")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 18
Role of Calcium Homeostasis in Alzheimer’s Disease
Authors Ge M , Zhang J, Chen S, Huang Y, Chen W, He L, Zhang Y
Received 24 November 2021
Accepted for publication 18 February 2022
Published 3 March 2022 Volume 2022:18 Pages 487—498
DOI https://doi.org/10.2147/NDT.S350939
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yuping Ning
Mengqian Ge,1 Jinghui zhang,2 Simiao Chen,2 Yanfen Huang,2 Weiyan Chen,3 Lan He,1 Yuyan Zhang2
1The Second School of Clinical Medicine, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 2School of Life Sciences, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 3School of Basic Medical Sciences, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China
Correspondence: Yuyan Zhang, Email [email protected]
Abstract: Alzheimer’s disease (AD) is a neurodegenerative disease associated with senile plaques (SP) and neurofibrillary tangles (NFTs) in the brain. With aging of the population, AD has become the most common form of dementia. However, the mechanisms leading to AD are still under investigation, and there are currently no specific drugs for its treatment. Therefore, further study on the pathogenesis of AD to develop new drugs for AD treatment remains a top priority. Several studies have suggested that intracellular calcium homeostasis is dysregulated in AD, and this has been implicated in the deposition of amyloid β (Aβ), hyperphosphorylation of tau protein, abnormal synaptic plasticity, and apoptosis, all of which are involved in the occurrence and development of AD. In addition, some based on pathways linking calcium homeostasis and AD have achieved results in AD treatment. This review comprehensively explores the relationship between calcium homeostasis and the pathogenesis of AD to provide a theoretical basis for the future exploration of AD and the development of novel therapeutic drugs.
Keywords: calcium homeostasis, Aβ, tau, apoptosis, synaptic plasticity, therapy
Introduction
Alzheimer’s disease (AD), the most common form of dementia, is characterized by memory loss, impairment of daily activities, and abnormal neurobehavior.1 AD is associated with low public awareness, low patient visit rate, lack of innovative and effective treatment methods, and high family and social care costs. In addition, the pathophysiology of AD is poorly understood and presents one of the biggest medical and social challenges. Despite the fact that AD was first discovered more than 100 years ago, there have been few innovative drugs for the treatment of AD worldwide. Currently, the most widely used clinical drugs include N-methyl-D-aspartate receptor (NMDAR) antagonists and acetylcholinesterase inhibitors (AChEIs), both of which have unsatisfactory effects.2 In addition, there are many defects in some other emerging treatment options, such as immunotherapy and β-secretase inhibitors.3,4 The Food and Drug Administration (FDA) approved a new drug for the treatment of AD on June 7, 2021. Biogen and Eisai’s anti-amyloid antibody aducanumab, now called Aduhelm, removes amyloid plaques from the brain, is still controversial, and its ability to slow cognitive decline has remained uncertain in trials to date.5 Therefore, the development of new drugs for AD treatment remains a top priority.
Calcium is known to play an essential role in our bodies, and can regulate a variety of activities, including neurotransmitter transmission, synaptic contact, cell proliferation, and apoptosis.6 Growing evidence in a variety of AD models indicates that dysregulation of intracellular calcium homeostasis is related to the neurobiology of AD. Calcium is closely associated with apoptosis, deposition of amyloid β (Aβ), hyperphosphorylation of tau protein, and abnormal synaptic plasticity, all of which are essential pathological hallmarks of AD. There has been a range of studies that indicate therapies targeting calcium-associated receptors can reverse AD-related pathological changes, including protecting neurons, ameliorating synaptic defects, amyloid and tau aggregation.6–11 For example, mitochondrial permeability transition pore (mPTP) work to maintain mitochondrial calcium homeostasis, and studies have shown that inhibiting mPTP can ameliorate cognitive deficiencies in AD transgenic mice.9 The ryanodine receptor (RyR) is a channel that exist in the endoplasmic reticulum (ER), previous studies have shown that cannabinoid-based therapies can modulate RyRs, reverse AD-related pathological changes, and mitigate cognitive deficits.7 The list goes on. As a result, it is believed that the development of drugs for the treatment of AD based on calcium homeostasis is feasible.
This review aims to provide a reference for more effective prevention and treatment of AD by studying the biological characteristics of calcium and its correlation with AD.
Biological Properties of Calcium Homeostasis
Formation and Maintenance of Calcium Homeostasis
Calcium is an indispensable substance for cell life. The precise regulation between the influx and removal of calcium ions generate calcium signals, then regulates a variety of activities in neurons.6 The increase in calcium concentration in cells occurs mainly in two ways: the inflow of extracellular calcium ions, and the release of calcium from the calcium pool.12 Both approaches are achieved through molecular tools with specific distributions and effects within cells, working together to maintain calcium homeostasis.13
The NMDAR is a glutamate receptor present on the plasma membrane (PM) that mediates the influx of calcium ions together with sodium and potassium ions.14 The voltage gated calcium channel (VGCC) is a class of receptors present on the cell membrane, which are activated by changes in membrane potential, and then increases the intracellular calcium concentration through persistent hyperpolarization, which may be mediated by NMDAR activation.15 Recent studies have predicted that reductions in the density of VGCC, increases in calcium buffer concentrations, and changes in the rate of calcium ion extrusion pumps might counteract the different forms of calcium imbalance. Moreover, it is predicted that calcium ion extrusion pump efficacy is reduced with aging, producing noticeable increases in resting calcium levels, explaining the calcium changes in AD.16
The inositol trisphosphate receptor (IP3R) and the ryanodine receptor (RyR) exist in the endoplasmic reticulum (ER), which is the main calcium pool, and its calcium concentration is much higher than that in the cytoplasm.15 IP3 and calcium ions interact with each other and act together on the IP3R channel, leading to the switching of the channel and the selective penetration of calcium ions.17 It is known that store-operated calcium entry (SOCE) regulation through transient receptor potential channel (TRPC) is critical for maintaining ER calcium levels in non-excitable cells.18 Although the evidence for SOCE in neurons remains contentious, TRPC1 is involved in regulating calcium homeostasis thus contributes to neuronal survival.19
Mitochondria play a fundamental role in maintaining intracellular calcium homeostasis.6 When the concentration of calcium in the cytosol increases the mitochondria take calcium ions through the mitochondrial calcium uniporter (MCU).13 If the mitochondrial calcium is overloaded, the mitochondrial permeability transition pore (mPTP) in the inner mitochondrial membrane will be opened.
The excretion of calcium ions is mainly achieved through the plasma membrane calcium ATPase (PMCA) and the sodium-calcium exchanger (NCX) (NCLX in mitochondria).
Calcium Homeostasis Disorders
Calcium dysregulation is often implicated in the pathogenesis of AD.20 Myriad studies have shown that presenilin (PS) mutations can cause changes in intracellular calcium homeostasis through different mechanisms,15 such as enhancing the functions of IP3R and RyR and TRPC21 or affecting CCE pathways,22 inhibiting the function of PS-mediated calcium release3,23–26 and encouraging lysosomal calcium release.27 Additional studies have shown that Aβ also causes intracellular calcium homeostasis disorders.12 Calcium homeostasis disorders are considered to be caused by generalized thinning of neuronal membranes caused by Aβ; these membranes work hard to maintain calcium concentrations, and Aβ changes the activity of calcium-related receptors,26 leads to the formation of pores that mediate calcium ion influx6 and induces oxidative stress, all of which lead to dysregulated calcium ion entry.2,28 The current evidence indicates that calcium homeostasis disorders are caused by Aβ and that PS mutations occur in the early stages of AD; however, the specific mechanism still requires further research.
PS1 Mutations in FAD are Related to Changes in Calcium Signaling
Sporadic AD (SAD) accounts for 90% of AD cases. Familial AD (FAD), which is characterized by early onset and inheritance, accounts for only a small component of AD cases. It is reasonable to use the experimental model of FAD to study AD due to the lack of a suitable SAD model and the overlapping clinical symptoms, including Aβ deposition and neuron loss.29
Presenilin1 (PS1) is involved in γ-secretase-mediated proteolytic cleavage of the C-terminal transmembrane fragments of APP after their generation by α- and β-secretase(s).30 PS1 mutation, accounting for more than 80% of the total known FAD-causing mutations,31 leads to increased intracellular calcium through different mechanisms, resulting in pathological changes in AD.15 As mentioned above, the calcium ions stored in the ER can be released into the cytoplasm through RyR and IP3R, whose function is enhanced when PS1 is mutated, increasing the cytoplasmic calcium concentration. PS1 mutation also heighten the cleavage of STIM1, decrease calcium ion influx, and disintegrate mature dendritic spines on the surface of neurons.21,32 Recent experiments have found that PS1 knockout or inactivation leads to a significant increase in capacitive calcium entry (CCE). However, the mutation of PS 1/2 in FAD weakens CCE, indicating that PS plays a valuable regulatory function in CCE, thereby affecting the intracellular calcium concentration. Nevertheless, Aβ has no effect on CCE; therefore, CCE may be an early event that leads to the production of Aβ associated with the PS mutation in FAD.22 Besides, PS itself functions to induce the release of calcium3 by forming passive calcium ion leak channels on the ER, thus implicating defective calcium signaling in AD.23,24 However, mutation of PS can inhibit this function and cause calcium overload in the ER.15,25,26 Moreover, PS1 is a key protein for lysosomal acidification, and previous experiments have found that defective lysosomal acidification in PS1 knocked out cells leads to the release of calcium ions in the lysosome and increases the intracellular calcium concentration.27
Aβ Aggregation Causes Calcium Homeostasis Disorder
The calcium hypothesis of AD posits that activation of the amyloidogenic pathway remodels neuronal calcium signaling, and alters normal calcium homeostasis and the mechanisms responsible for learning and memory. Many studies have demonstrated that Aβ upregulates calcium in neurons, causing intracellular calcium overload, which causes abnormal neuronal metabolism, neuronal apoptosis, and memory decline.12 Previous studies have shown that Aβ increases the intracellular calcium concentration instantaneously, followed by cell death.33 Calcium overload in neurons leads to dendrite disintegration, and changes in neuronal morphology depend on the existence and proximity of senile plaques, leading to dendrite disintegration.34 In transgenic mice, the mitochondrial calcium concentration has been shown to increase after plaque deposition, preceding neural death.35
There are some conjectures about the mechanism by which Aβ causes intracellular calcium overload. The first, is that Aβ causes generalized thinning of neuronal membranes, which have to work harder to maintain internal calcium concentrations. The second, is that Aβ alters the activity of receptors that respond to stimulatory signals.26 Indeed, research has shown that Aβ oligomers can directly activate NMDARs.36 APOE4 induces NMDAR-mediated calcium influx,37 which is amplified in the presence of Aβ.38,39 Third, toxic forms of Aβ can induce calcium ion influx into neurons by inducing membrane-associated oxidative stress.2 Aβ interacts with Fe2+ and Cu+ to generate dihydrogen monoxide and hydroxyl radicals, which cause membrane lipid peroxidation to generate toxic lipid aldehydes; damage to ATPase affects the transport of glutamate and glucose and causes calcium overload.40 Finally, Aβ forms calcium ion-permeable pores that lead to dysregulated calcium ion entry into the cytoplasm of brain cells, similar to tau. Notably, the ability of both proteins to form pores is determined by their structure and aggregation stage.6 Nonetheless, this proposed mechanism is controversial, although Aβ increases the permeability of cell membranes, it does not seem to depend on the formation of ion channels.26
Relationship Between Calcium Homeostasis and AD
The connection between calcium and AD was first observed several decades ago,41,42 but there is growing evidence supporting this hypothesis, strongly implicating a role for calcium in AD. To take a trivial instance, RNA expression of genes involved in calcium regulation is altered in AD brains.3 Changes in calcium homeostasis occur in the early stages of AD and are accompanied by changes in calcium-dependent proteases.43 The mechanism by which calcium influences AD progression can be divided into the following four pathways: (1) large amounts of calcium influx cause calcium homeostasis disorder, which causes neuronal structure damage, cell necrosis, and dysfunction; (2) increased intracellular calcium concentrations lead to the deposition of Aβ; (3) intracellular calcium overload causes abnormal phosphorylation of tau and inhibits its binding to microtubules, eventually resulting in neurofibrillary tangles;44 and (4) calcium homeostasis disorder leads to abnormal synaptic plasticity in the brain, which is associated with cognitive impairment in patients with AD.
Calcium Homeostasis Disorders Cause Apoptosis
Apoptosis is closely related to calcium homeostasis. For example, calcium activates PKC to modulate the downstream pathways involved in cell survival and division.15 Moderately increased intracellular calcium concentration promotes cell proliferation, but in the context of intracellular calcium overload, it can cause the mitochondrial calcium concentration to increase and then initiate the release of pro-apoptotic factors that promote cell death.45 Studies suggest that excessively increased mitochondrial calcium concentration stimulates the enzymes in the tricarboxylic acid cycle, ATP synthase, adenine nucleotide translocator, and oxidative phosphorylation in mitochondria to promote the synthesis of ATP and increase the generation of reactive oxygen species (ROS). Moreover, some researchers believe that calcium can change the three-dimensional conformation of the respiratory chain complex and induce the generation of ROS, the excessive generation of which can cause damage to the mitochondrial inner membrane, mitochondrial swelling, release of cytokines and activator-1, and activate caspases to induce apoptosis.46 Inversely, the regulation of intracellular calcium levels requires ATP consumption. The calcium hypothesis of AD suggests that the reduction of ATP production efficiency in elderly neurons weakens the calcium regulation mechanism, resulting in an increase in intracellular calcium concentration, which in turn eventually leads to cell death.47
Furthermore, not only is the mechanism of mitochondrial calcium efflux impaired, but NCX expression is lost in multiple AD experimental models.48 When neurons are exposed to repeated calcium stimulation, tau-induced NCLX damage leads to faster mitochondrial depolarization, which indicates an increased vulnerability to cell death induced by calcium. Additional studies have noted that MCU overexpression exacerbates excitotoxic cell death. Notably, there was no death in neighboring cells with low mitochondrial calcium levels, which once again emphasizes the promotion of apoptosis by mitochondrial calcium overload.6
Aβ oligomers can inhibit calcium transfer from the ER to the mitochondria in aging neurons (for example, TOM70 was found to impair IP3-related calcium transfer from the ER to mitochondria), thereby reducing mitochondrial potential, enhancing ROS production, and inducing cell apoptosis.7 Aβ has also been proposed to destroy brain cells by forming channels in neuronal membranes to deregulate calcium homeostasis, as mentioned earlier.26 Moreover, Aβ oligomers-stimulated calcium influx via the NMDAR is necessary for initiating cell cycle reentry (CCR), a prelude to neuronal death in AD.49 There has been speculation that calsenilin binds to PS and changes its endoproteolytic processing to disrupt intracellular calcium homeostasis and increase the susceptibility of neurons to apoptosis.50
Interestingly, degeneration in AD typically manifests as high levels of NMDARs, with relatively low levels of some calcium-binding proteins (CaBP) compared to resistant neurons, suggesting that calbindin buffers calcium ion loads and protects neurons against excitotoxicity.2 The age-related loss of the calcium-binding protein calbindin-D28K (CB) from the basal forebrain cholinergic neurons (BFCN) makes the BFCN vulnerable to calcium dysregulation that follows neurodegenerative insults, and results in the selective vulnerability of these neurons to degeneration.51
Calcium Homeostasis Disorder and Aβ Deposition
The amyloid cascade hypothesis postulates that the deposition of Aβ leads to senile plaques (SPs) and neurofibrillary tangles (NFTs), finally causing death of the cell and leading to AD.52 It is known that the activation of the amyloidogenic pathway remodels neuronal calcium signaling and subsequently alters normal calcium homeostasis, leading to Aβ production and/or alteration of its function.43 Exposure of cultured neurons to calcium ionophores increases Aβ production, while physiological calcium transients increase α-secretase cleavage of the amyloid precursor protein (APP), decreasing Aβ production.
Aβ is the result of proteolysis of APP by two proteases, β-secretases and γ-secretase.53 Due to the inaccurate cutting of γ-secretase, Aβ peptides ranging from 49 to 38 can eventually be produced, of which Aβ40 has the highest yield, while Aβ42 has a low yield due to its high hydrophobicity and easier oligomerization.54 Notably, mutation of APP in FAD is mainly located near the secretase cleavage site to promote the Aβ production pathway.13
BACE is a β-secretase that removes the N-terminal propeptide before its cleavage activity. The cleavage of BACE is mediated by furin protease, a calcium-dependent protease. Furin protease activity increases with the intracellular calcium concentration, thereby increasing active BACE and affecting the production of Aβ.55 In addition, increased calcium concentration can cause abnormal expression of downstream molecules and result in calcium signal disorder, and there is an inseparable connection between calcium signal disorder and Aβ. The high concentration of calcium in neurons activates calpain, which enhances p25/Cdk5 activity and phosphorylation and activation of the STAT3 (signal transducer and activator of transcription) transcription factor. STAT3 in the nucleus can regulate the mRNA levels of BACE1 and PS1, and directly or indirectly increase the production of Aβ, further promoting calcium signal disorder.56 P25/cdk5 molecules are also involved in neuronal death caused by DNA damage and play a role in the pathogenesis of AD.57 Previous studies have also shown that the cleavage of NCX3 is closely related to Aβ oligomers and is mediated by calpain, which in turn connects calcium with Aβ.58 It is important to note that CaMKII is a substrate of calpain.47 Experiments have found that the expression of CaMKIIα in neurons in the hippocampal CA1 region of AD increases. In brain slices of AD patients, CaMKIIα is deposited in SPs composed of Aβ, while no Aβ observed in the very small CaMKIIα deposition plaques only.59
Increased intracellular calcium concentration also activates AMPK in a CAMKKII-dependent manner. According to a previous study, the CAMKKII-AMPK kinase pathway is a critical mediator of the synaptotoxic effects of Aβ42 oligomers.60
Despite this evidence, not all increases in calcium concentration lead to an increase in Aβ production. Mutation of the CALHM1 gene causes the loss of CALHM1 protein function, and reduces the permeability of the cell membrane to calcium and the intracellular calcium concentration, which in turn promotes the production and aggregation of Aβ.61–63 The mechanism underlying this effect is not yet clear, but some scholars believe that it may involve a calcium-dependent effect on α-secretase.64 It has been speculated that the activation of CALHM1 can accelerate IDE secretion, promote the clearance of Aβ, and reduce extracellular Aβ levels.65 However, some studies have found no association between CALHM1 and AD, which is a research avenue that requires further exploration.66
S100B is a proinflammatory protein associated with SPs, which are chronically upregulated in AD. Research indicates that the combination of calcium and S100B is beneficial for S100B to lock Aβ42 aggregation.67
Calcium Homeostasis Disorder and Tau Protein Levels
In the early stages of AD, synaptic changes induced by Aβ are the main pathogenic mechanisms. However, in the later stages of AD, abnormal aggregation of tau protein promotes degeneration and loss of synapses, which are the decisive factors leading to the occurrence of dementia.68 Experiments have found that the overactivation of glutamate receptors causes calcium-mediated changes in tau and microtubules, similar to those seen in NFTs.2 Numerous studies have proven that calcium dysregulation plays a key role in the early stage of tau pathology and impaired physiology, and that reducing calcium leaks can normalize cognitive performance.8 There is a balance between phosphorylation and dephosphorylation of tau protein under normal circumstances. However, this balance is disrupted when the intracellular calcium concentration is too high to cause hyperphosphorylation of tau protein; this can reduce the aggregation ability of microtubules and depolymerize normal microtubules, thereby affecting axon transport and the transmission of cellular information.69
Moreover, although the expression of tau protein is not directly related to depolarization of the cell membrane, an increase in tau phosphorylation can be observed after the depolarization of the cell membrane for 30 min, which supports the conjecture that changes in calcium concentration affect tau phosphorylation.70
The phosphorylation and dephosphorylation of tau are catalyzed by kinases and phosphatases,71 respectively, which are mostly calcium-dependent proteases.72 It has been found that an increase in intracellular calcium homeostasis activates calcineurin (CaN) and promotes neuroinflammatory atrophy.73 In addition to causing memory loss, abnormal activation of CaN can also affect the double helix structure of the tau protein. Hyperphosphorylated tau protein (especially Ser262 located in its microtubule binding region) combines to form paired helical filaments (PHF), and then forms NTFs, in which calcium plays a role by regulating calcium-binding proteins (CaBP) and calcium-dependent protein kinase (CDPK).74 Mass spectrometry has revealed that CaMKII phosphorylates tau at multiple sites.75 Additional studies have noted that the expression of CaMKIIα is regulated by CYFIP2, the expression of which is reduced in AD.76
Calpain, whose inhibitors can block the process of tau fragmentation induced by Aβ, is also specifically dependent on calcium activation.77 Calpain affects the activities of glycogen synthase kinase 3 (GSK3) and protein phosphatase 2A (PP2A), resulting in abnormal hyperphosphorylation of tau, which occurs at multiple sites, mediates the truncation of tau protein monomers, and induces neurodegeneration.78 Research has shown that the activity of PP2A in the brain of patients with AD is significantly reduced at the gene and protein levels. The expression of PP2A is inhibited by calpain in the process of reducing hyperphosphorylation of tau.78 Calpain has two subtypes, calpain 1 and calpain 2, both of which have similar proteolytic effects on GSK3β. Calpain1 enhances the activity of GSK3β on tau phosphorylation by truncating the C-terminus of GSK3β, leading to hyperphosphorylation of tau and neurofibrillary degeneration in the brain of patients with AD. Conversely, phosphorylation at different sites of GSK3β can also affect calpain-mediated cleavage.78 In addition, p25, produced by calpain cleavage, has been found to accumulate in the brains of patients with AD, and it also activates CDK5, which leads to tau hyperphosphorylation.79 Furthermore, under conditions of calcium-induced neuronal injury, calpain can also activate the Erk1, 2 signaling pathway, which may be involved in the tau hyperphosphorylation process in the early stage of AD.77
Calcium Homeostasis and Synaptic Plasticity
Cognitive impairment is the main clinical feature of AD. Although the pathological mechanism of cognitive impairment is not fully understood, it has been confirmed that abnormal synaptic plasticity in the brain, especially in the hippocampus, is involved in cognitive impairment in patients with AD.80 Synaptic plasticity includes two main forms of long-term potentiation (LTP) and long-term depression (LTD), which are considered the basic mechanisms of learning, memory, and amnesia in the brain, and as a functional indicators of synaptic plasticity.81 Recent evidence suggests that calcium signaling regulates the energy production and neuronal metabolism necessary to sustain synaptic transmission.3
Research has shown that the increased calcium concentration of ER in the neurons of patients with AD causes compensatory changes in calcium signaling that break the balance between LTP and LTD synaptic mechanisms, and synaptic function is weakened by the LTD mechanism, leading to memory loss.82 Abnormal handling of calcium in the ER may weaken the mushroom-type postsynaptic spines associated with long-term memory. Several other studies have demonstrated that elevated cytosolic calcium caused by excessive calcium release impedes the induction of LTP.3,83 Of note, synaptic mitochondria can protect cells from damage caused by prolonged calcium stimulation because of its faster absorption of the instantaneous increase in calcium in the presynaptic terminal than in the ER.84
Calcium activates calmodulin (CaM) through conformational changes to activate CaN, CaMKII, and CaMKIV. In patients with AD, increased intracellular calcium activates CaN and participates in the induction of LTD; activation of CaMKII functions to strengthen synapses and plays a role in the induction of LTP.15
Studies have shown that the upregulation of glutamate receptors in patients with AD is accompanied by the downregulation of NMDAR and AMPAR, which have been proven to be the basic molecular mechanisms that mediate learning, memory, and cognition by mediating calcium homeostasis.85 The activation of NMDAR can participate in the induction of LTP, while the continuous activation of NMDAR can lead to the conversion of LTP to LTD, leading to mitochondrial membrane depolarization, free radical production, and even cell death. Many previous studies have suggested that extra-synaptic activation of NMDAR and intra-synaptic activation have completely different results; that is, intra-synaptic activation is beneficial to cell survival, while extra-synaptic activation may lead to cell death. Subsequent research has highlighted that separate extra- or intra-synaptic activation will not lead to cell death, but will upregulate the survival-related signal transduction system. In addition, a study at the 17th National Conference of Neurology demonstrated that the co-stimulation of the extra- and intra-synaptic activation led to cell death. Besides, Aβ oligomers caused a rapid decrease in membrane expression of NMDARs, followed by abnormal dendritic spine morphology and degeneration of spines.2
Furthermore, recent evidence suggests that Aβ can inhibit calcium signaling and synapse plasticity through t-type calcium channels and the Nogo receptors (NgRs), which may lead to learning difficulties in the early stages of AD progression.86
AD Treatment Based on Calcium Homeostasis
Comprehensive treatment and effective care can delay the development of AD, but relevant drugs have not yet been marketed. The most widely used clinical drugs include NMDAR antagonists and acetylcholinesterase inhibitors (AChEIs),68 which can be used in combination to achieve better results.87 Although both can affect the cytoplasmic calcium concentration, they cannot effectively reverse cognitive dysfunction. Moreover, approved cholinesterase inhibitors include oral donepezil, percutaneous rivastigmine, and oral galantamine, which have good efficacy in the treatment of mild to moderate AD. However, the effect obtained for patients with moderate to severe AD is very small, even if the dose is increased, and it will cause a certain degree of gastrointestinal adverse reactions.88 In addition to the above two types of therapeutic drugs, we propose some emerging treatment options, mainly immunotherapy and β-secretase inhibitors. The immunotherapy target the formation of Aβ plaques,89 some of which have recently been shown to have no effect on disease progression.3 Several immunotherapies target tau protein, whose clinical trials are only at the early stage, with few research reports on whether they can effectively improve clinical symptoms.90 The β-secretase inhibitors, which is currently undergoing early trials, also aims to achieve curative effects by inhibiting the formation of Aβ.91 At the same time, α-secretase inhibitors have failed clinically.3 Moreover, many defects in the previously reported anti-Aβ drugs for AD treatment are still in clinical trials.4 Therefore, the development of novel drugs for the treatment of AD is urgently required.
Based on the previous discussion, we know that calcium is inextricably linked to AD, and consequently, the development of drugs for the treatment of AD based on calcium homeostasis is feasible. At present, calcium ion antagonists are the main clinically developed AD therapeutic drugs based on calcium homeostasis, which can treat AD by inhibiting calcium overload. The most commonly used clinical calcium ion antagonists are dihydropyridine drugs such as nimodipine.92 Previous clinical trials have demonstrated that calcium ion antagonists have no positive effect in reducing the rate of cognitive decline in patients with AD. However, changes in calcium homeostasis occur in the early stages of AD, and the initial severity of the disease seems to affect the outcome.43 Several studies have shown that calcium ion antagonists reduces the risk of dementia in patients with hypertension.6 Curcumin is an intracellular calcium inhibitor that attenuates glutamate-induced neurotoxicity. There has been experiments showed that curcumin protect astrocytes from oxidative stress and white matter from hypoxia then inhibit apoptosis.93 Theracurmin, a very highly absorbable curcumin formulation, was shown to improve memory and attention in non-demented people. It seems to be a therapeutic option for elderly patients with AD via providing stabilization of the disease course by preventing progressive loss in cognitive functions.94 Aduhelm, a new drug approved by the FDA for the treatment of AD on June 7, 2021, removes amyloid plaques from the brain, also ameliorate calcium overload, which has been demonstrated by previous experiments.95
It is well known that mitochondria are essential organelles that link calcium homeostasis with AD. Therefore, many studies have attempted to improve AD by targeting mitochondrial calcium homeostasis. It is legitimate to propose reducing mitochondrial calcium overload by blocking the MCU as a novel potential therapeutic target for AD because mitochondrial calcium overload involves toxic extracellular Aβ oligomers and requires the mitochondrial calcium uniporter.35,96 TG-2112x, a novel compound, can protect neurons by partially inhibiting mitochondrial calcium uptake.6 In addition, studies have shown that inhibiting mPTP can ameliorate cognitive deficiencies in AD transgenic mice.9 More importantly, rescue of the NCLX gene was shown to completely restore cognitive decline and cellular pathology in AD mice.6 According to another recent study, urolithin A, which can maintain mitochondrial calcium and ROS homeostasis, is expected to be an effective treatment to prevent AD.97
In addition, restoring intracellular calcium homeostasis by regulating calcium-related receptors on the ER (especially RyRs) is a current research focus. Recent studies have found that post-translational remodeling of neuronal RYR2 channels can cause calcium ion leakage, which activates calcium-dependent signaling pathways, which are altered in AD. Oral Rycal S107 can repair calcium ion leakage and reduce neuropathological changes10 without side effects.8 Previous studies have shown that cannabinoid-based therapies can modulate RyRs, reverse AD-related pathological changes, and mitigate cognitive deficits.7
Previous research has also demonstrated that VGCC is upregulated in AD, and that the inhibition of L-VGCC could effectively improve memory loss and calcium disorders in patients with AD.11 Furthermore, polycyclic maleimide-based derivatives, which can bypass the cardiovascular side effects caused by peripheral L-channel blockade, synthesize dual calcium channels and GSK-3β modulators to treat AD.98
In summary, it is clear that calpain hyperactivation is a major player in tau hyperphosphorylation and Aβ deposition. Thus, it is worth investigating ways to treat AD by downregulating or inhibiting targeted calpain. It has been found that either calpain’s endogenous inhibitor, CAST, or synthetic inhibitors have achieved positive results in neurodegenerative animal and cell models.79 Moreover, AD transgenic mice overexpressing S100B exhibit elevated Aβ levels and are more susceptible to neuronal dysfunction; however, when the concentration of S100B in AD transgenic mice decreases, so does the concentration of Aβ, suggesting that targeting S100B can be used as a novel approach to ameliorate neurodegeneration in AD.67 In addition to all of the above, the various pathways that link calcium homeostasis and AD mentioned in Figure 1 deserve further investigation to determine more effective ways to prevent, diagnose, or treat AD.
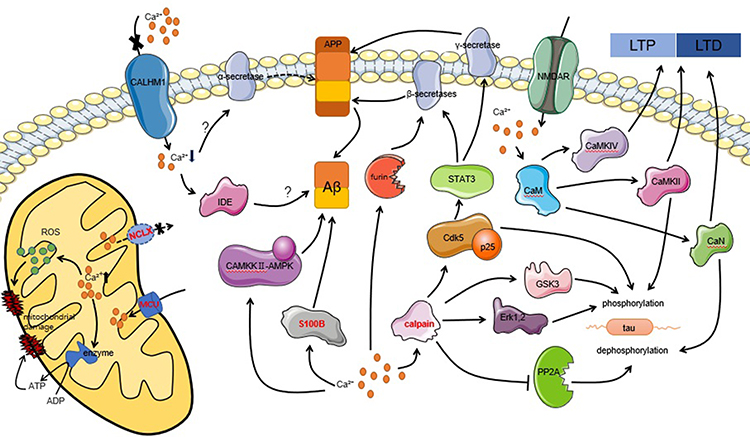 |
Figure 1 Intracellular calcium homeostasis is associated with the pathological features of AD, including abnormal synaptic plasticity (LTP and LTD), hyperphosphorylation of tau, Aβ deposition, and cell apoptosis, through multiple pathways. The word marked in red indicates new targets for therapy that have achieved positive results in experiments. |
Finally, recent studies point to the role of exercise, dietary energy restriction, and cognitive stimulation, in delaying the aging process and preventing AD, potentially by inducing the expression of neurotrophic factors.2 Hence, AD treatment should also focus on the above aspects.
Conclusion
In recent years, clinical trials of drugs for the treatment of AD have failed without exception. Aduhelm, a new drug approved by the FDA for the treatment of AD on June 7, 2021, removes amyloid plaques from the brain, is still controversial, and its ability to slow cognitive decline has remained uncertain in trials to date. The absence of any specific drug for the treatment of AD remains one of the most significant medical and social challenges. As detailed in the present review, calcium homeostasis plays a role in AD, and its disorder occurs in the early stages of AD. Both Aβ and PS mutations induce calcium deregulation through direct and indirect pathways. Importantly, calcium homeostasis disorders ultimately lead to cellular pathology in AD, including apoptosis, Aβ deposition, hyperphosphorylation of tau, and abnormal synaptic plasticity. Some therapeutic drugs based on calcium homeostasis have indeed achieved positive effects, such as reducing the risk of dementia in hypertensive patients. Curcumin, an intracellular calcium inhibitor, was shown to improve memory and attention in non-demented people. Inhibition of mPTP and NCLX gene rescue, cannabinoid-based therapies to modulate RyRs, the inhibition of L-VGCC, and downregulating or inhibiting targeted calpain to maintain intracellular calcium homeostasis have all achieved positive results in experiments. Therefore, we believe that is feasible. Moreover, it is legitimate to propose blocking the MCU and regulating IP3R to restore intracellular calcium homeostasis as novel potential therapeutic targets for AD. Of note, the other above-mentioned pathways linking calcium homeostasis and AD require further study. In summary, this review provides a comprehensive discussion of the relationship between calcium homeostasis and the pathogenesis of AD to provide theoretical help for the future exploration of AD and the development of novel therapeutic drugs.
Submission Declaration and Verification
This work described has not been published previously, that it is not under consideration for publication elsewhere, that its publication is approved by all authors and tacitly or explicitly by the responsible authorities where the work was carried out, and that, if accepted, it will not be published elsewhere in the same form, in English or in any other language, including electronically without the written consent of the copyright-holder.
Author Contributions
Mengqian Ge is the first author. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was jointly supported by grants from Zhejiang Provincial Natural Science Foundation of China (No.LY21H270012), the National Natural Science Foundation of China (No.81774230), and Zhejiang Science and Technology Plan of Traditional Chinese Medicine (No.2020ZB159).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Clive Ballard S, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377(9770):1019–1031. doi:10.1016/S0140-6736(10)61349-9
2. Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31(9):454–463. doi:10.1016/j.tins.2008.06.005
3. Ryan C, Ashkavand Z, Norman KR. The role of mitochondrial calcium homeostasis in Alzheimer’s and related diseases. Int J Mol Sci. 2020;21(23):9153. doi:10.3390/ijms21239153
4. Si-Jia L. Two-dimensional nanomaterials against β-amyloid in the treatment of Alzheimer’s disease. Chin J Biochem Mol Biol. 2021;37(05):e45.
5. Knopman S, Jones D, Greicius MD. Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021;17(4):696–701. doi:10.1002/alz.12213
6. Noemi Esteras N, Abramov AY. Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells. 2020;9(9):2135. doi:10.3390/cells9092135
7. Luísa Cortes L, Malva J, Rego AC, Pereira CF. Calcium signaling in aging and neurodegenerative diseases 2019. Int J Mol Sci. 2021;21(3):1125. doi:10.3390/ijms21031125
8. Dibyadeep Datta D, Leslie SN, Wang M. Age-related calcium dysregulation linked with tau pathology and impaired cognition in non-human primates. Alzheimer’s Dement. 2021;17(6):920–932. doi:10.1002/alz.12325
9. Starkov A, Beal FMB. Portal to Alzheimer’s disease. Nat Med. 2008;14(10):1020–1021. doi:10.1038/nm1008-1020
10. Alain Lacampagne A, Liu X, Reiken S. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017;134(5):749–767. doi:10.1007/s00401-017-1733-7
11. Gholami H, Pourbadie N. Preventing effect of L-type calcium channel blockade on electrophysiological alterations in dentate gyrus granule cells induced by entorhinal amyloid pathology. PLoS One. 2015;10(2):e117555.
12. Bei-Ling LU. Advances in calcium signaling and Alzheimer’s disease. Chin J Gerontol. 2017;37(10):2567–2570.
13. Luisa Galla L, Redolfi N, Pozzan T, Pizzo P, Greotti E. Intracellular calcium dysregulation by the Alzheimer’s disease-linked protein presenilin 2. Int J Mol Sci. 2020;21(3):770. doi:10.3390/ijms21030770
14. Li-ying JYLZ. Research progress of antidepressants targeting NMDAR. Chin J Med Chem. 2020;30(8):502–508.
15. Sushma S, Mondal AC. Role of GPCR signaling and calcium dysregulation in Alzheimer’s disease. Mol Cell Neurosci. 2019;101:103414. doi:10.1016/j.mcn.2019.103414
16. Angulo L, Henzi THSA, Neymotin SA. Amyloid pathology–produced unexpected modifications of calcium homeostasis in hippocampal subicular dendrites. Alzheimers Dement. 2020;16(2):251–261. doi:10.1016/j.jalz.2019.07.017
17. Jia-Yin JIN. Research progress on the involvement of inositol 1;4;5-trisphosphate receptors in the occurrence of neurodegenerative diseases. Prog Physiol Sci. 2020;51(06):453–458.
18. Cahalan MD. STIMulating store-operated Ca2+ entry. Nat Cell Biol. 2009;11(6):669–677. doi:10.1038/ncb0609-669
19. Senthil Selvaraj YSJA, Sun Y, Watt JA. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest. 2012;122(4):1354–1367. doi:10.1172/JCI61332
20. Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15(3):89–100. doi:10.1016/j.molmed.2009.01.001
21. Khachaturian ZS. Calcium Hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017;13(2):178–182. doi:10.1016/j.jalz.2016.12.006
22. Yoo AS, Cheng I, Chung S, et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27(3):561–572. doi:10.1016/S0896-6273(00)00066-0
23. Thinakaran GTAS, Sisodia SS. Presenilins and Alzheimer disease: the calcium conspiracy. Nat Neurosci. 2006;9(11):1354–1355. doi:10.1038/nn1106-1354
24. Tu H, Nelson O, Bezprozvanny A. Presenilins form ER Ca2+ leak channels. a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126(5):981–993. doi:10.1016/j.cell.2006.06.059
25. Lu GAI. The mechanism of calcium ion in regulating Aβ amyloid accumulation. Chin J Gerontol. 2016;36(5):1249–1251.
26. Marx J. Alzheimer’s disease. Fresh evidence points to an old suspect: calcium. Science. 2007;318(5849):384–385. doi:10.1126/science.318.5849.384
27. Jing NIE. The role of presenilin in the pathogenesis of Alzheimer’s disease. West China J Pharm Sci. 2017;32(01):99–103.
28. Ping SUN. Progress of neuronal calcium dyshomeostasis in the pathogenesis of Alzheimer’s disease. Prog Mod Biomed. 2009;9(06):1183–1185.
29. Lippa CF, Saunders AM, Smith TW. Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology. 1996;46(2):406–412. doi:10.1212/WNL.46.2.406
30. De Strooper B, Saftig P, Craessaerts K. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–390. doi:10.1038/34910
31. Ying Gao Y, Ren R-J, Zhong Z-L. Mutation profile of APP. PSEN1; and PSEN2 in Chinese Familial Alzheimer’s Disease. Neurobiol Aging. 2019;77:154–157. doi:10.1016/j.neurobiolaging.2019.01.018
32. Chun-Kit B, Tong CSLW. Familial Alzheimer’s disease-associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci Signal. 2016;9(444):a89.
33. Olga Simakova O, Arispe NJ. Early and late cytotoxic effects of external application of the Alzheimer’s Abeta result from the initial formation and function of Abeta ion channels. Biochemistry. 2006;45(18):5907–5915. doi:10.1021/bi060148g
34. Kuchibhotla KV, Goldman ST, Lattarulo CR, et al. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59(2):214–225. doi:10.1016/j.neuron.2008.06.008
35. Maria Calvo-Rodriguez M, Hou SS, Snyder AC. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat Commun. 2020;11(1):2146. doi:10.1038/s41467-020-16074-2
36. Laura Texidó L, Martín-Satué M, Alberdi E, Solsona C, Matute C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium. 2011;49(3):184–190. doi:10.1016/j.ceca.2011.02.001
37. Hartmann H, Eckert A, Muller WE. Apolipoprotein E and cholesterol affect neuronal calcium signalling: the possible relationship to beta-amyloid neurotoxicity. Biochem Biophys Res Commun. 1994;200(3):1185–1192. doi:10.1006/bbrc.1994.1576
38. LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3(11):862–872. doi:10.1038/nrn960
39. Ohm TG. Apolipoprotein E and beta A4-amyloid: signals and effects. Biochem Soc Symp. 2001;67:121–129.
40. Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631–639. doi:10.1038/nature02621
41. Peterson C. Altered calcium uptake in cultured skin fibroblasts from patients with Alzheimer’s disease. N Engl J Med. 1985;312(16):1063–1065.
42. Deary IJ, Hendrickson AE. Calcium and Alzheimer’s disease. Lancet. 1986;1(8491):1219. doi:10.1016/S0140-6736(86)91205-5
43. Green KN, Smith IF, Laferla FM. Role of calcium in the pathogenesis of Alzheimer’s disease and transgenic models. Subcell Biochem. 2007;45:507–521.
44. Yanyan YXLX. Experimental research progress on traditional Chinese medicine in treatment of Alzheimer’s disease by regulating and controlling calcium ions in steady state. Chin Arch Trad Chin Med. 2018;36(01):49–52.
45. Xi-Ju HE. Research progress of transient receptor potential channels in neurons. J Hubei Univ Med. 2014;33(01):81–84.
46. Xiaoyuan HOU. Interplay between calcium signaling and reactive oxygen species. Chin J Cell Biol. 2019;41(09):1837–1844.
47. Ru-Yi LIN. Progress of the relationship between Ca2+ dysregulation and Alzheimer’s disease development. Chin Bull Life Sci. 2012;24(2):297–303.
48. Pooja Jadiya P, Kolmetzky DW, Tomar D. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat Commun. 2019;10(1):3885. doi:10.1038/s41467-019-11813-6
49. Kodis J, Choi SCES. N-methyl-D-aspartate receptor–mediated calcium influx connects amyloid-b oligomers to ectopic neuronal cell cycle reentry in Alzheimer’s disease. Alzheimers Dement. 2018;14(10):1302–1312. doi:10.1016/j.jalz.2018.05.017
50. Tanzi RE. Of calcium, caspases, and cognitive decline. Nat Med. 1998;4(10):1127–1128. doi:10.1038/2622
51. David Riascos DDLA, de Leon D, Baker-Nigh A. Age-related loss of calcium buffering and selective neuronal vulnerability in Alzheimer’s disease. Acta Neuropathol. 2011;122(5):565–576. doi:10.1007/s00401-011-0865-4
52. Ling ZHANG. Research progress on the pathogenesis of Alzheimer’s disease and its immunotherapy. Mod pract med. 2019;31(02):147–148.
53. Min Y, Xu W, Xihan G. Progress on loss-of-function hypothesis of presenilin-1 mutations in Alzheimer diseases. J Zhejiang Univ. 2020;49(4):487–499.
54. Pei-Chang CYWY. Regulation of amyloidogenesis and clearance of β-amyloid in Alzheimer’s disease. Chin J Biochem Mol Biol. 2021;37(2):176–181.
55. Bennett BD, Denis P, Haniu M. A furin-like convertase mediates propeptide cleavage of BACE. the Alzheimer’s beta-secretase. J Biol Chem. 2000;275(48):37712–37717. doi:10.1074/jbc.M005339200
56. Li liu RMGK. Palmitate induces transcriptional regulation of BACE1 and presenilin by STAT3 in neurons mediated by astrocytes. Exp Neurol. 2013;248:482–490.
57. María florencia Ogara MF, Belluscio LM, de la Fuente V. CDK5-mediated phosphorylation of p19INK4d avoids DNA damage-induced neurodegeneration in mouse hippocampus and prevents loss of cognitive functions. Biochimica et Biophysica Acta (BBA) - Mol Cell Res. 2014;1843(7):1309–1324. doi:10.1016/j.bbamcr.2014.03.026
58. Joe Atherton J, Kurbatskaya K, Bondulich M. Calpain cleavage and inactivation of the sodium calcium exchanger-3 occur downstream of Aβ in Alzheimer’s disease. Aging Cell. 2014;13(1):49–59. doi:10.1111/acel.12148
59. Gui-hai CHEN. The expression of calcium/ calmodulin-dependent protein kinase II in the senile plaques in Alzheimer disease. Chin J Neurosci. 2004;20(6):441–445.
60. Georges Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Ab oligomers through tau phosphorylation. Neuron. 2013;78(1):94–108. doi:10.1016/j.neuron.2013.02.003
61. Wen-Ming YANG. Research progress on genes related to Alzheimer’s disease. Chin J Gerontol. 2013;33(2):455–459.
62. Tanzi E, Bertram LB. Alzheimer’s disease: the latest suspect. Nature. 2008;454(7205):706–708. doi:10.1038/454706a
63. Ute Dreses-Werringloer U, Lambert J-C, Vingtdeux V, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis. Ab levels. and Alzheimer’s disease risk. Cell. 2008;133(7):1149–1161. doi:10.1016/j.cell.2008.05.048
64. Green N, LaFerla FML. Linking calcium to Abeta and Alzheimer’s disease. Neuron. 2008;59(2):190–194. doi:10.1016/j.neuron.2008.07.013
65. Yang ZHOU. Research progress on genetics and signaling pathways of Alzheimer’s disease. Acta Univ Med Anhui. 2019;54(06):994–1000.
66. Lars Bertram L, Schjeide B-M-M, Hooli B. No association between CALHM1 and Alzheimer’s disease risk. Cell. 2008;135(6):993–994. doi:10.1016/j.cell.2008.11.030
67. Joana S, Cristóvão VKMI, Hugo M, Botelho CGRD, Tobias Madl GFBR. The neuronal S100B protein is a calcium-tuned suppressor of amyloid-aggregation. Sci Adv. 2018;4:1–13.
68. Meng-ting LI, Zhao-Yang Z, Wen-Rong H, et al. Research progress in the pathogenesis of Alzheimer’s disease. World Latest Med Info. 2020;20(76):
69. Ji-Yun WU. Effect of calcium signaling on Alzheimer diseases pathogenesis. J Jinggangshan Univ. 2010;31(04):107–109.
70. Nathalie Pierrot N, Santos SF, Feyt C, Morel M, Brion J-P, Octave J-N. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J Biol Chem. 2006;281(52):39907–39914. doi:10.1074/jbc.M606015200
71. Gong CX. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61(3):921–927. doi:10.1111/j.1471-4159.1993.tb03603.x
72. Hartigan JA, Johnson GVW. Transient increases in intracellular calcium result in prolonged site-selective increases in Tau phosphorylation through a glycogen synthase kinase 3beta-dependent pathway. J Biol Chem. 1999;274(30):21395–21401. doi:10.1074/jbc.274.30.21395
73. Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+] i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21(24):9744–9756. doi:10.1523/JNEUROSCI.21-24-09744.2001
74. Fei XU. The relationship between Tau protein. Aβ protein and Alzheimer’s disease and their effects. J Clin Med Pract. 2008;3:118–120.
75. Tiwari SS, Mizuno KMAG, Ghosh A. Alzheimer-related decrease in CYFIP2 links amyloid production to tau hyperphosphorylation and memory loss. Brain. 2016;139(10):2751–2765. doi:10.1093/brain/aww205
76. Anshua Ghosh A, Giese KP. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Ghosh Giese Mol Brain. 2015;8(1):78. doi:10.1186/s13041-015-0166-2
77. Rui DING. The role of calpain in Alzheimer’s disease. Foreign Med Sci. 2007;28(06):257–261.
78. Kai-wen GUO. Advance in mechanisms of calpain involved in hyperphosphorylation and truncation of tau protein. Chinese J Rehabilitation Theory Pract. 2020;26(10):1182–1185.
79. Mahaman Y, Huang F, Kessete Afewerky H, Maibouge TMS, Ghose B, Wang X. Involvement of calpain in the neuropathogenesis of Alzheimer’s disease. Med Res Rev. 2018;39(2):608–630. doi:10.1002/med.21534
80. Yi D. Irisin: anew target for exercise to improve decline in Alzheimer’s Disease. China Sport Sci Technol. 2020;9(56):30–36.
81. Zhao-Peng ZHOU. Research progress of synaptic plasticity after cerebral ischemia. Guangdong Med J. 2018;39(23):3573–3577.
82. Chengyu GUO. Research progress of calcium homeostasis in neurodegenerative diseases. J Pract Med. 2020;36(02):263–267.
83. Chun-Lei Z. New discovery of Alzheimer’s disease pathology——Mutated genes in patients may play an important role. World Sci. 2009;12:35.
84. Cassia Overk C, Masliah E. Perspective on the calcium dyshomeostasis hypothesis in the pathogenesis of selective neuronal degeneration in animal models of AD. Alzheimers Dement. 2017;13(2):183–185. doi:10.1016/j.jalz.2017.01.005
85. Na WJW. Research progress of learning and memory and neurotoxicity mediated by NMDA receptor. Chinese J Gen Pract. 2020;18(06):994–997.
86. Yanjun Zhao Y, Sivaji S, Chiang MC, Ali H. Amyloid beta peptides block new synapse assembly by nogo receptor-mediated inhibition of T-type calcium channels. Neuron. 2017;96(2):355–372. doi:10.1016/j.neuron.2017.09.041
87. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317–324. doi:10.1001/jama.291.3.317
88. Farlow R, Salloway SSPN, Tariot PN. Effectiveness and tolerability of high-dose (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: a 24-week randomized double-blind study. Clin Ther. 2010;32(7):1234–1251. doi:10.1016/j.clinthera.2010.06.019
89. Ross Penninkilampi R, Brothers HM, Eslick GD. Safety and efficacy of anti-amyloid-β immunotherapy in Alzheimer’s disease: a systematic review and meta analysis. J Neuroimmune Pharmacol. 2017;12(1):194–203. doi:10.1007/s11481-016-9722-5
90. Jian-liang HFF. Tau-targeting immunotherapies for Alzheimer ′s disease: research advances. Int J Pharm Res. 2020;47(10):774–780.
91. Moussa CE-H. Beta-secretase inhibitors in Phase I and Phase II clinical trials for Alzheimer’s disease. Expert Opin Investig Drugs. 2017;26(10):1131–1136. doi:10.1080/13543784.2017.1369527
92. Ji-Yu YANG. Alzheimers disease and its drug treatment progress. Sichuan J Physiol Sci. 2017;39(02):101–103.
93. Daverey A, Agrawal SK. Neuroprotective effects of Riluzole and Curcumin in human astrocytes and spinal cord white matter hypoxia. Neurosci Lett. 2020;738:135351. doi:10.1016/j.neulet.2020.135351
94. Dost FS, Kaya D, Ontan MS. Theracurmin supplementation may be a therapeutic option for older patients with Alzheimer’s disease: a 6-month retrospective follow-up study. Curr Alzheimer Res. 2021;18(14):1087–1092. doi:10.2174/1567205019666211221125255
95. Kastanenka V, Bussiere TBNS, Shakerdge N. Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J Neurosci. 2016;36(50):12549–12558. doi:10.1523/JNEUROSCI.2080-16.2016
96. Maria Calvo-Rodriguez M, Bacskai BJ. Mitochondria and calcium in Alzheimer’s disease: from cell signaling to neuronal cell death. Trends Neurosci. 2020;44(2):136–151. doi:10.1016/j.tins.2020.10.004
97. Hyun Jik Lee YHJG, Jung YH, Choi GE. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2020;28(1):184–202. doi:10.1038/s41418-020-0593-1
98. Alessandra Bisi RLAM, Arribas RL, Micucci M. Polycyclic maleimide-based derivatives as first dual modulators of neuronal calcium channels and GSK-3b for Alzheimer’s disease treatment. Eur J Med Chem. 2019;163:394–402. doi:10.1016/j.ejmech.2018.12.003
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.