")
Back to Journals » OncoTargets and Therapy » Volume 12
RNA m6A Methyltransferase METTL3 Promotes The Growth Of Prostate Cancer By Regulating Hedgehog Pathway
Authors Cai J , Yang F , Zhan H, Situ J, Li W, Mao Y, Luo Y
Received 11 August 2019
Accepted for publication 11 October 2019
Published 5 November 2019 Volume 2019:12 Pages 9143—9152
DOI https://doi.org/10.2147/OTT.S226796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Jiarong Cai,* Fei Yang,* Hailun Zhan, Jie Situ, Wenbiao Li, Yunhua Mao, Yun Luo
Department of Urology, The Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou 510630, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yun Luo
Department of Urology, The Third Affiliated Hospital of Sun Yat-sen University, 600 Tianhe Road, Guangzhou 510630, People’s Republic of China
Tel +86 20 8217 9727
Email [email protected]
Purpose: N6-methyladenosine (m6A) is the most abundant internal modification on eukaryotic mRNA and gained increasing attention recently. More and more evidence suggest that m6A methylation plays crucial role in tumor genesis and development. However, its role in prostate cancer remains largely unknown.
Methods: METTL3 expression status in prostate cancer was analyzed by using TCGA database and Western blotting. m6A content was analyzed by using RNA Methylation Quantification Kit. The role of METTL3 in prostate cancer cells was determined by proliferation, survival, colony formation, and invasion assays. The m6A level of GLI1 RNA was detected by methylated RNA immunoprecipitation (MeRIP) assay. In vivo role of METTL3 was studied on xenograft models.
Results: We found that m6A methyltransferase METTL3 was overexpressed in prostate cancer cell lines, together with increased m6A content. Functionally, silencing of METTL3 by shRNA in prostate cancer cell lines resulted in decreased m6A content, cell proliferation, survival, colony formation, and invasion. Interestingly, overexpression of wild-type METTL3 abrogated the repression effect of METTL3 depletion on m6A content, cell proliferation, survival, colony formation, and invasion, while the overexpression of m6A catalytic site mutant METTL3 was unable to rescue the inhibitory effect caused by METTL3 depletion. Further mechanism analysis demonstrated that METTL3 silence decreased the m6A modification and expression of GLI1, an important component of hedgehog pathway, which led to cell apoptosis. Moreover, depletion of METTL3 inhibited tumor growth in vivo.
Conclusion: Our results suggested that the m6A methyltransferase METTL3 promotes the growth and motility of prostate cancer cells by regulating hedgehog pathway.
Keywords: RNA methylation, METTL3, prostate cancer, hedgehog, GLI1
Introduction
Prostate cancer (PC), which occurs in the prostate, is one of the most common cancers in men.1 Among American men, PC is the second leading cause of death, with an estimated incidence of 174,650 new cases and 31,620 deaths according to the American Cancer Society statistic 2019. The majority of PC patients are treated with surgical removal of the prostate, radiation, chemotherapy, or hormonal therapy. Despite the improvement of these therapies, the five years’ recurrence for prostate cancer patients is still about 25%.2 Invasion is one of the most important factors responsible for lethal, the survival rate decreased significantly if tumor has metastasized since diagnose. Therefore, it is necessary to have a more in-depth understanding of molecular mechanism that regulates the growth and metastasis of PC.
In eukaryotes, in addition to the well-established epigenetic DNA modification, modifications on RNA gained more and more attention. N6-methyladenosine (m6A), one of the most abundant and reversible modification on messenger RNA, was identified more than forty years ago.3,4 The m6A network is composed of three classes of proteins: (1) the methyltransferase complex, which contains two catalytic components METTL3 (Methyltransferase-like 3, also known as MTA70), METTL14, and the accessory WTAP (Wilm’s tumor 1-associated protein); (2) two demethylases, FTO (Fat mass and obesity-associated protein) and ALKBH5 (alkB homolog 5, RNA demethylase); (3) the m6A reader protein.5–12 m6A plays important and diverse biological functions in mammals and several studies have demonstrated that deregulation of METTL3 was involved in various cancers.13–18 However, the roles of m6A and METTL3 in prostate cancer still need to be established.
In mammals, among the three hedgehog homologues, Sonic hedgehog (SHH), Desert hedgehog (DHH), and Indian hedgehog (IHH), SHH is the best studied. SHH pathway can be activated in two major ways, canonical signaling and non-canonical signaling. In canonical signaling, SHH binds and inactivates the 12-transmembrane protein Patched (Ptch1), which inhibits the activity of smoothened (Smo).19,20 Activation of Smo initiates the SHH downstream signaling cascade, which results in the translocation of GLI family proteins to the nucleus and begins the transcription of target genes, including Cyclin D1, Cyclin E, c-Myc, BCL-2, and SNAIL.21,22 Thus, three members of GLI family, GLI1, GLI2, and GLI3, are reliable markers for SHH activation. Inappropriate activation of the SHH-GLI pathway has been proved in several malignancies, including prostate cancer, pancreatic cancer, squamous lung cancer, and bladder cancer.23–28 More importantly, elevated SHH expression and activity was significantly correlated with more aggressive PC. Giving the importance of SHH in PC, it is urgent to understand the mechanism by which SHH pathway is regulated in PC.
In this study, we analyzed the roles of m6A and METTL3 in PC and found that METTL3 protein level and m6A methylated RNA level were increased in PC cell lines compared with normal cell line. Depletion of METTL3 inhibited PC cells proliferation, survival, colony formation, and invasion, and these regulatory effects are m6A catalytic activity-dependent. Further mechanism study demonstrated that SHH-GLI signaling is regulated by METTL3, which consequently induced cell apoptosis.
Materials And Methods
Reagents
Antibodies against METTL3 were purchased from Abnova (Taipei, Taiwan). Antibodies against Bak, Bax, Bcl-2, Bcl-xL, GLI1, GLI2, and PTCH1 were purchased from Cell Signaling Technology (Danvers, MA USA). Antibodies against GLI3 and Smo were purchased from Abcam (Cambridge, MA USA). Antibodies against α-Tubulin were purchased from Sigma Aldrich (St. Louis, MO USA).
Cell Culture
Human prostate cancer cell lines LNCaP, PC3, C4-2, C4-2B, and DU-145 (ATCC, Rockville, MD USA) were grown in RPMI (Roswell Park Memorial Institute) 1640 medium, which was supplemented with 10% FBS (fetal bovine serum) and 1% penicillin-streptomycin. Human normal prostate epithelial cell line RWPE-1 cells (ATCC, Rockville, MD USA) were grown in keratinocyte serum-free medium that contains bovine pituitary extract (0.05 mg/mL) and human recombinant epidermal growth factor (5ng/mL).
Plasmids And Lentiviral shRNA
Plasmids pCDNA3-FLAG and pCDNA3-FLAG-METTL3 were purchased from Addgene (Cambridge, MA USA). The catalytically dead mutant of METTL3 was conducted from pCDNA3-FLAG-METTL3 by using QuikChange II Site-Directed Mutagenesis Kit (California, CA USA). The residues 395–398, DPPW, were mutated to APPA. The mutant primers were synthesized as follows: forward 5ʹ-AGTTGTGATGGCTGCCCCACCCGCGGATATTCACATGGAACTG-3ʹ; reverse 5ʹ- CAGTTCCATGTGAATATCCGCGGGTGGGGCAGCCATCACAACT-3ʹ.
Lipofectamine 2000 (Invitrogen, Carlsbad, CA USA) was used for transit transfection according to the instruction. Scramble and METTL3 shRNAs were purchased from Sigma Aldrich (St. Louis, MO USA).
The lentiviruses were packaged in 293T cells through co-transfection with pLP1, pLP2, and VSVG. Supernatants containing lentiviral particles was collected and concentrated by addition of PEG, and cells were transduced and selected with puromycin.
Cell Proliferation, Survival, And Colony Formation Assay
Cell proliferation was assessed with the CCK-8 kit (Dojindo, Kumamoto, Japan). Briefly, 3000 cells per well were seeded into 96-well plates before 10 μL CCK-8 reagent was added into each well. Cell proliferation was determined by examining the absorbance at a wavelength of 450 nm after 2 hrs incubation at 37°C. Cell survival was assessed using trypan blue staining, in which dead cells were blue stained, and counted manually using hemocytometer. Colony formation was performed by seeding 3000 cells per well in 6-well plates. Following incubation at 37°C, 5% CO2 for 10–14 days, colonies were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for 10 mins at room temperature. After washed with PBS and dried, colonies consisting of more than 50 cells were counted in each well. Experiments were performed in triplicate.
Cell Invasion Assay
Invasion assay was conducted using transwell inserts (Corning) according to the instructions. Briefly, 1×105 cells were suspended in serum-free medium and seeded into the upper chamber of the inserts that were coated with matrigel. Inserts were then placed in 24-well plates fulfilled with complete growth medium in the lower chamber for 24 hrs. After washed twice with PBS, the invading cells on the lower side were fixed and stained with 0.5% crystal violet. Images of five randomly selected fields per filter-bottom surfaces were captured by using a phase-contrast microscope equipped with a digital camera, and cells in each image were counted by Image-Pro Plus software.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated using RNA isolation kit (Omega). cDNA was synthesized using the iScriptTM Reverse Transcription Super mix (Bio-Rad) before the samples were then analyzed using SYBR Green Master Mix on a real-time PCR system. Primers were synthesized as follows: c-Myc forward: 5ʹ GACGACGAGACCTTCATCAAAAAC 3ʹ, and c-Myc reverse:
5ʹ AGGCCAGCTTCTCTGAGAC 3ʹ; Cyclin D1 forward: 5ʹ ACACTTCCTCTCCAAAATGCC 3ʹ, and Cyclin D1 reverse: 5ʹ GAGGGCGGATTGGAAATGAAC 3ʹ; GAPDH forward: 5ʹ ATCACCATCTTCCAGGAGCG 3ʹ, and GAPDH reverse: 5ʹ CAAATGAGCCCCAGCCTTC 3ʹ. GAPDH was used as the endogenous calibrator control. Relative gene expression was calculated by the comparative CT method.
Western Blotting
For Western blotting, 20 μg protein was subjected to SDS-PAGE separation, which was then transferred to nitrocellulose membranes (Millipore, MA, USA). After blocking with 5% non-fat milk, the membranes were incubated with the primary antibodies at 4°C overnight. Then, the membranes were incubated with secondary antibody for 1 hr at room temperature. The signal was detected by Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific) and acquired on an LI-COR Odyssey Fc imaging system (LI-COR Biosciences, Lincoln, NE USA).
m6A RNA Methylation Quantification
The content of m6A in total RNA was determined using m6A RNA Methylation Quantification Kit (Abcam, Cambridge, MA USA) according to the manufacture’s instruction. In brief, negative and diluted positive control RNA, as well as 200 ng total RNA were bound to the wells using RNA high binding solution. m6A was then captured and detected using specific capture antibody and detection antibody. After adding enhancer and color developing solutions, m6A signal was quantified colorimetrically by reading the absorbance at a wavelength of 450 nm.
MeRIP Assay
The methylated RNA immunoprecipitation (MeRIP) assay was performed using anti-m6A antibodies as previously described.29 Briefly, total RNA was isolated from prostate cancer cells by using Trizol. Total RNA (100 μg) was incubated (4°C with mixing, overnight) with 2 μg of anti-m6A antibodies or anti-IgG and 20 μL of Dynabeads Protein G (4°C with mixing, 6 hrs). After immunoprecipitation (IP), RNA was eluted from the beads by incubating with 200 μL 0.5 mg/mL N6-methyladenosine 5-monophosphate sodium salt for 1 hr at 4°C. The eluted RNA was subjected to qRT-PCR analysis.
Caspase-3/7 Activity Assay
Caspase-3/7 activity was measured using Apo-ONE™ Homogeneous Caspase-3/7 Assay (Promega Corporation, Madison, WI, USA) according to the protocol provided by the manufacturer. In brief, 100 μL Apo-ONE Homogeneous Caspase-3/7 Reagent was added to each well of 96-well plate containing blank, control, and treatment. The plate was then placed on a shaker at 300–500rpm for 5 mins before incubating for 12 hrs at room temperature. The fluorescence of each well was measured using spectrofluorometer.
In-Vivo Xenograft Experiment
Six-week-old male NOD/SCID (Jackson Laboratory, Bar Harbor, ME USA) mice were randomly divided into two groups: Scr shRNA (n=5) and METTL3 shRNA (n=5). Equal number of PC-3 cells was injected subcutaneously into right flank. Tumor size and mice weight were measured at the indicated times. Tumor weight was measured at the end of the experiment (4 weeks). All animal experiments were conducted according to the NIH Guide for the Care and Use of Laboratory Animals and approved by Institutional Animal Care and Use Committee of Sun Yat-sen University (Guangzhou, China).
Statistical Analysis
Data were presented as mean± SD from three independent experiments. P value was determined using paired Student’s t-test, and a P value ˂ 0.05 was considered statistically significant.
Results
The Expression Of m6A Methyltransferase METTL3 Is Increased In Human Prostate Cancer Cell Lines
METTL3 has been shown to play an important role in various cancers, such as AML, lung cancer, bladder cancer, and melanoma. To explore the potential function of METTL3 in PC, we investigated the publicly available database TCGA (The Cancer Genome Atlas) and GEPIA (Gene Expression Profiling Interactive Analysis). We found that METTL3 mRNA is highly expressed in PC patients’ tissue sample (Figure 1A). The TCGA data indicated that the ratio of PC patients with genetic alterations could reach above 15% (Figure 1B). In addition, the data from GEPIA showed that the expression level of METTL3 in tumor (n=492) is higher than in normal tissue (n=52) (Figure 1C). Next, we examined the protein level of METTL3 in normal human prostate epithelial cells (RWPE-1) and PC cells (LNCaP, PC3, C4-2, C4-2B, and DU-145). The results indicated that METTL3 was highly expressed in PC cells compared with normal prostate epithelial cells (Figure 1D). Since METTL3 is one of the components of m6A methyltransferase complex, we sought to know the change of RNA m6A methylation. Consistent with the increase in METTL3 level, the RNA m6A methylation increased in PC cells (Figure 1E). These data suggested that METTL3 might potentially play an important role in PC.
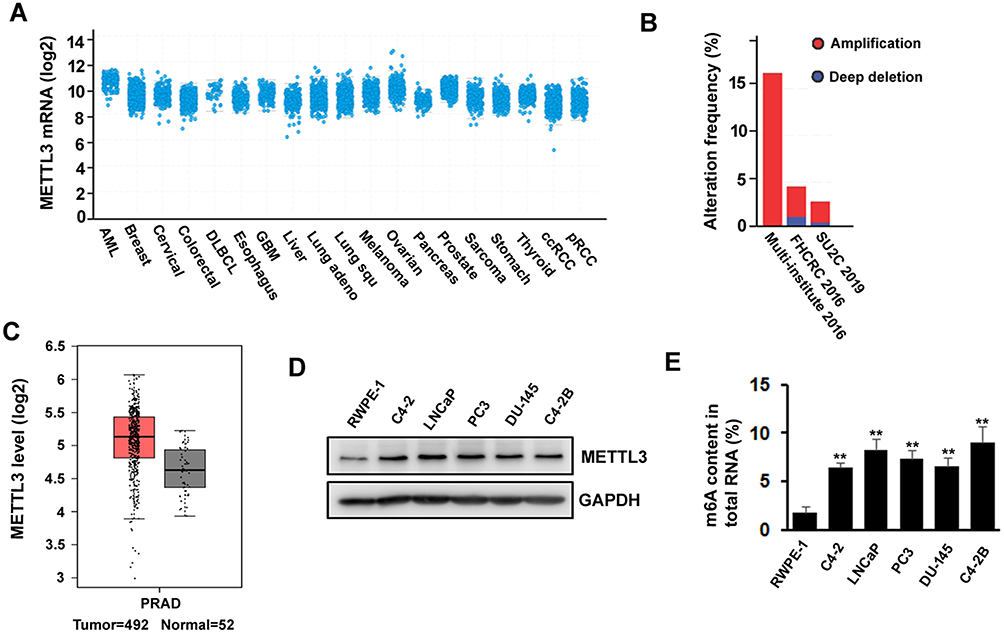 |
Figure 1 METTL3 is deregulated in prostate cancer. (A, B) As shown in TCGA database, expression of METTL3 is increased in several types of cancers including prostate cancer (A) and different studies showed different gene alteration frequencies of METTL3 (B). (C) Expression level of METTL3 in tumor is higher than in normal tissue, as seen on GEPIA database. (D) The protein level of METTL3 in human normal prostate epithelial cell and prostate cancer cell lines was detected by Western blotting. (E) The methylated RNA (m6A) level in human normal prostate epithelial cell and prostate cancer cell lines. **P ˂ 0.01. |
METTL3 Knockdown Inhibited PC Cells Proliferation, Survival, Colony Formation, And Invasion
To investigate the importance of METTL3 in PC, we used lentiviral-based scramble (Scr) or METTL3-specific shRNAs to knockdown METTL3 in LNCaP and PC3 cells. As presented by Western blotting analysis, more than 80% METTL3 was knockdown in both cell lines (Figure 2A). The RNA m6A methylation also decreased after METTL3 depletion in LNCaP and PC3 cells (Figure 2B). Then, we analyzed the effects of METTL3 knockdown on PC cells proliferation and survival. The results showed that METTL3 knockdown significantly represses LNCaP and PC3 cells proliferation and survival (Figure 2C and D). Consistent with the short-term growth and survival assays, our colony-forming unit assay also showed that METTL3 depletion inhibited the colony formation ability of LNCaP and PC3 cells (Figure 2E). Quantitative analysis showed a > 50% reduction in both cell lines (Figure 2E). To determine METTL3’s role in PC cell motility, we performed invasion assay. Interestingly, the invasion was greatly decreased in METTL3 silence cells compared with cells infected with Scr control in both LNCaP and PC3 cells (Figure 2F). These observations imply that METTL3 plays an important role in PC biology.
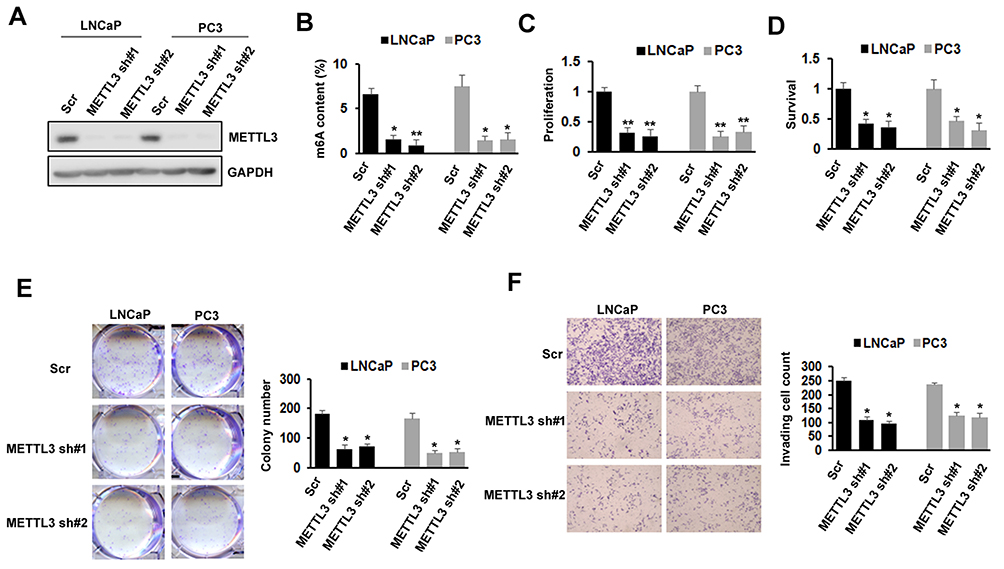 |
Figure 2 Effects of METTL3 depletion on cells proliferation, survival, colony formation, and invasion. (A) The knockdown efficiency of METTL3 in LNCaP and PC3 cells was detected by Western blotting. (B) The methylated RNA (m6A) level in LNCaP and PC3 cells after METTL3 depletion. *P ˂ 0.05, **P ˂ 0.01. (C) The proliferation of LNCaP and PC3 cells was analyzed by CCK-8 assay after METTL3 depletion. **P ˂ 0.01. (D) The survival of LNCaP and PC3 cells was analyzed by trypan blue staining after METTL3 depletion. *P ˂ 0.05. (E) LNCaP and PC3 cells were performed colony formation assay after transduced with control (Scr) or METTL3 shRNA. *P˂ 0.05. (F) The invasion ability of LNCaP and PC3 cells was analyzed by invasion assay after METTL3 depletion. *P ˂ 0.05. |
The Effects Of METTL3 On PC Cell Proliferation, Survival, Colony Formation, And Invasion Were m6A Catalytic Activity-Dependent
To further confirm the role of METTL3 in PC growth and motility, we studied the effect of exogenous expression of METTL3 in PC cells. First, we generated the METTL3 m6A catalytic site mutant (residues 395–398: DPPW-APPA) construct using site-directed mutagenesis kit (Figure 3A). We then infected LNCaP and PC3 cells with Scr or METTLE3 shRNA, followed with transfection of empty vector (EV), wild-typeMETTL3, and mutant METTL3 expression construct. Western blotting analysis showed that METTL3 was deleted by shRNA, overexpression with wild-type or mutant METTL3 constructs reversed the effect of METTL3 shRNA (Figure 3B). As expected, overexpression with wild-type METTL3 reversed the effect of METTL3 depletion on m6A content, whereas overexpression with mutant METTL3 did not (Figure 3C). Similar results were also obtained in cell proliferation, survival, colony formation, and invasion assay; overexpression with mutant METTL3 did not reverse the effects caused by METTL3 depletion (Figure 3D–G). All these data suggested that the regulatory effects of METTL3 on cell growth and motility are m6A catalytic activity-dependent.
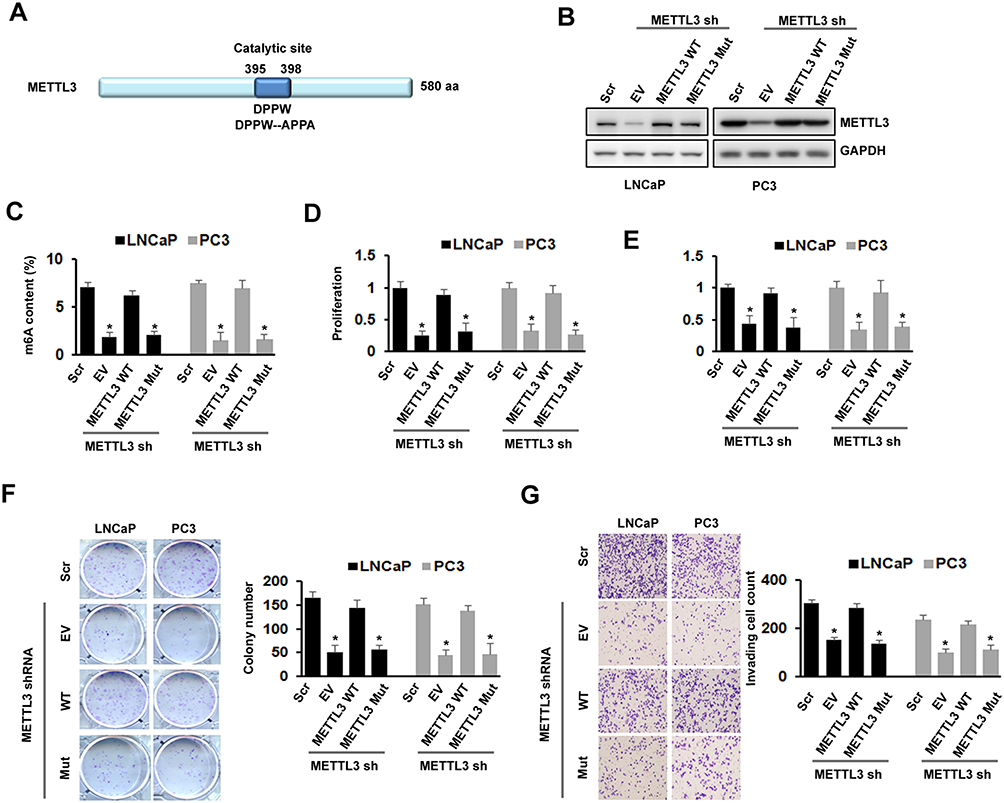 |
Figure 3 Effects of METTL3 depletion on cells proliferation, survival, colony formation, and invasion were m6A activity-dependent. (A) Protein structure of METTL3 showed location of catalytic domain and the mutation site for disruption of catalytic site. (B-G) LNCaP and PC3 cells were transduced with Scr or METTL3 shRNA for 48 hrs followed by overexpression with wild type or mutant METTL3 plasmid for 24 hrs. METTL3 protein level was detected by Western blotting (B); the m6A content was analyzed by kit (C); cell proliferation was analyzed by CCK-8 assay (D); cell survival was analyzed by trypan blue staining (E); colony formation ability was analyzed by colony formation assay (F); invasion ability was analyzed by invasion assay (G). *P ˂ 0.05. |
METTL3 Regulated PC Cells Apoptosis Via Sonic Hedgehog (SHH)-GLI Pathway
SHH signaling has been showed to play an important role in various cancers. To explore the underlying mechanism of METTL3, we analyzed whether SHH signaling was involved in METTL3 function in human PC cells. We found that the protein level of GLI1, an important component of SHH signaling, decreased after METTL3 depletion in LNCaP and PC3 cells. However, the protein level of other components, such as GLI2, GLI3, PTCH1, and Smo did not change obviously (Figure 4A). In addition, the mRNA level of SHH signaling downstream targets, such as c-Myc and cycling D1, decreased after METTL3 depletion (Figure 4B and C). Furthermore, the protein level of Bak and Bax (pro-apoptosis), PARP cleavage, and caspase 3/7activity increased after METTL3 depletion. In contrast, the protein level of Bcl-2 and Bcl-xL (anti-apoptotic) decreased after METTL3 depletion (Figure 4D–F). To investigate whether the regulatory effect of METTL3 on GLI1 is m6A catalytic activity-dependent, we infected the LNCaP and PC3 cells with METTL3 shRNA and then transfected with wild-type or mutant METTL3 constructs. Our results showed that only overexpression with wild-type METTL3 reversed the effect of METTL3 depletion on GLI1 protein and m6A methylation, but not mutant METTL3 (Figure 4G and H). Taken together, METTL3 regulated PC cell apoptosis through SHH/GLI pathway in an m6A catalytic activity-dependent manner.
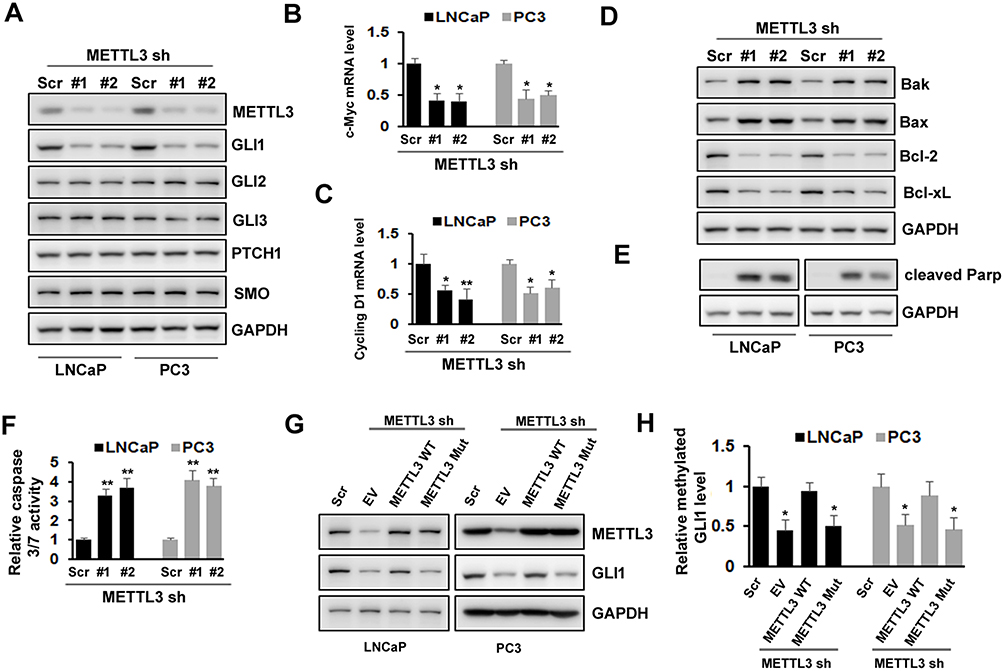 |
Figure 4 Effects of METTL3 depletion on SHH-GLI pathway and apoptosis. (A)The protein level of SHH-GLI pathway was analyzed by Western blotting after METTL3 depletion. (B, C) The mRNA level of c-Myc and cycling D1 was analyzed by qRT-PCR. *P ˂ 0.05, **P ˂ 0.01. (D, E) The protein level of Bak, Bax, Bcl-2, Bcl-xL, and cleaved PARP was analyzed by Western blotting after METTL3 depletion. (F) The relative caspase-3/7 activity was measured using Apo-One™ homogenous caspase-3/7 assay. **P ˂ 0.01. (G, H) LNCaP and PC3 cells were transduced with Scr or METTL3 shRNA for 48 hrs followed by overexpression with wild type or mutant METTL3 plasmid for 24 hrs. GLI1 protein level was detected by Western blotting (G) and the methylated GLI1 mRNA level was analyzed by Me-RIP assay (H). *P ˂ 0.05. |
METTL3 Depletion Inhibited Tumor Growth In Vivo
To investigate the possibility of METTL3 as a therapeutic target in prostate cancer, we tested the function of METTL3 on tumor growth in a mouse model. The mouse model was established by s. c. injection of PC-3 cells transduced with Scr or METTL3 shRNA into NOD/SCID mice. At the end of the experiments (4 weeks), we analyzed the tumor size and weight. As shown in Figure 5A–C, the tumor size and weight from METTL3 depletion group were significantly lower than that from control group. However, after 28 days of bearing tumor, the mice weight had no significant change (Figure 5D). These data indicated that METTL3 regulated tumor growth in vivo.
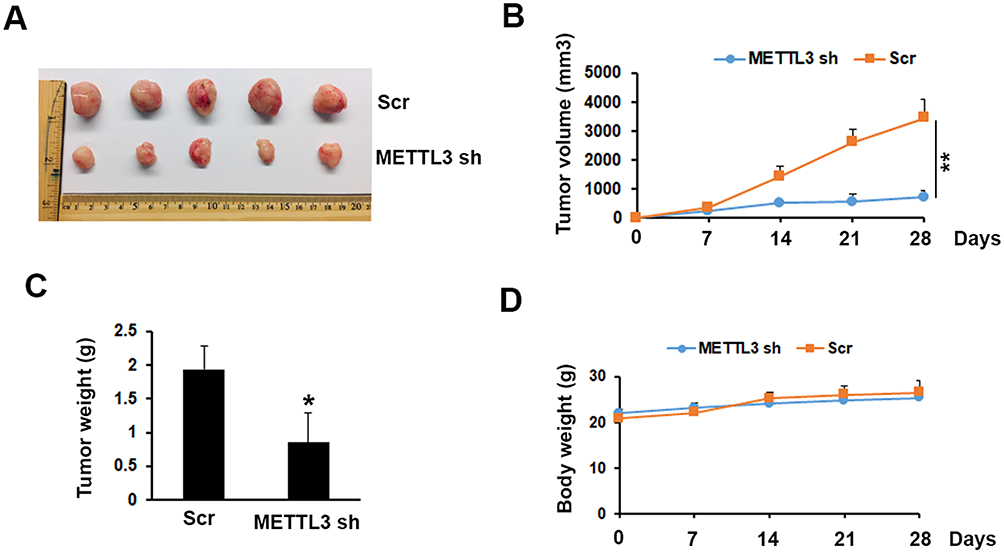 |
Figure 5 Effects of METTL3 depletion on tumor growth in vivo. (A) Typical photos of tumors on day 28 from Scr and METTL3 shRNA groups. (B, C) METTL3 depletion decreased tumor size and weight. *P ˂ 0.05, **P ˂ 0.01. (D) Mice body weight of Scr and METTL3 shRNA groups was measured at indicated time. |
Discussion
PC is one of the leading neoplasms in male population. Our results uncover a direct role of m6A methyltransferase METTL3 in promoting PC tumor growth and metastasis for the first time. Compared with normal human prostate epithelial cells, PC cell lines have higher METTL3 and m6A methylation level (Figure 1). A similar increase in METTL3 expression was also reported by other researchers in several cancers, such as AML, NSCLC, bladder cancer, melanoma, and osteosarcoma.13–18 In AML and NSCLC, depletion of METTL3 inhibited proliferation and colony formation, whereas apoptosis was increased.13,16 Similar results were also observed in bladder cancer and osteosarcoma.14,17 Interestingly, no significant effect of METTL3 depletion on cell proliferation and survival was seen in melanoma. However, the colony formation and invasion of melanoma cells were markedly inhibited after METTL3 knockdown.15 In addition, another m6A writer, Vir like m6A methyltransferase associated (VIRMA), showed significantly higher mRNA level in stage III/IV compared to stage II prostate cancers. Furthermore, VIRMA also showed positive correlation with m6A reader, YTHDF3, which indicated the importance of m6A modification in PC.30 Consistent with other’s reports, our study found that METTL3 depletion significantly inhibited PC cells proliferation, survival, colony formation, and invasion (Figure 2). Overexpression of wild type, but not m6A catalytic site mutant METTL3 rescued the effect on cell proliferation, survival, colony formation, and invasion that was caused by METTL3 depletion (Figure 3). All of these data suggested that METTL3 is a tumor supporter in PC, and its function is m6A catalytic activity-dependent.
Two PC cell lines, LNCaP and PC3, were used in our study. LNCaP is similar to human prostatic adenocarcinoma and androgen-dependent, whereas PC3 is similar to prostatic small cell neuroendocrine carcinoma (SCNC) and androgen-independent. Interestingly, similar results were obtained between LNCaP and PC3, which indicated that METTL3 might exert same function in both androgen-dependent and androgen-independent cell lines.
Aberrant activation of SHH-GLI signaling has been reported in PC.23,27,28 Multiple components contribute to the aberrant activation of SHH-GLI signaling in cancer cells. More importantly, activation of SHH-GLI signaling positively correlates the severity of PC, indicating that SHH-GLI signaling could be a potential therapeutic target.31–33 In addition, the correlation between SHH-GLI signaling activity and drug resistance has also been established.34 Although androgen deprivation therapy is the first choice for treating advanced PC, tumors often become androgen-independent in three years. GLI1 has been shown to contribute to the androgen-independent growth of PC by acting as a negative modulator for androgen receptor.23 All these studies indicated that SHH-GLI signaling plays an important role in PC progression. Here, we found that the expression level of GLI1, an important component of SHH-GLI signaling, decreased after METTL3 depletion. The mRNA levels of SHH signaling downstream targets, such as c-Myc and Cyclin D1, were also significantly inhibited, which consequently led to cell apoptosis. Previous studies have shown that METTL3 regulated gene expression in two distinct ways: m6A catalytic activity-dependent or -independent. Lin et al showed that EGFR and TAZ genes were regulated by METTL3 in an m6A catalytic activity-independent manner.35 In our study, we found that overexpression of m6A catalytic activity mutant METTL3 construct of METTL3 depletion on GLI1 protein and m6A methylation, which indicated that METTL3 regulates GLI1 expression rely on its catalytic activity (Figure 4). More importantly, the effect of METTL3 on prostate tumor growth was further confirmed by in vivo experiments (Figure 5), which suggested METTL3 as a potent therapeutic target in PC.
Conclusion
We demonstrated for the first time, to the best of our knowledge, that the m6A methyltransferase METTL3 is upregulated in human prostate cancer and contributes to the prostate cancer cells growth and invasion through SHH-GLI1 signaling.
Acknowledgment
This work was supported by grants from Natural Science Foundation of Guangdong Province no. 2018A030313459 (Yun Luo), Science and Technology Planning Project of Guangzhou no. 201610010016 (Yun Luo), Foundation of the 3rd Affiliated Hospital of Sun Yat-sen University (Yun Luo), Science and Technology Planning Project of Guangdong no. 2017A020215027 (Jie Situ), Medical Science and Technology Research Fund Project of Guangdong No. A2019560 (Fei Yang), Medical Science and Technology Research Fund Project of Guangdong No. C2019108 (Jiarong Cai), Higher Education Teaching Reform Project of Guangdong Province, No.82000-18842598 (Jiarong Cai) and Undergraduate Teaching Reform Research Project of Sun Yat-sen University, No.82000-18832601 (Jiarong Cai).
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflict of interest in this work.
References
1. Attard G, Parker C, Eeles RA, et al. Prostate cancer. Lancet. 2016;387(10013):70–82. doi:10.1016/S0140-6736(14)61947-4
2. Hudson SV, O’Malley DM, Miller SM. Achieving optimal delivery of follow-up care for prostate cancer survivors: improving patient outcomes. Patient Relat Outcome Meas. 2015;6:75–90. doi:10.2147/PROM.S49588
3. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71(10):3971–3975. doi:10.1073/pnas.71.10.3971
4. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–1200. doi:10.1016/j.cell.2017.05.045
5. Du H, Zhao Y, He J, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626. doi:10.1038/ncomms12626
6. Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95. doi:10.1038/nchembio.1432
7. Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–342. doi:10.1146/annurev-cellbio-100616-060758
8. Ping X-L, Sun B-F, Wang L. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–189. doi:10.1038/cr.2014.3
9. Schwartz S, Mumbach M, Jovanovic M, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5ʹ sites. Cell Rep. 2014;8(1):284–296. doi:10.1016/j.celrep.2014.05.048
10. Shi H, Wang X, Lu Z, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–328. doi:10.1038/cr.2017.15
11. Xiao W, Adhikari S, Dahal U, et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA splicing. Mol Cell. 2016;61(4):507–519. doi:10.1016/j.molcel.2016.01.012
12. Zhou J, Wan J, Gao X, et al. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526(7574):591–594. doi:10.1038/nature15377
13. Barbieri I, Tzelepis K, Pandolfini L, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552(7683):126–131. doi:10.1038/nature24678
14. Cheng M, Sheng L, Gao Q, et al. The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene. 2019;38(19):3667–3680. doi:10.1038/s41388-019-0683-z
15. Dahal U, Le K, Gupta M. RNA m6A methyltransferase METTL3 regulates invasiveness of melanoma cells by matrix metallopeptidase 2. Melanoma Res. 2019;29(4):382–389. doi:10.1097/CMR.0000000000000580
16. Du M, Zhang Y, Mao Y, et al. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys Res Commun. 2017;482(4):582–589. doi:10.1016/j.bbrc.2016.11.077
17. Miao W, Chen J, Jia L, Ma J, Song D. The m6A methyltransferase METTL3 promotes osteosarcoma progression by regulating the m6A level of LEF1. Biochem Biophys Res Commun. 2019;516(3):719–725. doi:10.1016/j.bbrc.2019.06.128
18. Vu LP, Pickering BF, Cheng Y, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–1376. doi:10.1038/nm.4416
19. Choudhry Z, Rikani AA, Choudhry AM, et al. Sonic hedgehog signalling pathway: a complex network. Ann Neurosci. 2014;21(1):28–31. doi:10.5214/ans.0972.7531
20. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel). 2016;8:2. doi:10.3390/cancers8020022
21. Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126(14):3089–3100.
22. Denef N, Neubuser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell. 2000;102(4):521–531. doi:10.1016/S0092-8674(00)00056-8
23. Chen G, Goto Y, Sakamoto R, et al. GLI1, a crucial mediator of sonic hedgehog signaling in prostate cancer, functions as a negative modulator for androgen receptor. Biochem Biophys Res Commun. 2011;404(3):809–815. doi:10.1016/j.bbrc.2010.12.065
24. Hidalgo M, Maitra A. The hedgehog pathway and pancreatic cancer. N Engl J Med. 2009;361(21):2094–2096. doi:10.1056/NEJMcibr0905857
25. Huang L, Walter V, Hayes DN, Onaitis M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin Cancer Res. 2014;20(6):1566–1575. doi:10.1158/1078-0432.CCR-13-2195
26. Islam SS, Mokhtari RB, Noman AS, et al. Sonic hedgehog (Shh) signaling promotes tumorigenicity and stemness via activation of epithelial-to-mesenchymal transition (EMT) in bladder cancer. Mol Carcinog. 2016;55(5):537–551. doi:10.1002/mc.22300
27. Karhadkar SS, Steven Bova G, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–712. doi:10.1038/nature02962
28. Karlou M, Lu J-F, Wu G, et al. Hedgehog signaling inhibition by the small molecule smoothened inhibitor GDC-0449 in the bone forming prostate cancer xenograft MDA PCa 118b. Prostate. 2012;72(15):1638–1647. doi:10.1002/pros.v72.15
29. Lichinchi G, Gao S, Saletore Y, et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol. 2016;1:16011. doi:10.1038/nmicrobiol.2016.11
30. Lobo J, Barros-Silva D, Henrique R, Jerónimo C. The emerging role of epitranscriptomics in cancer: focus on urological tumors. Genes (Basel). 2018;9(11):13. doi:10.1016/0006-2952(75)90009-x
31. Sanchez P, Clement V, Ruiz i Altaba A. Therapeutic targeting of the Hedgehog-GLI pathway in prostate cancer. Cancer Res. 2005;65(8):2990–2992. doi:10.1158/0008-5472.CAN-05-0439
32. Sanchez P, Hernandez AM, Stecca B, et al. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci U S A. 2004;101(34):12561–12566. doi:10.1073/pnas.0404956101
33. Sheng T, Li C, Zhang X, et al. Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer. 2004;3:29. doi:10.1186/1476-4598-3-29
34. Statkiewicz M, Maryan N, Lipiec A, et al. The role of the SHH gene in prostate cancer cell resistance to paclitaxel. Prostate. 2014;74(11):1142–1152. doi:10.1002/pros.22830
35. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–345. doi:10.1016/j.molcel.2016.03.021
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.