Back to Journals » Infection and Drug Resistance » Volume 13
Role of Monocytes/Macrophages in Covid-19 Pathogenesis: Implications for Therapy
Authors Gómez-Rial J ![]() , Rivero-Calle I
, Rivero-Calle I ![]() , Salas A, Martinón-Torres F
, Salas A, Martinón-Torres F ![]()
Received 18 April 2020
Accepted for publication 26 June 2020
Published 22 July 2020 Volume 2020:13 Pages 2485—2493
DOI https://doi.org/10.2147/IDR.S258639
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Jose Gómez-Rial,1,2 Irene Rivero-Calle,1,3 Antonio Salas,1,4 Federico Martinón-Torres1,3
1Genetics, Vaccines, Infectious Diseases Research Group (GENVIP), Health Research Institute Santiago (IDIS), Hospital Clínico Universitario Santiago de Compostela (SERGAS), Galicia 15706, Spain; 2Immunology Laboratory, Clinical Laboratory, Hospital Clínico Universitario Santiago de Compostela (SERGAS), Galicia 15706, Spain; 3Translational Pediatrics and Infectious Diseases, Department of Pediatrics, Hospital Clínico Universitario de Santiago De Compostela, Galicia 15706, Spain; 4Unidade de Xenética, Instituto de Ciencias Forenses (INCIFOR), Facultade de Medicina, Universidade de Santiago de Compostela, and GenPoB Research Group, Instituto de Investigaciones Sanitarias (IDIS), Hospital Clínico Universitario de Santiago (SERGAS), Galicia, 15706, Spain
Correspondence: Jose Gómez-Rial Tel +34 981 950 379
Email [email protected]
cov-Abstract: Emerging studies from SARS-CoV-2-infected patients indicate a preponderant role of monocytes/macrophages in the pathogenesis of this viral infection, in a similar way to that previously observed in other coronavirus outbreaks (SARS and MERS). The clinical presentation of severe patients resembles viral-associated hemophagocytic syndrome, a rare condition previously seen during lethal influenza pandemics and during previous SARS and MERS coronavirus outbreaks. SARS-CoV-2 infection triggers an over-exuberant inflammatory response due to the development of a cytokine storm and the depletion of the adaptative immune compartment, which may prelude sepsis in many cases. The present review summarizes past evidence on the role of monocytes/macrophages in previous coronavirus outbreaks and the emerging knowledge on their role in COVID-19 pathogenesis. Treatment strategies incorporating the blockade of migration and differentiation of monocyte-macrophage, such as granulocyte macrophage-colony stimulating factor inhibitors, might enhance the promising results seen so far with selective cytokine blockade.
Keywords: monocyte/macrophage, COVID-19 infection, anti-GM-CSF, coronavirus
Introduction
Novel coronavirus infection (SARS-Cov-2) produces a severe respiratory syndrome, similar to the severe acute respiratory syndrome (SARS) observed in previous coronavirus outbreaks. This syndrome is associated to intensive care unit (ICU) admission and high lethality rates.1 Emerging studies on SARS-CoV-2 pathogenesis point to infiltration of pro-inflammatory monocytes as key mediators of the hyper-inflammatory response produced during viral shedding in the infectious phase, and mainly involved in the cytokine storm observed during the inflammatory phase in severe cases. Zhang et al observed that during SARS-CoV-2 infection, there are morphological and inflammation-related phenotypic changes in peripheral blood monocytes that correlate with the patient’s outcome, suggesting that an excessive monocyte-macrophage activation may lead to the subsequent respiratory failure in severe patients.2 Characterization of lung immune microenvironment through bronchoalveolar lavage fluid in first SARS-CoV-2 patients also showed a predominant monocyte-derived macrophage infiltration in the severely damaged lungs and a highly expanded clonal CD8+T cell in mild patients; suggesting that a robust adaptive immune response is connected to a successful control of SARS-CoV-2 infection.3
Dysregulation of the immune response is one of the hallmarks of severe SARS-CoV-2 infection, with lower lymphocytes counts and an increased neutrophil–lymphocyte ratio. Acute phase reactants levels (C-reactive protein (CRP) and ferritin) are highly increased along with inflammatory cytokines such as IL-6.4 This stimulates the production of an excessive, non-effective host immune response by innate cells, which is associated with severe lung injury. Hyperinflammation observed in severe infection results in an immunosuppression status with a clinical course that resembles hemophagocytic syndrome, triggering the production of fatal hypercytokinaemia and consequently multiorgan failure.5
Lessons Learned from SARS and MERS Coronavirus Outbreaks
Other coronaviruses have caused major outbreaks of severe respiratory infections in the 21st century. In 2002, SARS-Cov-1 was identified as the cause of severe acute respiratory syndrome (SARS) in the Guangdong province of China, which subsequently spread to more than 30 countries with more than 8000 cases reported worldwide and a 10% case fatality rate.6 Ten years later, an outbreak of another coronavirus in the Arabian Peninsula was identified, called Middle East respiratory syndrome (MERS), provoking a severe, acute respiratory illness similar to SARS with nearly of 2500 cases in at least 27 countries.7
These previous highly pathogenic human respiratory coronaviruses showed close similitudes with the current SARS-CoV-2 outbreak, triggering an exuberant inflammatory response leading to respiratory failure and lung damage. Studies of these previous coronavirus outbreaks showed that a robust viral replication was accompanied by a delayed type I interferon (IFN-I) signalling and a lung immunopathology with very low survival.8,9 This delayed activation of the IFN-I pathway and promoted the accumulation of inflammatory monocytes infiltrating from peripheral blood, resulting in a cytokine storm and the absence of a virus-specific T cell response. A characteristic of patients who developed severe SARS was an increased number of macrophages in the lungs10,11 signalling this cell as the key mediator to lung destruction.
Furthermore, human coronaviruses have showed their ability to infect human leukocytic cell lines and peripheral blood mononuclear cells.12 Moreover, coronavirus infection of a primary monocytic cell line leads to cell activation and an aberrant production of pro-inflammatory mediators with increased chemoattraction. This observation suggests that, for some human coronavirus, monocytes/macrophages could serve as a reservoir which, consequently, would work as vectors for viral dissemination to other tissues.13,14 This fact has so far not been proven for SARS-CoV-2. Infection of macrophages might be enhanced by an antibody-dependent effect, where previous non-neutralizing antibodies against coronavirus facilitate binding to Fc receptors in monocytes and macrophages.15 In vitro infection of human macrophages by SARS-CoV-1 induced high expression of chemokines such as CXCL10 and CCL2 and a poor induction of Interferon-β.16 In another study, strong up-regulation of several inflammatory chemokines (MIP-1α, RANTES, IP-10 and MCP-1) linked to a low expression of the antiviral cytokines interferon-α/β/γ and IL-12 in SARS-infected monocytes was interpreted as a escape mechanism for coronavirus.17
Differences in the transcriptional profile of monocytic cells infected with different coronaviruses were also observed. SARS-CoV-1 induced down-regulation of interferon-α/β and cathepsin/proteasome genes, while hypoxia/hyperoxia related genes were up-regulated. However, coronavirus 229E (Cov-229E), causing common cold, does not induce these changes, showing that regulation of immune-related genes in monocyte/macrophage might be important for differences in pathogenesis between coronavirus strains.18 Modulation of intrinsic functions of monocyte-derived macrophages and dendritic cells by coronavirus-induced lung epithelial cytokines (IL-6 and IL-8) was responsible for the exacerbated pathogenesis during SARS-CoV-1 infection of human lung epithelial cells in culture.19 Given the importance of monocyte/macrophage cells during the immune response, it is possible that infection by coronaviruses and alteration of their function may be an important clue to unravel the course of infection in humans.
SARS-CoV-1 also demonstrated potential neurotropism, as several patients experienced central nervous symptoms during the course of the illness. Moreover, the brain tissue revealed the presence of high amounts of the monokine-induced by interferon-γ (MIG) produced by monocytic cells, and infiltration of CD68+ monocytes/macrophages and CD3+ T lymphocytes in the brain mesenchyme20 during pathologic examination.
Cytokine Storm and Immunopathology
A large number of clinical data collected from SARS-CoV-2 patients suggest the presence of a mild-to-severe cytokine storm in patients requiring admission, which has also been argued as an important cause of death.5,21 Cytokine storm or cytokine release syndrome (CRS) is a form of systemic inflammatory response usually triggered by a variety of factors such as infections and drugs.22 CRS occurs when large numbers of leucocytes are activated and release pro-inflammatory cytokines, which recruits and activates more leucocytes in a positive feedback loop of hyperinflammation.23 Recently, a key role for macrophages in driving CRS was revealed by analysing CRS produced following chimeric antigen receptor-modified T (CAR T) therapy.24,25 These studies identified human monocytes as the major source of IL-1, IL-6 and nitric oxide – the main hallmarks of CRS. Monocyte depletion or therapeutic depletion of IL-6 and IL-1 prevented CRS in this model. Similarly, in two clinically relevant animal models, depletion of macrophages at disease onset was shown to decrease lethality and limit the cytokine storm, pointing to macrophage-based therapies for CRS.26
CRS is closely linked to the macrophage activation syndrome (MAS), a life-threatening complication of several autoimmune diseases related to hemophagocytic lymphohistiocytosis (HLH). MAS is characterized by pancytopenia, liver insufficiency, hyperferritinemia, coagulopathy and neurologic symptoms due to uncontrolled proliferation of well-differentiated macrophages; leading to widespread hemophagocytosis and cytokine overproduction.27,28
Severe COVID-19 patients developing pneumonia exhibit features of the systemic hyper-inflammation that is characteristic of MAS.29,30 This overexuberant immune response associated with MAS may be driving SARS-CoV-2 related adult respiratory distress syndrome (ARDS), as typical of severe SARS pathogenesis.
At the same time, viral infection can trigger MAS and HLH, in what is known as virus-associated hemophagocytic syndrome (VAHS), a severe complication of various viral infections often resulting in multiorgan failure and death.31 These phenomena have been observed during lethal influenza pandemics, such as 2009 influenza A H1N1, 1998 avian influenza H5N1, and 1918 Spanish flu H1N1.32,33 VAHS clinical course presented as an aggressive, life-threatening condition, analogous to HLH, and associated with massive pro-inflammatory cytokine release, elevated plasma levels of acute phase reactants, and the accumulation of activated T-lymphocytes and macrophages in various organs. Further, VAHS has been linked to the high fatality rate in MERS-Cov outbreak and SARS-CoV,34 and is now linked to the current SARS-CoV-2.5,35
Also, MAS and VAHS are linked to thrombotic events and coagulopathy36,37 highlighting the key role of monocyte-macrophages in the regulation of coagulation events.38 Monocytes and macrophages can act as procoagulant factors, and interact with blood coagulation mechanisms resulting in thrombus formation and extravascular fibrin accumulation39 COVID-19 is linked to a microvascular vessel obstructive thrombo-inflammatory syndrome in lung and other vital organs, leading to multiple organ failure and death.40,41
Action of IL-6 and GM-CSF on Monocyte-Macrophage Differentiation
IL-6 and granulocyte macrophage-colony stimulating factor (GM-CSF) are cytokines closely linked to regulation of monocyte activation and differentiation to macrophages. Both cytokines increased during the cytokine storm in SARS-CoV-2 patients requiring ICU29 and are involved in the immunopathology of autoimmunity and the inflammatory condition; they were identified as therapeutic blockade targets of the cytokine storm during systemic inflammatory responses, as observed in MAS and HLH.42,43 In this sense, emerging reports indicate that SARS-CoV-2 infection precedes the onset of various autoimmune and autoinflammatory diseases, including the newly described paediatric inflammatory multisystemic syndrome.44 This new evidence reinforces the link between infectious diseases and the triggering of autoimmune and autoinflammatory sequelae.45
IL-6 is an essential factor in the molecular control of monocyte activation and differentiation. It is secreted by a variety of cellular types, including macrophages, fibroblasts, T-cells and endothelial cells46 in response to infection, and it sends out an early warning signal to the entire body, activating inflammatory events and immunity. IL-6 regulates the differentiation of monocytes to macrophages, up-regulating expression of functional GM-CSF receptors on monocytes, and switching the differentiation of monocytes from dendritic cells with antigen-presenting functions to inflammatory macrophages.47 IL-6 is the chief stimulator of the production of most acute phase proteins, and critical for the febrile response. During systemic inflammation such as in MAS, IL-6 is pyrogenic by binding to IL-6 receptors on brain endothelial cells to induce prostaglandin synthesis and aggravate inflammation;48 furthermore, it dictates the transition from acute to chronic inflammation by changing the nature of the leucocyte infiltrate from polymorphonuclear neutrophils to monocyte/macrophages.49
Obesity and diabetes, both risk factors for greater severity in COVID-19 patients, are closely related to perturbation of the monocyte compartment, with a marked shift toward a pro-inflammatory phenotype which might contribute to the development of low-grade inflammation observed in obesity.50 IL-6 has been highlighted as a driver of this metabolic inflammation51 and therefore increased risk for severe COVID-19 condition.
Moreover, during CRS, IL-6 is an important member of the cytokine network and plays a key role in acute inflammation. In SARS-CoV-2 infection, blockade of IL-6 signal transduction with Tocilizumab has become an important therapeutic weapon in a small series of severe cases with early reports of success; this evidence demonstrates that blockade of cytokine storm is an effective therapeutic option.21
GM-CSF, another monocyte/macrophage-related cytokine, has an important effect under inflammatory conditions, promoting neutrophil and monocyte migration, proliferation and maturation. GM-CSF stimulates stem cells to produce granulocytes and monocytes, and promotes migration of monocytes to the inflammatory tissues and differentiation to inflammatory macrophages.52 In autoimmune inflammatory diseases, such as rheumatoid arthritis and multiple sclerosis, GM-CSF has a pathogenic role; several inhibitory drugs are currently undergoing clinical trials.53 Both autoimmune conditions are frequently associated with viral infections, suggesting a key role for viruses in the dysregulation of tolerance immune mechanisms and the triggering of autoimmune diseases.54
In a similar way, GM-CSF blockade has been demonstrated to inhibit the cytokine release syndrome and neuroinflammation in the management of CART cell therapy-associated toxicities,55 thus arising as a promising drug for SARS-CoV-2 patients.
Monocytes, ACE2 and RAAS
It has been reported that ACE2 is the main host cell receptor for human pathogenic coronaviruses (SARS-CoV-1, MERS and SARS-CoV-2) and that it plays a key role in the entry of the virus into the cell and viral spreading and pathogenesis, interfering in the renin-angiotensin-aldosterone system (RAAS).56 ACE2 is widely distributed in various human tissues and may be a determinant factor of activity in severe disease pathogenesis57 Several immune cells are involved in the RAAS, playing a pivotal role in the regulation of vascular inflammation and hypertension.58 Human monocyte subsets exhibit different expression on angiotensin-converting enzyme type 1 and 2 (ACE1 and ACE2) and may be directly involved in the regulation of vascular homeostasis via the RAAS mechanisms59 Therefore, SARS-CoV-2 activated-monocytes could also be critically involved in the pathogenic effect on the vascular homeostasis, through perturbation of ACE2 function.
RAAS activation in monocytes through its physiological effectors plays a key role in promoting and maintaining inflammation. Monocytes have been demonstrated to be actively involved in all key stages of acute coronary syndromes and constitute an important part of the coagulation system60 Therefore, SARS-CoV-2 activation of monocytes and perturbation of RAAS through ACE2-monocyte activation may trigger acute coronary syndromes in predisposed patients, as shown in COVID-19 infection.61
Antibody-Dependent Enhancement of Macrophages
Antibody-dependent enhancement (ADE) of virus infection is a phenomenon whereby virus-specific non-neutralizing antibodies enhance the entry of the virus, and in some cases the replication, into monocytes/macrophages through interaction with Fc receptors;62 the result is that a normally mild viral infection becomes life-threatening. This phenomenon has been described as mediator of acute lung injury in both SARS and MERS outbreaks63,64 and demonstrated in animal models. This evidence suggests that people exposed to MERS-CoV who failed to develop a neutralizing antibody response may be at risk of severe lung disease on re-exposure to MERS-CoV.65 Similarly, evaluation of SARS vaccines in mice induced protection against infection but challenged animals exhibited an exacerbated immunopathologic-type lung disease.66 Anti-spike viral protein antibodies were identified as mediators of the infection of immune cells15,67 causing severe acute lung injury by skewing macrophage responses during acute SARS-CoV-1 infection.34 Recent studies have revealed complex roles for antibodies in viral entry, describing an ADE effect with neutralizing antibody to the surface spike protein of coronaviruses, and molecular mechanisms for ADE in coronavirus entry.68 Binding of these antibodies to the viral spike triggers a conformational change that mediates viral entry into Fc receptor-expressing cells. This evidence reveals the impact of antibody dosages on viral entry and serves as a guide for future vaccine design and antibody-based drug therapies69 The potential danger of these suboptimal antibody responses has been also noted for COVID-19 highlighting the importance of safety evaluation of candidate vaccines.70
ADE effect on monocytes is also currently proposed as an explanation for the geographic discrepancy in the pathogenesis of the current SARS-CoV-2 epidemic around the world. Prior exposure to similar antigenic epitopes from local circulating coronaviruses might be a possible explanation for the geographic differences of lethality rates; it may also provide an explanation to the high death rates among elderly people, more exposed to previous coronaviruses.71
This ADE mechanism is not exclusive to coronaviruses and was also observed during influenza pandemics,72 in dengue73 and VIH74 viral infections, leading to enhanced pathology.
Targeting IL-6 and GM-CSF as a Therapeutic Approach in SARS-CoV-2 Patients
Several biological agents targeting pro-inflammatory cytokines have demonstrated effectiveness in SARS-CoV-2 severe pathology75 Tocilizumab is a recombinant human IL-6 monoclonal antibody, which binds to soluble and membrane-bound IL-6 receptors (IL-6R). Blocking IL-6 signalling is highly effective in the attenuation of inflammatory responses76 according to the experience gained in rheumatic and other inflammatory diseases, and the prevention of CRS caused by CART immunotherapy.
Other drugs proposed for avoiding harmful CRS syndrome in SARS-CoV-2 patients are JAK inhibitors drugs, which inhibit signalling cascades for a variety of inflammatory cytokines.77 However, one of the most significant concerns about JAK inhibitors is the inhibition of IFN-α, with anti-viral properties and theoretically useful against SARS-Cov2; further, JAK inhibitors also inhibit IL-10, which is the main regulator cytokine of the immune system.
An interesting alternative, in combination with cytokine blocking drugs, could be the use of GM-CSF inhibitors to inhibit monocyte/macrophage differentiation and migration to pulmonary tissues. Several GM-CSF inhibitor drugs are currently undergoing clinical trials for rheumatic arthritis (RA) and other inflammatory conditions. Otilimab (GSK3196165) is a fully human antibody against GM-CSF, currently in Phase III in patients with RA, and it has shown promising results in initial developmental phases. Namilumab (MT203) is currently being tested for application in RA and psoriatic arthritis. Lenzilumab (KB003), is a humanized monoclonal antibody that targets GM-CSF originally designed for the treatment of chronic myelomonocytic leukaemia; it is currently under clinical trial for refractory large B-cell lymphoma and has demonstrated to be effective in vitro to avoid CRS in CART-associated toxicities.55 Very recently, GM-CSF neutralization with lenzilumab showed positive results related to improved clinical outcomes, oxygen requirement and ameliorated cytokine storm in a single-centre study78 Mavrilimumab is a human monoclonal antibody that inhibits human GM-CSF receptor (GM-CSF-R) and is also an investigational drug for the treatment of rheumatic diseases79 Also, very recently, mavrilimumab treatment has shown improved clinical outcomes in non-mechanically ventilated patients with severe COVID-19 pneumonia and systemic hyperinflammation in a single-centre study80 The use of this monocyte-macrophage differentiation inhibitor drugs could be employed at initial phases of CRS to avoid massive migration of inflammatory monocytes to pulmonary tissues, preventing the cytokine storm and immunopathology associated to SARS-CoV-2 infection. A combination of GM-CSF inhibitor drugs and selective cytokine blockers could act synergistically to ameliorate lung injury (Figure 1) in patients at high risk of ICU admission and mechanical ventilation support. Monitorization of pro-inflammatory cytokines and acute phase reactants levels would help identify patients who can benefit from this therapy. Results from ongoing clinical trials will provide more extensive knowledge about indications and potential limitations of this therapy.
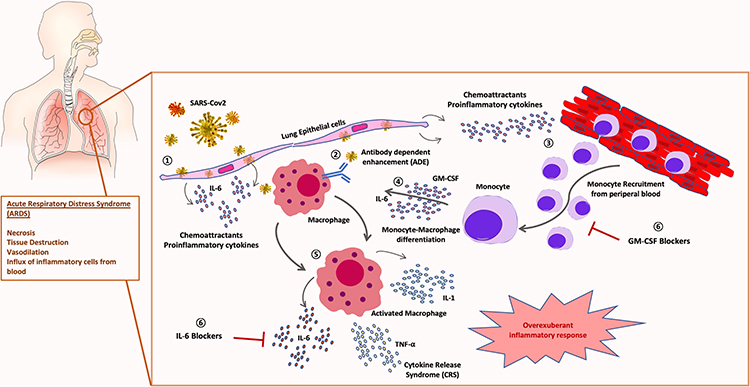 |
Figure 1 Role of monocyte-macrophage in SARS-CoV-2 pathogenesis: 1) SARS-CoV-2 infection of lung tissue induces release of chemoattractant proinflammatory cytokines by epithelial cells and fibroblasts. 2) Viral antibody-dependent enhancement (ADE) of macrophages could trigger SARS-CoV-2 infection of leucocytes. 3) Chemoattractant gradient induces massive recruitment of inflammatory monocytes from peripheral blood. 4) Monocyte-macrophage activation and differentiation is triggered by GM-CSF and IL-6. 5) An overexuberant activation of macrophages produces the cytokine release syndrome (CRS) responsible for the acute respiratory distress syndrome (ARDS) typical of severe patients. 6) Therapeutical blockade of IL-6 and GM-CSF combination could avoid severe lung immunopathology in COVID-19 patients. |
Future Directions
Undoubtedly, monocyte-macrophage cells have a preponderant role in the immunopathology associated to severe outcome in SARS-CoV-2 infection. Given that these cells are considered drivers of CRS, future studies should focus on monitoring them, aiming to early detect individuals initiating uncontrolled and ineffective immune responses and developing targeted therapeutic approaches. This could shed light on strategies to prevent or at least decrease lung injury and the high lethal rates associated to SARS-CoV-2. Treatment strategies that encompass blocking migration and differentiation of these cells, such as GM-CSF inhibitors, might enhance the promising results seen with selective cytokine blockade.
Acknowledgments
This study received support from the Instituto de Salud Carlos III: project GePEM (Instituto de Salud Carlos III(ISCIII)/PI16/01478/Cofinanciado FEDER), DIAVIR (Instituto de Salud Carlos III(ISCIII)/DTS19/00049/Cofinanciado FEDER; Proyecto de Desarrollo Tecnológico en Salud) and Resvi-Omics (Instituto de Salud Carlos III(ISCIII)/PI19/01039/Cofinanciado FEDER) and project BI-BACVIR (PRIS-3; Agencia de Conocimiento en Salud (ACIS)—Servicio Gallego de Salud (SERGAS)—Xunta de Galicia; Spain) given to A.S.; and project ReSVinext (Instituto de Salud Carlos III(ISCIII)/PI16/01569/Cofinanciado FEDER), and Enterogen (Instituto de Salud Carlos III(ISCIII)/PI19/01090/Cofinanciado FEDER) given to F.M.-T.
Disclosure
Jose Gomez-Rial reports personal fees from and being a speaker in scientific meetings and advisor for Merck Sharp and Dohme, GlaxoSmithkline, and Pfizer, outside the submitted work. Irene Rivero-Calle reports trials fees paid to the institution from Ablynx, Jansen, and Mediummune, grants, personal fees, other from GSK, grants and personal fees (research grants and honoraria as an advisor and speaker, and for attending conferences and practical courses; trials fees paid to the institution) from Pfizer, MSD, Sanofi Pasteur, outside the submitted work. Federico Martinón-Torres has received honoraria from GSK, Pfizer, Sanofi Pasteur, MSD, Seqirus, and Janssen for taking part in advisory boards and expert meetings, and for acting as speaker in congresses outside the scope of the submitted work. FM-T has also acted as principal investigator in RCTs of the above-mentioned companies as well as Ablynx, Regeneron, Roche, Abbott, Novavax, and MedImmune, with honoraria paid to his institution. The authors report no other possible conflicts of interest in this work.
References
1. Lin YC, Chen SC, Huang CM, et al. Clinical features and diagnosis of new malignancy in patients with acute pulmonary embolism and without a history of cancer. J Chin Med Assoc. 2020;83(3):245–250.
2. AlBshabshe A, Al-Asmary M, Al-Harthi M, et al. Usage of venous thromboembolism prophylaxis at a tertiary care hospital in Aseer region of Saudi Arabia. Saudi Med J. 2015;36(11):1367–1368. doi:10.15537/smj.2015.11.12231
3. Almasood A, Al Ahmari S, El-Shurafa H, et al. The change in mitral regurgitation severity after trans-catheter aortic valve implantation. J Saudi Heart Assoc. 2015;27(1):10–17. doi:10.1016/j.jsha.2014.05.002
4. Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. 2020.
5. Mehta P, McAuley DF, Brown M, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. doi:10.1016/S0140-6736(20)30628-0
6. Peiris JS, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med. 2004;10(12 Suppl):S88–97. doi:10.1038/nm1143
7. Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet. 2015;386(9997):995–1007. doi:10.1016/S0140-6736(15)60454-8
8. Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–193. doi:10.1016/j.chom.2016.01.007
9. Lau SKP, Lau CCY, Chan KH, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol. 2013;94(Pt 12):2679–2690. doi:10.1099/vir.0.055533-0
10. Franks TJ, Chong PY, Chui P, et al. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol. 2003;34(8):743–748. doi:10.1016/S0046-8177(03)00367-8
11. Nicholls J, Dong XP, Jiang G, Peiris M. SARS: clinical virology and pathogenesis. Respirology. 2003;8(s1):S6–8. doi:10.1046/j.1440-1843.2003.00517.x
12. Yilla M, Harcourt BH, Hickman CJ, et al. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005;107(1):93–101. doi:10.1016/j.virusres.2004.09.004
13. Desforges M, Miletti TC, Gagnon M, Talbot PJ. Activation of human monocytes after infection by human coronavirus 229E. Virus Res. 2007;130(1–2):228–240. doi:10.1016/j.virusres.2007.06.016
14. Zhou J, Chu H, Li C, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209(9):1331–1342. doi:10.1093/infdis/jit504
15. Yip MS, Leung NH, Cheung CY, et al. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J. 2014;11:82. doi:10.1186/1743-422X-11-82
16. Cheung CY, Poon LL, Ng IH, et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. 2005;79(12):7819–7826. doi:10.1128/JVI.79.12.7819-7826.2005
17. Law HK, Cheung CY, Ng HY, et al. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106(7):2366–2374. doi:10.1182/blood-2004-10-4166
18. Hu W, Yen YT, Singh S, Kao CL, Wu-Hsieh BA. SARS-CoV regulates immune function-related gene expression in human monocytic cells. Viral Immunol. 2012;25(4):277–288. doi:10.1089/vim.2011.0099
19. Yoshikawa T, Hill T, Li K, Peters CJ, Tseng CT. Severe acute respiratory syndrome (SARS) coronavirus-induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J Virol. 2009;83(7):3039–3048. doi:10.1128/JVI.01792-08
20. Xu J, Zhong S, Liu J, et al. Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin Infect Dis. 2005;41(8):1089–1096. doi:10.1086/444461
21. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int J Antimicrob Agents. 2020;55(5):105954. doi:10.1016/j.ijantimicag.2020.105954
22. Shimabukuro-Vornhagen A, Godel P, Subklewe M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. doi:10.1186/s40425-018-0343-9
23. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729
24. Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi:10.1038/s41591-018-0036-4
25. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–738. doi:10.1038/s41591-018-0041-7
26. Sumaili H, Al-Kofide A, Al-Seraihi A, et al. Outcome of pediatric patients with lymphoma following stem cell transplant: a single institution report. Leuk Lymphoma. 2015;56(5):1327–1334. doi:10.3109/10428194.2014.951846
27. Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002;14(5):548–552. doi:10.1097/00002281-200209000-00012
28. Lerkvaleekul B, Vilaiyuk S. Macrophage activation syndrome: early diagnosis is key. Open Access Rheumatol. 2018;10:117–128. doi:10.2147/OARRR.S151013
29. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi:10.1016/S0140-6736(20)30183-5
30. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. 2020;19(6):102537. doi:10.1016/j.autrev.2020.102537
31. Beutel G, Wiesner O, Eder M, et al. Virus-associated hemophagocytic syndrome as a major contributor to death in patients with 2009 influenza A (H1N1) infection. Crit Care. 2011;15(2):R80. doi:10.1186/cc10073
32. Hsieh YC, Wu TZ, Liu DP, et al. Influenza pandemics: past, present and future. J Formos Med Assoc. 2006;105(1):1–6. doi:10.1016/S0929-6646(09)60102-9
33. Henter JI, Chow CB, Leung CW, Lau YL. Cytotoxic therapy for severe avian influenza A (H5N1) infection. Lancet. 2006;367(9513):870–873. doi:10.1016/S0140-6736(06)68232-9
34. Liu L, Wei Q, Lin Q, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019;4(4). doi:10.1172/jci.insight.123158.
35. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect. 2020;80(6):607–613. doi:10.1016/j.jinf.2020.03.037
36. Chiang WC, Wu MS, Tsai CC, Lin SL, Tsai TJ, Hsieh BS. Thrombotic microangiopathy in hemophagocytic syndrome: a case report. J Formos Med Assoc. 2002;101(5):362–367.
37. Valade S, Mariotte E, Azoulay E. Coagulation disorders in hemophagocytic lymphohistiocytosis/macrophage activation syndrome. Crit Care Clin. 2020;36(2):415–426. doi:10.1016/j.ccc.2019.12.004
38. Tracy PB, Allen DH, Worfolk LA, Lawler RR. Monocyte/macrophage regulation of coagulant events. Haemostasis. 1996;26(Suppl 1):6–11. doi:10.1159/000217230
39. Helin H. Macrophage procoagulant factors–mediators of inflammatory and neoplastic tissue lesions. Med Biol. 1986;64(4):167–176.
40. Ciceri F, Beretta L, Scandroglio AM, et al. Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis. Crit Care Resusc. 2020;15.
41. Klok FA, Kruip M, van der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–147. doi:10.1016/j.thromres.2020.04.013
42. Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016;8(8):959–970. doi:10.2217/imt-2016-0020
43. Spath S, Komuczki J, Hermann M, et al. Dysregulation of the cytokine GM-CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity. 2017;46(2):245–260. doi:10.1016/j.immuni.2017.01.007
44. Whittaker E, Bamford A, Kenny J, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA. 2020. doi:10.1001/jama.2020.10369
45. Galeotti C, Bayry J. Autoimmune and inflammatory diseases following COVID-19. Nat Rev Rheumatol. 2020. doi:10.1038/s41584-020-0448-7
46. Akira S, Taga T, Kishimoto T. Interleukin-6 in biology and medicine. Adv Immunol. 1993;54:1–78.
47. Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol. 2000;1(6):510–514. doi:10.1038/82763
48. Eskilsson A, Mirrasekhian E, Dufour S, Schwaninger M, Engblom D, Blomqvist A. Immune-induced fever is mediated by IL-6 receptors on brain endothelial cells coupled to STAT3-dependent induction of brain endothelial prostaglandin synthesis. J Neurosci. 2014;34(48):15957–15961. doi:10.1523/JNEUROSCI.3520-14.2014
49. Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3. doi:10.1186/ar1917
50. Friedrich K, Sommer M, Strobel S, et al. Perturbation of the monocyte compartment in human obesity. Front Immunol. 2019;10:1874. doi:10.3389/fimmu.2019.01874
51. Sindhu S, Thomas R, Shihab P, Sriraman D, Behbehani K, Ahmad R. Obesity is a positive modulator of IL-6R and IL-6 expression in the subcutaneous adipose tissue: significance for metabolic inflammation. PLoS One. 2015;10(7):e0133494. doi:10.1371/journal.pone.0133494
52. Hamilton JA. GM-CSF in inflammation and autoimmunity. Trends Immunol. 2002;23(8):403–408. doi:10.1016/S1471-4906(02)02260-3
53. Lotfi N, Thome R, Rezaei N, et al. Roles of GM-CSF in the pathogenesis of autoimmune diseases: an update. Front Immunol. 2019;10:1265. doi:10.3389/fimmu.2019.01265
54. Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM. Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses. 2019;11(8):762. doi:10.3390/v11080762
55. Sterner RM, Sakemura R, Cox MJ, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697–709. doi:10.1182/blood-2018-10-881722
56. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi:10.1016/S0140-6736(20)30251-8
57. Bourgonje AR, Abdulle AE, Timens W, et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol. 2020;251(3):228–248. doi:10.1002/path.5471
58. Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol. 2019;19(8):517–532. doi:10.1038/s41577-019-0160-5
59. Rutkowska-Zapala M, Suski M, Szatanek R, et al. Human monocyte subsets exhibit divergent angiotensin I-converting activity. Clin Exp Immunol. 2015;181(1):126–132. doi:10.1111/cei.12612
60. Shantsila E, Lip GY. Monocytes in acute coronary syndromes. Arterioscler Thromb Vasc Biol. 2009;29(10):1433–1438. doi:10.1161/ATVBAHA.108.180513
61. Bangalore S, Sharma A, Slotwiner A, et al. ST-segment elevation in patients with Covid-19 - a case series. N Engl J Med. 2020;382(25):2478–2480. doi:10.1056/NEJMc2009020
62. Tirado SM, Yoon KJ. Antibody-dependent enhancement of virus infection and disease. Viral Immunol. 2003;16(1):69–86. doi:10.1089/088282403763635465
63. Yang ZY, Werner HC, Kong WP, et al. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc Natl Acad Sci U S A. 2005;102(3):797–801. doi:10.1073/pnas.0409065102
64. Ho MS, Chen WJ, Chen HY, et al. Neutralizing antibody response and SARS severity. Emerg Infect Dis. 2005;11(11):1730–1737. doi:10.3201/eid1111.040659
65. Houser KV, Broadbent AJ, Gretebeck L, et al. Enhanced inflammation in New Zealand white rabbits when MERS-CoV reinfection occurs in the absence of neutralizing antibody. PLoS Pathog. 2017;13(8):e1006565. doi:10.1371/journal.ppat.1006565
66. Tseng CT, Sbrana E, Iwata-Yoshikawa N, et al. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS One. 2012;7(4):e35421. doi:10.1371/journal.pone.0035421
67. Jaume M, Yip MS, Cheung CY, et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcgammaR pathway. J Virol. 2011;85(20):10582–10597. doi:10.1128/JVI.00671-11
68. Wan Y, Shang J, Sun S, et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J Virol. 2020;94(5).
69. Wan Y, Shang J, Graham R, Baric RS, Li F, Gallagher T. Receptor recognition by the Novel Coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS Coronavirus. J Virol. 2020;94(7). doi:10.1128/JVI.00127-20
70. Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol. 2020;20(6):339–341. doi:10.1038/s41577-020-0321-6
71. Tetro JA. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020;22(2):72–73. doi:10.1016/j.micinf.2020.02.006
72. Gotoff R, Tamura M, Janus J, Thompson J, Wright P, Ennis FA. Primary influenza A virus infection induces cross-reactive antibodies that enhance uptake of virus into Fc receptor-bearing cells. J Infect Dis. 1994;169(1):200–203. doi:10.1093/infdis/169.1.200
73. Boonnak K, Slike BM, Burgess TH, et al. Role of dendritic cells in antibody-dependent enhancement of dengue virus infection. J Virol. 2008;82(8):3939–3951. doi:10.1128/JVI.02484-07
74. Gras GS, Dormont D. Antibody-dependent and antibody-independent complement-mediated enhancement of human immunodeficiency virus type 1 infection in a human, Epstein-Barr virus-transformed B-lymphocytic cell line. J Virol. 1991;65(1):541–545. doi:10.1128/JVI.65.1.541-545.1991
75. Zhang W, Zhao Y, Zhang F, et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. Clin Immunol. 2020;214:108393. doi:10.1016/j.clim.2020.108393
76. Luo P, Liu Y, Qiu L, Liu X, Liu D, Li J. Tocilizumab treatment in COVID-19: a single center experience. J Med Virol. 2020;92(7):814–818. doi:10.1002/jmv.25801
77. Stebbing J, Phelan A, Griffin I, et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis. 2020;20(4):400–402. doi:10.1016/S1473-3099(20)30132-8
78. Temesgen Z, Assi M, Vergidis P, et al. First clinical use of Lenzilumab to neutralize GM-CSF in patients with severe COVID-19 pneumonia. medRxiv. 2020.
79. Burmester GR, Feist E, Sleeman MA, Wang B, White B, Magrini F. Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-alpha, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann Rheum Dis. 2011;70(9):1542–1549. doi:10.1136/ard.2010.146225
80. De Luca G, Cavalli G, Campochiaro C, Della-Torre E, Angelillo P, Tomelleri A. GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia systemic hyperinflamation: a single-centre, prospective cohort study. Lancet Rheumatol. 2020. doi:10.1016/S2665-9913(20)30170-3
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.