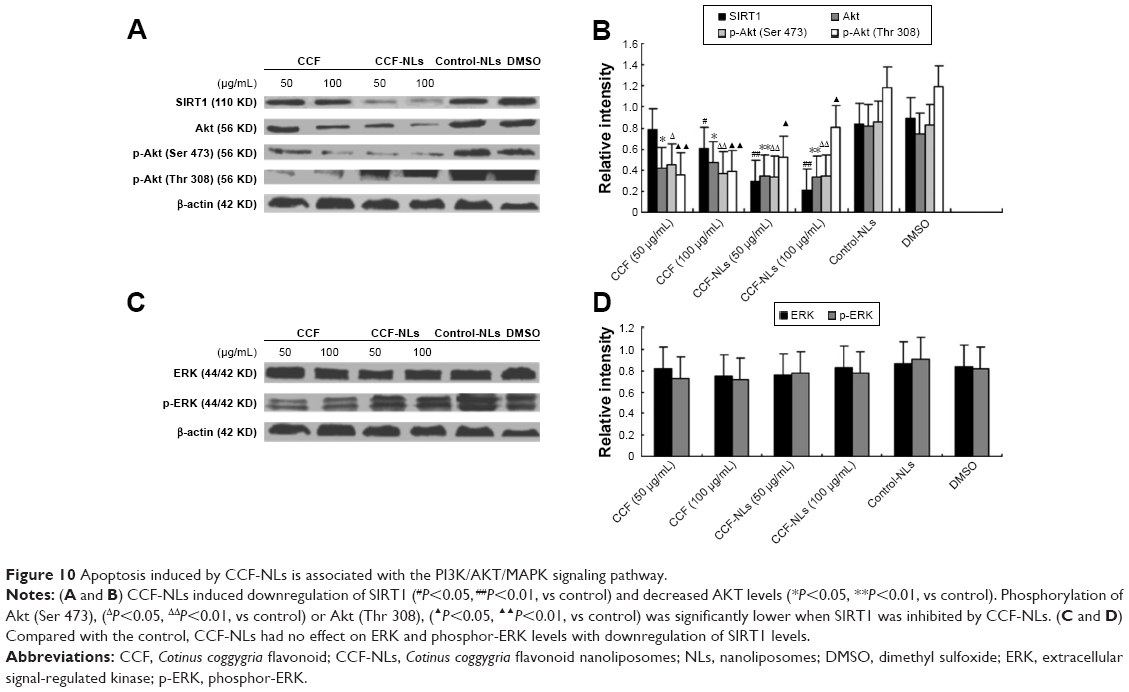 | Figure 10 Apoptosis induced by CCF-NLs is associated with the PI3K/AKT/MAPK signaling pathway. |
Altogether, our work identified the downregulation of SIRT1 in DBTRG-05MG GBM cells by CCF-NLs, resulting in the inhibition of growth and proliferation and the promotion of apoptosis. It was proposed that the SIRT1-mediated activities of Akt and p-AKT (Ser 473) were regulated by CCF-NLs. Several publications have reported tumor cell apoptosis or growth arrest after transient knockdown of SIRT1 or treatment with SIRT1 inhibitors, such as sirtinol, splitomycin, and cambinol.36 Our study results support these conclusions.
Discussion
At present, GBM is still being treated using radical surgery followed by radiotherapy and chemotherapy; likewise, the conventional drug used in chemotherapy is temozolomide because of its therapeutic benefits in prolonging the survival of GBM patients. Nevertheless, the effort to develop targeted therapy against a genetic anomaly using specific small-molecule inhibitors are now in clinical trials, and some of them have shown promising results. However, multiple genetic changes in GBM have suggested that a single drug or agent is unlikely to offer an effective and complete solution to the problem. Therefore, combination therapy with two or more therapeutic agents may provide better management of the disease. Instead of using many drugs together in combination therapy, it would be ideal to make drugs that can simultaneously act on many of the aberrant genetic pathways. In searching for such compounds, one of the aims of this study was to evaluate the effect of flavonoids extracted from the common ornamental plant CC.
The use of CC for plant medicinal purposes has been widespread across all ethnic groups since ancient times. Furthermore, the chemical compounds present in this plant are termed phytochemicals. Those that contain at least one aromatic ring bearing one or more hydroxyl group are called phenolic compounds, such as flavonoids, which are abundant in fruits, vegetables, and plants. Notably, many of these flavonoids are used as chemopreventive agents,42 while others are in clinical trials for treating various ailments, including cancers.43 Flavonoids exhibit a wide range of biological and pharmacological activities, including anti-inflammatory, antimicrobial, and anticancer properties; moreover, the latter include anti-invasive and antimetastatic activities.44 In a previously funded study, we found that the total flavonoid extract of CC Scop. was responsible for anticancer effects on GBM cell growth via the induction of apoptosis and identified CCFs as the major activity constituents, which played a critical role in anti GBM. In addition, an in vivo study has found that CCF decreased tumor growth and inhibited migration in orthotopic mouse models of DBTRG-05MG GBM. However, CCFs have limitations regarding their antibrain tumor effects because of their limited water solubility, short storage time, and low brain targeting. Therefore, a continued need exists for the use of CCFs in antibrain tumor DDSs. Notably, several PEGylated nanomaterials have been shown to successfully sequester hydrophobic compounds, for example, SDS-based PVP-K30 and PEGylated layers and oil phase (cholesterol) can be made into nanoparticles, which are modified with liposomes to encapsulated the flavonoids inside the nanoparticles.45 Phospholipid nanoliposomes represent a classical example of fully degradable DDSs – particularly, cholesterol is known to enhance the rigidity of the soy lipid bilayer; consequently, the drugs incorporated into these carriers were fully or partially morphous. Furthermore, the solubility of the drugs in the PVP-K30 showed improved dissolution rates.46 To enhance the circulation properties of carriers in the bloodstream, the typical course of action is to coat a carrier with PEG, for example, PEGylation of liposomes increased their half-life from <30 minutes to ~5 hours in mice.47 Other benefits, such as reduced interactions with the immune system, have also been found. Importantly, due to their dense PEG surface brush, polymersomes and filomicelles are compatible with the blood48 because they: i) remain suspended and flexible in the plasma; ii) do not adhere to red blood cells and leukocytes in the blood; iii) do not fix opsonins or activate complement;49 and iv) do not cause hemolysis.50
Because CCFs are weak water-soluble compounds,51 in this study, we loaded CCFs onto the hydrophobic surfaces of SDS-based PVP-K30 and PEGylated liposomal carriers and characterized their physicochemical properties. Additionally, we prepared SDS-based PVP-K30 and PEG-coated drug delivery carriers of CCF to further investigate the anticancer effects on apoptotic death in GBM cells by CCF-NLs. The CCF-NL-induced apoptotic changes in DBTRG-05MG cells and cell viability were found to be decreased in a dose- and time-dependent manner. This study also showed that CCFs mainly induced the necrotic population in DBTRG-05MG glioma cells exposed to different concentrations of CCF (25 μg/mL and 100 μg/mL), most likely due to necrosis occurring shortly after exposure of the cells to CCF. In this study, we found that rutin, fisetin, quercetin, and myricetin are the primary flavonoid components of CC, and a synergetic effect was likely produced by multiple flavonoids acting together to induce accidental, normal physiological, and programed necrosis.52,53 Several studies have also shown that certain types of cell death share both apoptotic and necrotic mechanisms in a process referred to as “necrapoptosis.”54 Our studies have shown that several common points that induce the necrotic and apoptotic pathways exist in DBTRG-05MG GBM cells. Therefore, CCF-NLs modulate apoptotic and necrotic cell death signaling simultaneously. In the case of conventional chemical therapy, tumor cells have been reported to undergo apoptosis.55 Histological analysis of human tumor specimens has also shown a necrotic change as a result of high-dose chemical agents.56 To the best of our knowledge, this is the first report concerning the detailed molecular mechanism of CCF-NL-induced glioma cell death, including the type of cell death involved. Particularly, the novel involvement of the molecular/metabolic transducer SIRT1 that mediates p53-induced arrest/death by CCF-NLs in GBM cells was recognized, and the efficacy of SIRT1-mediated p53 gene expression following treatment with CCF-NLs has also already been shown. SIRT1 has been found to target p53 for deacetylation and to attenuate p53-mediated transcriptional activity,2,57 and p53 functions, most importantly, to induce cell cycle arrest, apoptosis, and DNA repair. Therefore, inhibition of SIRT1 activity, which leads to elevated p53 acetylation and transactivation and results in enhanced apoptosis and cytostasis, would be beneficial for cancer treatment.
In this study, CCF-NLs abrogated SIRT1 activation downstream of the mitochondrial pathway. Therefore, through SIRT1-mediated p53 and mitochondrial pathways and the induction of caspase-3-dependent apoptosis in DBTRG-05MG GBM cells, CCF-NLs could be a more effective method for treating chemical-resistant GBM. Additionally, the levels of Bcl-2 proteins were downregulated by CCF-NLs. Most researchers agree that the antiapoptotic protein Bcl-2 is associated with cancer cell survival.58 Recent work has also indicated that SIRT1 binds directly to the p53 protein59 and enhances or downregulates p53 expression. The p53 protein, a nonhistone substrate of SIRT1, activates the transcription of proapoptotic factors and suppresses the transcription of antiapoptotic genes, such as Bcl-2.60 There are protein–protein interactions between SIRT1 and p53 that are crucial for the proper function of cells, and the SIRT1/p53 interactions are regulated by posttranslational modifications.7 Moreover, the effects of SIRT1 proteins on p53 may be particularly different in various cell death types because SIRT1 proteins themselves are distributed in a cell-specific manner.61 Many oncogenic signals can trigger the constitutive activation of mitochondria, and SIRT1 has dual functions of both promoting cytoprotection and apoptosis, depending on the nature and severity of the stress.62,63 In this study, our data indicated that the downregulation of p53 protein levels by CCF-NLs is associated with inhibited SIRT1 expression. The SIRT1-mediated p53 signaling pathway may be one of the signal pathways regulated by CCF-NLs that engage the Bcl-2 family and mitochondrial activity through ROS independent SIRT1-mediated p53 signal pathway mechanisms, either via the necrotic pathway through ROS (ROS-mediated p53) by CCF regulation of Bcl-2 expression or via mitochondrial mRNA mechanisms. As a distinct mechanism of CCF-NL-induced apoptosis at the mitochondrial level, high ROS can direct cytosolic p53 to induce apoptosis via SIRT1-induced targeting of p53 to this organelle. In this way, deacetylated p53 binds to the outer membrane of the mitochondria and outcompetes Bax for binding to Bcl-2 proteins. Consequently, the subsequent activation of Bax leads to the release of cytochrome c from the mitochondria and the induction of apoptosis (Figure 11).64
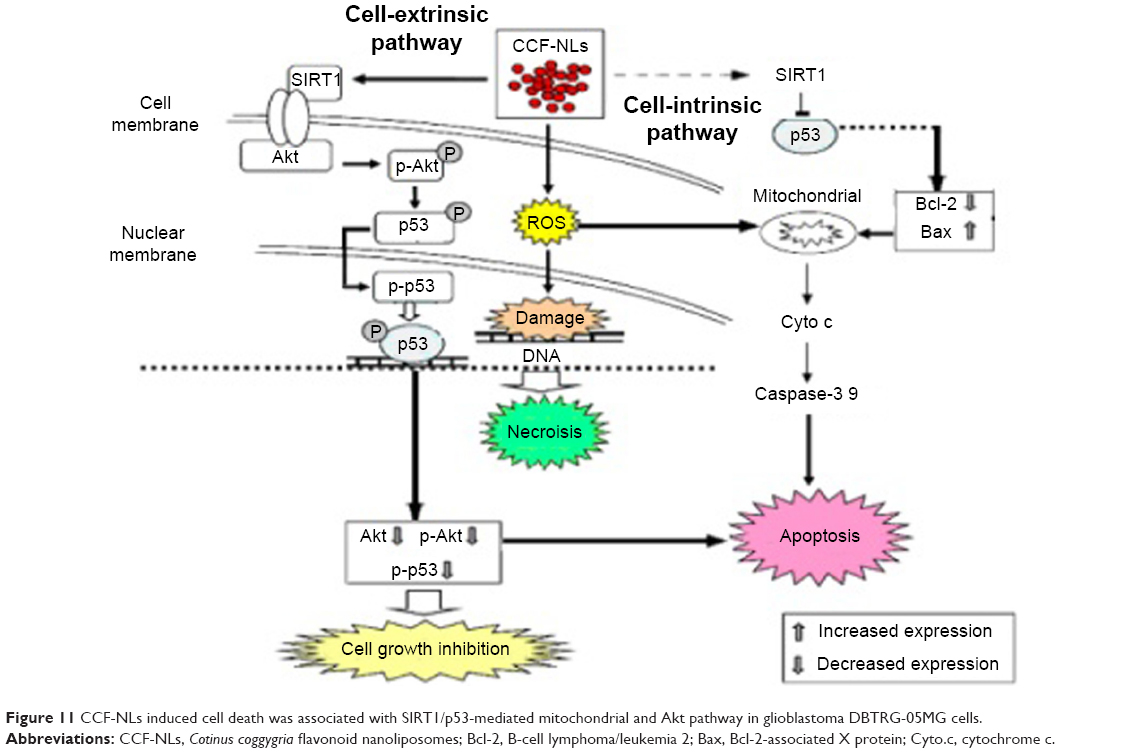 | Figure 11 CCF-NLs induced cell death was associated with SIRT1/p53-mediated mitochondrial and Akt pathway in glioblastoma DBTRG-05MG cells. |
It has long been suggested that the regulation of p53 activity sensitizes cells to the effects of several anticancer drugs.65,66 More recently, the development of effective SIRT1-specific antagonists or inhibitors of SIRT family proteins and the need for potent and selective inhibitors, particularly those of SIRT1, remain to be fulfilled.67 Among these antagonists was salermide, one of the first-discovered sirtinol analogs, which has a strong in vitro inhibitory effect on SIRT1 and SIRT2 and was shown to selectively induce apoptosis in cancer cells.68 It has been reported that p53 is essential for salermide-induced apoptosis, and the apoptotic effect was ascribed to the activation of proapoptotic genes that are repressed in cancer cells by SIRT1. An independent study suggested that salermide treatment leads to increased histone and tubulin acetylation and that apoptosis induction is p53 dependent.61 Our findings indicated that the SIRT1 protein was inhibited to downregulate phosphorylated p53 activity during CCF-NL-induced apoptosis, which explains the mechanisms above, thereby enhancing the cytotoxic effects of anticancer drugs. Thus, a novel mechanism involving SIRT1-mediated regulation of p-p53 levels to induce apoptosis by CCF-NLs was discovered by this finding, which might help to develop new anticancer strategies.
The imbalance between cell proliferation and cell death is considered to be an early and important event in the carcinogenic process, and SIRT1 regulated apoptosis in various mammalian cells. Although the role of SIRT1 in metabolism is relatively well defined,69,70 the function of SIRT1 in cancer is complex, and whether SIRT1 serves as a tumor suppressor or a tumor promoter is still debatable. In mammalian cells, SIRT1 decreases PTEN acetylation and inactivates the Akt pathway in a SIRT1 deacetylase-dependent manner. However, the function of SIRT1 in GBM was unknown. Huang and coworkers found that miR-34a acts as a tumor suppressor in p53-mutant U251 cells,59 partially through the regulation of SIRT1. Our results indicated that SIRT1 activation was suppressed constitutively during CCF-NL-induced apoptosis in DBTRG-05MG cells via the downregulation of both Akt levels and phosphorylation, and SIRT1 has been implicated as a deacetylase for the tumor suppressor for the PI3K/Akt pathway, a key oncogenic pathway that promotes cell growth and survival. Our findings suggested that SIRT1 inactivated the Akt pathway in a SIRT1 deacetylase-dependent manner by CCF-NLs. These results showed that suppression of SIRT1 by CCF-NLs might affect the tumorigenesis of GBM through Akt-mediated regulation of the p53 pathway. However, CCF-NLs had no effect on the levels of ERK and p-ERK proteins. Our results identified that SIRT1 was downregulated significantly by CCF-NLs in DBTRG-05MG GBM cells, inhibited their growth and proliferation, and promoted their apoptosis; mechanistically, CCF-NLs regulated the SIRT1 mediation of the expression of levels of Akt and p-Akt (Ser 473). Several publications have reported tumor cell apoptosis or growth arrest after the transient knockdown of SIRT1 or treatment with SIRT1 inhibitors, such as sirtinol, splitomycin, and cambinol.71 Our results supported these conclusions; in addition, the different genetic models also suggest that SIRT1 has the properties of an atypical tumor suppressor. SIRTs appear to be the molecular/metabolic transducer for the adaptation to several external stimuli, representing a natural and quite obvious strategy for new therapeutical interventions. The modulation of their activity may show beneficial effects on preventing, retarding, and curing cardiovascular diseases, tumors, metabolic diseases, and aging. In addition, SIRTs seem to play an important role in cancers that are associated with the mitochondrial pathway and metabolism levels, findings that could be extended to the SIRT function in glioma. Several laboratories are working to bridge the beneficial effects of SIRTs in cancers, suggesting that compounds and their derivatives that affect SIRT activity may be new pharmacological agents that are naturally useful in cancer therapy and prevention.
Conclusion
In conclusion, we revealed that SIRT1 is a promoting factor in the tumorigenesis of glioma and acts through the Akt signaling pathway. SIRT1 might function as a novel target for selectively killing glioma. This study might provide insight into possible SIRT1 inhibition in the clinical treatment of GBM. In summary, our studies provided rational evidence for CCF-NLs use in the further preclinical development of liposomal formulations that preferentially target alternative cell death pathways to improve therapy in glioma treatment and achieve better preclinical outcomes.
Disclosure
The authors report no conflicts of interest in this work.
References
Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65:10457–10463. | ||
Vaziri H, Dessain SK, Ng Eaton E. hSIR2 (SIRT1) functions as an NAD dependent p53 deacetylase. Cell. 2001;107:149–159. | ||
Lim CS. Human SIRT1: a potential biomarker for tumorigenesis? Cell Biol Int. 2007;31:636–637. | ||
Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489–5504. | ||
Choi HN, Bae JS, Jamiyandor U. Expression and role of SIRT1 in hepatocellular carcinoma. Oncol Rep. 2011;26:503–510. | ||
Ikenoue T, Inoki K, Zhao B, Guan KL. PTEN acetylation modulates its interaction with PDZ domain. Cancer Res. 2008;68:6908–6912. | ||
Qu Y, Zhang J, Wu S, Li B, Liu S, Cheng J. SIRT1 promotes proliferation and inhibits apoptosis of human malignant glioma cell lines. Neurosci Lett. 2012;525(2):168–172. | ||
Wang J, Kim TH, Ahn MY, Lee J, Jung JH. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int J Oncol. 2012;41:1101–1109. | ||
Ota H, Tokunaga E, Chang K, Hikasa M. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006;25:176–185. | ||
Jin KL, Park JY, Noh EJ, Hoe KL. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. J Gynecol Oncol. 2010;21:262–268. | ||
Kojima K, Ohhashi R, Fujita Y, Hamada N. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun. 2008;373:423–428. | ||
Peck B, Chen CY, Ho KK, Di Fruscia P. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. 2010;9:844–855. | ||
Antal DS, Schwaiger S, Ellmerer-Müller EP, Stuppner H. Cotinus coggygria wood: novel flavanone dimer and development of an HPLC/UV/MS method for the simultaneous determination of fourteen phenolic constituents. Planta Med. 2010;76(15):1765–1772. | ||
Matić S, Stanić S, Bogojević D, et al. Extract of the plant Cotinus coggygria Scop. attenuates pyrogallol-induced hepatic oxidative stress in Wistar rats. Can J Physiol Pharmacol. 2011;89(6):401–411. | ||
Matić S, Stanić S, Bogojević D, et al. Methanol extract from the stem of Cotinus coggygria Scop., and its major bioactive phytochemical constituent myricetin modulate pyrogallol-induced DNA damage and liver injury. Mutat Res. 2013;755(2):81–89. | ||
Marčetić M, Božić D, Milenković M, Malešević N, Radulović S, Kovačević N. Antimicrobial, antioxidant and anti-inflammatory activity of young shoots of the smoke tree, Cotinus coggygria Scop. Phytother Res. 2013;27(11):1563–1658. | ||
Ratnam DV, Ankola DD, Bhardwaj V, Sahana DK, Ravi Kumar MNV. Role of antioxidants in prophylaxis and therapy: a pharmaceutical perspective. J Control Release. 2006;113:189–207. | ||
Svenson S. Dendrimers as versatile platform in drug delivery applications. Eur J Pharm Biopharm. 2009;71:445–462. | ||
Mahmud A, Xiong XB, Aliabadi HM, Lavasanifar A. Polymeric micelles for drug targeting. J Drug Target. 2007;15:553–584. | ||
Sarker DK. Engineering of nanoemulsions for drug delivery. Curr Drug Deliv. 2005;2:297–310. | ||
Bansal T, Mustafa G, Khan ZI, Ahmad FJ, Khar RK, Talegaonkar S. Solid self-nanoemulsifying delivery systems as a platform technology for formulation of poorly soluble drugs. Crit Rev Ther Drug Carrier Syst. 2008;25:63–116. | ||
Shah JC, Sadhale Y, Chilukuri DM. Cubic phase gels as drug delivery systems. Adv Drug Deliv Rev. 2001;47:229–250. | ||
Murakami T, Tsuchida K. Recent advances in inorganic nanoparticle-based drug delivery systems. Mini Rev Med Chem. 2008;8: 175–183. | ||
Junghanns JU, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomedicine. 2008;3:295–309. | ||
Prato M, Kostarelos K, Bianco A. Functionalized carbon nanotubes in drug design and discovery. Acc Chem Res. 2008;41:60–68. | ||
Misra RD. Quantum dots for tumor-targeted drug delivery and cell imaging. Nanomedicine. 2008;3:271–274. | ||
Fang JY, Hung CF, Hwang TL, Huang YL. Physicochemical characteristics and in vivo deposition of liposome-encapsulated tea catechins by topical and intratumor administrations. J Drug Target. 2005;13:19–27. | ||
Siddiqui IA, Adhami VM, Bharali DJ, et al. Introducing nanochemoprevention as a novel approach for cancer control: proof of principle with green tea polyphenol epigallocatechin-3-gallate. Cancer Res. 2009;69:1712–1716. | ||
Pazos M, Torres JL, Andersen ML, Skibsted LH, Medina I. Galloylated polyphenols efficiently reduce α-tocopherol radicals in a phospholipid model system composed of sodium dodecyl sulfate (sds) micelles. J Agric Food Chem. 2009;57:5042–5048. | ||
Lapenna S, Bilia AR, Morris GA, Nilsson M. Novel artemisinin and curcumin micellar formulations: drug solubility studies by NMR spectroscopy. J Pharm Sci. 2009;98:3666–3675. | ||
Wang G, Wang JJ, Chen XL, et al. JAK2/STAT3 and mitochondrial pathway is essential for quercetin nanoliposomes induced cell death in C6 glioma cells. Cell Death Dis. 2013;4:e746. | ||
Jang KY, Hwang SH, Kwon KS, et al. SIRT1 expression is associated with poor prognosis of diffuse large B-cell lymphoma. Am J Surg Pathol. 2008;32:1523–1531. | ||
Lu HF, Hsueh SC, Ho YT, et al. ROS mediates baicalin-induced apoptosis in human Promyelocytic leukemia HL-60 cells through the expression of the Gadd153 and mitochondrial dependent pathway. Anticancer Res. 2007;27:117–125. | ||
Lee JH, Li YC, Ip SW, et al. The role of Ca2+ in baicalein-induced apoptosis in human breast MDA-MB-231 cancer cells through mitochondria- and caspase-3-dependent pathway. Anticancer Res. 2008;28:1701–1711. | ||
Tomiyama A, Serizawa S, Tachibana K, Sakurada K, Samejima H, Kuchino Y. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J Natl Cancer Inst. 2006;98:1462–1473. | ||
Yuan H, Wang Z, Li L, Zhang H. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 2012;119:1904–1914. | ||
Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–427. | ||
Kuwabara M, Asanuma T, Niwa K, Inanami O. Regulation of cell survival and death signals induced by oxidative stress. J Clin Biochem Nutr. 2008;43:51–57. | ||
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. | ||
Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. | ||
Tomiyama A, Serizawa S, Tachibana K, et al. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J Natl Cancer Inst. 2006;98:1462–1473. | ||
Bommareddy A, Eggleston W, Prelewicz S, et al. Chemoprevention of prostate cancer by major dietary phytochemicals. Anticancer Res. 2013;33:4163–4174. | ||
Tollefsbol TO. Dietary epigenetics in cancer and aging. Cancer Treat Res. 2014;159:257–267. | ||
Weng CJ, Yen GC. Flavonoids, a ubiquitous dietary phenolic subclass, exert extensive in vitro anti-invasive and in vivo anti-metastatic activities. Cancer Metastasis Rev. 2012;31:323–325. | ||
Seguin J, Brullé L, Boyer R, et al. Liposomal encapsulation of the natural flavonoid fisetin improves bioavailability and antitumor efficacy. Int J Pharm. 2013;444(1–2):146–154. | ||
Thybo P, Kristensen J, Hovgaard L. Characterization and physical stability of tolfenamic acid-PVP K30 solid dispersions. Pharm Dev Technol. 2007;12(1):43–53. | ||
Siepmann J, Gopferich A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv Drug Deliv Rev. 2001;48(2–3): 229–247. | ||
Lee JC, Wong DT, Discher DE. Direct measures of large, anisotropic strains in deformation of the erythrocyte cytoskeleton. Biophys J. 1999;77(2):853–864. | ||
Photos PJ, Bacakova L, Discher B, Bates FS, Discher DE. Polymer vesicles in vivo: correlations with PEG molecular weight. J Control Release. 2003;90(3):323–334. | ||
Bermudez H, Brannan AK, Hammer DA, Bates FS, Discher DE. Molecular weight dependence of polymersome membrane structure, elasticity, and stability. Macromolecules. 2002;35(21):8203–8208. | ||
Seguin J, Brullé L, Boyer R, et al. Liposomal encapsulation of the natural flavonoid CCF improves bioavailability and antitumor efficacy. Int J Pharm. 2013;444(1–2):146–154. | ||
Lemasters JJV. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol. 1999;276:G1–G6. | ||
Syntichaki P, Tavernarakis N. Death by necrosis. Uncontrollable catastrophe, or is there order behind the chaos? EMBO Rep. 2002;3:604–609. | ||
Proskuryakov SY, Gabai VL, Konoplyannikov AG. Necrosis is an active and controlled form of programmed cell death. Biochemistry. 2002;67:387–408. | ||
Belka C, Jendrossek V, Pruschy M, Vink S, Verheij M, Budach W. Apoptosis-modulating agents in combination with radiotherapy-current status and outlook. Int J Radiat Oncol Biol Phys. 2004;58:542–554. | ||
Perry A, Schmidt RE. Cancer therapy-associated CNS neuropathology: an update and review of the literature. Acta Neuropathol. 2006;111:197–212. | ||
Luo J, Nikolaev AY, Imai S. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. | ||
Cherbonnel-Lasserre C, Dosanjh MK. Suppression of apoptosis by overexpression of Bcl-2 or Bcl-xL promotes survival and mutagenesis after oxidative damage. Biochimie. 1997;79:613–617. | ||
Luan S, Sun L, Huang F. MicroRNA-34a: a novel tumor suppressor in p53-mutant glioma cell line U251. Arch Med Res. 2010;41(2):67–74. | ||
Benchimol S. p53-dependent pathways of apoptosis. Cell Death Differ. 2001;8:1049–1051. | ||
Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. 2010;9:844–855. | ||
Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–251. | ||
Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol. 2005;17:631–636. | ||
Alaimo A, Gorojod RM, Kotler ML. The extrinsic and intrinsic apoptotic pathways are involved in manganese toxicity in rat astrocytoma C6 cells. Neurochem Int. 2011;59(2):297–308. | ||
Park CM, Park MJ, Kwak HJ, et al. Induction of p53- mediated apoptosis and recovery of chemosensitivity through p53 transduction in human glioblastoma cells by cisplatin. Int J Oncol. 2006;28:119–125. | ||
Jane EP, Premkumar DR, Pollack IF. AG490 influences UCN-01-induced cytotoxicity in glioma cells in a p53-dependent fashion, correlating with effects on BAX cleavage and BAD phosphorylation. Cancer Lett. 2007;257(1):36–46. | ||
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. | ||
Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P. Salermide, a sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009;28:781–791. | ||
Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. | ||
Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer. Nat Rev Cancer. 2009;9:123–128. | ||
Heltweg B, Gatbonton T, Schuler AD, Posakony J. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66:4368–4377. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.