")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 9
Role and inhibition of GLI1 protein in cancer
Authors Mastrangelo E , Milani M
Received 16 November 2017
Accepted for publication 22 January 2018
Published 27 March 2018 Volume 2018:9 Pages 35—43
DOI https://doi.org/10.2147/LCTT.S124483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ignatius Ou
Eloise Mastrangelo, Mario Milani
CNR – Biophysics Institute, c/o Dipartimento di Bioscienze, Università degli Studi di Milano, Milano, Italy
Abstract: GLI1 is a transcriptional regulator involved in the development of different types of cancer. GLI1 transcriptional activity is regulated within the Hedgehog pathway (canonical activity), but can also be controlled independently (non-canonical activity) in the context of other signaling pathways. Experimental evidences show GLI1 involvement in both small- and non–small-cell lung cancers. Direct inhibition of the protein, in combination with other chemotherapeutic agents, represents a promising strategy for the treatment of different malignancies.
Keywords: Hedgehog pathway, transcriptional regulator, lung cancer, inhibitors
Introduction
GLI1 is a transcriptional factor component of the canonical and non-canonical Hedgehog (Hh) pathway. Hh signaling is evolutionarily conserved in Drosophila melanogaster and superior vertebrates. In mammals, three different Hh genes have been described as initiators of the signaling pathway: SHH, IHH, and DHH – each with a characteristic role and distribution.1,2 IHH and DHH have been shown to play important roles in normal tissue development, including bone formation and the pancreas, respectively. The SHh signaling pathway (initiated by the SHH glycoprotein) is the most studied, and it plays an essential role in the embryonic development required for appropriate cell differentiation and maintenance of tissue polarity.3
SHh-mediated transduction is activated through the binding of SHH to the PTCH.4 PTCH, in the absence of the SHH ligands, inhibits the activity of seven-transmembrane G-protein-coupled receptor like Smoothened (SMO). After SSH binding, PTCH inhibition of SMO is released, with resultant phosphorylation and nuclear translocation of the zinc finger (ZF) glioma-associated transcription factors GLI1, GLI2, and GLI3, which are terminal effectors of SHh signaling (Figure 1).
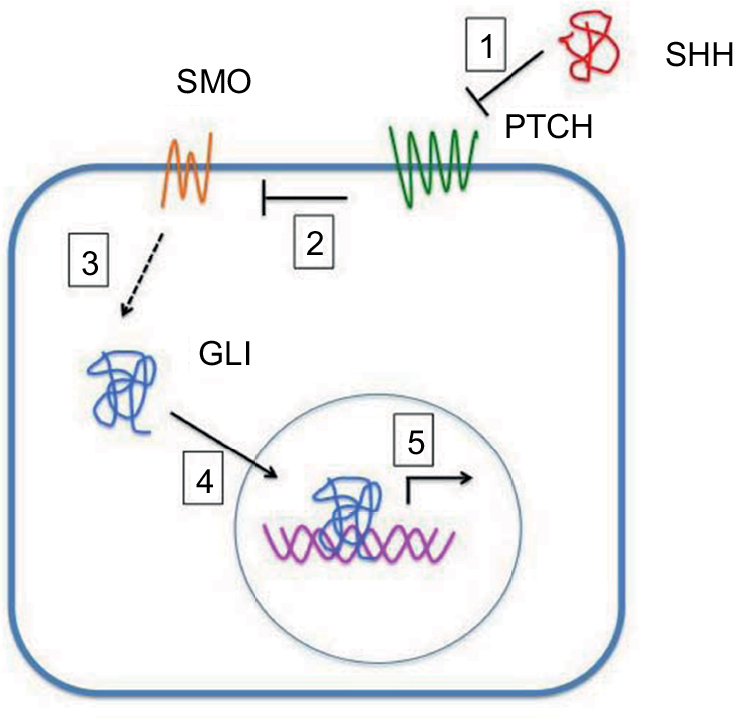 | Figure 1 SHH protein binds to the PTCH receptor (1). In the absence of the ligand, PTCH inhibits SMO, a downstream protein in the pathway (2). The binding of SHH relieves SMO inhibition, leading to activation of the GLI transcription factors: the activators GLI1 and GLI2 and the repressor GLI3 (3). Activated GLI accumulates in the nucleus (4) and controls the transcription of Hh target genes (5). |
Of these proteins, GLI1 and GLI2 mainly have a transcriptional activator function, whereas GLI3 acts as a transcriptional repressor.5 GLI targets include specific genes that regulate differentiation, proliferation, and survival. In particular, GLI1 can activate many downstream proteins such as SHH, PTCH, GLI1, and GLI2 as well as cell-cycle-regulating proteins, including the proto-oncogene N-myc and cyclin D.
The Hh pathway plays an important role in cell proliferation, differentiation, apoptosis, and migration, and it has been shown to cross talk with other signaling pathways such as MAPK/ERK,6 PI3K/AKT/mTOR,7 EGFR,8 and NOTCH.9
It is not difficult to imagine that deregulation of the Hh pathway could contribute to tumorigenesis or accelerate the rate of tumor growth.10 Anomalous activation of the SHh pathway has been shown in many human cancers, including basal cell carcinoma (BCC),11 malignant gliomas,12 medulloblastoma,13 leukemias,14 and carcinomas of the breast, lung, pancreas, and prostate.15 Moreover, activated Hh signaling has been identified in cancer stem cells (CSCs) of numerous solid tumors (eg, glioblastoma, breast, colon, pancreatic, hepatocellular) and hematologic malignancies, and it has been shown to increase tumor-initiating populations and contribute to cell migration, clonogenicity, growth, and survival. Hh signaling has, furthermore, been shown to promote tumor metastasis and recurrence, likely through the induction of genes involved in the epithelial–mesenchymal transition (EMT).
SHh deregulation can occur by somatic mutations in upstream pathway elements (ligand independent), such as SMO and PTCH, or can be induced in a ligand-dependent manner, through enhanced autocrine or paracrine signaling.
Moreover, multiple mechanisms have been involved in enhancing GLI protein activity in the absence of SHH ligands.16 Such non-canonical activation can occur through PTCH acting as a dependence receptor independently of SMO through regulation of cyclin D117 and caspase 9.18 Non-canonical activation of GLI1 and GLI2 by pathways orthogonal to the classical SHh pathway has been reported in a number of tumors,19 including loss of SMARCB1 in malignant rhabdoid tumors,20 K-ras and tumor growth factor beta (TGFβ) in pancreatic cancer,21 and PI3K in glioblastoma.22 Thus, GLI1 and GLI2 can also act as terminal effectors for numerous other oncogenic pathways.
In breast cancer, TGFβ signaling can activate GLI2 target genes, thereby causing enhanced bone metastasis.23 In BCC, mutated PTCH expression prevents a cellular response to the cell-cycle checkpoint cyclin B1 and promotes GLI activation.24 In Burkitt lymphoma, the oncogene c-MYC regulates GLI1 expression independently of SMO, PTCH, or the presence of Hh ligands.25 In a pancreatic cancer model, the Hh pathway is activated in a paracrine manner.21 Other signaling pathways, such as PI3K/AKT7,26,27 and RAS/ERK, can activate GLI proteins in different cancer types.28,29
It is generally thought that specific inhibitors of the SHh pathway may provide an efficient therapy for a wide range of malignancies. Several Hh inhibitors have been developed so far,15 and their use has been integrated into the treatment of human carcinomas.
GLI1 transcription factor
GLI1 (1106 amino acids; MW 117.9 kDa) is a C2-H2-type ZF transcription factor, harboring five ZF domains (ZF1–5, amino acids 234–388). ZF4 and ZF5 bind specifically to consensus sequence 5′-GACCACCCA-3′, whereas ZF1–3 interact with the phosphate backbone (Figure 2) and contribute to binding stability and recruitment of co-regulatory factors.30 The GLI1 C-terminal region (amino acids 1020–1091) has transactivating function through modulation of chromatin remodeling and is able to interact with histone acetyltransferase (HAT), histone deacetylase (HDAC),31,32 SWI-SNF5,20 and SWI/SNF-like Brg/Brm-associated factor.33 Such a region includes an acidic α-helix (amino acids 1037–1054) sharing 50% similarity with the herpes simplex viral protein 16 (VP16) transcription activation domain and hosts the three residues (Asp1040, Phe1048, and Leu1052) binding to the TFIID TATA box-binding protein-associated factor, TAFII31.34,35
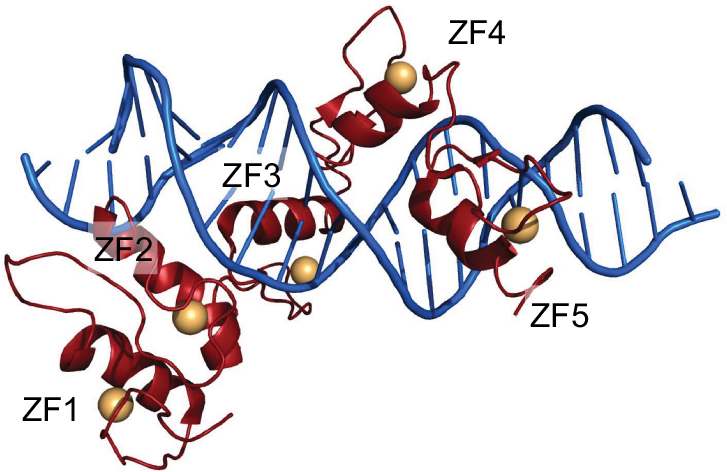 | Figure 2 GLI1 ZF1–5 in red cartoons (Zn atoms as orange spheres) bound to dsDNA (blue cartoon). Notes: Figure generated using the program Pymol and the crystal structure with pdb-id 2GLI. Data from Pavletich and Pabo.86 |
Transcription of GLI proteins is regulated through phosphorylation, which occurs through the intermediary molecules IGUANA,36 protein kinase A (PKA),37 glycogen synthase kinase 3 beta (GSK3B), and casein kinase 1 alpha (CK1α), among others. Moreover, GLI transcription factors are inhibited by suppressor of fused (SUFU),38 which sequesters GLI proteins in the cytoplasm and abolishes their nuclear entry and transcriptional effects.
GLI1-binding proteins
The activity of GLI transcription factors can be limited by acetylation, via acetyltransferase p300.31 Indeed, GLI function can be restored by endogenous HDACs. In particular, HDAC1 and HDAC2 can form a complex with GLI1, as demonstrated by co-immunoprecipitation experiments.31 β-Arrestin1 (Arrb1) can suppress the transcriptional activity of GLI1 by potentiating its p300-mediated acetylation, and GLI1 protein levels sharply decrease in CSCs overexpressing Arrb1.39
Overexpression of GLI1, either canonical or non-canonical, is very common in lung cancers. WW45 is a component of Hippo signaling pathway.40 Subcellullar localization analysis of exogenously expressed WW45 and GLI1 (using immunofluorescence staining) demonstrated the co-localization of these two proteins. Moreover, overexpression of WW45 promotes the ubiquitination of GLI1 in a dose-dependent manner, suggesting that WW45 is able to inhibit Hh signaling by promoting the degradation of GLI1.41 Most importantly, overexpression of WW45 not only inhibits the growth and migration of lung cancer cells but also impairs de novo tumorigenesis of lung cancer that is driven by loss of liver kinase B1 (LKB1) and mutated Kras.41
GLI1 is an effector of TGFβ signaling in the regulation of gene expression in cancer cells. Activation of the TGFβ pathway induces the formation of a complex between GLI1 and the transcription factor Sma- and Mad-related family member 4 (SMAD4) that is able to bind to the BCL2 promoter. SMAD4 depletion impairs complex formation and GLI1-mediated transcription of BCL2, leading to cell apoptosis.42
Arginine N-methyltransferase 1 (PRMT1) methylates GLI1 at residue Arg597, promoting its transcriptional activity by enhancing its binding to target gene promoters. Abolishing GLI1 methylation attenuates its oncogenic functions and sensitizes pancreatic ductal adenocarcinoma (AD) cells to gemcitabine treatment.43
USP21 is a centrosome-associated deubiquitylase (DUB) that is implicated in the formation of primary cilia – crucial organelles for the regulation of the Hh signaling pathway. USP21 is able to interact with GLI1, thereby suppressing GLI1-dependent transcription. In fact, USP21 recruits GLI1 to the centrosome, where it is negatively regulated through phosphorylation by PKA.44
GLI1 binds to the C-terminal domain of the pluripotency factor NANOG. The N-terminus of NANOG, that contains a transcriptional repressor motif, inhibits GLI1-mediated transcriptional activation.45
GLI1 interacts with 14-3-3ϵ – a member of a family of evolutionarily conserved regulatory proteins capable of modulating the activity of the binding partner(s). These 14-3-3ϵ proteins bind to phosphoserine/threonine-containing motifs through Lys49 – a critical residue in the phosphosubstrate-binding pocket. Only the wild-type 14-3-3ϵ, but not the Lys49Glu mutant, is able to co-precipitate with GLI1, demonstrating that the interaction is phosphorylation dependent. Ser640 of GLI1 is crucial for the interaction with 14-3-3ϵ, as proved by the Ser → Ala mutation. The sequence around Ser640 – RRXS – is the consensus phosphorylation sequence for PKA. Accordingly, PKA downregulates Hh signaling by inducing an interaction between GLI and 14-3-3ϵ.46
Finally, GLI1 can interact with the kinesin-like protein KIF7,47,48 together with serine/threonine-protein kinase 36 STK3649 and with ZIC1,50 that enhances transcriptional activation.
Hh pathway in cancer
Unusual activation of the Hh pathway is related to the development of many types of cancer, and three different mechanisms have been proposed: type I, ligand-independent signaling due to protein mutations (eg, in BCC and medulloblastoma); type II, ligand-dependent signaling in autocrine or juxtacrine (mediated by cell–cell adhesion) way, dependent on overexpression of the SHH ligand (eg, in ovarian, colorectal, and pancreatic cancers); type III, ligand-dependent signaling in a paracrine manner, driven by overexpression of the SHH ligands by tumor cells that are received by the distant cells in the stroma and emit signals (like vascular endothelial growth factor [VEGF], insulin-like growth factor [IGF], etc.) back to the tumor to promote its growth and survival (eg, pancreatic, prostate, and colon cancers). Moreover in Type III, there is reverse paracrine signaling where the SHH ligand secreted by the stromal cells is received by the tumor cells. Type II or III patients may show effectiveness to antagonistic drugs that target the SHH ligand.
Glioblastoma multiforme (GBM) is the most common and a deadly brain cancer. It has been shown that the SMO inhibitor cyclopamine (Cyp)51 can potentiate the effect of chemotherapy in GBM, reducing cell proliferation through attenuated GLI1 expression.52 The combination of Cyp with the chemotherapeutic agent temozolomide (TMZ) is effective in killing tumor parenchyma (TMZ effect) as well as the CSCs (Cyp effect) to resist tumor recurrence.52
The SHh pathway can be regulated by ubiquitination. The deubiquitinase USP48 is able to interact directly with GLI1, cleaving off its ubiquitin and thus enhancing its lifetime.53 In GBM cells, knockdown of USP48 inhibits cell proliferation and expression of GLI1’s downstream targets, leading to repressed tumorigenesis. Such a mechanism is part of a positive feedback loop because USP48 expression is induced through GLI1-mediated transcriptional activation.
Cutaneous melanoma is the most aggressive form of skin cancer. GLI1 knockdown (KD) melanoma cell lines – established by using GLI1-targeting lentiviral short hairpin RNA – exhibit a markedly reduced invasion ability. GLI1 KD melanoma cells undergo a mesenchymal-to-epithelial-like transition, demonstrating that GLI1 is important for maintaining the invasive and mesenchymal-like properties of melanoma cells – most likely, by modulating EMT-inducing transcription factors.54
Melanoma cell lines resistant to vemurafenib (a competitive kinase inhibitor) show increased levels of GLI1 and GLI2 as compared with naïve cells, with activation of the non-canonical Hh pathway, involving TGFβ/SMAD signaling. Knockdown of GLI1 and GLI2 as well as treatment with the GLI inhibitor GANT6155 leads to a decreased invasion capability of melanoma cells and restores their sensitivity to vemurafenib.56
In breast cancer models, EMT increases the metastatic capability of tumor cells in a paracrine way – in part, by non-cell autonomous activation of the GLI transcription factor. Treatment with the GLI inhibitor GANT61,55 but not with the SMO inhibitor IPI-926, inhibits growth in patient-derived xenograft (PDX) models.57
These results clearly show that all Hh inhibitors may act against tumors with canonical Hh/GLI signaling, but only inhibitors directly targeting the GLI effectors can be useful against non-canonical EMT-induced GLI activation. Therefore, GLI antagonists are expected to more effectively inhibit Hh signaling in the context of heterogeneous breast cancers, restraining both primary tumor growth and metastasis.
Human embryonal tumors with multilayered rosette (ETMR) cells transplanted into host mice are susceptible to the inhibitor arsenic trioxide (ATO), demonstrating the driving role of the SHh pathway activation in the growth of ETMRs.58
Truncated GLI1 isoform (tGLI1) in cancer
A novel alternative splice variant of GLI1, named the truncated GLI1 (tGLI1),59 originates from a deletion of 123 bases covering the entire exon 3 and part of exon 4 of the GLI1 gene. Expression of tGLI1 is undetectable in normal cells, but is high in GBM and other cancer cells. tGLI1 undergoes nuclear translocalization and is associated with increased motility and invasiveness of GBM cells. Moreover, tGLI1 expression is positively correlated with VEGF-C, tumor endothelial marker 7 (TEM7), and microvessel density,60 suggesting that tGLI1 could be a more potent transcriptional regulator than GLI1.61
GLI1 in lung cancers
Lung cancers are malignancies that arise from epithelial cells (carcinomas). Currently, no effective treatment options exist for lung cancer, and the 5-year survival rate is ~14% for patients who receive the available treatment. Lung cancers are classified according to histologic type and therapeutic purposes into two broad classes: non-small-cell lung carcinoma (NSCLC; accounting for ~85% of all lung cancer cases), and small-cell lung carcinoma (SCLC; ~15%). The three main subtypes of NSCLC are AD (~40%), squamous cell carcinoma (SCC; ~30%), and large-cell carcinoma (~10%).
Non-small-cell lung carcinoma
In NSCLC, the deregulated SHh pathway with reactivation of GLI transcription factors facilitates the initiation, progression, and metastasis as well as confers drug resistance. Accordingly, inhibition of SHh signaling induces a significant decrease in the proliferation of NSCLC cells.62,63
The role of SHh in NSCLC proliferation has been evaluated using A549 AD and H520 SCC cells (with AD and SCC being the first and second most frequent types of lung cancer, respectively). Both cells secrete the SHH ligand, which induces fibroblast proliferation, survival, migration, invasion, and collagen synthesis. Furthermore, SHH secreted by NSCLC stimulates the production of proangiogenic and metastatic factors in lung fibroblasts, demonstrating its important role in mediating epithelial/mesenchymal cross talk.62
Accordingly, the blockage of the Hh pathway through SMO inhibition by cyclopamine reduces NSCLC proliferation and viability. Furthermore, upon silencing of the three human GLI factors, GLI1 was demonstrated to be the major regulator of NSCLC cell proliferation, whereas GLI2 displays only a modest effect, and GLI3 slightly increases proliferation.62 Such evidences suggest that GLI1 and GLI2 have a redundant role in NSCLC cells, as observed in mice, where the absence of GLI2 can be compensated by GLI1.64 The role of GLI1 in NSCLC proliferation may be related to its main function as an activator of transcription.65
High GLI1 mRNA expression correlates with worse overall survival of NSCLC patients (in particular, AD and SCC patients).66 Knockdown of GLI1 significantly inhibits anchorage-dependent liquid colony-forming ability in both the SCC cells – HCC95 and HCC2814 – compared with scrambled siRNA controls, suggesting that GLI1 is critical for lung SCC clonogenicity. Subcutaneous xenograft tumors of shGLI1 HCC95 cells grow significantly slower, exhibiting decreased expression of GLI1 mRNA. In contrast, the addition of SMO antagonists or other Hh pathway agonists does not affect GLI1 expression in lung SCC cells, indicating that GLI1 expression is independent of SHH ligands and SMO. In this case, GLI1 expression is regulated by the PI3K and MAPK pathways. Thus, a combinatorial therapeutic strategy capable of targeting the PI3K/mTOR pathway and GLI1 together may lead to effective outcomes for PI3K–pathway-dependent cancers, in contrast to recent results of human trials with a single PI3K antagonist.66 SCC cells treated with BEZ235 (PI3K/mTOR dual inhibitor), LY294002 (PI3K inhibitor), and U0126 (MEK1/2 inhibitor) show diminished levels of protein kinase B and pERK, together with decreased GLI1 protein levels. Indeed, BEZ235 significantly inhibits clonogenicity and proliferation of lung SCC cells. Moreover, CC95 cells treated with ATO (a Food and Drug Administration [FDA]-approved drug with sub-micromolar potency against GLI1/267) displays significantly reduced GLI1 mRNA and protein expression. In addition, through inhibition of GLI1, ATO strongly inhibits HCC95 colony-forming efficiency and cell proliferation as compared with controls.66 GLI1 expression in these PIK3CA-amplified SCC cell lines is critical for tumor growth, being regulated primarily by the PI3K pathway68 instead of the classical Hh pathway, and combinatorial inhibition of PI3K and GLI1 leads to tumor regression in vitro and in vivo.66
NSCLC progression can be contrasted by the Chinese herb Scutellariabarbata D. Don (SBE). SBE can repress SHh signaling, thereby arresting cell-cycle progression and sensitizing lung cancer cells to the chemotherapeutic agent cisplatin in vitro and in vivo. Mechanistic investigations indicate that SBE specifically downregulates SMO and consequently attenuates the activities of GLI1 and its downstream targets, representing a promising novel drug candidate for NSCLC treatment.69
Several homeobox-related gene (HOX) transcription factors such as mesenchyme HOX-2 (MEOX2) are associated with cancer drug resistance. MEOX2 can occupy the GLI1 gene promoter region from −2192 to −109, accompanied by transcriptionally active RNA Pol II, and is epigenetically linked to the active histones H3K27Ac and H3K4me3. MEOX2-dependent GLI1 protein expression is associated with disease progression and reduced survival rates.70 The overexpressed MEOX2–GLI1 axis is clinically correlated with cancer resistance to platinum-based drugs and tyrosine kinase inhibitors in patients with NSCLC.70
GLI1 is expressed in ~76% of AD; however, in roughly half of these tumors, the activator of the canonical Hh pathway displays low expression levels.71 This feature is explained by the non-canonical activation of GLI1 by MAPK/ERK signaling.21 In this case, direct suppression of GLI1 – by RNA silencing or small-molecule inhibitors – prevents AD cell proliferation, attenuates stem-like properties, and increases their susceptibility to apoptosis in vitro and in vivo.71
GLI1 expression in SCC – evaluated by immunohistochemistry in tissue specimens from patients – is significantly correlated with T-stage lymph node metastasis and reduced overall survival.72
Small-cell lung carcinoma
The SCLC is an aggressive neuroendocrine subtype of lung cancer for which there is no effective treatment. The SHh pathway is activated in both neuroendocrine progenitor cells and SCLC cells in mice.73,74
The crucial role of Hh signaling in the development and maintenance of SCLC suggests that Hh pathway inhibition could be a valuable therapeutic strategy to slow disease progression and delay cancer recurrence.74 The activation of Hh signaling has been reported in a subset of human SCLC cell lines and tumors without changes in Hh pathway gene copy numbers or no evidence of recurrent mutations. The Hh pathway is activated in SCLC cells by the constitutive activation of SMO independently of the lung microenvironment, promoting the clonogenicity of human SCLC in vitro and the initiation and progression of mouse SCLC in vivo. Treatment of mouse SCLC cell lines with inhibitors of GLI proteins HPI-175 and GANT6176 reduces GLI1 levels and cell survival as compared to that with vehicle treatment, indicating that the Hh pathway is required for the maintenance of the disease. In particular, the Hh pathway is active in mouse SCLC cells through an autocrine–juxtacrine loop, and one crucial function of the pathway is to enhance survival.
A mouse model of SCLC demonstrates that the SHH ligand is crucial for disease progression. SHH overexpression activates canonical Hh signaling and markedly accelerates tumor progression, supporting an autocrine model of Hh signaling in SCLC pathogenesis.77 Furthermore, the non-canonical activation of Hh signaling is the cause of the chromosomal instability observed in SCLC.77
GLI1 inhibitors
The most common way to target SHh is to modulate SMO. However, the inhibition of the downstream effector GLI1 has been used to inhibit cell growth and promote apoptosis. A SHh inhibitor of particular importance is a monoclonal antibody directed at PTCH,78 but also small molecules such as GDC-0449 (Genentech), BMS-833923/XL139 (Exelixixs/Bristol–Myers Squibb), LDE-225 (Novartis), IPI-926 (Infinity Pharmaceuticals), and SANT (Sigma Aldrich), as well as AZD8542 (AstraZeneca) are able to inhibit SMO activation.
Several small-molecule inhibitors have been developed to suppress Hh signaling by blocking SMO, and nine SMO antagonists have already been evaluated in clinical trials.79 Cyclopamine – a natural steroid alkaloid derived from the plant Veratrum californicum – was the first identified SMO inhibitor. Many derivatives of cyclopamine have been developed to enhance specificity and pharmacologic potency. To date, two SMO inhibitors (LDE225/sonidegib and GDC-0449/vismodegib80) have received FDA approval for treating BCC, and many clinical trials are being conducted to evaluate the efficacy of this class of inhibitors in different cancers.
Despite the encouraging clinical results in patients with medulloblastoma or BCC, in patients with extensive-stage SCLC, the addition of SMO inhibitors to standard chemotherapy does not appear to offer any additional benefit in terms of overall survival. Such results are related to the non-canonical, SMO-independent activation of GLI-mediated transcription by other signaling pathways,81 such as the neuropilin-2/ERK cascade (NRP2/ERK),82 K-Ras/ERK signaling,21 and the mTOR/S6K1 pathway.83
Because the GLI proteins are the final effectors of SHh and other important molecular pathways, the development of a GLI-targeted approach might be useful to downregulate many pro-proliferative, pro-survival, and pro-angiogenic genes, thereby inhibiting tumor growth and therapeutic resistance.
The library of GLI1 antagonists is not as extensive as that for SMO. Among GLI antagonists, there are GANT58 and GANT61 (GLI-ANTagonist).15 Both compounds have been identified in a cell-based screen for small-molecule inhibitors of GLI1-mediated transcription using HEK293 cells transiently expressing GLI1 and a GLI-dependent luciferase reporter.76 These two compounds belong to different chemical classes, with GANT61 being a hexahydropyrimidine derivative and GANT58 possessing a thiophene core with four pyridine rings. GANT61 is more specific toward GLI proteins and effectively reduces GLI1 and GLI2 DNA-binding ability, inhibiting the Hh pathway with a half maximal effective concentration (EC50) of 5 μM in GLI1-expressing HEK293T cells. GANT61 binds to the GLI1 protein between ZF2 and ZF3, by interacting with Glu119 and Glu167, as demonstrated by in silico docking on the crystal structure of the ZF domain of GLI1 bound to DNA (PDB-ID 2GLI; Figure 2). Experimental analysis shows that mutation of the predicted binding sites significantly reduces GANT61-GLI binding affinity. The GANT61 binding site is different from the GLI DNA-binding region, and the inhibitor is not able to bind to other ZF transcription factors such as KLF4 or TFIIβ.55
Cyclopamine, GANT58, and GANT61 are effective in suppressing tumor cell growth in nude mice injected with GLI1-positive 22Rv1 prostate cancer cells. Treatment with GANT58 inhibits additional growth of xenografted tumor size and with GANT61 induces complete tumor regression. During the 18-day treatment time, no adverse side effects were observed with either of the GANT compounds.76 No clinical trials are currently ongoing with either GANT58 or GANT61 to treat any type of cancer.
ATO is an FDA-approved inhibitor of GLI1 and GLI2 transcription factors, currently tested in several clinical trials ranging from Phase I to Phase IV for both solid tumors and hematologic malignancies, including lung cancers. Despite its in vitro cytotoxicity, in a recent clinical trial, ATO failed to show any in vivo efficacy against SCLC tumor growth.84
Numerous reports have demonstrated a tumor inhibitory effect of polyunsaturated fatty acids (PUFAs). The PUFA arachidonic acid (AA) is able to repress GLI1 expression by stimulating the nuclear translocation of nuclear factor of activated T cells 1 (NFATc1), which then binds the GLI1 promoter to repress its transcription.85
Glabrescione B (GlaB) – a natural isoflavone present in the seeds of Derris glabrescens – was identified on the basis of the crystal structure of the ZF domain of GLI1 in complex with DNA.86 GlaB is able to bind the GLI1 ZF domain, interfering with its interaction with DNA87 and inhibiting GLI-dependent tumors and CSCs in vitro and in vivo. Such features indicate that direct interference of GLI/DNA interaction can be a valuable therapeutic strategy to control Hh pathway activation in cancer.
HPI-1 was identified with a high-throughput screening for compounds capable of abolishing the Hh target gene expression induced by the SMO agonist SAG.88 HPI-1 can suppress Hh pathway activation, likely through targeting a posttranslational modification of the GLI proteins and/or an interaction between the transcription factor and a co-factor.75
Finally, solasonine – a glycoalkaloid found in Solanaceous plants – is able to inhibit the GLI-luciferase activity induced by ectopic expression of GLI1 or GLI2 with half-maximal inhibitory concentration (IC50) values of 2.5 and 2.3 μM, respectively.89
Acknowledgment
This work has been conducted with the support of the Italian Association for the Cancer Research (AIRC): “My First AIRC Grant (MFAG),” project code: 17083.
Disclosure
The authors report no conflicts of interest in this work.
References
Kumar S, Balczarek KA, Lai ZC. Evolution of the hedgehog gene family. Genetics. 1996;142(3):965–972. | ||
McMahon AP. More surprises in the Hedgehog signaling pathway. Cell. 2000;100(2):185–188. | ||
Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. | ||
Ryan KE, Chiang C. Hedgehog secretion and signal transduction in vertebrates. J Biol Chem. 2012;287(22):17905–17913. | ||
Ruiz i Altaba A. Catching a Gli-mpse of Hedgehog. Cell. 1997;90(2):193–196. | ||
Seto M, Ohta M, Asaoka Y, et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol Carcinog. 2009;48(8):703–712. | ||
Zhou J, Zhu G, Huang J, et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: a novel potential therapeutic target. Cancer Lett. 2016;370(2):313–323. | ||
Kasper M, Schnidar H, Neill GW, et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol. 2006;26(16):6283–6298. | ||
Schreck KC, Taylor P, Marchionni L, et al. The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistance. Clin Cancer Res. 2010;16(24):6060–6070. | ||
Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9(7):873–886. | ||
Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH Jr, Scott MP. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276(5313):817–821. | ||
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17(2):165–172. | ||
Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277(5329):1109–1113. | ||
Dierks C, Beigi R, Guo GR, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14(3):238–249. | ||
Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel). 2016;8(2):pii: E22 | ||
Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer. 2016;15:24. | ||
Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14(7):416–429. | ||
Fombonne J, Bissey PA, Guix C, Sadoul R, Thibert C, Mehlen P. Patched dependence receptor triggers apoptosis through ubiquitination of caspase-9. Proc Natl Acad Sci U S A. 2012;109(26):10510–10515. | ||
Gu D, Xie J. Non-canonical Hh signaling in cancer-current understanding and future directions. Cancers (Basel). 2015;7(3):1684–1698. | ||
Jagani Z, Mora-Blanco EL, Sansam CG, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. 2010;16(12):1429–1433. | ||
Nolan-Stevaux O, Lau J, Truitt ML, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23(1):24–36. | ||
Filbin MG, Dabral SK, Pazyra-Murphy MF, et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: new therapeutic opportunities. Nat Med. 2013;19(11):1518–1523. | ||
Johnson RW, Nguyen MP, Padalecki SS, et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 2011;71(3):822–831. | ||
Yu FY, Hong YY, Qu JF, Chen F, Li TJ. The large intracellular loop of ptch1 mediates the non-canonical Hedgehog pathway through cyclin B1 in nevoid basal cell carcinoma syndrome. Int J Mol Med. 2014;34(2):507–512. | ||
Yoon JW, Gallant M, Lamm ML, et al. Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma. Mol Cancer Res. 2013;11(6):604–615. | ||
Ramaswamy B, Lu Y, Teng KY, et al. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012;72(19):5048–5059. | ||
Kern D, Regl G, Hofbauer SW, et al. Hedgehog/GLI and PI3K signaling in the initiation and maintenance of chronic lymphocytic leukemia. Oncogene. 2015;34(42):5341–5351. | ||
Stecca B, Mas C, Clement V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007;104(14):5895–5900. | ||
Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014;40(6):750–759. | ||
Kinzler KW, Vogelstein B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol. 1990;10(2):634–642. | ||
Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12(2):132–142. | ||
Malatesta M, Steinhauer C, Mohammad F, Pandey DP, Squatrito M, Helin K. Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res. 2013;73(20):6323–6333. | ||
Zhan X, Shi X, Zhang Z, Chen Y, Wu JI. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc Natl Acad Sci U S A. 2011;108(31):12758–12763. | ||
Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL. Induced alpha helix in the VP16 activation domain upon binding to a human TAF. Science. 1997;277(5330):1310–1313. | ||
Yoon JW, Liu CZ, Yang JT, Swart R, Iannaccone P, Walterhouse D. GLI activates transcription through a herpes simplex viral protein 16-like activation domain. J Biol Chem. 1998;273(6):3496–3501. | ||
Wolff C, Roy S, Lewis KE, et al. iguana encodes a novel zinc-finger protein with coiled-coil domains essential for Hedgehog signal transduction in the zebrafish embryo. Genes Dev. 2004;18(13):1565–1576. | ||
Ferretti E, De Smaele E, Di Marcotullio L, Screpanti I, Gulino A. Hedgehog checkpoints in medulloblastoma: the chromosome 17p deletion paradigm. Trends Mol Med. 2005;11(12):537–545. | ||
Pham A, Therond P, Alves G, et al. The Suppressor of fused gene encodes a novel PEST protein involved in Drosophila segment polarity establishment. Genetics. 1995;140(2):587–598. | ||
Miele E, Po A, Begalli F, et al. β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC cancer. 2017;17(1):488. | ||
Lee JH, Kim TS, Yang TH, et al. A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J. 2008;27(8):1231–1242. | ||
Li X, Zhou X, Fan Y, et al. WW45, a Gli1 binding protein, negatively regulated Hedgehog signaling in lung cancer. Oncotarget. 2016;7(42):68966–68975. | ||
Nye MD, Almada LL, Fernandez-Barrena MG, et al. The transcription factor GLI1 interacts with SMAD proteins to modulate transforming growth factor β-induced gene expression in a p300/CREB-binding protein-associated factor (PCAF)-dependent manner. J Biol Chem. 2014;289(22):15495–15506. | ||
Wang Y, Hsu JM, Kang Y, et al. Oncogenic functions of Gli1 in pancreatic adenocarcinoma are supported by its PRMT1-mediated methylation. Cancer Res. 2016;76(23):7049–7058. | ||
Heride C, Rigden DJ, Bertsoulaki E, et al. The centrosomal deubiquitylase USP21 regulates Gli1 transcriptional activity and stability. J Cell Sci. 2016;129(21):4001–4013. | ||
Li Q, Lex RK, Chung H, et al. The pluripotency factor NANOG binds to GLI proteins and represses hedgehog-mediated transcription. J Biol Chem. 2016;291(13):7171–7182. | ||
Asaoka Y, Kanai F, Ichimura T, et al. Identification of a suppressive mechanism for Hedgehog signaling through a novel interaction of Gli with 14-3-3. J Biol Chem. 2010;285(6):4185–4194. | ||
Maurya AK, Ben J, Zhao Z, et al. Positive and negative regulation of Gli activity by Kif7 in the zebrafish embryo. PLoS Genet. 2013;9(12):e1003955. | ||
Cheung HO, Zhang X, Ribeiro A, et al. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci Signal. 2009;2(76):ra29. | ||
Murone M, Luoh SM, Stone D, et al. Gli regulation by the opposing activities of fused and suppressor of fused. Nat Cell Biol. 2000;2(5):310–312. | ||
Mizugishi K, Aruga J, Nakata K, Mikoshiba K. Molecular properties of Zic proteins as transcriptional regulators and their relationship to GLI proteins. J Biol Chem. 2001;276(3):2180–2188. | ||
Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16(21):2743–2748. | ||
Liu YJ, Ma YC, Zhang WJ, et al. Combination therapy with micellarized cyclopamine and temozolomide attenuate glioblastoma growth through Gli1 down-regulation. Oncotarget. 2017;8(26):42495–42509. | ||
Zhou A, Lin K, Zhang S, et al. Gli1-induced deubiquitinase USP48 aids glioblastoma tumorigenesis by stabilizing Gli1. EMBO Rep. 2017;18(8):1318–1330. | ||
Gunarta IK, Li R, Nakazato R, et al. Critical role of glioma-associated oncogene homolog 1 in maintaining invasive and mesenchymal- like properties of melanoma cells. Cancer Sci. 2017;108(8):1602–1611. | ||
Agyeman A, Jha BK, Mazumdar T, Houghton JA. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget. 2014;5(12):4492–4503. | ||
Faião-Flores F, Alves-Fernandes DK, Pennacchi PC, et al. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib-resistant human melanoma cells. Oncogene. 2017;36(13):1849–1861. | ||
Neelakantan D, Zhou H, Oliphant MUJ, et al. EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells. Nat Commun. 2017;8:15773. | ||
Neumann JE, Wefers AK, Lambo S, et al. A mouse model for embryonal tumors with multilayered rosettes uncovers the therapeutic potential of Sonic-hedgehog inhibitors. Nat Med. 2017;23(10):1191–1202. | ||
Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009;69(17):6790–6798. | ||
Carpenter RL, Paw I, Zhu H, et al. The gain-of-function GLI1 transcription factor TGLI1 enhances expression of VEGF-C and TEM7 to promote glioblastoma angiogenesis. Oncotarget. 2015;6(26):22653–22665. | ||
Carpenter RL, Lo HW. Hedgehog pathway and GLI1 isoforms in human cancer. Discov Med. 2012;13(69):105–113. | ||
Bermudez O, Hennen E, Koch I, Lindner M, Eickelberg O. Gli1 mediates lung cancer cell proliferation and Sonic Hedgehog-dependent mesenchymal cell activation. PLoS One. 2013;8(5):e63226. | ||
Yuan Z, Goetz JA, Singh S, et al. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene. 2007;26(7):1046–1055. | ||
Bai CB, Joyner AL. Gli1 can rescue the in vivo function of Gli2. Development. 2001;128(24):5161–5172. | ||
Stecca B, Ruiz I Altaba A. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J Mol Cell Biol. 2010;2(2):84–95. | ||
Kasiri S, Shao C, Chen B, et al. GLI1 blockade potentiates the antitumor activity of PI3K antagonists in lung squamous cell carcinoma. Cancer Res. 2017;77(16):4448–4459. | ||
Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010;107(30):13432–13437. | ||
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. | ||
Du J, Chen W, Yang L, et al. Disruption of SHH signaling cascade by SBE attenuates lung cancer progression and sensitizes DDP treatment. Sci Rep. 2017;7(1):1899. | ||
Armas-López L, Piña-Sánchez P, Arrieta O, et al. Epigenomic study identifies a novel mesenchyme homeobox2-GLI1 transcription axis involved in cancer drug resistance, overall survival and therapy prognosis in lung cancer patients. Oncotarget. 2017;8(40):67056–67081. | ||
Po A, Silvano M, Miele E, et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene. 2017;36(32):4641–4652. | ||
Cui Y, Cui CA, Yang ZT, Ni WD, Jin Y, Xuan YH. Gli1 expression in cancer stem-like cells predicts poor prognosis in patients with lung squamous cell carcinoma. Exp Mol Pathol. 2017;102(2):347–353. | ||
Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422(6929):313–317. | ||
Park KS, Martelotto LG, Peifer M, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011;17(11):1504–1508. | ||
Hyman JM, Firestone AJ, Heine VM, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci U S A. 2009;106(33):14132–14137. | ||
Lauth M, Bergström A, Shimokawa T, Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104(20):8455–8460. | ||
Szczepny A, Rogers S, Jayasekara WSN, et al. The role of canonical and non-canonical Hedgehog signaling in tumor progression in a mouse model of small cell lung cancer. Oncogene. 2017;36(39):5544–5550. | ||
Maun HR, Wen X, Lingel A, et al. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J Biol Chem. 2010;285(34):26570–26580. | ||
Khatra H, Bose C, Sinha S. Discovery of hedgehog antagonists for cancer therapy. Curr Med Chem. 2017;24(19):2033–2058. | ||
Fellner C. Vismodegib (erivedge) for advanced Basal cell carcinoma. P T. 2012;37(12):670–682. | ||
Lauth M, Toftgård R. Non-canonical activation of GLI transcription factors: implications for targeted anti-cancer therapy. Cell Cycle. 2007;6(20):2458–2463. | ||
Goel HL, Pursell B, Chang C, et al. GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol Med. 2013;5(4):488–508. | ||
Wang Y, Ding Q, Yen CJ, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012;21(3):374–387. | ||
Owonikoko TK, Zhang G, Kim HS, et al. Patient-derived xenografts faithfully replicated clinical outcome in a phase II co-clinical trial of arsenic trioxide in relapsed small cell lung cancer. J Transl Med. 2016;14(1):111. | ||
Comba A, Almada LL, Tolosa EJ, et al. Nuclear factor of activated T cells-dependent down-regulation of the transcription factor glioma-associated protein 1 (GLI1) underlies the growth inhibitory properties of arachidonic acid. J Biol Chem. 2016;291(4):1933–1947. | ||
Pavletich NP, Pabo CO. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science. 1993;261(5129):1701–1707. | ||
Infante P, Mori M, Alfonsi R, et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015;34(2):200–217. | ||
Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A. 2002;99(22):14071–14076. | ||
Yang J, Huang W, Tan W. Solasonine, A natural glycoalkaloid compound, inhibits Gli-mediated transcriptional activity. Molecules. 2016;21(10): pii: E1364. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.