Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
RNA Sequencing and Related Differential Gene Expression Analysis in a Mouse Model of Emphysema Induced by Tobacco Smoke Combined with Elastin Peptides
Authors Feng X, Deng J, Li X ![]() , Zhang H, Wei X, Ma T, Tang S, Zhang J
, Zhang H, Wei X, Ma T, Tang S, Zhang J
Received 13 November 2022
Accepted for publication 30 May 2023
Published 3 October 2023 Volume 2023:18 Pages 2147—2161
DOI https://doi.org/10.2147/COPD.S397400
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Xin Feng,1,2,* Jiehua Deng,1,2,* Xiaofeng Li,2 Hui Zhang,2 Xuan Wei,2 Tingting Ma,3 Shudan Tang,2 Jianquan Zhang1
1Department of Respiratory and Critical Medicine, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, Guangdong, 518000, People’s Republic of China; 2Department of Respiratory and Critical Medicine, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, 530021, People’s Republic of China; 3Department of Respiratory and Critical Medicine, Zhuhai People’s Hospital, Zhuhai, Guangdong, 519099, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianquan Zhang, Department of Respiratory and Critical Medicine, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, Guangdong, 518000, People’s Republic of China, Tel +8613978123845, Fax +860755-23482484, Email [email protected]
Objective: To establish a model of emphysema induced by tobacco smoke combined with elastin peptides (EP), explore the biochemical metabolic processes and signal transduction pathways related to emphysema occurrence and development at the transcriptional level, and identify new targets and signaling pathways for emphysema prevention and treatment.
Methods: Mice were randomly divided into the air pseudoexposure group (NORMAL group) and the tobacco smoke + EP group (EP group). The differentially expressed genes (DEGs) in lung tissue between the two groups were identified by RNA-seq, and functional annotation and Gene Ontology (GO)/ Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed. The differential expression of the selected genes were verified using qRT‒PCR and immunohistochemistry (IHC).
Results: EP group mice showed emphysema-like changes. The expression levels of 1159 genes in the EP group differed significantly (529 up-regulated and 630 down-regulated) from those in the NORMAL group. GO enrichment analysis showed that the DEGs were significantly enriched in the terms immune system, adaptive immune response, and phosphorylation, while KEGG pathway enrichment analysis showed that the DEGs were enriched mainly in the pathways cytokine‒cytokine receptor interaction, T-cell receptor signaling pathway, MAPK signaling pathway, Rap1 signaling pathway, endocytosis, chemokine signaling pathway, Th17 cell differentiation, and Th1 and Th2 cell differentiation. The differential expression of the selected DEGs were verified by qRT‒PCR and IHC, and the expression trends of these genes were consistent with those identified by RNA-seq.
Conclusion: Emphysema may be related to the inflammatory response, immune response, immune regulation, oxidative stress injury, and other biological processes. The Bmp4-Smad-Hoxa5/Acvr2a signaling pathway may be involved in COPD/ emphysema occurrence and development.
Keywords: emphysema, tobacco smoke, elastin peptide, RNA-seq, DEGs
Introduction
Chronic obstructive pulmonary disease (COPD) is a condition that is characterized by persistent respiratory symptoms and airflow limitations that are not fully reversible. The severe complications of the disease may adversely affect its morbidity and mortality.1 According to World Health Organization (WHO) statistics, over 3 million people per year die from COPD, which is the third-leading cause of death worldwide.2–4 The state of COPD prevention and control is particularly grave. Previous studies have suggested that smoking is the main risk factor for COPD.1 However, we have not thoroughly studied the molecular mechanism underlying the immunological regulation of the exacerbated inflammation caused by respiratory stimuli such as tobacco smoke, and the reasons that chronic airway inflammation and airflow limitation persistently progress after smoking cessation remain unclear. Thus, performing a systematic and comprehensive mechanistic study of COPD occurrence and development and identifying effective therapeutic targets are highly important for the early prevention and treatment of COPD.
Elastin is an extracellular matrix protein that plays an important role in maintaining reversible contraction and dilation of the lungs during breathing,5,6 and its degraded peptide fragment, elastin peptides (EP), has endogenous antigenicity.7 Evidence indicates that EP induces immune imbalance by inducing the release of proinflammatory cytokines and the differentiation of many kinds of immune cells (Type 1 T helper (Th1) cells, Type 17 T helper (Th17) cells, Type 1 CD8+ T(Tc1) cells, myeloid dendritic cells (mDCs), CD4+ T cells, etc.), thus promoting the occurrence and development of chronic inflammation in COPD/emphysema.8–12 Under the stimulation of tobacco smoke, EP has a chemotactic effect on neutrophils and macrophages, which can not only result in the accumulation and release of many inflammatory factors to induce airway inflammation but also cause activated neutrophils and macrophages to secrete neutrophil elastase, MMP-9, and MMP-12, resulting in an imbalance in the protease-antiprotease system, destruction of the pulmonary interstitium and rupture of the lining septum.13 Several studies have reported that the autoimmune response caused by EP is related to tobacco smoke exposure. Under stimulation such as smoking/environmental smoke inhalation, the immune response is triggered, various inflammatory factors and proteases are released, and elastin in the lungs is destroyed, resulting in bioactive EP. EP acts as a chemoattractant for various inflammatory cells, promotes cell activation, releases many inflammatory factors, and further aggravates the inflammatory response in COPD.5,13–18
The animal models of emphysema currently used to study the pathogenesis of COPD are complex and diverse. Given the complex pathogenesis of COPD, we referred to a newly reported autoimmune model19 of tobacco smoke exposure combined with elastin and established an emphysema model by treatment with tobacco smoke combined with EP. RNA-seq of lung tissue was performed to provide a more complete gene map for COPD research. In addition, we identified the differentially expressed genes (DEGs) and then performed functional annotation and Gene Ontology (GO) /Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the DEGs to determine the biological and biochemical metabolic pathways and signal transduction pathways related to COPD pathogenesis. Moreover, we further studied the genes of airway inflammation that cause oxidative stress (OS) and immunity to COPD. To this end, we selected DEGs screened by RNA-seq for verification at the mRNA and protein levels by qRT‒PCR and immunohistochemistry (IHC), respectively, to further understand the pathogenesis of COPD and search for additional biomarkers of disease risk or new targets for chemoprophylaxis.
Materials and Methods
Animals
Six- to eight-week-old male C57BL/6 mice (Guangxi Medical University Laboratory Animal Center, China) were housed individually in standard laboratory cages on a 12 h light-dark cycle. All experimental protocols were approved by the Animal Research Care Committee of Guangxi Medical University. Ethics review of the animal study was performed following the Guiding Opinions on the Treatment of Laboratory Animals issued by the Ministry of Science and Technology of the People’s Republic of China and the Laboratory Animal Guideline for Ethical Review of Animal Welfare issued by National Standard GB/T35892-2018 of the People’s Republic of China.
Models
Based on the protocol used by our previous research and by Zhou et al8,19–22 with slight modifications, tobacco smoke combined with EP was used to establish the emphysema model. Mice in the model group were exposed to smoke from 12 cigarettes (Nanning Zhen long unfiltered cigarettes: 12 mg of tar and 0.9 mg of nicotine) in a closed 90×55×40 cm chamber. The smoke-to-air ratio was 1:6. Cigarette smoke exposure took 40 min per session and occurred 8 times a day and 5 days a week for a total of two weeks. Mice in the control group were exposed to room air (NORMAL group), and mice in the EP group were intranasally administered 50 µL of an EP suspension (VGVAPG, EPC company, America, dissolved in phosphate buffered saline, 100 µg/mL) at 14-day intervals after being anesthetized. On the 33rd day, the mice were anesthetized and sacrificed by cervical dislocation (Figure 1). The right upper lung lobes were isolated and sent to BGI (Wuhan, China) for RNA-seq. The rest of the lung tissue was used for histopathological analysis or RNA extraction or was stored at −80 °C.
 |
Figure 1 Animal experimental protocol. Abbreviation: EP, elastin peptides. Notes: The NORMAL group was exposed to room air from day 1 to day 33. The EP group was exposed to tobacco smoke from day 1 to day 14 and then intranasally administered 50 µL of an EP suspension (day 29 to day 31). Mice in the NORMAL and EP groups were sacrificed on day 33. |
Histopathological Analysis
The left upper lung lobes of mice were fixed with 4% paraformaldehyde for 24–48 h and embedded in paraffin. After sectioning, they were stained with hematoxylin-eosin (H&E) to observe and quantify the alveolar airspace enlargement and inflammatory infiltration associated with emphysema. The mean linear intercept (MLI) was used to assess alveolar airspace enlargement.20,22 The MLI reflects the average alveolar diameter of mice and was compared with that of mice in the NORMAL group. All procedures were performed in a double-blind manner by two pathologists.
RNA-Seq
Sample Collection
A total of 8 samples from the EP group (n=4) and the NORMAL group (n=4) were quickly transported to BGI for RNA-seq. BGI performed RNA extraction, quality assessment, mRNA purification, mRNA fragmentation, and cDNA reverse transcription.
Total RNA Extraction and Quality Assessment
The TRIzol method was used to extract total RNA, and then a fragment analyzer was used for the assessment of quality, including measurements of the total RNA concentration, RNA integrity number (RIN) or RNA quality number (RQN), 28S/18S ratio, and fragment size. The integrity of the RNA was determined by agarose gel electrophoresis. Total RNA samples meeting the quality thresholds (RQN or RIN≥7.0, 28S/18S≥1.0) were selected (one sample in the NORMAL group was rejected).
RNA-Seq Library Construction and Sequencing
After the library was constructed and its quality was verified, the single-stranded circular DNA molecules were copied by rolling circle amplification to form DNA nanoballs (DNBs). A sequence read length of 150 bp was obtained by sequencing on the BGISEQ-500 platform powered by combinatorial probe-anchor synthesis (cPAS).
Bioinformatics Analysis
Data Quality Control
The sequencing data obtained on the BGISEQ500 platform were raw reads or raw data. The raw reads were quality controlled, and the raw data was filtered using the filtering software SOAPnuke (v1.5.2), which was independently developed by BGI.23 The data was saved in FASTQ format for subsequent bioinformatics analysis.
Gene Alignment
The filtered clean reads were aligned to the reference genome sequence using HISAT2 software (v2.0.4).24 The statistical alignment rate, distribution of reads on the reference sequence (base content distribution statistics and base quality distribution), saturation, etc., were used to determine whether the data passed the second quality control step for use in subsequent data interpretation.
Gene Expression Quantification
After passing the second quality control step, Bowtie225 software was used to align the clean reads to the reference gene sequence, and RSEM26 was used to calculate the gene expression levels in each sample. Fragments per kilobase of exon model per million mapped fragments (FPKM) values were used to estimate gene expression levels, and the genes in each group with a mean FPKM value greater than 0.5 were considered to be expressed in the group and incorporated into downstream statistical analysis.
Identification of DEGs
The DEGs in different samples were analyzed. The DEGs between the two groups were identified with DESeq2 (v1.4.5).27 Differences in gene expression between the two groups were calculated, and the significant DEGs were identified as those meeting the screening criteria of | log2(FoldChange)| > 0 and Q-value<0.05. A volcano plot of the DEGs was generated with the R package ggplot2 to show the distribution of the data and the situation of the differentially expressed genes. In addition, the R package pheatmap was used for hierarchical cluster analysis to draw a heatmap showing the union of the DEGs.
GO and KEGG Pathway Enrichment Analyses
The main biochemical and biological functions in which the identified DEGs were involved and the main related biochemical metabolic pathways and signal transduction pathways were determined through GO database28 (http://geneontology.org/) and KEGG database29 (http://www.kegg.jp/) analyses.
Quantitative Real-Time PCR
Selected DEGs (Homeobox A5(Hoxa5), Human hedgehog interacting protein (Hhip), Activin receptor IIA (Acvr2a), Bone morphogenetic protein-4(Bmp4), Family with sequence similarity 13 member A (Fam13a), Cytotoxic T-lymphocyte-associated protein 4(Ctla4), aryl hydrocarbon receptor nuclear translocator like (Arntl), and Glia maturation factor gamma (Gmfg)) enriched in the KEGG pathways that differed between the EP group and NORMAL group were selected for qRT‒PCR to validate the RNA-seq results. qRT‒PCR was performed on an Agilent AriaMx Real-Time PCR System according to the reagent manufacturer’s protocol (Takara, China). Relative gene expression was calculated using the 2−ΔΔCt method. The primer sequences are listed in Table 1.
 |
Table 1 The Primer Sequence of Quantitative PCR Required for This Experiment |
IHC Staining
Lung tissue sections were stained with anti-Bmp4 (Servicebio, China) and anti-Hoxa5/Arntl/Ctla4/Gmfg (Bioss, China) antibodies. The samples were observed and imaged with an Olympus microscope (10x40 magnification). Ten visual fields were randomly selected, and 100 cells were counted. Cells with brown staining in the cytoplasm or nucleus were considered positive, and the percentage of positive cells was calculated accordingly. The above process was performed in a double-blind manner by two pathologists.
Statistical Analysis
Data was shown as the mean ± standard deviation (x ± s). Since our data was normally or approximately normally distributed, comparisons between the two groups were performed with an independent samples t-test. All statistical tests were performed using SPSS 25.0 software (SPSS, Chicago, IL, USA). P values <0.05 were considered statistically significant. The statistical data were plotted with GraphPad Prism 7.0 (GraphPad Software, San Diego, USA).
Results
The Use of Tobacco Smoke Combined with EP to Establish an Emphysema Model
In the NORMAL mice, we observed minimal inflammatory infiltration and alveolar wall destruction (Figure 2A-NORMAL). Lung sections from EP mice showed emphysema-like changes: the alveolar walls exhibited thinning, the alveolar lining was significantly enlarged and disrupted, and bronchiolar cilia were lost and misarranged. Disordered structures, massive inflammatory cell infiltration, vessel wall destruction, alveolar cavity fusion, and pulmonary bullae were observed (Figure 2A-EP). Accordingly, with alveolar enlargement, the MLI values in the EP group were higher than those in the NORMAL group (P<0.001, Figure 2B).
 |
Figure 2 Pathological changes in the lungs of each group of mice. Abbreviations: H&E, hematoxylin-eosin; MLI, mean linear intercept. Notes: (A) Representative H&E sections of lung tissues from the group NORMAL and EP mice, original magnification, ×200; (B) Comparison of MLI in the NORMAL group and the EP group. Data are expressed as x±s (n=15). The comparisons were determined by an independent sample t-test on ranks. *P<0.001. |
RNA-Seq Analysis
Each sample yielded 11.19 G of data, and 18,020 genes were detected. The Q30 level of each sample was above 90%, and the sequence of clean reads was compared with the reference genome (Mus musculus GCF_000001635.26_GRCm38.p6). The alignment rate of the reads in each sample with the reference genome was greater than 80%.
DEG Mapping
The gene expression profiles in the NORMAL group and EP group were compared, and 1159 DEGs were identified (Q-value < 0.05, 529 up-regulated genes and 630 down-regulated genes, Figure 3). The FPKM values of the DEGs were used as the expression levels, and hierarchical cluster analysis was performed (Figure 4).
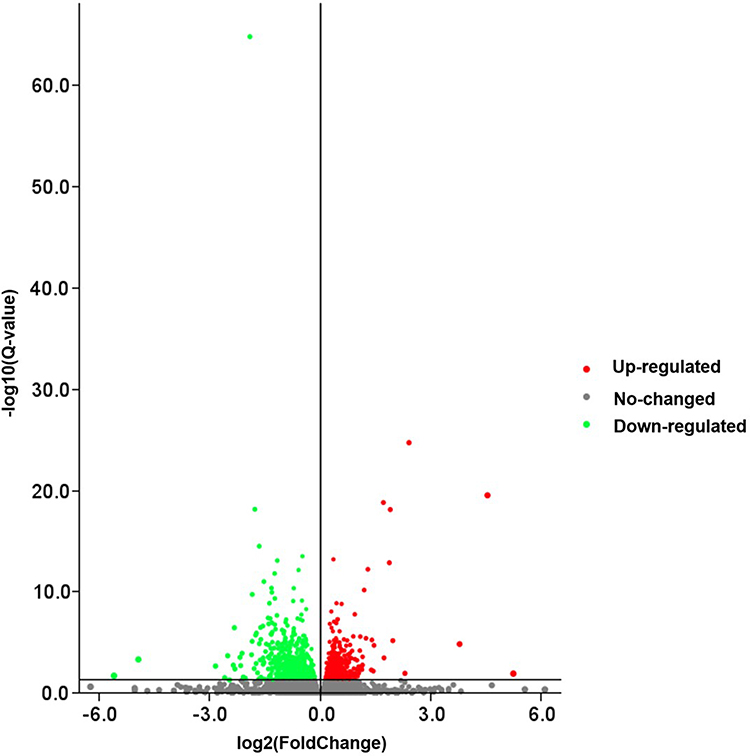 |
Figure 3 Volcano map of DEGs between the NORMAL group and EP group. Abbreviation: DEGs, differentially expressed genes. Notes: The X-axis represents the fold change of the difference after conversion to log2, and the Y-axis represents the significance value after conversion to-log10. Red points represent up-regulation, green dots represent down-regulation, and gray points represent non-DEGs. |
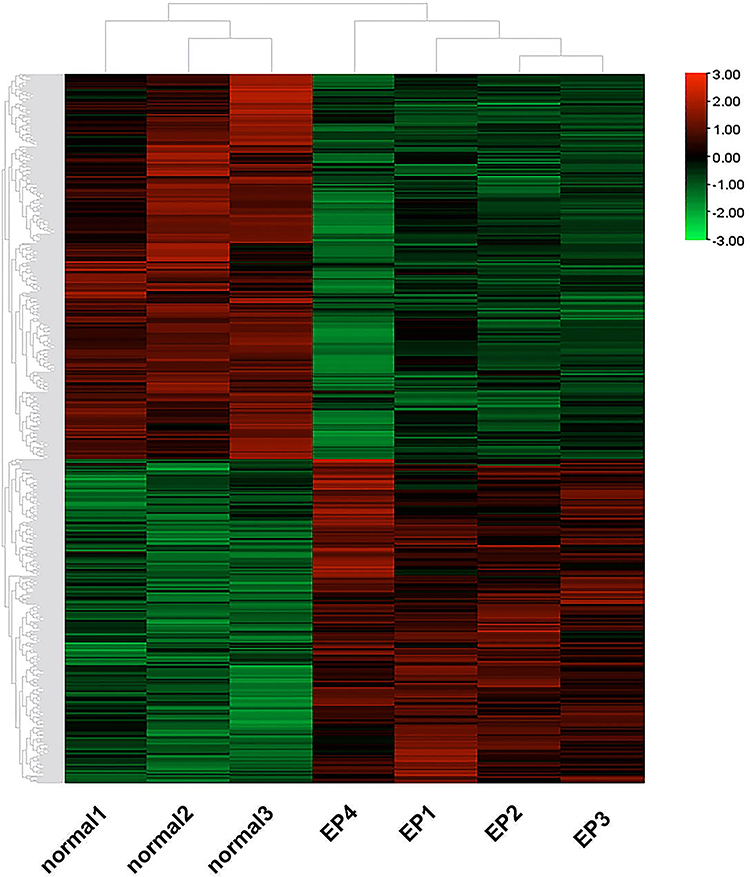 |
Figure 4 Clustering heatmap of the DEGs. Abbreviation: DEGs, differentially expressed genes. Notes: Red indicates high expression; green indicates low expression in the two groups. |
GO Functional Annotation and Enrichment Analysis
The DEGs were enriched in 26, 15, and 13 biological process (BP), cellular component (CC), and molecular function (MF) functional categories, respectively. As indicated above, more DEGs were enriched in the BP category, with the highest enrichment in terms of cellular process, biological regulation, regulation of the biological process, response to stimulus, and metabolic process. According to the annotation results, the DEGs were subjected to GO enrichment analysis, and the results are shown in bubble plots (Q-value <0.05, Figure 5). The BP terms significantly enriched with the DEGs included immune system process, adaptive immune response, immune response, phosphorylation, ERK1/ERK2 signaling regulation pathway, inflammatory response, and positive regulation of GTPase activity.
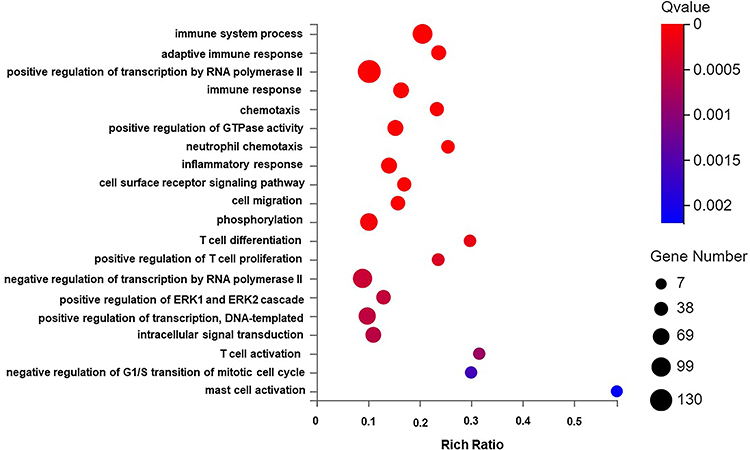 |
Figure 5 GO biological process enrichment analysis graph of DEGs. Abbreviations: DEGs, differentially expressed genes; GO, Gene Ontology. Notes: The X-axis is the enrichment ratio (the ratio of the number of genes annotated to an entry in the selected gene set to the total number of genes annotated to the entry in the species, calculated as Rich Ratio = Term Candidate Gene Num/Term Gene Num) It is GO Term. The size of the bubble represents the number of DEGs annotated to a GO Term. The color represents the enriched significance. The redder the color, the smaller the significance value. The items displayed on the Y-axis are as follows from top to bottom:1. immune system process; 2. adaptive immune response; 3. positive regulation of transcription by RNA polymerase II; 4. immune response; 5. chemotaxis; 6. positive regulation of GTPase activity; 7. neutrophil chemotaxis 8. inflammatory response; 9. cell surface receptor signaling pathway; 10. cell migration; 11. phosphorylation; 12. T cell differentiation; 13. positive regulation of T cell proliferation; 14. negative regulation of transcription by RNA polymerase II; 15. positive regulation of ERK1 and ERK2 cascade; 16. positive regulation of transcription, DNA-templated; 17. intracellular signal transduction; 18. T cell activation; 19. negative regulation of G1/S transition of mitotic cell cycle; 20. mast cell activation. |
KEGG Pathway Enrichment Analysis
The DEGs were then subjected to KEGG pathway enrichment analysis, and the results are shown in bubble plots (Q-value <0.05, Figure 6). The DEGs were mainly enriched in cytokine‒cytokine receptor interaction, endocytosis, MAPK signaling pathway, Rap1 signaling pathway, chemokine signaling pathway, Th17 cell differentiation, Th1 and Th2 cell differentiation, T-cell receptor T-cell receptor signaling pathway, etc.
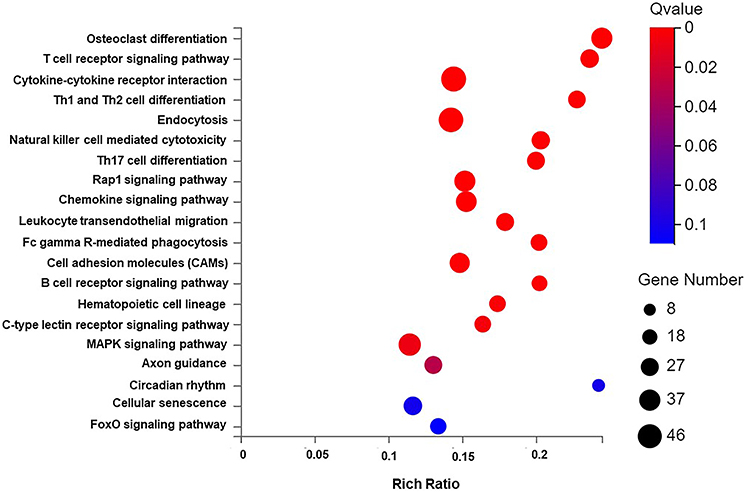 |
Figure 6 Bubble chart of DEGs KEGG pathway enrichment results. Abbreviations: DEGs, differentially expressed genes; KEGG, Kyoto Encyclopedia of Genes and Genomes. Notes: The X-axis is the enrichment ratio (the ratio of the number of genes annotated to an entry in the selected gene set to the total number of genes annotated to the entry in the species, calculated as Rich Ratio = Term Candidate Gene Num/Term Gene Num), Y-axis is KEGG Pathway. The size of the bubble represents the number of genes annotated to KEGG Pathway. The color represents the enriched significance. The redder the color, the smaller the significance value. The items displayed on the Y-axis are as follows from top to bottom: 1. Osteoclast differentiation; 2. T cell receptor signaling pathway; 3. Cytokine-cytokine receptor interaction; 4. Th1 and Th2 cell differentiation; 5. Endocytosis; 6. Natural killer cell mediated cytotoxicity; 7. Th17 cell differentiation; 8. Rap1 signaling pathway; 9. Chemokine signaling pathway; 10. Leukocyte transendothelial migration; 11. Fc gamma R-mediated phagocytosis; 12. Cell adhesion molecules (CAMs); 13. B cell receptor signaling pathway; 14. Hematopoietic cell lineage; 15. C-type lectin receptor signaling pathway; 16. MAPK signaling pathway; 17. Axon guidance; 18. Circadian rhythm; 19. Cellular senescence; 20. FoxO signaling pathway. |
qRT‒PCR Analysis
Some genes with high abundance and statistically significant fold changes were selected from the RNA-seq results (Table 2). The expression levels of Hoxa5, Hhip, Acvr2a, Bmp4, and Fam13a were significantly up-regulated and those of Ctla4, Arntl, and Gmfg were significantly down-regulated in the EP group compared with the NORMAL group (all P<0.05, Figure 7). The above results show that the expression trends of the candidate genes were consistent with those identified by RNA-seq, thus confirming the reliability of the sequencing results. This consistency supports our subsequent use of the sequencing analysis results to further study the pathogenesis of emphysema.
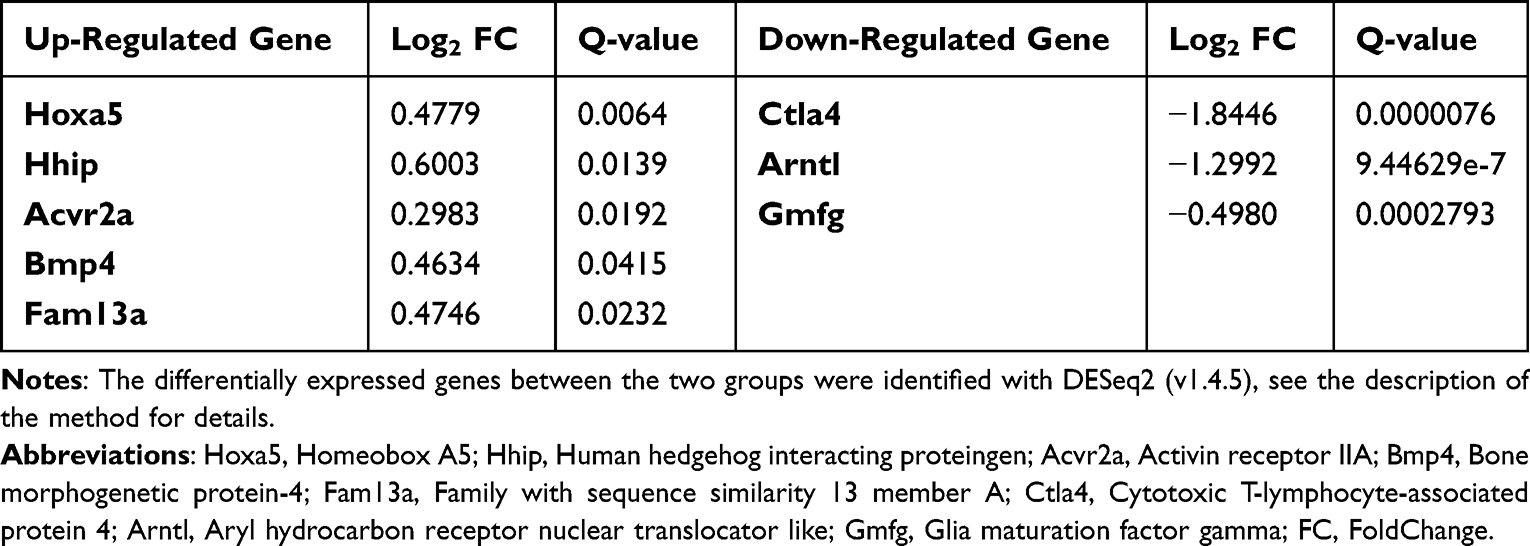 |
Table 2 Selected Differentially Expressed Genes |
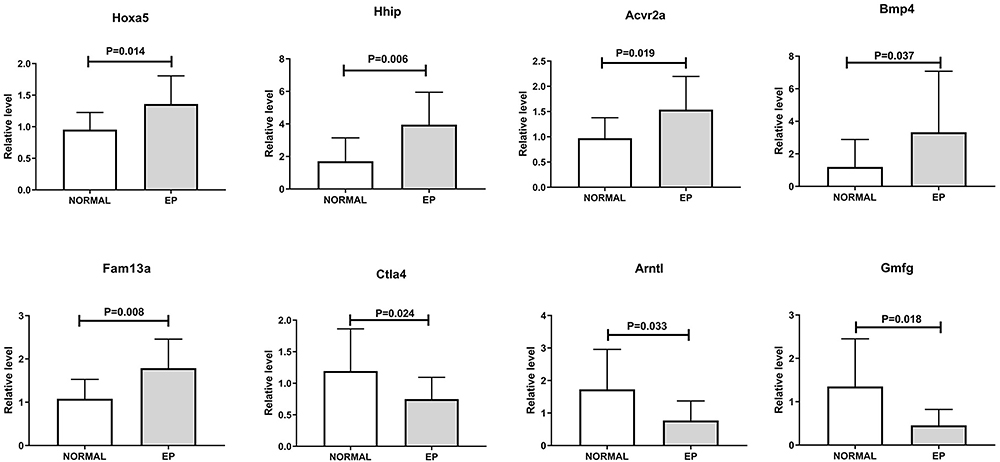 |
Figure 7 Results of RT‒PCR analysis of DEGs. Abbreviations: DEGs, differentially expressed genes; Hoxa5, homeobox A5; Hhip, human hedgehog interacting protein; Acvr2a, activin receptor IIA; Bmp4, bone morphogenetic protein-4; Fam13a, family with sequence similarity 13 member A; Ctla4, cytotoxic T-lymphocyte-associated protein 4; Arntl, aryl hydrocarbon receptor nuclear translocator like; Gmfg, glia maturation factor gamma. Notes: Horizontal bars indicate x±s, and the comparisons were determined by independent sample t-test between the two groups (n=15). |
IHC Staining Results
The percentages of Bmp4+ and Hoxa5+ cells were significantly higher and those of Arntl+, Ctla4+, and Gmfg+ cells were significantly lower in the EP group than in the NORMAL group (P<0.05, Table 3, Figures 8, 9).
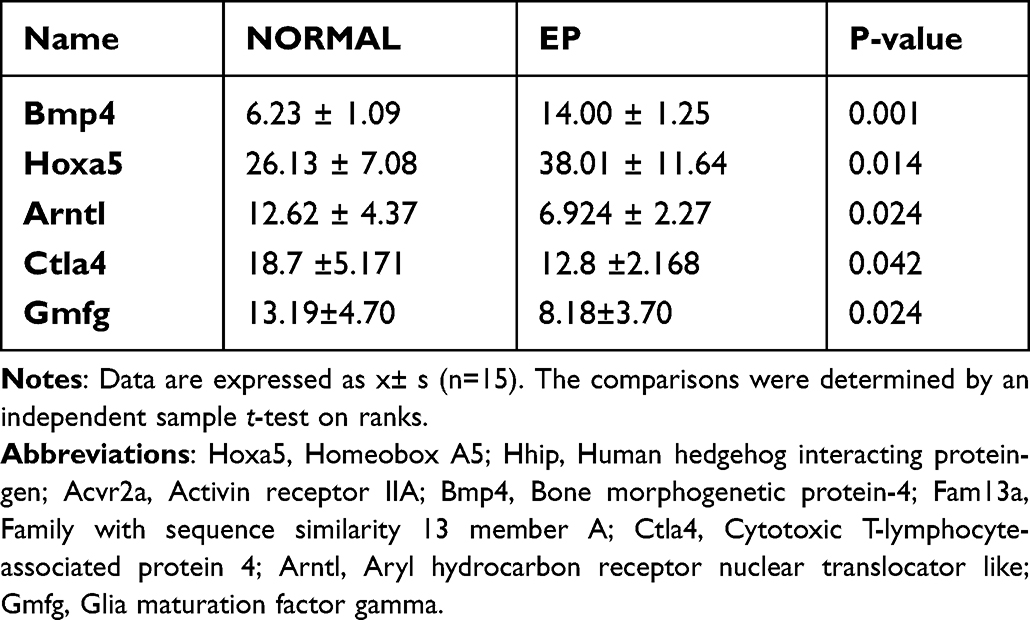 |
Table 3 The Percentage of Positive Cells in Each Indicator of Mouse Lung Tissue (%, x±s) |
 |
Figure 8 The percentage of each indicator-positive cell in the lung in the NORMAL and EP groups. Abbreviations: Bmp4, bone morphogenetic protein-4; Hoxa5, homeobox A5; Arntl, Aryl hydrocarbon receptor nuclear translocator like; Ctla4, cytotoxic T-lymphocyte-associated protein 4; Gmfg, glia maturation factor gamma. |
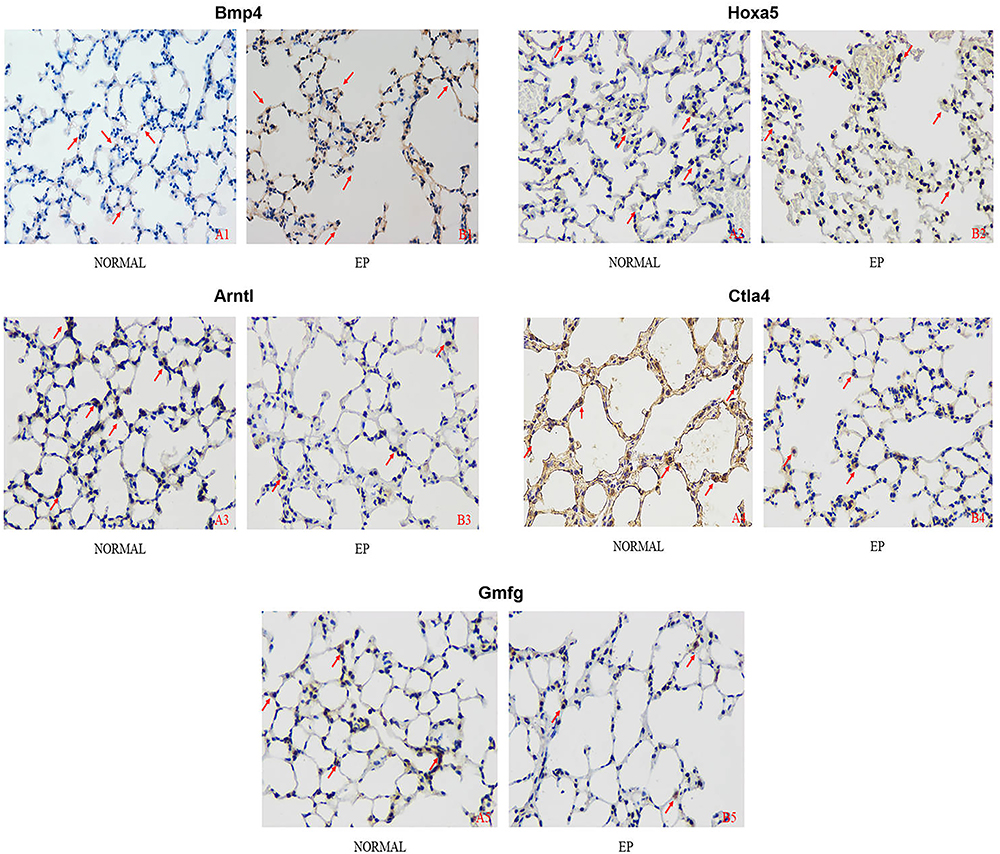 |
Figure 9 The immunohistochemical results of Bmp4/Hoxa5/Arntl/Ctla4/Gmfg staining in the lung tissue of each group (×400). Abbreviations: Bmp4, bone morphogenetic protein-4; Hoxa5, homeobox A5; Arntl, aryl hydrocarbon receptor nuclear translocator like; Ctla4, cytotoxic T-lymphocyte-associated protein 4; Gmfg, glia maturation factor gamma. Notes: The arrow marks where the positive expression of the index is obvious. A1/B1: Bmp4; A2/B2: Hoxa5; A3/B3: Arntl; A4/B4: Ctla4; A5/B5: Gmfg. |
Discussion
Cigarette smoke is a major risk factor for COPD.1 Quitting may reduce chronic cough, sputum symptoms, shortness of breath, and wheezing in the short term.30 However, smoking cessation cannot completely reverse pulmonary inflammation or emphysema.1 To date, some studies have indicated that COPD is a chronic inflammatory disease with autoimmune characteristics triggered by smoking8 and that foreign substances or autoantigens are the keys to activating the acquired immune response. Smoking can induce nonspecific immune responses to release proteases to degrade pulmonary elastin, which is then processed into polypeptides with antigenic activity in COPD-susceptible people, thus stimulating the activation of adaptive immune responses that lead to lung tissue destruction and COPD development.13 Polosukhin VV showed that persistent airflow restriction in COPD is most closely related to the loss of radial alveolar attachment in small airways, and the destruction of alveolar attachments may be mediated by neutrophilic inflammation.31 OS not only causes endothelial and mitochondrial dysfunction, lipid peroxidation, and apoptosis to produce a large number of proinflammatory factors32 but also damages the function of antiproteases such as α1-antitrypsin and secretory leukocyte protease inhibitors, resulting in the accelerated decomposition of lung elastin, carbonyl modification to form highly immunogenic new antigens, and the stimulation of autoantibody production in patients with COPD. These events may lead to the persistence and amplification of the inflammatory response, which leads to the production of more oxides, further aggravating OS damage.33 RNA-seq has been widely used in many fields, such as disease mechanism discovery. Currently, most researchers mainly obtain abundant transcriptome data from peripheral blood and the airway epithelial cells of human COPD patients and identify the related differential genes and regulatory mechanisms. However, few transcriptomic studies have been conducted with lung tissue. According to previous literature and our previous studies, endogenous elastin peptide may play an important role in the occurrence and development of human COPD. To determine the possible targets of the elastin peptide and explore the possibility of its application in humans in the future, we established an emphysema model by combining tobacco smoke exposure and EP treatment, identified the DEGs between mice with and without emphysema by RNA-seq, and verified the differential expression of selected genes by qRT‒PCR and IHC staining to further explore the mechanism of pulmonary inflammation and destruction at the transcriptional level in mice with emphysema. Thus, new therapeutic methods and targets for the early prevention and treatment of COPD were identified.
In this study, we found that the expression of Bmp4, Hoxa5, Hhip, Acvr2a, and Fam13a was up-regulated but that of Ctla4, Arntl, and Gmfg was down-regulated in the EP group compared with the NORMAL group. The related literature shows that different genes maintain important biological functions through interactions, which may be the key factor in promoting chronic airway inflammation in COPD. Among these interactions, the Bmp4-Smad-Hoxa5/Acvr2a signaling pathway attracted our attention. Bmp4, a member of the TGF-β superfamily, is a multifunctional growth factor that is expressed mainly in airway epithelial cells and the pulmonary interstitium. It plays an important role in pulmonary differentiation and morphogenesis.34–36 Bmp4 is considered to be a pro-inflammatory gene in systemic circulation and can trigger and activate inflammatory responses.37 By establishing a model of acute airway inflammation, Li38 found that the expression of Bmp4 was increased, suggesting that Bmp4 may participate as an anti-inflammatory factor in the feedback regulation of innate immunity and preventing lung injury caused by excessive airway inflammation by inhibiting the release of proinflammatory factors. However, the role of Bmp4 in chronic airway inflammation is still controversial, and it has been observed to be either upregulated or downregulated in animal models of allergic airway inflammation or tobacco smoke-induced chronic airway inflammation.34,39 Studies have shown that Modulation of BMP4 or the downstream receptors and signaling decades in human airway basal stem/progenitor cells may be a potential therapeutic approach to cure or prevent the pathogenesis of smoking induced airway disease.40 Currently, studies can only show that Bmp4 is involved in the damage or repair process of lung inflammation or chronic airway inflammation, the Smad signaling pathway, the p38-MAPK-ERK1/2 pathway, the activation of JNK and ID1 and blocking the anti-inflammatory effect of the NF-κB pathway.41–44 Hoxa5 reduces inflammation and enhances adipose tissue browning by negatively regulating TNC/TLR4/NF-κB-mediated inflammatory signaling and activating the Bmp4/Smad1 pathway.45 Hoxa5 is involved in cell proliferation, differentiation and apoptosis; is required for embryonic airway morphogenesis, and is most abundantly expressed in adult lung tissue. It may also reduce airway inflammation by promoting M2 macrophage polarization.46,47 In addition, Acvr2a has been reported to participate in cell differentiation, proliferation, and inflammation through the SMAD signaling pathway by binding to activin.48 In BMP type II receptor-deficient pulmonary artery smooth muscle cells, BMP ligands perform signal transduction by activating Acvr2a.49 In this study, we found that Bmp4 was up-regulated in mice with emphysema in the EP group, suggesting that targeting Bmp4 signaling in the airway may constitute a new target for the prevention/treatment of COPD-related airway diseases and that the Bmp4-Smad-Hoxa5/Acvr2a signaling pathway may be involved in the occurrence and development of COPD.
COPD is often associated with abnormal inflammatory responses in the airways and/or alveoli caused by smoking or heavy exposure to toxic particles or gases.1 Cigarette smoking extracts can induce the expression of many interleukins, leading to lung injury.50 We found that the DEGs were significantly enriched in BP terms such as immune system process, protein kinase/kinase activity, adaptive immune response, phosphorylation, and inflammation and identified significant changes in important pathways such as cytokine receptor interaction, T-cell receptor, Th17 cell differentiation, Th1 and Th2 cell differentiation, Rap1 signaling pathway and MAPK signaling pathway in the EP group. These observations are consistent with previous studies on the relationship between COPD occurrence and development and OS and immune-inflammatory responses. These pathways play an important role in the immune response, immune-inflammatory defense, cell growth and differentiation, and apoptosis. Further study of these pathways will help elucidate the mechanism underlying the continuous amplification of chronic airway inflammation and the development of emphysema and will provide a theoretical basis for the development of new therapeutic drugs for COPD.
We used RNA-seq to preliminarily explore the transcriptomic changes, oxidation–antioxidation response, and immune response in emphysema induced by tobacco smoke combined with EPs and identified the key factors and signaling pathways. Although we performed preliminary verification of the findings regarding these genes at the molecular and/or protein levels, the number of sequencing samples in each group was small, which is a limitation. In addition, the mechanisms related to or affecting these changes remain controversial or unclear, and verification of gene expression levels and functions is needed.
Conclusion
(1) KEGG pathway enrichment analysis showed that the interaction of cytokines and cytokine receptors, Th-cell differentiation, the Rap1 signaling pathway, the MAPK signaling pathway, and other signaling pathways may be involved in the development of emphysema. GO enrichment analysis indicated that emphysema may be mainly related to the inflammatory response, immune response, immune regulation, OS injury, apoptosis, and other biological processes. These findings provide a transcriptomic basis and a more complete gene map for studying the mechanism of COPD occurrence and development.
(2) The upregulated genes Hoxa5, Hhip, Acvr2a, Bmp4, and Fam13a and the downregulated genes Ctla4, Arntl, and Gmfg are related to OS and immunity, and the Bmp4-Smad-Hoxa5/Acvr2a signaling pathway may mediate the occurrence and development of COPD.
Acknowledgments
We are grateful for the assistance from the Experimental Center of Guangxi Medical University (Nanning, Guangxi, People’s Republic of China) and The Eighth Affiliated Hospital, Sun Yat-sen University (Shenzhen, Guangdong, People’s Republic of China).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the Natural Science Foundation of China (NSFC81760010 and 82060364), the Science and Technology Department of Guangxi Zhuang Autonomous Foundation of Guangxi Key Research and Development Program (No. GuikeAB20238025), Guangxi Natural Science Foundation (NO. 2021GXNSFBA220064), the Shenzhen Science Technology Program (NO. JCYJ20210324115000002) and Futian Healthcare Research Project (No: FTWS2021004).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Committee GE. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2022 report); 2021. Available from: https://goldcopd.org/2022-gold-reports/.
2. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940. doi:10.1016/S0140-6736(17)31222-9
3. Labaki WW, Rosenberg SR. Chronic obstructive pulmonary disease. Ann Intern Med. 2020;173(3):ITC17–ITC32. doi:10.7326/AITC202008040
4. World Health Organization. The top 10 causes of death; 2020. Available from: https://wwwwhoint/news-room/fact-sheets/detail/the-top-10-causes-of-death.
5. Gaggar A, Weathington N. Bioactive extracellular matrix fragments in lung health and disease. J Clin Invest. 2016;126(9):3176–3184. doi:10.1172/JCI83147
6. Rauscher S, Pomès R. The liquid structure of elastin. Elife. 2017;6:e26526. doi:10.7554/eLife.26526
7. Turino GM, Lin YY, He J, et al. Elastin degradation: an effective biomarker in COPD. COPD. 2012;9(4):435–438. doi:10.3109/15412555.2012.697753
8. Sellami M, Meghraoui-Kheddar A, Terryn C, et al. Induction and regulation of murine emphysema by elastin peptides. Am J Physiol Lung Cell Mol Physiol. 2016;310(1):L8–L23. doi:10.1152/ajplung.00068.2015
9. Meghraoui-Kheddar A, Pierre A, Sellami M, et al. Elastin receptor (S-gal) occupancy by elastin peptides modulates T-cell response during murine emphysema. Am J Physiol Lung Cell Mol Physiol. 2017;313(3):L534–L547. doi:10.1152/ajplung.00465.2016
10. Baranek T, Debret R, Antonicelli F, et al. Elastin receptor (spliced galactosidase) occupancy by elastin peptides counteracts proinflammatory cytokine expression in lipopolysaccharide-stimulated human monocytes through NF-kappaB down-regulation. J Immunol. 2007;179(9):6184–6192. doi:10.4049/jimmunol.179.9.6184
11. Tingting M, Caimei T, Haiguang X, et al. Differences in expression of cell surface costimulatory molecules and secretion of inflammatory factors by bone marrow-derived dendritic cells in response to elastin peptide. J Guangxi Med Univ. 2018;35(6):751–755.
12. Lemaire F, Audonnet S, Perotin J-M, et al. The elastin peptide VGVAPG increases CD4 T-cell IL-4 production in patients with chronic obstructive pulmonary disease. Respir Res. 2021;22(1):14. doi:10.1186/s12931-020-01609-4
13. Rovina N, Koutsoukou A, Koulouris NG. Inflammation and immune response in COPD: where do we stand? Mediators Inflamm. 2013;2013:413735. doi:10.1155/2013/413735
14. Taraseviciene-Stewart L, Scerbavicius R, Choe K-H, et al. An animal model of autoimmune emphysema. Am J Respir Crit Care Med. 2005;171(7):734–742. doi:10.1164/rccm.200409-1275OC
15. Houghton AM, Quintero PA, Perkins DL, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116(3):753–759. doi:10.1172/JCI25617
16. Lee S-H, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13(5):567–569. doi:10.1038/nm1583
17. Wen L, Krauss-Etschmann S, Petersen F, et al. Autoantibodies in chronic obstructive pulmonary disease. Front Immunol. 2018;9:66. doi:10.3389/fimmu.2018.00066
18. Shan M, Cheng H-F, Song L-Z, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. 2009;1(4):4ra10. doi:10.1126/scitranlsmed.3000154
19. Zhou JS, Li ZY, Xu XC, et al. Cigarette smoke-initiated autoimmunity facilitates sensitisation to elastin-induced COPD-like pathologies in mice. Eur Respir J. 2020;56(3):2000404. doi:10.1183/13993003.00404-2020
20. Tang S, Ma T, Zhang H, et al. Erythromycin prevents elastin peptide-induced emphysema and modulates CD4T cell responses in mice. Int J Chron Obstruct Pulmon Dis. 2019;14:2697–2709. doi:10.2147/COPD.S222195
21. Gu BH, Sprouse ML, Madison MC, et al. A novel animal model of emphysema induced by anti-elastin autoimmunity. J Immunol. 2019;203(2):349–359.
22. D’Hulst AI, Vermaelen KY, Brusselle GG, et al. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J. 2005;26(2):204–213. doi:10.1183/09031936.05.00095204
23. Cock PJA, Fields CJ, Goto N, et al. The sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38(6):1767–1771. doi:10.1093/nar/gkp1137
24. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi:10.1038/nmeth.3317
25. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi:10.1038/nmeth.1923
26. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12(1):323. doi:10.1186/1471-2105-12-323
27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
28. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Gene Ontol Consortium Nat Genet. 2000;25(1):25–29.
29. Kanehisa M, Goto S, Sato Y, et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–D114. doi:10.1093/nar/gkr988
30. Pezzuto A, Stellato M, Catania G, et al. Short-term benefit of smoking cessation along with glycopirronium on lung function and respiratory symptoms in mild COPD patients: a retrospective study. J Breath Res. 2018;12(4):046007. doi:10.1088/1752-7163/aad0a8
31. Polosukhin VV, Gutor SS, Du RH, et al. Small airway determinants of airflow limitation in chronic obstructive pulmonary disease. Thorax. 2021;76(11):1079–1088. doi:10.1136/thoraxjnl-2020-216037
32. Liu Q, Gao Y, Ci X. Role of Nrf2 and its activators in respiratory diseases. Oxid Med Cell Longev. 2019;2019:7090534. doi:10.1155/2019/7090534
33. Kirkham PA, Caramori G, Casolari P, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184(7):796–802. doi:10.1164/rccm.201010-1605OC
34. Rosendahl A, Pardali E, Speletas M, et al. Activation of bone morphogenetic protein/Smad signaling in bronchial epithelial cells during airway inflammation. Am J Respir Cell Mol Biol. 2002;27(2):160–169. doi:10.1165/ajrcmb.27.2.4779
35. Anderson L, Lowery JW, Frank DB, et al. Bmp2 and Bmp4 exert opposing effects in hypoxic pulmonary hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298(3):R833–R842. doi:10.1152/ajpregu.00534.2009
36. Weaver M, Dunn NR, Hogan BL. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development. 2000;127(12):2695–2704. doi:10.1242/dev.127.12.2695
37. Yin W, Jo H, Voit EO. Systems analysis of the role of bone morphogenic protein 4 in endothelial inflammation. Ann Biomed Eng. 2010;38(2):291–307. doi:10.1007/s10439-009-9822-y
38. Li Z, Wang J, Wang Y, et al. Bone morphogenetic protein 4 inhibits liposaccharide-induced inflammation in the airway. Eur J Immunol. 2014;44(11):3283–3294. doi:10.1002/eji.201344287
39. Kariyawasam HH, Xanthou G, Barkans J, et al. Basal expression of bone morphogenetic protein receptor is reduced in mild asthma. Am J Respir Crit Care Med. 2008;177(10):1074–1081. doi:10.1164/rccm.200709-1376OC
40. Zuo WL, Yang J, Strulovici-Barel Y, et al. Exaggerated BMP4 signalling alters human airway basal progenitor cell differentiation to cigarette smoking-related phenotypes. Eur Respir J. 2019;53(5):1702553. doi:10.1183/13993003.02553-2017
41. Güngör N, Pennings JLA, Knaapen AM, et al. Transcriptional profiling of the acute pulmonary inflammatory response induced by LPS: role of neutrophils. Respir Res. 2010;11(1):24. doi:10.1186/1465-9921-11-24
42. Bhavsar TM, Patel SN, Lau-Cam CA. Protective action of taurine, given as a pretreatment or as a posttreatment, against endotoxin-induced acute lung inflammation in hamsters. J Biomed Sci. 2010;17(Suppl 1):S19. doi:10.1186/1423-0127-17-S1-S19
43. Maruyama H, Dewachter C, Belhaj A, et al. Endothelin-Bone morphogenetic protein type 2 receptor interaction induces pulmonary artery smooth muscle cell hyperplasia in pulmonary arterial hypertension. J Heart Lung Transplant. 2015;34(3):468–478. doi:10.1016/j.healun.2014.09.011
44. Wang D, Prakash J, Nguyen P, et al. Bone morphogenetic protein signaling in vascular disease: anti-inflammatory action through myocardin-related transcription factor A. J Biol Chem. 2012;287(33):28067–28077. doi:10.1074/jbc.M112.379487
45. Csiszar A, Labinskyy N, Jo H, et al. Differential proinflammatory and prooxidant effects of bone morphogenetic protein-4 in coronary and pulmonary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295(2):H569–H577. doi:10.1152/ajpheart.00180.2008
46. Bazzan E, Turato G, Tinè M, et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir Res. 2017;18(1):40. doi:10.1186/s12931-017-0522-0
47. Le Y, Cao W, Zhou L, et al. Infection of promotes both M1/M2 polarization and MMP production in cigarette smoke-exposed macrophages. Front Immunol. 2020;11:1902. doi:10.3389/fimmu.2020.01902
48. Xia Y, Schneyer AL. The biology of activin: recent advances in structure, regulation and function. J Endocrinol. 2009;202(1):1–12. doi:10.1677/JOE-08-0549
49. Yu PB, Beppu H, Kawai N, et al. Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J Biol Chem. 2005;280(26):24443–24450. doi:10.1074/jbc.M502825200
50. Pezzuto A, Citarella F, Croghan I, et al. The effects of cigarette smoking extracts on cell cycle and tumor spread: novel evidence. Future Sci OA. 2019;5(5):FSO394. doi:10.2144/fsoa-2019-0017
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.