")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Role of Alpelisib in the Treatment of PIK3CA-Mutated Breast Cancer: Patient Selection and Clinical Perspectives
Authors Chang DY, Ma WL, Lu YS
Received 1 December 2020
Accepted for publication 16 February 2021
Published 5 March 2021 Volume 2021:17 Pages 193—207
DOI https://doi.org/10.2147/TCRM.S251668
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Deyun Wang
Dwan-Ying Chang,1 Wei-Li Ma,1,2 Yen-Shen Lu1
1Department of Oncology, National Taiwan University Hospital, Taipei, Taiwan; 2Graduate Institute of Oncology, National Taiwan University College of Medicine, Taipei, Taiwan
Correspondence: Yen-Shen Lu
Department of Oncology, National Taiwan University Hospital, Taipei, Taiwan
Email [email protected]
Abstract: The PI3K/AKT/mTOR pathway has long been known to play a major role in the growth and survival of cancer cells. Breast tumors often harbor PIK3CA gene alterations, which therefore constitute a rational drug target. However, it has taken many years to demonstrate clinically-relevant efficacy of PI3K inhibition and eventually attain regulatory approvals. As data on PI3K inhibitors continue to mature, this review updates and summarizes the current state of the science, including the prognostic role of PIK3CA alterations in breast cancer; the evolution of PI3K inhibitors; the clinical utility of the first-in-class oral selective PI3Kα inhibitor, alpelisib; PIK3CA mutation detection techniques; and adverse effect management. PIK3CA-mutated breast carcinomas predict survival benefit from PI3K inhibitor therapy. The pan-PI3K inhibitor, buparlisib and the beta-isoform-sparing PI3K inhibitor, taselisib, met efficacy endpoints in clinical trials, but pictilisib did not; moreover, poor tolerability of these three drugs abrogated further clinical trials. Alpelisib is better tolerated, with a more manageable toxicity profile; the principal adverse events, hyperglycemia, rash and diarrhea, can be mitigated by intensive monitoring and timely intervention, thereby enabling patients to remain adherent to clinically beneficial treatment. Alpelisib plus endocrine therapy shows promising efficacy for treating postmenopausal women with HR+/HER2– advanced breast cancer. Available evidence supporting using alpelisib after disease progression on first-line endocrine therapy with or without CDK4/6 inhibitors justifies PIK3CA mutation testing upon diagnosing HR+/HER2– advanced breast cancer, which can be done using either tumor tissue or circulating tumor DNA. With appropriate toxicity management and patient selection using validated testing methods, all eligible patients can potentially benefit from this new treatment. Further clinical trials to assess combinations of hormone therapy with PI3K, AKT, mTOR, or CDK 4/6 inhibitors, or studies in men and women with other breast subtypes are ongoing.
Keywords: HR+/HER2− advanced/metastatic breast cancer, PIK3CA mutation test, alpelisib PI3K alpha-selective inhibitor, prognosis, survival benefit, toxicity management
Introduction
Breast cancer is the most common malignancy in women and the second most common cancer-related cause of death among women in the United States (US).1 The phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway is involved in cell survival and human cancers.2 The PIK3CA gene, which encodes the p110α (catalytic subunit alpha) of PI3K protein, has often mutated in breast cancer, especially in hormone receptor positive/human epidermal growth factor receptor 2 negative (HR+/HER2−) breast cancer, also termed luminal subtype.3 Alpelisib is an oral α-specific PI3K inhibitor approved by the US Food and Drug Administration (FDA) for use in combination with fulvestrant to treat postmenopausal women with HR+/HER2−, PIK3CA-mutated, advanced or metastatic breast cancer that has progressed following treatment with an endocrine-based regimen.4 This review focuses on drug development, patient selection, and clinical perspectives on using PI3K inhibitors to treat breast cancer.
Biochemistry of the PI3K Signaling Pathway
Three PI3K classes have been discovered, of which class IA PI3K, a heterodimer comprising a p85 regulatory subunit and a p110 catalytic subunit,5 is clearly implicated in human cancers.6 The genes PIK3CA, PIK3CB, and PIK3CD encode three homologous class IA catalytic isoforms: p110α, p110β, and p110δ, respectively.7 In usual state, the PI3K p85 regulatory subunit interacts with p110 and inhibits its kinase activity, upon the receptor tyrosine kinase activation; the p110 is relieved,8,9 then phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) to generate phosphatidylinositol (3,4,5)-trisphosphate (PIP3) at the plasma membrane. The second messenger PIP3 enables phosphorylation of the serine/threonine kinase AKT and of 3-phosphoinositide-dependent protein kinase-1 (PDK-1).10–12 Moreover, PDK-1 also activates AKT, which phosphorylates many downstream kinases, including the mammalian target of rapamycin (mTOR) complex.13,14 This phosphorylation cascade regulates cell survival and metabolism (Figure 1).15 On the other hand, the tumor suppressor phosphatase and tensin homolog (PTEN) dephosphorylates PIP3, counteracting PI3K signaling.16
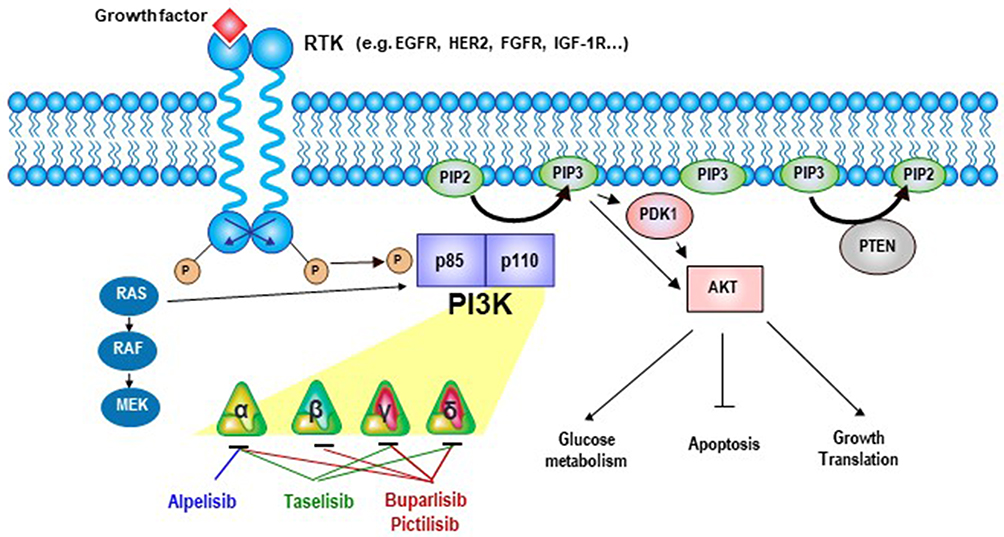 |
Figure 1 Signaling by the phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway. |
PI3K Signaling Pathway Alterations in Breast Cancer
In breast cancer, the PI3K signaling pathway can be activated in two ways: one involves alterations (amplification or activating mutations) in genes that encode molecules which participate in the PI3K pathway; the other involves activation of upstream receptor tyrosine kinases. Table 1 summarizes numerous studies that have revealed different frequencies of alterations in PI3K pathway components.17 PIK3CA activating mutations are the most common type, with rates of 28–47%, 23–33%, and 8–25% in HR+/HER2−, HER2+, and triple-negative breast cancer subtypes, respectively.18–24 PIK3CB amplification or PIK3R1 inactivating mutations have been reported but are uncommon.25,26 Loss-of-function mutations or reduced PTEN expression hyperactivate PI3K signaling, and occur at frequencies of 29–44%, 22%, and 67% in HR+/HER2−, HER2+, and triple-negative subtypes, respectively.18,19,27,28 Furthermore, inositol polyphosphate-4-phosphatase type IIB (encoded by INPP4B) can inhibit PI3K signaling, and reduced INPP4B expression and genomic loss have been reported in breast cancer.29,30 Amplification or mutations of AKT1, AKT2, and PDK1 hyperactivate AKT and its downstream proteins.19,31–33 Amplification of the ribosomal protein S6 kinase 1 gene (RPS6K1) and activation of RAS are infrequent mechanisms known to activate the PI3K pathway.34–36 Moreover, alterations that enhance signaling from receptor tyrosine kinases, such as HER2,37,38 epidermal growth factor receptor (EGFR),39 insulin-like growth factor-1 receptor (IGF-1R),40,41 and fibroblast growth factor receptor (FGFR),42,43 can also activate the PI3K pathway. In HR+/HER2− subtype breast cancer, overactive PI3K signaling pathway not only promotes cell proliferation and survival but also plays an important role in endocrine resistance by activating estrogen receptor in an estrogen-independent manner.44,45
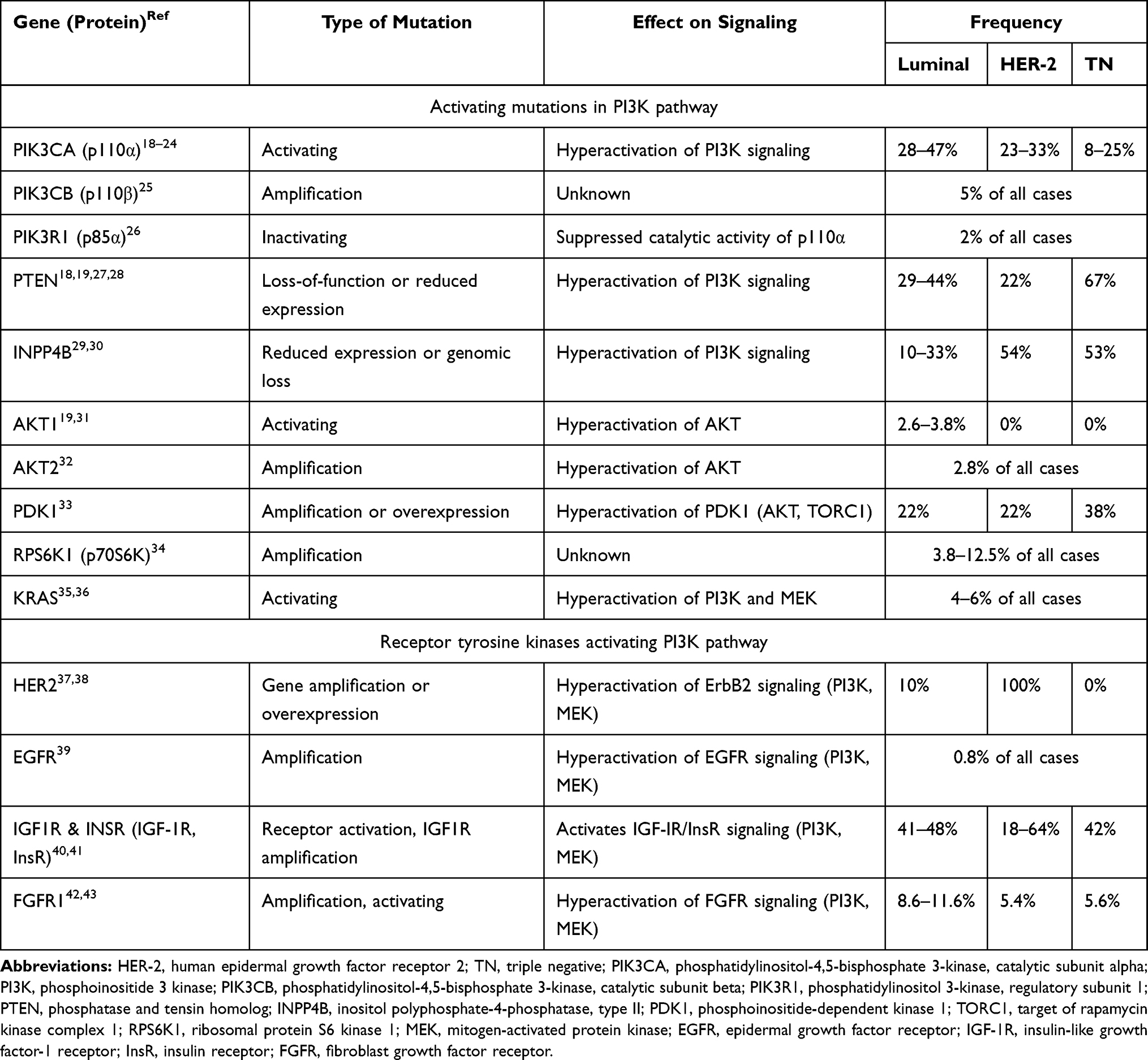 |
Table 1 Phosphatidylinositol 3-Kinase Pathway Alterations in Human Breast Cancers by Molecular Subtypes |
The Prognosis of Breast Cancer Patients with PIK3CA Mutations
Investigations of the prognostic role of PIK3CA mutation in early breast cancer have produced conflicting results.46–58 Zardavas et al conducted the largest pooled analysis of data from 10,319 patients in 19 studies of early breast cancer;59 32% of patients had PIK3CA mutations, with prevalence of 37%, 22%, and 18% in the HR+/HER2−, HER2+, and HR−/HER2− breast cancers, respectively. PIK3CA mutations were significantly associated with advanced age, estrogen receptor positivity (ER+), lower grade, and smaller tumors. In multivariate analysis, PIK3CA mutations remained significant for better invasive disease-free survival, but not for distant disease-free survival or overall survival (OS).
However, PIK3CA mutation has different clinical implications in metastatic breast cancer compared with early breast cancer. Table 2 summarizes the effect of PIK3CA mutations on treatment outcomes of metastatic breast cancer in pivotal Phase III clinical trials (only FERGI was phase II) that evaluated endocrine therapy with or without modern targeted agents.4,60–67 With few exceptions, progression-free survival (PFS) among cohorts using non-PI3K inhibitor-based therapies was numerically shorter in PIK3CA-mutated versus non-mutated patients. Signorovitch et al did a meta-regression analysis of the association between PIK3CA mutation and PFS, which focused on non-PI3K inhibitor therapies in ER+/HER2− metastatic breast cancer;68 PIK3CA mutation was consistently associated with significantly shorter PFS for patients with ER+/HER2− metastatic breast cancer who received non-PI3K inhibitor therapy. In addition, subgroup analysis of the SAFIR02 study provided information about the effect of PIK3CA mutations in ER+/HER2− metastatic breast cancer treated with chemotherapy.69 Among 364 patients, 337 (93%) had received hormone therapy and 104 (28%) had PIK3CA mutations, among whom 51% had stable disease or objective response after induction chemotherapy, compared to 69% in patients with wild-type PIK3CA. Patients with PIK3CA mutations also had worse OS, with median of 19.6 months versus 23.5 months for those without PIK3CA mutations (p = 0.04). Multivariate analysis confirmed that PIK3CA mutation was associated with chemoresistance and poor OS.69
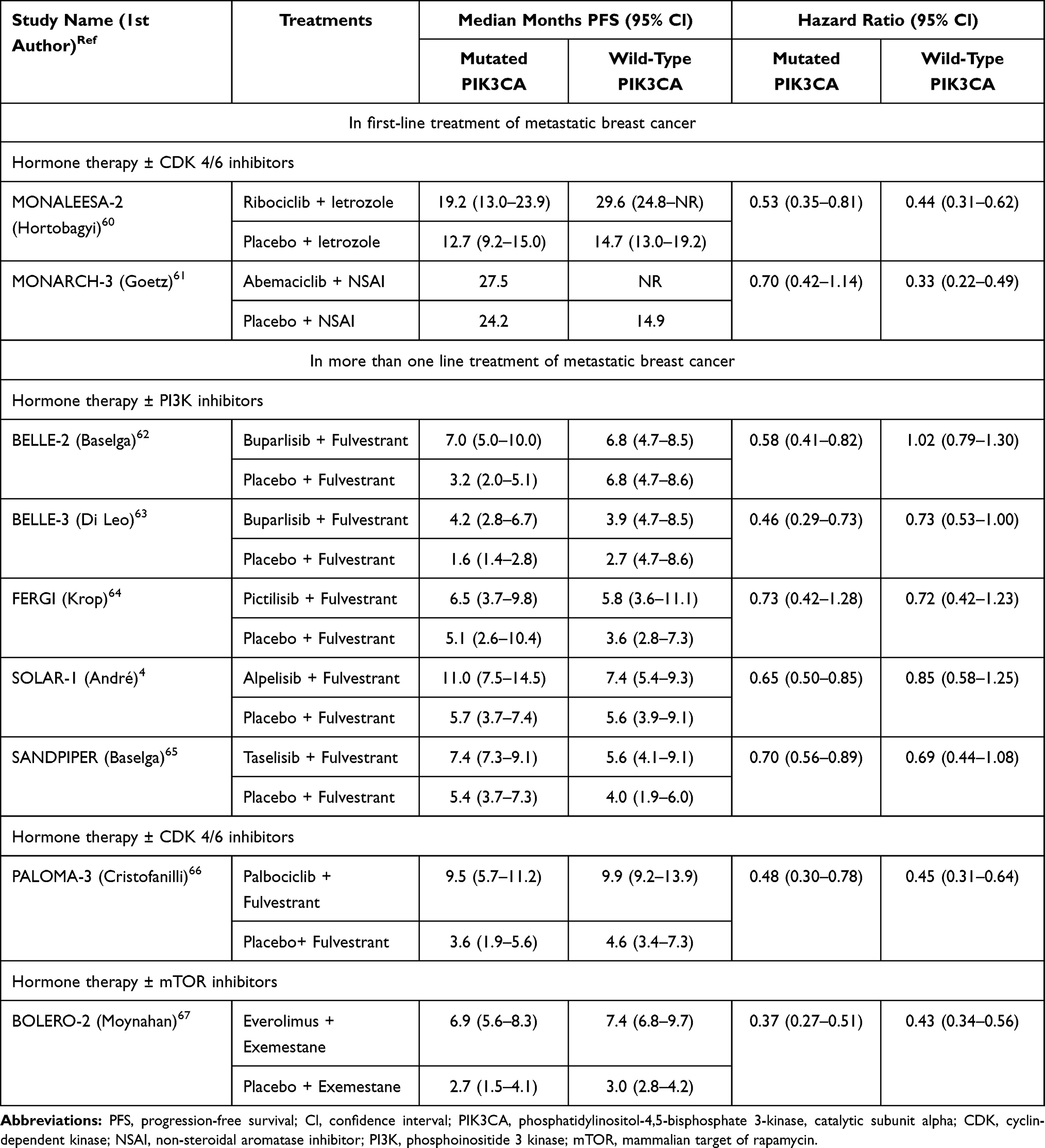 |
Table 2 Prognosis of PIK3CA Mutation Status and Treatment Outcomes in Luminal-Type, Metastatic Breast Cancer |
On the other hand, among cohorts that used PI3K-inhibitor based therapies, patients with PIK3CA mutations had a significantly better median PFS compared with non-mutated patients, highlighting that PIK3CA mutation is a predictive marker of PI3K inhibitor benefit (Table 2).
Clinical Trial Results of PI3K Inhibitors
Pan-PI3K Inhibitors and Beta Isoform-Sparing PI3K Inhibitor
Buparlisib
Buparlisib (BKM120; Novartis Pharmaceuticals, Basel, Switzerland) is an oral pan-PI3K inhibitor. Safety and efficacy of buparlisib combined with fulvestrant to treat patients with HR+/HER2− metastatic breast cancer were assessed in two large phase III randomized clinical trials (RCTs), BELLE-2 and BELLE-3, in 2nd/3rd-line settings or beyond, respectively.62,63 BELLE-2 enrolled postmenopausal women with HR+/HER2− metastatic breast cancer that progressed during/after aromatase inhibitor (AI) treatment and up to one previous line of chemotherapy for advanced disease.62 BELLE-3 differed in requiring participants to have HR+/HER2− metastatic breast cancer treated unsuccessfully by prior endocrine therapy and mTOR inhibitors.63 Patients in both trials were randomly assigned to receive fulvestrant plus either buparlisib, or placebo. Both trials met their primary efficacy endpoint, but buparlisib was associated with significantly more grade 3/4 adverse events (AEs), which included hepatic aminotransferase elevations, hyperglycemia, and rash. Many patients discontinued buparlisib prematurely due to poor tolerability, resulting in very short treatment exposure (median 1.9 months in BELLE-2). Moreover, central nervous system penetration of buparlisib proved disadvantageous – some patients suffered from depression, anxiety, and even rare suicide ideation. The study team decided to cease further development owing to the toxicity profile and proposed the investigation of more selective PI3K inhibitors, such as an α-specific PI3K inhibitor, to further improve the safety and benefits in this setting.
Nonetheless, the clinical trials of buparlisib provided valuable information. In an exploratory analysis of BELLE-2, in subsets (587 out of 1147 submitted plasma, and 34% [n=200] PIK3CA mutation tested positive) with detectable PIK3CA mutation in their circulating tumor DNA (ctDNA), the median PFS was 7.0 months in the buparlisib group and 3.2 months in the placebo group (HR 0.58; p = 0.001).62 Likewise, in also exploratory analyses of the BELLE-3 trial, adding buparlisib to fulvestrant significantly increased PFS in patients who had PIK3CA mutation, either using tumor tissue or ctDNA to determine mutation status. The median PFS was 4.2 months vs. 1.6 months; (HR 0.46 [0.29–0.73]; p = 0.00031) by ctDNA testing, and 4.7 months vs. 1.4 months (HR 0.39 [0.23–0.65], p<0.001) by tissue real-time polymerase chain reaction (RT-PCR).63 All of these observations support the rationale for PI3K inhibition plus endocrine therapy in ER+/HER2− patients with PIK3CA mutations.
Pictilisib
Another oral pan-PI3K inhibitor, pictilisib (GDC-0941; Genentech, San Francisco, CA, USA), was evaluated in the Phase II FERGI RCT of fulvestrant plus pictilisib versus placebo, which enrolled postmenopausal women with HR+/HER2− metastatic breast cancer resistant to treatment with AI in adjuvant or metastatic settings.64 Part 1 enrolled 168 patients irrespective of PIK3CA mutation status, whereas Part 2 excluded patients without PIK3CA mutations. In part 1, there was no significant difference in median PFS between pictilisib (6.6 months) versus placebo (5.1 months) (HR 0.74 95% CI 0.52–1.06; p = 0.096). In addition, no difference was observed when patients were stratified by presence of PIK3CA mutations (pictilisib 6.5 months vs. placebo 5.1 months; HR 0.73 95% CI 0.42–1.28; p = 0.268) or their absence (pictilisib 5.8 months vs. placebo 3.6 months; HR 0.72 95% CI 0.42–1.23; p = 0.230). Grade 3 or worse AEs occurred in 54 (61%) of 89 patients in the pictilisib group and in 22 (28%) of 79 patients in the placebo group; 19 pictilisib-related serious AEs were reported in 14 (16%) of 89 patients. Substantial toxicities caused 45% of patients to discontinue pictilisib or require dose reduction. The protocol in Part 2 (n = 61) was amended to reduce the starting dose of pictilisib from 340 mg to 260 mg; there was still no between-group difference in median PFS (pictilisib 5.4 months vs. placebo 10.0 months; HR 1.07 95% CI 0.53–2.18; p = 0.840). The authors concluded that the toxicity of pictilisib limited its tolerability, potentially limiting its efficacy.64 Similar to buparlisib, they suggested that future development should investigate agents with higher selectivity for specific PI3K isoforms or mutants, thus improving tolerability and affording a more robust therapeutic index to realize clinical benefit.
Taselisib
Taselisib (GDC-0032; Genentech) is not only beta-isoform sparing but also potently inhibits PI3K, exhibiting greater sensitivity for mutant PI3Kα compared with the wild-type isoform. In the phase III SANDPIPER RCT, postmenopausal patients with disease recurrence or progression during/following AI treatment were randomized 2:1 to taselisib versus placebo combined with fulvestrant.65 Participants were enriched for PIK3CA mutations (80% of total), and the primary endpoint was investigator-assessed PFS in patients with PIK3CA-mutated tumors. Among 516 participants with PIK3CA mutations, 340 received taselisib plus fulvestrant and 176 placebo plus fulvestrant; median PFS was 7.4 months in the taselisib arm versus 5.4 months in the placebo arm (HR 0.70 95% CI 0.56–0.89; p = 0.004). Meanwhile, the taselisib combination arm also had a significantly higher objective response rate: 28% versus 11.9%, p < 0.001. Among 120 participants without PIK3CA mutations, median PFS did not differ significantly between taselisib versus placebo (5.6 vs. 4.0 months; HR 0.69 95% CI 0.44–1.08; p = 0.106). Although SANDPIPER was a positive study and demonstrated that PIK3CA mutation could be targeted, the clinical benefit was modest and the tolerability was questionable. The most common grade ≥3 AEs in the taselisib/fulvestrant arm were diarrhea (12%), hyperglycemia (10%), colitis (3%), and stomatitis (2%). AEs led to more taselisib discontinuations (17% vs. 2%) and dose reductions (37% vs. 2%) compared with placebo. Concerns over safety and efficacy have halted further investigation of taselisib.65
PI3K Alpha-Selective Inhibitor
Alpelisib
Alpelisib (BYL719; Novartis Pharmaceuticals, Basel, Switzerland) is the first selective PI3Kα inhibitor. In preclinical models, it not only potently inhibited the two most common (hotspot) PIK3CA mutations (H1047R and E545K) at an IC50 of ~4 nmol/L,70 but also demonstrated a dual mechanism of action by inhibiting PI3K and inducing degradation of p110α in a dose-dependent manner in certain ER+/PIK3CA mutant breast cancer cell lines.71 Notably, PIK3CA mutation and/or amplification were mandatory inclusion criteria of the first-in-human Phase I study conducted by Juric et al,72 which enrolled 134 patients across cancer types, who all received alpelisib monotherapy. Frequent treatment-related AEs of all grades included hyperglycemia (51.5%), nausea (50.0%), decreased appetite (41.8%), diarrhea (40.3%), and vomiting (31.3%). Objective tumor responses were only observed at once-daily doses of ≥270 mg, with an overall response rate (ORR) of 6.0%. Stable disease was achieved in 70 (52.2%) patients and maintained for >24 weeks in 13 (9.7%) patients. Thirty-six patients had breast cancer, and among 22 with ER+/HER2− advanced breast cancer who were treated with ≥270 mg alpelisib once daily, the median PFS was 5.5 (95% CI 3.0–7.0) months.72
The subsequent phase Ib study (ClinicalTrials.gov: NCT01219699) assessed the maximum tolerated dose, safety, and efficacy of alpelisib combined with fulvestrant in 87 women with ER+/HER2– metastatic breast cancer.73 Patients in the dose escalation cohort were required to have PIK3CA-altered tumors and received fixed-dose fulvestrant 500 mg plus once-daily doses of alpelisib (300 mg, n = 9; 350 mg, n = 8; 400 mg, n = 11). In the dose-expansion cohort, 59 patients received 400 mg alpelisib once-daily plus fulvestrant, irrespective of PIK3CA status. Similar to single-agent alpelisib, the maximum tolerated dose in combination with fulvestrant was 400 mg once daily; the recommended Phase 2 dose was 300 mg once daily. The most frequent grade 3/4 AEs (≥10% of patients) during treatment with 400 mg alpelisib once daily were hyperglycemia (22%) and maculopapular rash (13%); nine patients permanently discontinued due to AEs. The median PFS of patients with PIK3CA-altered tumors (treated with 300–400 mg alpelisib once-daily plus fulvestrant) was longer (9.1 months 95% CI, 6.6–14.6) than those with PIK3CA -wild-type tumors, who all received 400 mg alpelisib once-daily plus fulvestrant (4.7 months; 95% CI, 1.9–5.6). The ORR among patients with PIK3CA alterations was 29% (95% CI, 17–43%) compared to no objective tumor response in the PIK3CA wild-type group.73
A small pilot phase Ib study (B-YOND) tested alpelisib or buparlisib combined with tamoxifen plus goserelin as first-line endocrine therapy in Asian premenopausal patients with HR+/HER2– advanced breast cancer.74 The recommended phase 2 doses of alpelisib and buparlisib in combination with tamoxifen and goserelin were 350 mg and 100 mg, respectively. No unexpected safety findings were reported. Treatment emergent AEs led to discontinuation in 18.8% and 53.8% of patients treated with alpelisib (n = 16) or buparlisib (n = 13), respectively. The most common grade 3/4 treatment emergent AEs were hypokalemia (12.5%), hyperglycemia (6.3%), and rash (6.3%) in the alpelisib group, and increased alanine aminotransaminase (30.8%), increased aspartate aminotransaminase (23.1%), and anxiety (15.4%) in the buparlisib group. Either the higher recommended phase 2 dose of 350 mg or the significantly lower incidence of hyperglycemia suggests that premenopausal women (younger, with a lower probability of glucose intolerance/insulin resistance) tolerate alpelisib better than postmenopausal women do. The observed median PFS was 25.2 months in the alpelisib group and 20.6 months in the buparlisib group. Notably, tumor PIK3CA mutation status was not an inclusion criteria in the B-YOND study; although the patient number was limited, these data suggest that alpelisib plus endocrine therapy may be a potentially efficacious treatment for premenopausal patients with HR+/HER2– advanced breast cancer.
The large phase III SOLAR-1 RCT compared fulvestrant plus alpelisib (300 mg once daily) versus placebo in 572 men or postmenopausal women with HR+/HER2– advanced breast cancer that recurred or progressed on AI treatment.4 Patients were stratified according to centrally assessed PIK3CA mutation status based on tumor tissue RT-PCR (11 hotspot mutations on exons 7, 9, and 20), with 341 in the PIK3CA-mutant cohort and 231 in the non-mutant cohort. The primary endpoint was investigator-assessed PFS in the PIK3CA-mutant cohort. Median PFS of patients with PIK3CA mutations was 11.0 months in the alpelisib/fulvestrant arm versus 5.7 months in the placebo/fulvestrant arm (HR 0.65 95% CI 0.50–0.85; p < 0.001). The ORR was also higher with alpelisib/fulvestrant versus placebo/fulvestrant (26.6% vs. 12.8%); the corresponding ORR rates among patients with measurable disease were 35.7% versus 16.2%. In contrast, there was no meaningful between-treatment difference in median PFS in the PIK3CA-non-mutant cohort; 7.4 months vs. 5.6 months (HR 0.85 95% CI 0.58–1.25). In 2019, the positive results of SOLAR-1 prompted the US FDA to approve alpelisib plus fulvestrant for treating men and postmenopausal women with ER+/HER2–, PIK3CA-mutated advanced or metastatic breast cancer. André et al reported OS results of the PIK3CA-mutant cohort;75 with median follow-up of 30.8 months, median OS was 39.3 months (95% CI 34.1–44.9) with alpelisib/fulvestrant and 31.4 months (95% CI 26.8–41.3) with placebo/fulvestrant (HR 0.86 95% CI 0.64–1.15; p = 0.15). In patients with lung and/or liver metastases, median OS was 37.2 months (95% CI 28.7–43.6) with alpelisib/fulvestrant and 22.8 months (95% 19.0–26.8) with placebo/fulvestrant (HR 0.68 95% CI 0.46–1.00). Median time to chemotherapy was 23.3 months (95% CI 15.2–28.4) with alpelisib/fulvestrant and 14.8 months (95% CI 10.5–22.6) with placebo/fulvestrant (HR 0.72 95% CI 0.54–0.95). Although the OS result did not meet the prespecified criterion for statistical significance, the absolute between-treatment difference of 8 months was clinically relevant and valuable. The PFS benefit was not only maintained but even enhanced in terms of OS outcome.
When SOLAR-1 was designed, CDK 4/6 inhibitors were not yet approved; however, combined hormonal plus CDK4/6 inhibitor therapy has now become the standard first-line treatment for HR+/HER2– advanced breast cancer. In the SOLAR-1 PIK3CA-mutant cohort, only 20 patients (5.9%) had previously received CDK4/6 inhibitors. To assess the efficacy and safety of combining alpelisib with endocrine therapy (fulvestrant or letrozole) in PIK3CA-mutated, HR+/HER2– advanced breast cancer after CDK4/6 inhibition failed, Rugo et al conducted the BYLieve phase 2 study (ClinicalTrials.gov: NCT03056755).76 Cohort A results were presented at ASCO 2020: 127 patients who had received AI and CDK 4/6 inhibitors as the last prior therapy line were enrolled and received alpelisib plus fulvestrant. The primary endpoint of the proportion of patients surviving without disease progression at 6 months, was 50.4%. The secondary endpoint of median PFS was 7.3 months.76 The result was consistent with SOLAR-1 subgroup analysis: of 9/20 patients receiving fulvestrant plus alpelisib, 44% were alive without disease progression at 6 months and median PFS was 5.5 months.4 These results support using alpelisib plus fulvestrant after CDK4/6 inhibitors;4,76 data on other BYLieve cohorts are anticipated.76
Since high incidence of hyperglycemia was observed in early-phase studies,72,73 SOLAR-1 excluded patients with type 1 diabetes or uncontrolled type 2 diabetes (fasting plasma glucose [FPG] >140 mg/dL [7.7 mmol/L] or glycosylated hemoglobin [HbA1c] level >6.4%). Nevertheless, hyperglycemia was still the most frequent grade 3/4 AE in the overall population (36.6% in the alpelisib/fulvestrant group vs. 0.7% in the placebo/fulvestrant group), followed by rash (9.9% vs. 0.3%) and diarrhea (6.7% vs. 0.3%). Compared to the placebo group, more patients treated with alpelisib discontinued due to AEs (25.0% vs. 4.2%).4
Clinical Practice Considerations
Hyperglycemia, diarrhea, and rash were the most common toxicities associated with PI3K inhibitors, and appropriate monitoring and management are essential, especially regarding hyperglycemia. Because PI3K/AKT/mTOR plays a key role in the insulin signaling pathway, hyperglycemia is a mechanism-based, on-target effect of PI3K/AKT/mTOR inhibitors.77 Although the RCT excluded patients with diabetes, in real world practice it would be impossible to disallow the use of effective anti-cancer drugs by patients with diabetes. According to the World Health Organization, the global prevalence of diabetes in adult is around 8.5%,78 and physicians will inevitably treat patients with breast cancer who have comorbid diabetes. The minimal requirement is to follow the approved label instructions to assess FPG weekly for at least 2 weeks and every 4 weeks thereafter (plus HbA1c).79 However, longer duration of weekly follow-up may be required for those with higher risk of developing grade 3/4 hyperglycemia. In SOLAR-1, in addition to the prediabetic (FPG, 5.6 to <7.0 mmol/L and HbA1c, 5.7 to <6.5%) and diabetic (FPG, ≥7.0 mmol/L or HbA1c, ≥6.5%) status, obesity (defined as body mass index ≥30) and advanced age (≥75 years old) were also associated with higher incidence rates of grade 3/4 hyperglycemia.80 In the earliest phase of SOLAR-1, before the protocol was amended to exclude patients with diabetes, one patient whose HbA1c was ~8% was enrolled; this patient did have grade 4 hyperglycemia, but after proper dose reduction and supervision by an endocrinologist, she was able to continue study treatment for more than 2 years. Although there are insufficient data regarding the safety of alpelisib in patients with diabetes, they should not be prohibited from using this treatment provided that careful monitoring is implemented and early endocrinologist consultation is sought. There is some pertinent indirect evidence: in SOLAR-1, 6.3% of patients discontinued alpelisib due to hyperglycemia compared with only 1.6% in the BYLieve study.4,76 Intensive monitoring and early interventions, such as metformin and diet education, all help to keep patients on treatment with tolerable toxicities.
Rugo et al reported the time course and management of the key AEs in SOLAR-1 and provided valuable clinical guidance.80 Eighty-six participants had received prophylactic (before the onset of skin rash) anti-rash medication, among whom, 70% received anti-histamines. Compared with those (n = 198) who did not receive preventive medication, these patients experienced lower rates of any-grade rash (26.7% versus 64.1%) and grade 3 rash (11.7% versus 22.7%).80 Therefore, prophylactic antihistamine has been recommended to reduce the frequency and severity of skin rash and is also endorsed by the ABC-5 consensus.81 Nonetheless, systemic corticosteroid use and treatment interruption are necessary for higher grade events, and it is highly desirable to include a dermatologist in the team. In SOLAR-1, most bouts of diarrhea were grade 1 and 2. And like other anti-cancer drugs, the incidence usually decreased with education and preemptive anti-diarrheal agents and can be satisfactorily handled. We believe that to keep patients adherent to alpelisib treatment by proper toxicity management may potentially increase clinical benefit. An indirect supportive evidence was observed in SOLAR-1. In PIK3CA mutation cohort, the median dose intensity of alpelisib was 248mg/day; and the PFS was longer in patients who received higher dose intensity compared with lower dose intensity (12.5 months versus 9.6 months).80
Both SANDPIPER and SOLAR-1 showed that the benefit of PI3K inhibitors is limited to PIK3CA-mutant tumors;4,65 therefore, patient selection is crucially important. Several questions arise: What sample should be tested? When should the test be ordered? What method should be used to detect mutations? Do PI3K inhibitors benefit different PIK3CA mutations to the same degree?
Unlike ESR1 mutations, which are acquired under the selective pressure of estrogen deprivation, PIK3CA mutations are initiating truncal events, supported by the high percentage in the TCGA dataset,3 or the tumor evolution analysis by Turajlica et al.82 In paired primary tumor and metastatic tumor mutation analyses, PIK3CA status was highly concordant, and mutation rates were not always increased upon metastases.83,84 The SOLAR-1 protocol determined PIK3CA mutation status using tissue-based testing (therascreen® PCR kit, Qiagen), thus definitively endorsing tissue-testing as standard. Meanwhile, a secondary endpoint was PFS in patients with PIK3CA mutations detected by plasma ctDNA, and the results also supported the value of ctDNA-detected PIK3CA mutations in predicting benefit from alpelisib. In the ctDNA-defined PIK3CA-mutant cohort, the median PFS for the alpelisib plus fulvestrant arm was 10.9 months versus 3.7 months for the fulvestrant only arm (HR 0.55).4 This finding led to US FDA approval of liquid biopsy testing as an alternative to tissue testing. Nevertheless, we have some concerns. First, the ctDNA titer is typically quite low and may be undetectable unless the method is highly sensitive. Indeed, by applying the same PCR platform in SOLAR-1, only 186 patients were defined as PIK3CA-mutated by plasma ctDNA compared to 341 patients by tissue testing. The much lower detection rate compared with tissue testing owed primarily to low ctDNA titer. Therefore, although the US FDA approves plasma ctDNA testing, it also reminds us to repeat tissue testing once ctDNA testing becomes negative for PIK3CA mutations. Second, some may favor ctDNA testing because the ctDNA-determined PIK3CA-mutant cohort appeared to get even larger PFS benefit with alpelisib, suggested by a lower HR of 0.55 compared to 0.65 in the primary cohort.4 We would like to reemphasize that ctDNA originates from DNA fragments shed by cancer cells, primarily from apoptosis, and less from necrosis, or even secretory processes.85 Therefore, detectable ctDNA correlates with larger tumor burden or aggressive tumors with active proliferation, which are usually accompanied by stress and consequent apoptosis. Based on this theory, researchers tracked serial ctDNA to successfully predict disease relapse,86 or treatment response.87 Hence, compared to ctDNA-non-mutant patients, those with ctDNA-detected PIK3CA-mutant status would be expected to have had disease that is more serious and was PIK3CA activating mutation driven. This may explain why the small ctDNA-detected PIK3CA mutation subset in SOLAR-1 seemed to derive larger benefit from adding on alpelisib compared with the primary cohort.4 Among patients with advanced breast cancer that progressed on endocrine therapy who harbored PIK3CA-mutant tumors, those with high disease burden certainly get most benefit from PI3K inhibition. Meanwhile, OS subgroup analysis showed that the absolute difference of 37.2 versus 22.8 months in patients with visceral (lung or liver) metastases was more pronounced than that in the entire PIK3CA-mutant cohort (39.3 vs. 30.8 months), which also supports this hypothesis.75 Despite the lack of level I evidence, the preferred strategy endorsed by current international guidelines is to treat HR+/HER2− advanced breast cancer with upfront endocrine-based therapies.81,88 The use of chemotherapy is delayed until failing consecutive 2 to 3 lines of endocrine therapies (with or without targeted therapies); unless the patients have visceral crisis, aggressive disease status, or endocrine therapy refractory diseases.81,88 Recently, a large network meta-analysis comparing chemotherapies and endocrine-based therapies provided supportive evidences for these recommendations.89 Based on trial evidence demonstrating the efficacy of alpelisib after failing first-line endocrine therapy with or without CDK4/6 inhibitors, it is justifiable to test PIK3CA mutation status upon diagnosing HR+/HER2– advanced breast cancer.
We also know that the ctDNA mutation detection rate varies with different methodologies. For instance, in BELLE-3,63 the BEAMing assay (a kind of droplet digital PCR) was used to detect PIK3CA mutations in ctDNA. Droplet digital PCR has very high sensitivity, with a lower detection limit as low as 0.01%. In BELLE-3, tumor tissue analysis, the PIK3CA mutation rates were 34% by RT-PCR and 39% in ctDNA analysis using BEAMing technology, although not within the same subsets; the concordance rate was 83%. Both PIK3CA mutation-testing methods were shown to be a valid predictive marker of buparlisib benefit. Notably, in BELLE-3, using ctDNA analysis to define PIK3CA mutation status did not predict greater PFS benefit: HR 0.46 (95% CI 0.29–0.73) for ctDNA analysis versus HR 0.39 (95% CI 0.23–0.65) for tissue analysis. Consequently, tissue testing is appropriate unless no specimen is readily available.
Although the companion diagnostic Qiagen kit (therascreen® assay) detects 11 PIK3CA mutations over exons 7 (C420R), 9 (E542K, E545K/A/D/G, Q546E/R), and 20 (H1047R/Y/L), most mutations occur in exon 9 (helical domain) and exon 20 (kinase domain). In SOLAR-1, H1047X was the most prevalent locus (n = 193; 57%), followed by E545X (n = 106; 31%), and E542K (n = 60; 18%). Exon 7 C420R (n = 6; 2%) and exon 9 Q546X (n = 5; 1%) were much less frequent.90 The rarity of non-hotspot PIK3CA mutations makes it difficult to demonstrate alpelisib benefit; more data will be required to validate the efficacy.
Next generation sequencing (NGS) is another promising technology, since NGS covers almost all exons (depending on the panel), and goes beyond PIK3CA point mutations, to also detect insertion/deletion and gene copy number changes. In an exploratory analysis of SOLAR-1, retrospective tissue NGS results (tested with FoundationOne CDx 324-gene panel) correlated with median PFS.91 Valid NGS results were available in 404 (71%) of all participants, among whom, 31 patients had tumor PIK3CA alterations detected by NGS but not by RT-PCR-based testing. Despite some discordance, patients whose tumors harbored PIK3CA alterations detected by NGS also benefited from alpelisib; therefore, the US FDA has approved FoundationOne CDx as a companion diagnostic.
We all acknowledged that PIK3CA mutations are heterogeneous and the full spectrum is far beyond the therasceen® 11 gene panels. Martínez-Sáez et al conducted a pooled NGS database analysis and found that the therascreen® gene panels can capture as low as 72% of all the NGS identified PIK3CA mutations.24 It implied that in clinical practice, as NGS testings become more and more popular, it would not be uncommon to identify PIK3CA mutations that have not been validated in SOLAR-1 study. That is a difficult situation; nevertheless, based on available clinical trial results, we should only give alpelisib to patients with one of the aforementioned 11 mutations, detected by whichever tools we use. Hopefully, the efficacy of alpelisib in other PIK3CA mutations (beyond the therascreen® 11 gene panels) or even other PI3K pathway alterations can be validated in future clinical trials.
Perspective
Many preclinical studies have investigated the potential mechanisms of PI3K inhibitor resistance. Although PI3K inhibitors can decrease AKT signaling, AKT inhibitors can sensitize breast cancer cell lines resistant to PI3K inhibitors. A combinational drug screen of PIK3CA mutant cell lines revealed that synergistic inhibition of CDK4/6 and PI3K inhibitors may overcome the resistance of PI3K inhibitor monotherapy.92 PDK-1 signaling can activate the mTOR complex without activating AKT,93 and IGF-1 and other growth factors can also activate mTOR signaling and mediate resistance to PI3K inhibitors.94 Thus, hormone therapy in combination with more than one class of PI3K, AKT, mTOR, or CDK 4/6 inhibitors can be developed to enhance the efficacy of treatments for HR+/HER2– breast cancer. However, tolerability will remain a concern due to the different toxicity profiles of these drugs.
Table 3 lists ongoing clinical trials of alpelisib in various indications. In subgroup analyses of SOLAR-1, patients with PTEN loss and non-altered PIK3CA also seemed to benefit from alpelisib treatment (HR: 0.5 95% CI 0.13–1.89).91 It is unlikely that any future clinical trials will specifically aim at this population. However, a phase III RCT (EPIK-B3; ClinicalTrials.gov: NCT04251533) focused on patients with triple negative breast cancer and with PIK3CA mutations or PTEN loss is ongoing; patients will be randomized to nab-paclitaxel with or without alpelisib.
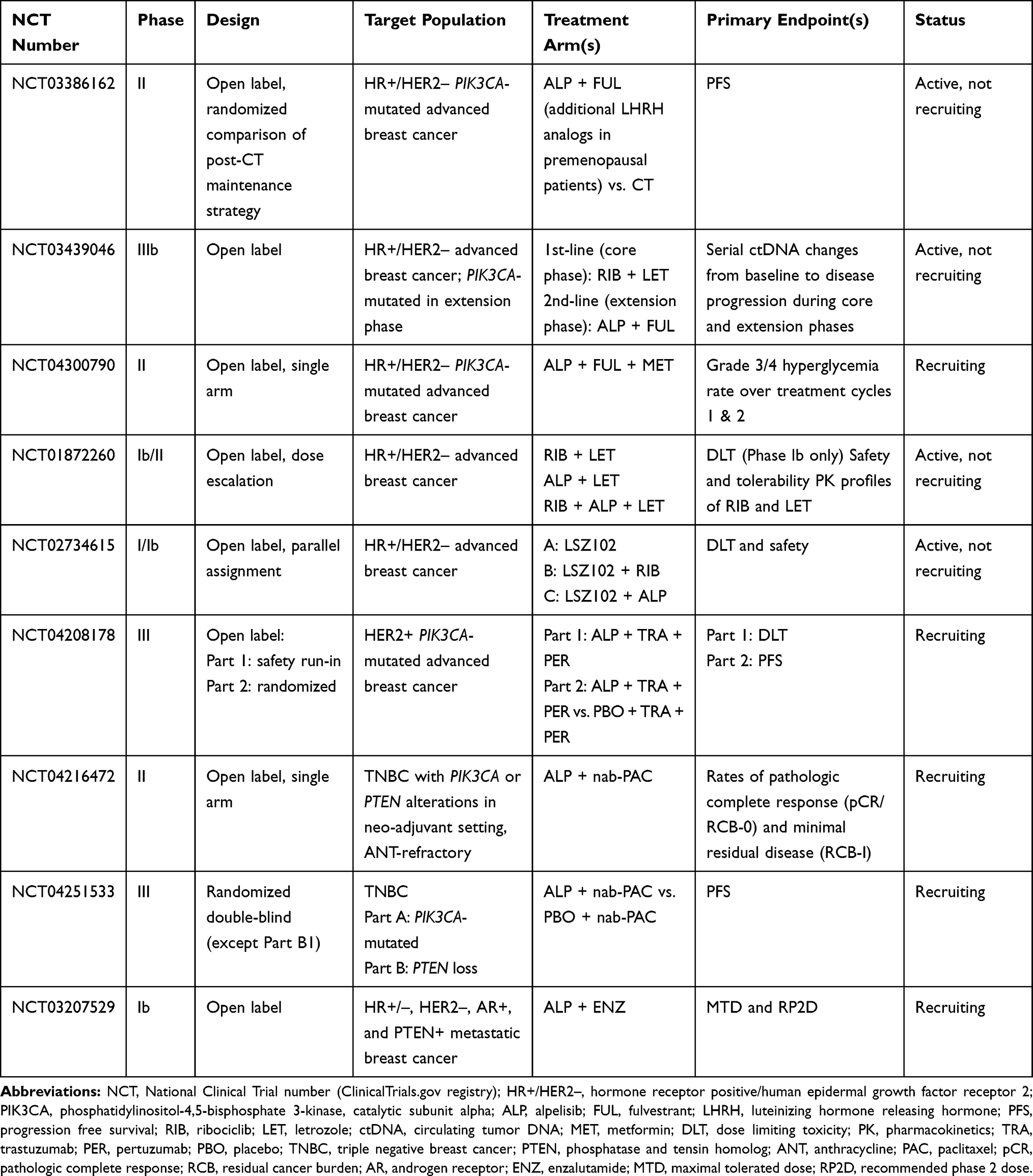 |
Table 3 Current/Future Clinical Trials Involving Alpelisib Treatment in Various Combinations and Settings |
On the other hand, PIK3CA mutations contribute to resistance to trastuzumab, which is an anti-HER2 monoclonal antibody.95,96 In the BOLERO-3 study, women with HER2+, trastuzumab-resistant advanced breast cancer were randomly assigned to receive everolimus plus trastuzumab and vinorelbine versus placebo plus trastuzumab and vinorelbine. The median PFS was 7.0 months in patients treated with everolimus and 5.8 months in patients treated with placebo (HR 0.78; p = 0.007). Although BOLERO-3 met the primary endpoint, the benefit was not clinically meaningful. Thus, it is intriguing to hypothesize that direct targeting of PI3K with specific inhibitors may give more clinical benefit than targeting the downstream mTOR complex. A phase III clinical trial (EPIK-B2; ClinicalTrials.gov: NCT04208178) specifically targeting HER2+ breast cancer is ongoing (Table 3).
In conclusion, alpelisib is the first oral α-specific PI3K inhibitor approved by the US FDA, and regulatory authorities in Europe and Australia for use in men and postmenopausal women with HR+/HER2–, PIK3CA-mutated, advanced or metastatic breast cancer that progresses on an endocrine therapy. Although isoform selectivity made alpelisib more tolerable than previous pan-PIK3 inhibitors, there were still substantial toxicities, especially hyperglycemia, rash, and diarrhea. With appropriate AE management and patient selection by validated testing methods, we hope that all eligible patients can benefit from receiving this new treatment. Further clinical trials to assess combinations of hormone therapy with PI3K, AKT, mTOR, or CDK 4/6 inhibitors, or studies in other breast subtypes are ongoing.
Acknowledgments
David Neil, PhD, of Dr. Word Ltd., Taiwan, provided professional editorial services, supported by funding from National Taiwan University Hospital.
Disclosure
Dr Chang reports consultancy/speaker fees from Novartis, Pfizer, AstraZeneca, and Roche, outside the submitted work. Dr Lu discloses clinical study grants/fees and/or consultancy/speaker fees from Novartis, Pfizer, Roche, Eisai, Eli Lily, MSD, and AstraZeneca. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. doi:10.1126/science.296.5573.1655
3. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi:10.1038/nature11412
4. André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–1940. doi:10.1056/NEJMoa1813904
5. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. doi:10.1038/nrg1879
6. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. doi:10.1038/onc.2008.245
7. Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507.
8. Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3ʹ-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18(3):1379–1387. doi:10.1128/MCB.18.3.1379
9. Yu J, Wjasow C, Backer JM. Regulation of the p85/p110 alpha phosphatidylinositol 3ʹ-kinase. Distinct roles for the N-terminal and C-terminal SH2 domains. J Biol Chem. 1998;273(46):30199–30203. doi:10.1074/jbc.273.46.30199
10. Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275(5300):665–668. doi:10.1126/science.275.5300.665
11. Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17(1):338–344. doi:10.1128/MCB.17.1.338
12. Mora A, Komander D, van Aalten DMF, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15(2):161–170. doi:10.1016/j.semcdb.2003.12.022
13. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi:10.1126/science.1106148
14. Wang L, Lawrence JC
15. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi:10.1016/j.cell.2012.03.017
16. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–13378. doi:10.1074/jbc.273.22.13375
17. Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224. doi:10.1186/bcr3039
18. Pérez-Tenorio G, Alkhori L, Olsson B, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007;13(12):3577–3584. doi:10.1158/1078-0432.CCR-06-1609
19. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–6091. doi:10.1158/0008-5472.CAN-07-6854
20. Ellis MJ, Lin L, Crowder R, et al. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast Cancer Res Treat. 2010;119(2):379–390. doi:10.1007/s10549-009-0575-y
21. Campbell IG, Russell SE, Choong DY, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64(21):7678–7681. doi:10.1158/0008-5472.CAN-04-2933
22. Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, et al. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther. 2011;10(6):1093–1101. doi:10.1158/1535-7163.MCT-10-1089
23. Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69(10):4116–4124. doi:10.1158/0008-5472.CAN-08-3441
24. Martínez-Sáez O, Chic N, Pascual T, et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020;22:45. doi:10.1186/s13058-020-01284-9
25. Crowder RJ, Phommaly C, Tao Y, et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009;69(9):3955–3962. doi:10.1158/0008-5472.CAN-08-4450
26. Jaiswal BS, Janakiraman V, Kljavin NM, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16(6):463–474. doi:10.1016/j.ccr.2009.10.016
27. Saal LH, Johansson P, Holm K, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007;104(18):7564–7569. doi:10.1073/pnas.0702507104
28. Shoman N, Klassen S, McFadden A, Bickis MG, Torlakovic E, Chibbar R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod Pathol. 2005;18(2):250–259. doi:10.1038/modpathol.3800296
29. Gewinner C, Wang ZC, Richardson A, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16(2):115–125. doi:10.1016/j.ccr.2009.06.006
30. Fedele CG, Ooms LM, Ho M, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A. 2010;107(51):22231–22236. doi:10.1073/pnas.1015245107
31. Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–444. doi:10.1038/nature05933
32. Bellacosa A, de Feo D, Godwin AK, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64(4):280–285. doi:10.1002/ijc.2910640412
33. Maurer M, Su T, Saal LH, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009;69(15):6299–6306. doi:10.1158/0008-5472.CAN-09-0820
34. Monni O, Barlund M, Mousses S, et al. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc Natl Acad Sci U S A. 2001;98(10):5711–5716. doi:10.1073/pnas.091582298
35. Rochlitz CF, Scott GK, Dodson JM, et al. Incidence of activating ras oncogene mutations associated with primary and metastatic human breast cancer. Cancer Res. 1989;49(2):357–360.
36. Di Nicolantonio F, Arena S, Tabernero J, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120(8):2858–2866. doi:10.1172/JCI37539
37. Arpino G, Green SJ, Allred DC, et al. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a Southwest Oncology Group study. Clin Cancer Res. 2004;10(17):5670–5676. doi:10.1158/1078-0432.CCR-04-0110
38. Ellis MJ, Tao Y, Young O, et al. Estrogen-independent proliferation is present in estrogen-receptor HER2-positive primary breast cancer after neoadjuvant letrozole. J Clin Oncol. 2006;24(19):3019–3025. doi:10.1200/JCO.2005.04.3034
39. Al-Kuraya K, Schraml P, Torhorst J, et al. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res. 2004;64(23):8534–8540. doi:10.1158/0008-5472.CAN-04-1945
40. Law JH, Habibi G, Hu K, et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008;68(24):10238–10246. doi:10.1158/0008-5472.CAN-08-2755
41. Fu P, Ibusuki M, Yamamoto Y, et al. Insulin-like growth factor-1 receptor gene expression is associated with survival in breast cancer: a comprehensive analysis of gene copy number, mRNA and protein expression. Breast Cancer Res Treat. 2011;130(1):307–317. doi:10.1007/s10549-011-1605-0
42. Elbauomy Elsheikh S, Green AR, Lambros MBK, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007;9(2):R23. doi:10.1186/bcr1665
43. Turner N, Pearson A, Sharpe R, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70(5):2085–2094. doi:10.1158/0008-5472.CAN-09-3746
44. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. doi:10.1146/annurev-med-070909-182917
45. Schettini F, Buono G, Trivedi M, et al. PI3K/mTOR inhibitors in the treatment of luminal breast cancer. why, when and to whom? Breast Care. 2017;12:290–294. doi:10.1159/000481657
46. Loi S, Michiels S, Lambrechts D, et al. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst. 2013;105(13):960–967. doi:10.1093/jnci/djt121
47. Sabine VS, Crozier C, Brookes CL, et al. Mutational analysis of PI3K/AKT signaling pathway in tamoxifen exemestane adjuvant multinational pathology study. J Clin Oncol. 2014;32(27):2951–2958. doi:10.1200/JCO.2013.53.8272
48. Azim HA
49. Boyault S, Drouet Y, Navarro C, et al. Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res Treat. 2012;132(1):29–39. doi:10.1007/s10549-011-1518-y
50. Barbareschi M, Buttitta F, Felicioni L, et al. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res. 2007;13(20):6064–6069. doi:10.1158/1078-0432.CCR-07-0266
51. Papaxoinis G, Kotoula V, Alexopoulou Z, et al. Significance of PIK3CA mutations in patients with early breast cancer treated with adjuvant chemotherapy: a Hellenic Cooperative Oncology Group (HeCOG) Study. PLoS One. 2015;10(10):e0140293. doi:10.1371/journal.pone.0140293
52. Li SY, Rong M, Grieu F, Iacopetta B. PIK3CA mutations in breast cancer are associated with poor outcome. Breast Cancer Res Treat. 2006;96(1):91–95. doi:10.1007/s10549-005-9048-0
53. Jensen JD, Knoop A, Laenkholm AV, et al. PIK3CA mutations, PTEN, and pHER2 expression and impact on outcome in HER2-positive early-stage breast cancer patients treated with adjuvant chemotherapy and trastuzumab. Ann Oncol. 2012;23(8):2034–2042. doi:10.1093/annonc/mdr546
54. Gallardo A, Lerma E, Escuin D, et al. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br J Cancer. 2012;106(8):1367–1373. doi:10.1038/bjc.2012.85
55. Kalinsky K, Jacks LM, Heguy A, et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009;15(16):5049–5059. doi:10.1158/1078-0432.CCR-09-0632
56. Maruyama N, Miyoshi Y, Taguchi T, Tamaki Y, Monden M, Noguchi S. Clinicopathologic analysis of breast cancers with PIK3CA mutations in Japanese women. Clin Cancer Res. 2007;13(2 Pt 1):408–414. doi:10.1158/1078-0432.CCR-06-0267
57. López-Knowles E, O’Toole SA, McNeil CM, et al. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer. 2010;126(5):1121–1131. doi:10.1002/ijc.24831
58. Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65(7):2554–2559. doi:10.1158/0008-5472-CAN-04-3913
59. Zardavas D, Te Marvelde L, Milne RL, et al. Tumor PIK3CA genotype and prognosis in early-stage breast cancer: a pooled analysis of individual patient data. J Clin Oncol. 2018;36(10):981–990. doi:10.1200/JCO.2017.74.8301
60. Hortobagyi GN, Stemmer SM, Burris HA, et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol. 2018;29:1541–1547. doi:10.1093/annonc/mdy155
61. Goetz MP, Beck JT, Campone M, et al. Abstract PD2-06: efficacy of abemaciclib based on genomic alterations detected in baseline circulating tumor DNA from the MONARCH 3 study of abemaciclib plus nonsteroidal aromatase inhibitor. Cancer Res. 2020;80(4 Suppl):P02–P06.
62. Baselga J, Im SA, Iwata H, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet Oncol. 2017;18(7):904–916. doi:10.1016/S1470-2045(17)30376-5
63. Di Leo A, Johnston S, Lee KS, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19(1):87–100. doi:10.1016/S1470-2045(17)30688-5
64. Krop IE, Mayer IA, Ganju V, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17(6):811–821. doi:10.1016/S1470-2045(16)00106-6
65. Baselga J, Dent SF, Cortés J, et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): primary analysis from SANDPIPER. J Clin Oncol. 2018;36(18Suppl):LBA1006. doi:10.1200/JCO.2018.36.18_suppl.LBA1006
66. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425–439. doi:10.1016/S1470-2045(15)00613-0
67. Moynahan ME, Chen D, He W, et al. Correlation between PIK3CA mutations in cell-free DNA and everolimus efficacy in HR+, HER2− advanced breast cancer: results from BOLERO-2. Br J Cancer. 2017;116(6):726–730. doi:10.1038/bjc.2017.25
68. Signorovitch J, Andre F, Wang R, et al. PIK3CA mutation status and progression-free survival in advanced hormone receptor positive (HR+)/human endocrine receptor negative (HER2−) metastatic breast cancer (mBC): a meta-analysis of published clinical trials. J Clin Oncol. 2020;38(15 Suppl):1069. doi:10.1200/JCO.2020.38.15_suppl.1069
69. Mosele F, Stefanovska B, Lusque A, et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann Oncol. 2020;31(3):377–386. doi:10.1016/j.annonc.2019.11.006
70. Fritsch C, Huang A, Chatenay-Rivauday C, et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117–1129. doi:10.1158/1535-7163.MCT-13-0865
71. Fritsch C, Pfister E, Ebel N, et al. Abstract 3934: determination of the PI3Kα selective inhibitor alpelisib mechanism of action and efficacy in ER+/PIK3CA mutant breast cancer pre-clinical models. Cancer Res. 2018;78(13 Suppl):Abstract 3934.
72. Juric D, Rodon J, Tabernero J, et al. Phosphatidylinositol 3-kinase α–selective inhibition with alpelisib (BYL719) in PIK3CA -altered solid tumors: results from the First-in-Human Study. J Clin Oncol. 2009;69(9):1291–1299. doi:10.1200/JCO.2017.72.7107
73. Juric D, Janku F, Rodón J, et al. Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol. 2019;5(2):e184475. doi:10.1001/jamaoncol.2018.4475
74. Lu YS, Lee KS, Chao TY, et al. A phase ib study of alpelisib or buparlisib combined with tamoxifen plus goserelin in premenopausal women with HR-positive HER2-negative advanced breast cancer. Clin Cancer Res. 2020. doi:10.1158/1078-0432.CCR-20-1008
75. André F, Ciruelos EM, Juric D, et al. Overall survival (os) results from SOLAR-1, a phase III study of alpelisib (ALP) + fulvestrant (FUL) for hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2–) advanced breast cancer (ABC). Ann Oncol. 2020;31(Suppl 4):S1150–S1151.
76. Rugo HS, Lerebours F, Ciruelos E, et al. Alpelisib (ALP) + fulvestrant (FUL) in patients (pts) with PIK3CA-mutated (mut) hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2–) advanced breast cancer (ABC) previously treated with cyclin-dependent kinase 4/6 inhibitor (CDKi) + aromatase inhibitor (AI): bYLieve study results. J Clin Oncol. 2020;38(15 Suppl):1006.
77. Busaidy N, Farooki A, Dowlati A, et al. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J Clin Oncol. 2012;30(23):2919–2928. doi:10.1200/JCO.2011.39.7356
78. World Health Organization. Global Report on Diabetes. Geneva: World Health Organization; 2016. Available from https://apps.who.int/iris/handle/10665/204871.
79. Novartis Pharmaceuticals Corporation. Piqray (Alpelisib) [Full Prescribing Information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; September, 2020. Available from: https://www.novartis.us/sites/www.novartis.us/files/piqray.pdf.
80. Rugo HS, André F, Yamashita T, et al. Time course and management of key adverse events during the randomized phase III SOLAR-1 study of PI3K inhibitor alpelisib plus fulvestrant in patients with HR-positive advanced breast cancer. Ann Oncol. 2020;31(8):P1001–P1010. doi:10.1016/j.annonc.2020.05.001
81. Cardoso F, Paluch-Shimon S, Senkus E, et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol. 2020;31(12):1623–1649. doi:10.1016/j.annonc.2020.09.010
82. Turajlica S, McGranahana N, Swanton C. Inferring mutational timing and reconstructing tumour evolutionary histories. Biochim Biophys Acta. 2015;1855(2):264–275. doi:10.1016/j.bbcan.2015.03.005
83. Bertucci F, Finetti P, Guille A, et al. Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget. 2016;7(19):27208–27219. doi:10.18632/oncotarget.8349
84. Jeselsohn R, Yelensky R, Buchwalter G, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20(7):1757–1767. doi:10.1158/1078-0432.CCR-13-2332
85. Alix-Panabières C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6(5):479–491. doi:10.1158/2159-8290.CD-15-1483
86. Garcia-Murillas I, Schiavon G, Weigelt B, et al. Mutation tracking in circulating tumor DAN predicts relapse in early breast cancer. Sci Transl Med. 2016;7(302):302ra133. doi:10.1126/scitranslmed.aab0021
87. O’Leary B, Hrebien S, Morden JP, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun. 2018;9(1):896. doi:10.1038/s41467-018-03215-x
88. National Comprehensive Cancer Network. Breast cancer (version 6.2020). Available from: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf.
89. Giuliano M, Schettini F, Rognoni C, et al. Endocrine treatment versus chemotherapy in postmenopausal women with hormone receptor-positive, HER2-negative, metastatic breast cancer: a systematic review and network meta-analysis. Lancet Oncol. 2019;20(10):1360–1369. doi:10.1016/S1470-2045(19)30420-6
90. Rugo HS, Mayer I, Conte P, et al. Abstract CT142: prevalence of PIK3CA mutations in patients with hormone receptor-positive, human epidermal growth factor-2-negative advanced breast cancer from the SOLAR-1 trial. Cancer Res. 2019;79(13Suppl):CT142.
91. Juric D, Andre F, Singer CF, et al. Abstract P4-10-04: clinical outcomes of alpelisib in hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer by next-generation sequencing-detected PIK3CA alteration status and phosphatase and tensin homolog loss: biomarker analysis from the SOLAR-1 study. Cancer Res. 2020;80(4Suppl):
92. Vora SR, Juric D, Kim N, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26(1):136–149. doi:10.1016/j.ccr.2014.05.020
93. Castel P, Ellis H, Bago R, et al. PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell. 2016;30(2):229–242. doi:10.1016/j.ccell.2016.06.004
94. Elkabets M, Vora S, Juric D, et al. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5(196):196ra99. doi:10.1126/scitranslmed.3005747
95. Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi:10.1016/j.ccr.2007.08.030
96. Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, Minami H. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2010;21(2):255–262. doi:10.1093/annonc/mdp304
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.