")
Back to Journals » OncoTargets and Therapy » Volume 10
RKIP reduction enhances radioresistance by activating the Shh signaling pathway in non-small-cell lung cancer
Authors Xie SY, Li G , Han C, Yu YY, Li N
Received 15 August 2017
Accepted for publication 20 October 2017
Published 23 November 2017 Volume 2017:10 Pages 5605—5619
DOI https://doi.org/10.2147/OTT.S149200
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Shi-Yang Xie, Guang Li, Chong Han, Yang-Yang Yu, Nan Li
Department of Radiation Oncology, The First Affiliated Hospital of China Medical University, Shenyang, China
Abstract: Non-small-cell lung cancer (NSCLC) is exceptionally deadly because the tumors lack sensitive early-stage diagnostic biomarkers and are resistant to radiation and chemotherapy. Here, we investigated the role and mechanism of Raf kinase inhibitory protein (RKIP) in NSCLC radioresistance. The clinical data showed that the RKIP expression level was generally lower in radioresistant NSCLC tissues than in radiosensitive tissues. Reduced RKIP expression was related to NSCLC radioresistance and poor prognosis. In vitro experiments showed that RKIP knockdown increased radioresistance and metastatic ability in NSCLC cell lines. Mechanistically, RKIP reduction activated the Shh signaling pathway by derepressing Smoothened (Smo) and initiating glioma-associated oncogene-1 (Gli1)-mediated transcription in NSCLC. In addition, the inappropriately activated Shh–Gli1 signaling pathway then enhanced cancer stem cell (CSC) expression in the cell lines. The increasing quantity of CSCs in the tumor ultimately promotes the radiation resistance of NSCLC. Together, these results suggest that RKIP plays a vital role in radiation response and metastasis in NSCLC. RKIP reduction enhances radioresistance by activating the Shh signaling pathway and initiating functional CSCs. This role makes it a promising therapeutic target for improving the efficacy of NSCLC radiation treatment.
Keywords: RKIP, radiosensitivity, Shh signaling pathway, CSCs, NSCLC
Introduction
According to WHO’s International Agency for Research on Cancer criteria, lung cancer is the leading cause of cancer-related morbidity, and non-small-cell lung cancer (NSCLC) accounts for 80%–85% of all lung cancer cases.1 Approximately 2/3 of the patients were diagnosed with an advanced stage of cancer and the main therapy was radiation treatment combined with chemotherapy.2 Among these patients, however, only 10% achieved a complete response, and the total 5-year survival rate remained a dismal 15%.3 Radiation resistance is the major biological factor that affects the efficacy of radiotherapy.4,5
Raf kinase inhibitory protein (RKIP), also known as PEBP-1, was first found in the bovine brain in 1984 and is a member of the phosphatidylethanolamine-binding protein (PEBP) family.6 RKIP is an inhibitory protein in the Raf/mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway and acts via direct interaction with Raf-1 kinase. Therefore, RKIP is a crucial modulator in the MAPK signaling pathway that controls many cellular functions such as cellular proliferation, cell differentiation, cell survival and angiogenesis.7 RKIP also plays an important role in suppressing tumor progression and metastasis through the coordination of intracellular signals such as the phosphoinositide 3-kinase-AKT pathway or the nuclear factor nuclear factor κB pathway.3,8 A substantial amount of previous research evidence suggested that RKIP acts as a metastasis suppressor in multiple types of cancers such as squamous cell lung cancer, breast cancer, melanoma, squamous cell esophageal cancer, colorectal cancer and hepatocellular carcinoma.9,10 In addition to modulating various cellular functions, RKIP also proves to be a good prognostic marker for disease-free survival in cancer patients. Survival curves suggest that low RKIP expression is associated with worse prognosis and easier relapse of the tumor focus.11 In addition to enhancing metastasis and affecting prognosis, RKIP depletion is also closely related to tumor chemotherapy sensitivity and radiosensitivity.12–16 For example, in the study of nasopharyngeal carcinoma, low RKIP expression is significantly correlated with clinical stage, lymph node metastasis and radiosensitivity.17,18 Some researchers believe that the overexpression of RKIP could increase the radiosensitivity of nasopharyngeal carcinoma.11
The sonic hedgehog (Shh) signaling pathway is highly conserved in the human genome and plays an important role in tissue patterning, cell proliferation and differentiation.19 It also contributes to tumorigenesis when it is mutated or misregulated.20,21 When the Shh ligand binds to its transmembrane protein receptor, known as Patched-1 (PTC1), the receptor is inactivated and loses its catalytic inhibition of G-protein-coupled receptors such as the signal transducer Smoothened (Smo). Once activated, Smo will trigger the transcription of the Shh target gene, a zinc finger transcription factor known as glioma-associated oncogene-1 (Gli1).22 Gli1 is a transcriptional activator that induces the expression of numerous target genes. Its targets regulate proliferation, differentiation, extracellular matrix interactions and stem cell activation.23,24 Increasing research evidence has demonstrated that the Shh signaling pathway plays a key role in multiple types of cancers including basal cell carcinoma,25 pancreatic cancer, colon carcinoma, gastric cancer, breast cancer and esophageal cancer.20,26,27 Notably, recent evidence suggests that Hedgehog signaling may contribute to NSCLC.28 Gli1 expression is associated with poor overall survival,29 and the depletion of Gli1 significantly inhibits the growth of stem-like cells from NSCLC.30 Cancer stem cells (CSCs) are a subset of cells within the tumor that have been proposed to be responsible for the initiation and progression of various cancers.30 CSCs from tumor cell lines, tissue samples and xenograft models have been isolated based on different markers such as ALDH1, CD133 positivity and side population.31 CSCs are highly undifferentiated cells that are resistant to both drugs and irradiation. It has become clear that targeting the CSCs in the tumor would be indispensable in curing cancer. Effective therapy has been lacking because of the complexity of CSCs, and a profound understanding of the regulatory mechanism of CSCs is urgently required. The hedgehog pathway has been implicated in the regulation of CSCs in various cancers and is known to increase tumor invasiveness and chemoresistance involving EGFRs.32,33 Our hypothesis, which is consistent with the RKIP-regulated changes in radiosensitivity, is that RKIP silencing inappropriately activates the Shh–Gli1 pathway in the stem cells of NSCLC and that such cells, with almost unlimited replicative potential, would significantly improve tumor resistance to irradiation.
Our research found that RKIP expression was elevated in NSCLC samples that were more sensitive to radiotherapy and that the silencing of RKIP could increase the radioresistance of cancer cells and could promote tumor cell migration and invasion in vitro. Moreover, we found that RKIP expression is related to the Gli1 levels in NSCLC cell lines. Gli1 activation and further functions such as increasing CSC expression were required for the increased radioresistance in RKIP silencing cell lines. RKIP depletion in NSCLC cell lines activates Smo, a crucial regulatory protein in the Shh signaling pathway. To summarize, our research strongly recommends RKIP as a highly promising drug target for increasing radiosensitivity and improving the efficacy of radiotherapy in NSCLC.
Materials and methods
Patients and specimens
Specimens were obtained from 93 NSCLC patients of the radiation oncology department of the First Affiliated Hospital, China Medical University, between 2011 and 2016. The patients were all enrolled with no surgical indications, and the Karnofsky Performance Status scores were above 70. Each of the patients underwent a spiral contrast-enhanced computed tomography (CT) simulation before the treatment began, and radical radiotherapy was administered at 2 Gy per fraction per day, 5 days per week, for a total dose of 60 Gy. When the therapeutic dose reached 40 Gy (20 times), the patient underwent another CT scan, which was combined with the initial one on the treatment planning system to compare the tumor volume and evaluate the radiation response. If the reduction rate of the tumor volume was <30%, then the tumor was considered low sensitivity; 30%–60% was considered moderate sensitivity; and >60% was considered high sensitivity. All the patients who received radiotherapy underwent follow-up for a maximum period of 64 months and a median period of 14 months. The overall survival was defined as the interval from the initiation of primary radiotherapy to the date of cancer-related death or to the latest date recorded if the patient was still alive.
Immunohistochemistry
Immunohistochemical staining of RKIP was performed on formalin-fixed and paraffin-embedded tissue sections. Antigen retrieval tissue sections were incubated with 1:400 dilutions of anti-RKIP (ab76582, Abcam, Cambridge, MA, USA) overnight at 4°C and then incubated with biotinylated secondary antibody followed by streptomycin biotin peroxidase solution. Finally, the sections were stained with diaminobenzidine and counterstained with hematoxylin. Primary antibodies were omitted in the negative controls.
Immunohistochemical staining was examined under high magnification (400×). For each slice, 5 fields of view were selected randomly, and 200 cells were counted in each field of view. Cells with brownish yellow particles in the cytoplasm were considered positive. The percentage of stained cells was categorized as no positive cells =0, <25% positive cells =1, 26%–50% positive cells =2, 51%–75% positive cells =3, and >75% positive cells =4. Staining intensity was categorized as absent staining =0, light yellow =1, brownish yellow =2, and brown =3. The staining score (ranging from 0 to 7) for each tissue was calculated by adding the quantity score and the intensity score. A combined staining score of 0–2 was considered low expression (−), 3–5 was moderate expression (+), and 6–7 was high expression (++). A score of >3 was considered positive.
Ethics approval and consent to participate
This study was approved by the ethics committee of China Medical University. Written informed consent was obtained from all participants in the study.
Cell lines and cell culture
The human lung adenocarcinoma cell line A549 and the human squamous cell lung carcinoma cell line SK-MES-1 were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai Institute of Cell Biology, Chinese Academy of Sciences. These two cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (HyClone, LA, USA) or minimum essential medium (HyClone, LA, USA) supplemented with 10% fetal bovine serum (FBS) (Clark, USA) in a humidified incubator containing 5% CO2.
Construction of shRNA expression lentivirus
For RNA interference experiments, four shRNA targets of RKIP were designed and separately cloned into the pGFP-puro-RKIP lentiviral vector. The infectious lentivirus was produced by transfecting the lentivector and packaging vectors into 293T cells. The puromycin resistance gene (pac) and luciferase full-length cDNA were cloned into the expressing construct and used for the lentivirus package. In addition, a negative control plasmid with scrambled shRNA and a positive pac shRNA plasmid were constructed. All shRNA plasmids were synthesized by GenePharma (Shanghai, China).
Transfection and isolation of stable cell clones
To generate NSCLC cell lines with RKIP KD, lentiviral pGFP271-puro-RKIP shRNA and control lentiviral pGLV3-puro-control shRNA were used to infect A549 and SK-MES-1 cell lines, respectively, according to the manufacturer’s instructions. The efficiency of transfection was assessed by fluorescence microscopy. Cells were selected using puromycin for 2 weeks, and NSCLC cell lines with stable KD of RKIP and control cell lines with empty vector were obtained.
Western blot
Proteins were exacted from cells and tissues using RIPA buffer supplemented with the protease inhibitor PMSF. The protein concentrations were measured using the bicinchoninic acid Protein Assay Kit (Beyotime, China). An equal amount of protein in each sample was separated on a sodium dodecyl sulfate–polyacrylamide gel and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat dry milk in PBS for 1 h at room temperature and incubated with primary antibodies at 4°C overnight. After incubation with a secondary antibody, the blots were visualized using enhanced chemiluminescent substrate detection reagent (Beyotime, China). Anti-RKIP, anti-SMO, anti-Gli1 and anti-CD133 (1:1,000) were purchased from Abcam (Cambridge, UK). As a loading control, the expression of β-actin was detected using monoclonal mouse anti-β-actin antibody (1:5,000) from Santa Cruz Biotechnology (CA, USA) according to the manufacturer’s instructions.
Irradiation conditions
The NSCLC cells were irradiated using a 6-MV X-ray linear accelerator (model: Varian Medical System, CA, USA) at a dose rate of 300 cGy/min. The radiation doses were 0, 2, 4, 6, 8, 12 and 16 Gy. Cells were placed in the incubator, and samples were collected at the indicated time points (0, 1, 12 and 24 h).
Cell proliferation assay
The cell viability of the RKIP-KD A549 and SK-MES-1 cells was determined by the MTT assay. Cells (2,000 cells per well) were seeded in 96-well plates. After treatment with different radiation doses (0, 2, 4, 6 and 8 Gy), the cells were incubated for another 24 h. Each radiation dose was applied to three wells of cells. The cell viability assay was performed using the MTT Cell Proliferation and Cytotoxicity Assay Kit (Beyotime, China) according to the manufacturer’s instructions. First, 10 μL of MTT (5 mg/mL) was added to each well. The cells were incubated at 37°C for an additional 4 h. At the end of the incubation, 100 μL of formazan solution was added to each well. The absorbance of each well was monitored by a spectrophotometer at 570 nm (A570). The experiments were performed in triplicate.
Colony formation assay
Cells were seeded in triplicate in six-well plates at a density of 1×103 cells per well. After overnight attachment, the plates were exposed to 0, 2, 4, 6 or 8 Gy of irradiation. After incubation for 12 days, the colonies were washed twice with PBS, fixed with methanol and stained with 0.5% crystal violet. The colonies containing more than 50 cells were counted as surviving clones. The experiments were performed in triplicate.
TUNEL assays
The TUNEL method was used to label the apoptotic cells using a TUNEL Apoptosis Detection Kit (US Everbright, Suzhou, China) according to the manufacturer’s protocol. After treatment with 0 and 4 Gy of irradiation, cells were incubated at 37°C for 24 h in six-well plates. The cells were washed twice with PBS and fixed in 1 mL of 4% paraformaldehyde for 10 min at 4°C, then permeabilized with 0.1% Triton X-100 at 25°C for 5 min. The cells were washed twice with PBS and then stained with the TUNEL mixture for 1 h at 37°C in the dark, stained with streptavidin-TRITC for 30 min at 37°C in the dark, and finally stained with 4′,6-diamidino-2-phenylindole for 10 min. The cells were washed with PBS. Afterward, the cells were counted and examined under a fluorescence microscope (Olympus IX71, Tokyo, Japan). Apoptotic cells were identified by the karyopyknosis and karyolysis of the nucleus. The apoptotic ratio was obtained by the following calculation: apoptotic ratio = apoptotic cell number/seeded cell number ×100%.
Apoptosis analysis
Apoptosis was determined by double staining with Annexin V-PE and 7AAD according to the manufacturer’s instructions (Keygene, China). After treatment with 0, 4 or 8 Gy of irradiation, cells were incubated at 37°C for 24 h, harvested, suspended in 500 μL of binding buffer and labeled with Annexin V-PE/7AAD reagent for 15 min in the dark at room temperature. The stained cells were immediately analyzed by FACScan flow cytometry. All of the samples were assayed in triplicate, and the cell apoptosis rate was calculated as follows: apoptosis rate = (apoptotic cell number/total cell number) ×100%.
Cell cycle analysis
After treatment with 4 Gy irradiation, the cells were incubated for an additional 24 h, harvested and resuspended in 500 μL of cell cycle rapid detection solution (DAKEWE, China) at room temperature. Samples were immediately analyzed by FACScan flow cytometry. The distribution of cell cycle phases was determined using FlowJo software. All of the samples were assayed in triplicate, and the fraction of every cell cycle phase was calculated.
Wound-healing assay
Cells were seeded in six-well plates and grown to confluence. The plates were washed twice with PBS, and serum-free medium was added to the plates. Three parallel “wounding” lines were scratched into the cell monolayer with a separate sterile 1,000-μL pipette tip used for each plate. The width of the wound area was photographed and measured under the inverted phase contrast microscope (40× magnification, Nikon, Japan) to assess cell migration at 0, 6, 12, 24 and 48 h after scratching.
Cell migration and invasion assay
For Transwell migration assays, 200 μL containing 2×104 infected cells was plated into each top chamber with an 8-mm pore filter insert (Corning, NY, USA). For invasion assays, 200 μL containing 2×104 cells was plated into each top chamber with the insert covered with Matrigel. In both assays, cells were plated in medium without serum, and 500 μL of serum-containing medium (20% FBS) was used in the lower chamber as a chemoattractant. After 48 h of incubation, the cells on the upper surface of the filter were removed using a cotton swab, and cells that had invaded through the bottom surface of the filter were fixed with methanol, stained with crystal violet, and imaged and counted in 10 randomly selected viewing fields under the microscope (100× magnification).
Other drugs used
Commercial N-Shh (R&D Systems, MN, USA) was used at 50 nM. Cyclopamine (Selleck, Houston, TX, USA) was used at 8 μM. Treated cells were incubated for an additional 24 h before further experiments.
Statistical analysis
Statistical analysis was performed using GraphPad Prism and the SPSS 20.0 software, and the data were represented as the mean ± SD from at least three independent experiments. The data were tested for normal distribution. Student’s t-test was used for comparisons between groups and one-way analysis of variance (ANOVA) used for comparisons in the same group at different doses or time points. The Spearman’s rank correlation coefficient was used to determine the correlation between two parameters. P-values <0.05 were considered to be statistically significant. The choices of methods are listed below.
The Western blot results used Student’s t-test to make comparisons between different cell lines and one-way ANOVA to test for significant differences between different drug treatments in the same cell line.
The cell proliferation assay and the colony formation assay used Student’s t-test to make comparisons between different cell lines and one-way ANOVA to test for significant differences between different doses in the same cell line.
The apoptosis assay used Student’s t-test to make comparisons between different cell lines and one-way ANOVA to test for significance differences between different doses in the same cell line. The cell cycle analysis used Student’s t-test to compare the differences in the G2-M phase changes between different cell lines.
The cell migration and invasion assay used Student’s t-test to compare the results in different cell lines. The wound-healing assays used Student’s t-test to compare the results in different cell lines and one-way ANOVA to test for significance in the same cell line at different time points.
Results
RKIP reduction is associated with NSCLC response to irradiation and poor prognosis
Immunohistochemistry was performed in 44 radiosensitive NSCLC tissues and 49 radioresistant ones. We observed that RKIP expression was significantly reduced in the radioresistant NSCLC tissues compared with that in the radiosensitive ones (Figure 1A). The relationships between RKIP expression level and clinicopathological features are summarized in Table 1. As shown in the table, the expression levels of RKIP were positively correlated with NSCLC radiosensitivity (r=0.378, P<0.001), and tumors with high RKIP expression were frequently radiosensitive. In addition, the expression level of RKIP was related to the T stage of the tumor. As radioresistance is a major cause of the poor prognosis of NSCLC patients, we observed the ability of RKIP to predict overall survival in clinical practice. Kaplan–Meier analysis revealed that lower RKIP expression in NSCLC tissues is significantly correlated with clearly decreased overall survival (Figure 1B). A univariate Cox proportional hazards regression analysis indicated that both clinical T stage and RKIP expression level significantly affected the overall survival of NSCLC patients (Table 2). A multivariate Cox proportional hazards regression analysis has confirmed that a low expression level of RKIP is an independent predictor for decreased patient overall survival (Table 2). Taken together, the result indicates the importance of the RKIP expression level in reflecting NSCLC radiosensitivity and prognosis.
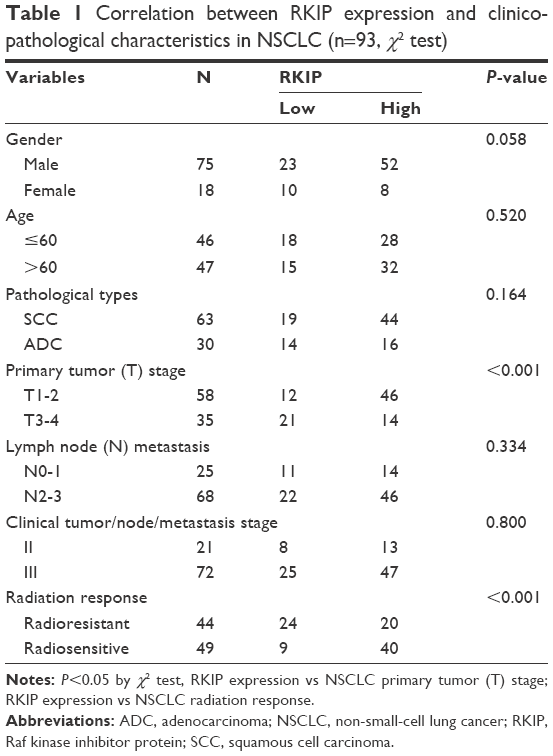 | Table 1 Correlation between RKIP expression and clinicopathological characteristics in NSCLC (n=93, χ2 test) |
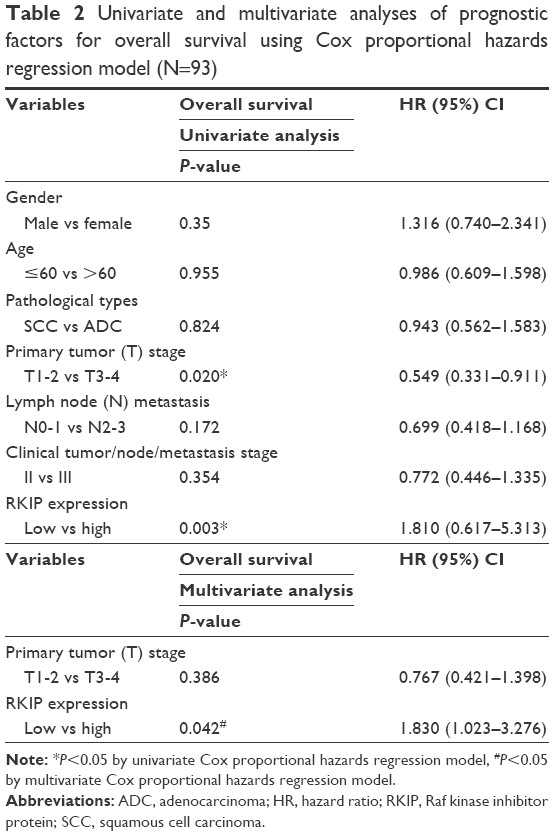 | Table 2 Univariate and multivariate analyses of prognostic factors for overall survival using Cox proportional hazards regression model (N=93) |
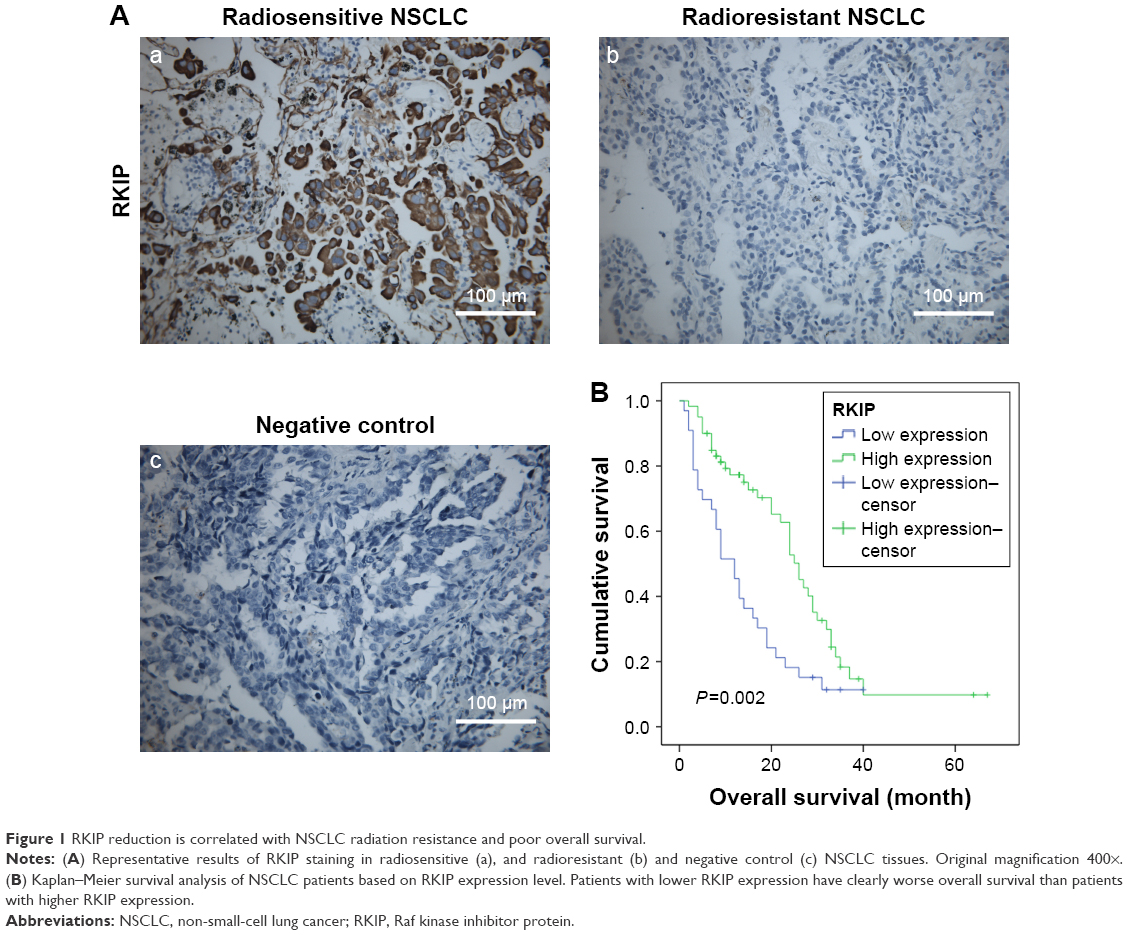 | Figure 1 RKIP reduction is correlated with NSCLC radiation resistance and poor overall survival. |
RKIP reduction increases the radioresistance of NSCLC cell lines in vitro
To explore the effect of RKIP reduction on the radioresistance of NSCLC cells, we regulated RKIP expression by stably transfecting an antisense RKIP short hairpin RNA (shRNA) expression lentivirus into the human lung adenocarcinoma cell line A549 and the squamous cell carcinoma cell line SK-MES-1. We thereby obtained two specific cell lines with low expression of RKIP (A/RKIP-knockdown [KD] and SK/RKIP-KD) and their corresponding control cell lines (A-EV and SK-EV). Western blotting was used to verify the effects of the transfection (Figure 2A), and the expression levels of RKIP in A/RKIP-KD and SK/RKIP-KD were significantly lower than in the corresponding control lines (Figure 2B) (P<0.05). The radiosensitivity of the cell lines was also detected. The surviving fraction of NSCLC cell lines after irradiation is shown in Figure 3A. The radiobiological parameters of A-EV were D0=2.161, Dq=0.533, SF2=0.476, k=0.463, and N=1.280, while those of A/RKIP-KD were D0=2.433, Dq=2.656, SF2=0.822, k=0.411, and N=2.979. Meanwhile, the radiobiological parameters of SK-EV were D0=2.588, Dq=0.015, SF2=0.464, k=0.386, and N=1.006, while those of SK/RKIP-KD were D0=2.108, Dq=3.395, SF2=0.914, k=0.474, and N=5.005. The results showed that NSCLC cell lines with downregulated RKIP expression had a higher survival fraction at 2 Gy irradiation and an obvious increase in quasi-threshold dose compared to RKIP-positive NSCLC cell lines (P<0.05). Furthermore, a methylthiazol tetrazolium (MTT) assay was performed to determine the impact of RKIP depletion on the viability of NSCLC cells under irradiation. As shown in the data (Figure 3B), the cell viability rate was clearly higher in cell lines with low RKIP expression subjected to varying degrees of radiotherapy (P<0.05). Taken together, the results revealed that RKIP reduction enhances NSCLC radioresistance in vitro.
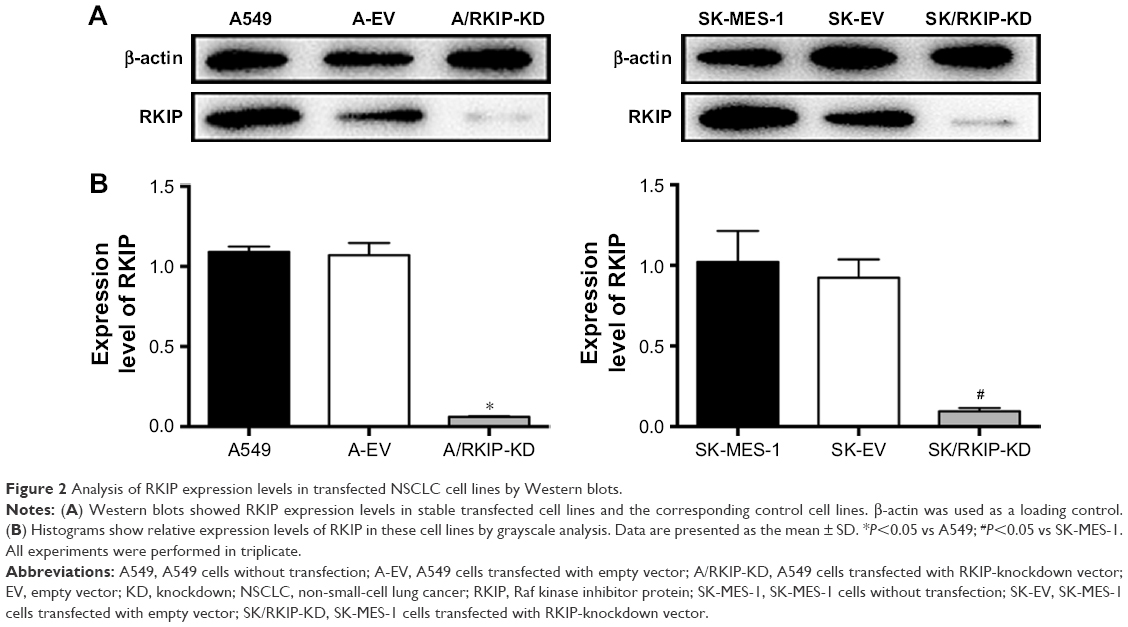 | Figure 2 Analysis of RKIP expression levels in transfected NSCLC cell lines by Western blots. |
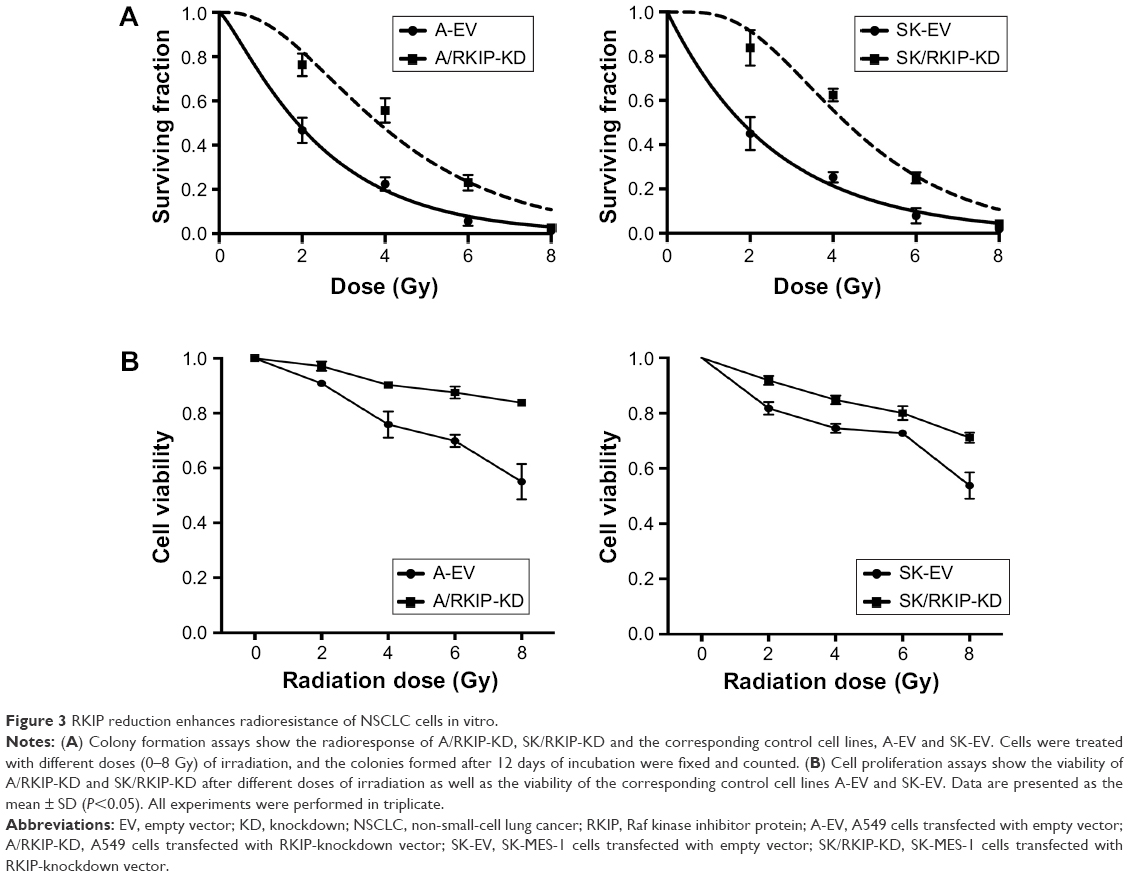 | Figure 3 RKIP reduction enhances radioresistance of NSCLC cells in vitro. |
RKIP reduction enhances the migration and invasion of NSCLC cells
Metastatic progression can accelerate in cancer cells during treatment with drugs or irradiation,34 and thus metastasis in NSCLC can seriously affect the efficiency of radiotherapy. To explore how RKIP reduction could affect the metastatic behaviors of NSCLC cell lines, Transwell chambers were applied to test the migration and invasion abilities of the cancer cells. As shown in the data, cell lines with low RKIP expression have stronger migration and invasion abilities (Figure 4A and B) (P<0.05). Furthermore, a wound-healing assay was conducted to determine the influence of RKIP reduction on the metastatic ability of cancer cells. The results revealed that reduced RKIP expression clearly increased the metastatic abilities of NSCLC cells (Figure 4C). Overall, RKIP reduction enhanced the migration and invasion of NSCLC cell lines, which means that NSCLC cells with low RKIP expression will exhibit a less satisfactory response to radiotherapy.
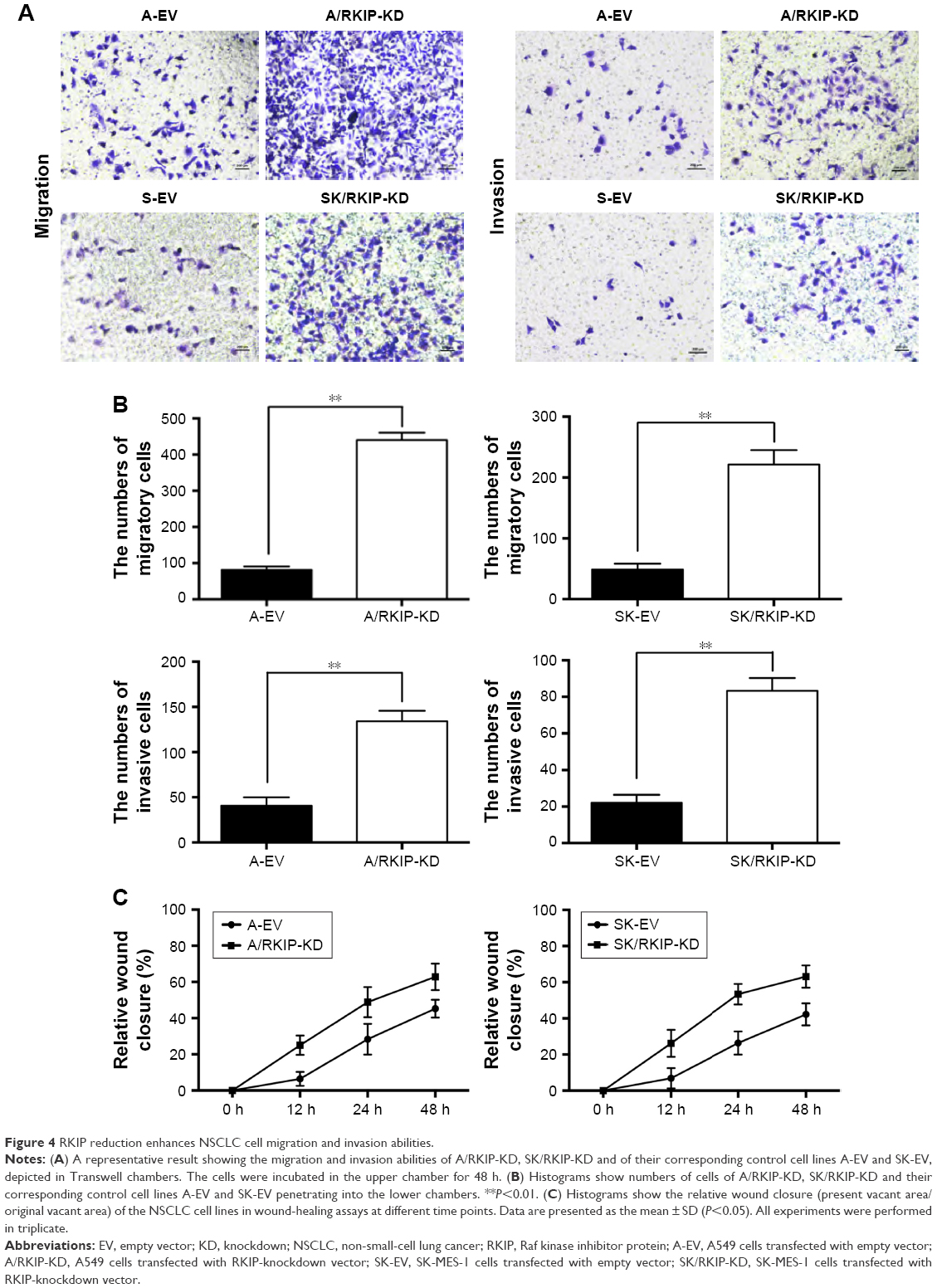 | Figure 4 RKIP reduction enhances NSCLC cell migration and invasion abilities. |
RKIP reduction decreases radiotherapy-mediated apoptosis
Radiation-mediated apoptosis, to some extent, could be considered an aspect of radiosensitivity.35 Therefore, we measured the radiation-mediated apoptotic rates of NSCLC cell lines with different expression levels of RKIP using both flow cytometry and the TUNEL assay system. The flow cytometry data showed significantly lower radiation-induced apoptosis in the A/RKIP-KD and SK/RKIP-KD cell lines than in the corresponding control cell lines (A-EV and SK-EV) under different doses of irradiation (Figure 5A). The TUNEL assay results were consistent with the flow cytometry data. Fewer apoptotic cells were observed in RKIP-KD cell lines under 4 Gy irradiation (Figure 5C). These experiments showed that reduced RKIP expression could protect NSCLC cells from apoptosis induced by irradiation, thus promoting the radioresistance of NSCLC cells in vitro.
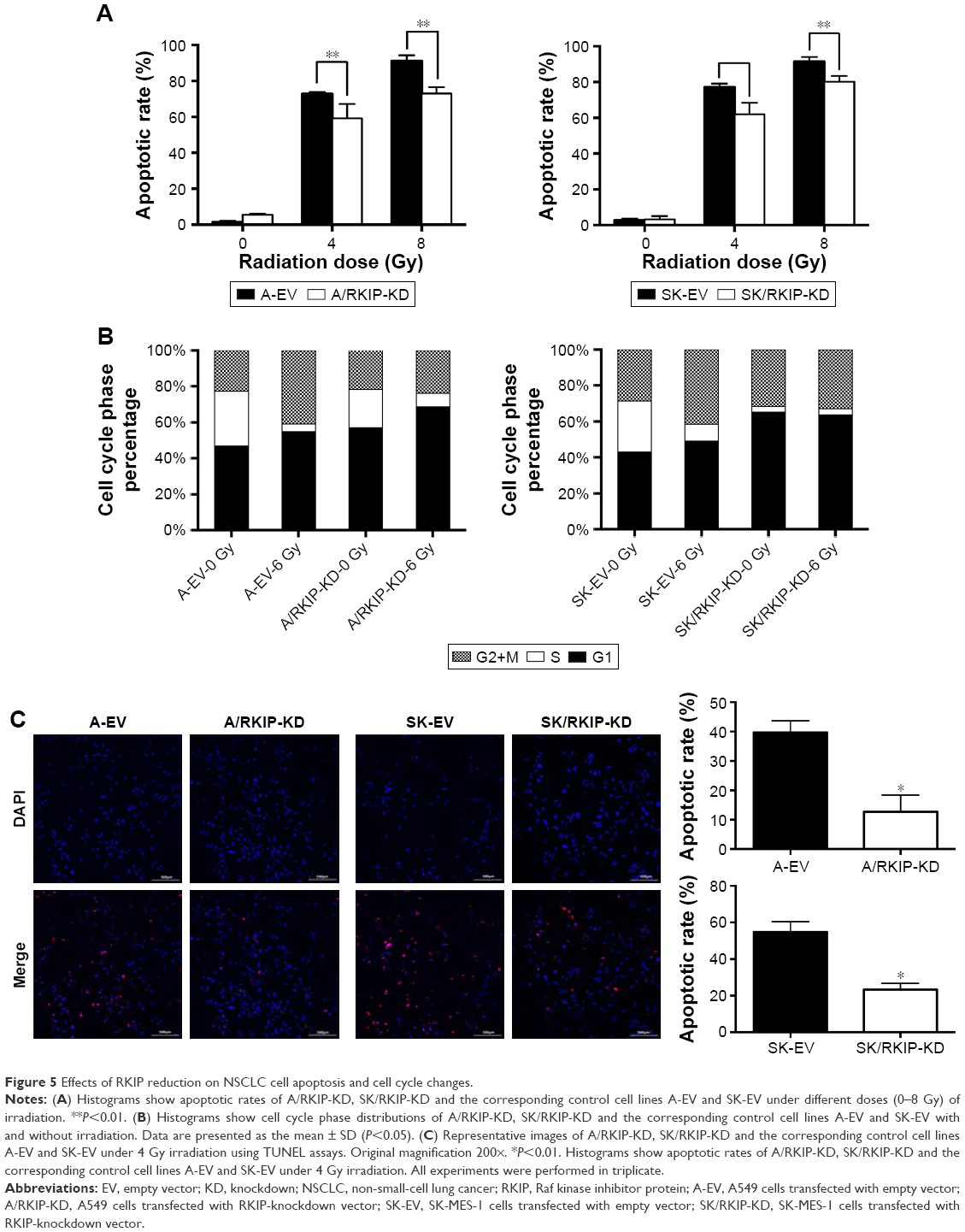 | Figure 5 Effects of RKIP reduction on NSCLC cell apoptosis and cell cycle changes. |
RKIP reduction enhances radioresistance by affecting cell cycle progression
The cell cycle distributions of four cell lines were analyzed by flow cytometry. As shown in Figure 5B, compared with the A-EV and SK-EV cell lines with high RKIP expression, distribution trends such as the cell cycle arrest of the G2-M phase and the G1 phase depletion caused by irradiation were reversed in the cell lines with reduced RKIP expression. In conclusion, RKIP reduction could promote the radioresistance of NSCLC by preventing cancer cells from entering the G2-M phase, which is relatively sensitive to irradiation.
RKIP reduction enhances the radioresistance of NSCLC cell lines through activation of the Shh–Gli1–CD133 signaling pathway
To explore the signaling mechanism of RKIP in NSCLC radioresistance, we examined whether the RKIP-regulated variation in the radiosensitivity of NSCLC cells is mediated through the Shh–Gli1–CD133 signaling pathway. The Western blot results showed that the expression levels of Smo and Gli1 in the RKIP-KD cell lines A/RKIP-KD and SK/RKIP-KD were clearly higher than in their corresponding control cell lines as well as that of CD133, which indicates that the number of CSCs was elevated in these cell lines (Figure 6A). Moreover, to thoroughly understand the pattern of RKIP and Shh pathway interaction, we introduced the agonist cytokine Recombinant Human Sonic Hedgehog (N-shh) and the antagonist cyclopamine of the Shh signaling pathway into the experiments. When the Shh signaling pathway status was altered by N-shh and cyclopamine, the RKIP expression level of the cell lines did not change correspondingly, which indicated that RKIP occupied an upstream position in the pathway (Figure 6A). Furthermore, when N-shh was added to SK-EV and A-EV, SMO, Gli1 and CD133 all increased, as they did in RKIP-KD cell lines A/RKIP-KD and SK/RKIP-KD (Figure 6A). This result shows that RKIP reduction could activate the Shh–Gli1–CD133 signaling pathway just as N-shh did. Similarly, when cyclopamine was added to the RKIP-KD cell lines A/RKIP-KD and SK/RKIP-KD, the activated Shh–Gli1–CD133 pathway went silent again, as in the RKIP-positive cell lines A-EV and SK-EV (Figure 6A). All these Western blot results were verified multiple times and are displayed in a histogram (Figure 6B) (P<0.05). These results indicated that RKIP was most likely functioning in the same way as cyclopamine, interacting with SMO and keeping it inactivated in the cytoplasm.36 Taken together, these data suggested that RKIP regulates the radiation sensitivity by activating the Shh–Gli1 signaling pathway and increasing the number of CSCs among NSCLC cells.
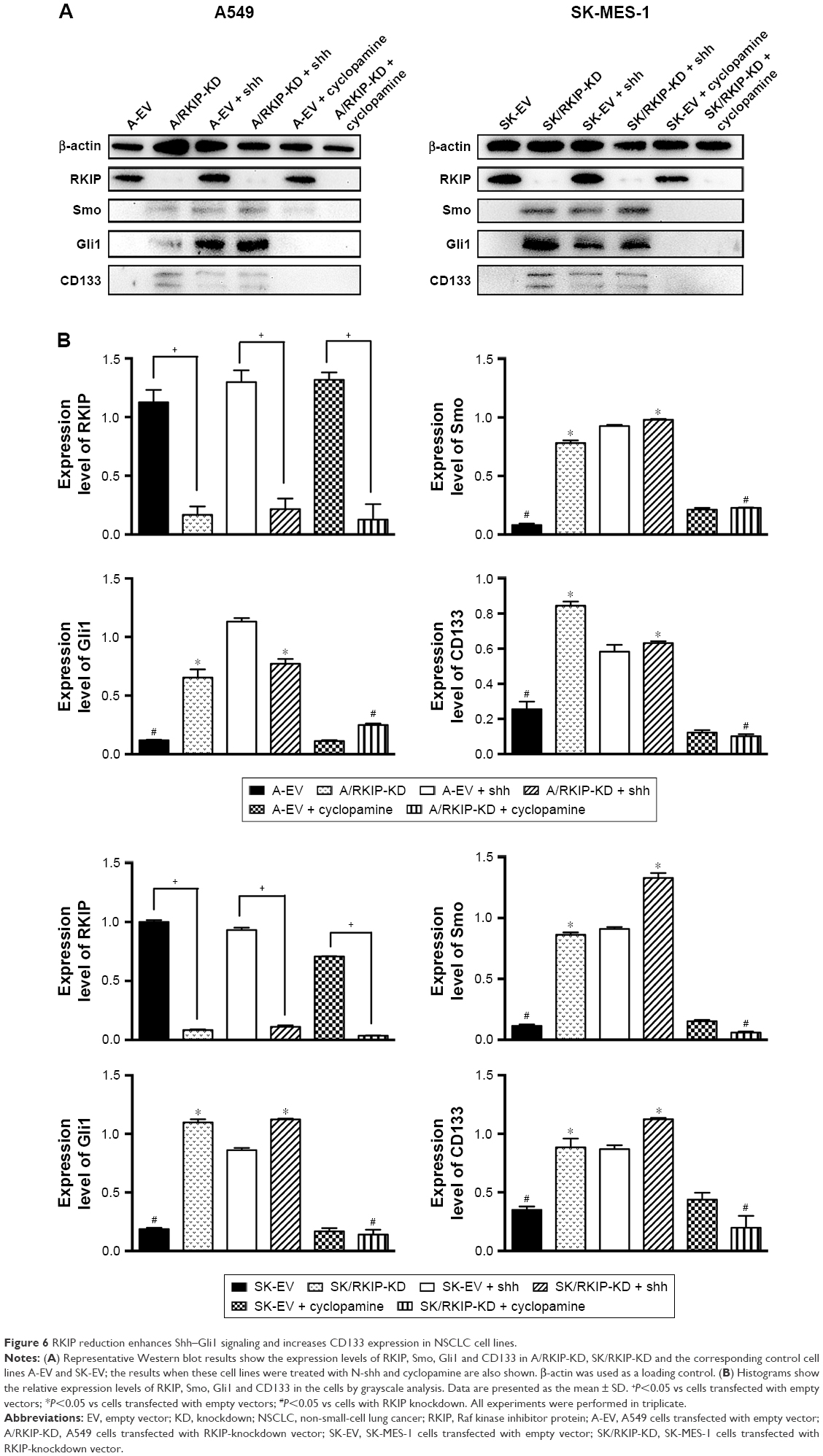 | Figure 6 RKIP reduction enhances Shh–Gli1 signaling and increases CD133 expression in NSCLC cell lines. |
Discussion
During the study, we discovered that RKIP depletion activated the Shh signaling pathway, significantly increased the production of Gli1 and subsequently enhanced the radioresistance of NSCLC cells. Our experiments suggested that RKIP is a prominent negative regulator of the Shh signaling pathway, which plays a vital role in cancer tumorigenesis, metastasis and chemo/radiosensitivity.37 RKIP has been massively reported to be dysregulated in several types of cancers, such as prostate, cervical, colonic, nasopharyngeal and gastric cancers.38 The RKIP expression level is ~50% lower in prostate carcinoma in situ than in nonmalignant cells.38 The loss of RKIP is also relevant to clinical stage, pathological differentiation and highly malignant behaviors in NSCLC.39 Despite all the links between RKIP depletion and enhanced resistance of cancers in the clinical background, the molecular mechanism of how the RKIP expression level results in enhanced radioresistance, poor prognosis and malignant progression in cancers still remains to be found.
RKIP was initially identified as PEBP in the bovine brain and was later identified as an inhibitory protein in the Raf–MEK–ERK signaling pathway via its interaction with Raf1 kinase and renamed RKIP by Granovsky and Rosner.40 In recent years, RKIP has been identified as a member of a novel class of molecules that suppress the metastatic spread of tumors. In addition, RKIP is now well established to be involved in MAPK signal transduction. This signaling machinery evolved to rapidly activate nuclear transcription factors in response to extracellular stimuli and can influence diverse cellular functions including cell proliferation, differentiation and apoptosis. As signaling proteins, especially those from the MAP kinase superfamily, are often linked with cancer and as RKIP is a well-known inhibitor of the MAPK pathway, the links between molecules such as ERK, AKT and PKC and the expression of RKIP has been extensively studied. Reduced levels of RKIP enhance ERK and AKT activity and increase the survival ability of tumor cells in nasopharyngeal carcinoma.41 RKIP also inhibits the invasion and metastasis of hepatocellular carcinoma by inhibiting the ERK/MAPK signaling pathway.42 Based on previous studies, we concluded that the interaction of RKIP with the MAPK signaling pathway can affect the survival of multiple kinds of tumor cells. In addition to its suppressive role in cancer metastasis and tumorigenesis, RKIP expression is also related to the sensitivity of tumors to chemotherapy and radiotherapy.14,16 Some studies have found that RKIP overexpression could increase the sensitivity of breast cancer cells to chemotherapy.15 In our experiments, we observed that RKIP depletion correlates with changes in radiation-induced apoptosis, migration and invasion ability and the cell cycle in NSCLC cell lines. Furthermore, we found that the malignant biological behaviors and enhanced radioresistance caused by RKIP depletion were induced through activation of the Shh signaling pathway. A substantial amount of evidence suggests that the Shh pathway is involved in NSCLC, but the molecular mechanism remains unclear.43 Gli1 is a vital target gene of the Shh signaling pathway, and the expression level of Gli1 is a significant indicator of Shh activity. We observed that RKIP depletion could promote the expression of Gli1 in NSCLC cell lines and that the increased malignant behaviors of the tumors were associated with low expression levels of RKIP, which activated the Shh signaling pathway. When the Shh ligand binds to PTC1, Smo moves from the cytoplasm and changes into an active form on the cell membrane.44 The activated Smo then triggers the transcriptional function of Gli1.45 Our present data indicate that RKIP inhibits the Shh signaling pathway through interacting with Smo, preventing Smo from reaching the cell membrane to perform its function in radiation-sensitive cancer cells. Conversely, in radiation-resistant NSCLC cells, RKIP depletion decreases its inhibition of cytoplasmic Smo and promotes the effect of Smo on Gli1, which fully activates the Shh signaling pathway.
Our analysis has established an oncogenic role for the depletion of RKIP and identified the activated Shh signaling pathway as the mechanism of the resulting enhanced radiation resistance in NSCLC cells. However, the question remains of how the activated Shh signaling pathway increases the radiation resistance of cancer cells. We suspect that the CSC theory is the answer. CSCs were initially defined as cells with self-renewal ability that are capable of producing heterogeneous tumor cells.46 Recent studies have shown that CSCs might be responsible for tumorigenesis, invasion and distant metastasis.47 In addition to the malignant behaviors above, CSCs display particular resistance to drugs and irradiation, leading to tumor suppression failure and relapse.48,49 It is well known that many cancers are resistant to radiotherapy, and the existence of CSCs within hypoxic niches is clearly one possible cause of lower levels of reactive oxygen species and enhanced free radical scavenging.33 Moreover, the Shh–Gli1 pathway is involved in several types of tumors50 and plays a vital role in CSC differentiation.51,52 Direct effects of inhibiting the Shh pathway on CSCs have been observed. For instance, CML stem cells resistant to tyrosine kinase inhibitors were efficiently eliminated by combined treatment with cyclopamine and imatinib.53 Vismodegib is also effective against CD133+ pancreatic CSCs and operates by enhancing apoptosis.54 The above evidence led us to propose that the Shh pathway activated by RKIP depletion enhances NSCLC radioresistance through increasing the ratio of CSCs in the tumor cell population. To test this theory, human CD133+ (AC133+) was used as a specific marker to identify cells highly enriched in CSCs.55,56 CD133+ CSCs have been proved to be radioresistant because of the upregulation of the DNA damage response in glioblastomas.57 In addition, we discovered that the CSC biological marker CD133 was positive in RKIP-deficient NSCLC cell lines with the Shh–Gli1 pathway activated. Overall, we strongly believe that RKIP depletion activates the Shh–Gli1 pathway and then increases the quantity of CSCs in the tumor niche, thereby enhancing the radiation resistance of the NSCLC cell lines.
In the near future, our further studies may focus on in vivo experiments on nude mice. Experiments with xenograft mouse models have provided striking evidence that tumor radioresistance is coupled with RKIP expression and that RKIP expression affects tumorigenesis. Metastasis has been shown in xenograft mouse models for multiple types of cancers. RKIP-inhibited cells revealed a significant increase in blood vessel recruitment in cervical cancer.58 In NSCLC, nude mice loaded with RKIP-overexpressing A549 cells through veil injection showed inhibition of both tumor growth and metastasis.59 RKIP overexpression also enhanced metastatic ability in colorectal cancer60 and increased tumor volume in pancreatic cancer.61 From the evidence above, we can see a strong connection between RKIP expression and malignant tumor behaviors in vivo. We plan to explore the connection between NSCLC radioresistance and RKIP expression using tumor-bearing nude mice in our subsequent studies. Furthermore, the theory that Shh–Gli1 signaling and CSCs are involved in RKIP expression-regulated radioresistance will also be tested in the xenograft mouse models. We hope that the planned experiments on xenograft mouse models will support our proposed mechanism for RKIP expression-regulated radioresistance in NSCLC.
Conclusion
These studies indicate an oncogenic role of RKIP depletion in NSCLC. In addition to its well-known character as a potential biomarker for metastasis and prognosis, RKIP is a promising drug target for improving the radiosensitivity of NSCLC in clinical practice by affecting the Shh–Gli1 pathway and CSCs in cancer cells.
Acknowledgments
Funding from the Department of Science and Technology of Shenyang City is gratefully acknowledged. This study was supported by funding from the Department of Science and Technology of Shenyang City (No F13-316-1-70).
Author contributions
GL and SYX conceived and designed the study. SYX, CH and YYY performed the experiments. NL provided the pathological sections. SYX wrote the paper. GL reviewed and edited the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Pore MM, Hiltermann TJ, Kruyt FA. Targeting apoptosis pathways in lung cancer. Cancer Lett. 2013;332(2):359–368. | ||
Kodym E, Kodym R, Reis AE, Habib AA, Story MD, Saha D. The small-molecule CDK inhibitor, SNS-032, enhances cellular radiosensitivity in quiescent and hypoxic non-small cell lung cancer cells. Lung Cancer. 2009;66(1):37–47. | ||
Schuurbiers OC, Kaanders JH, van der Heijden HF, Dekhuijzen RP, Oyen WJ, Bussink J. The PI3-K/AKT-pathway and radiation resistance mechanisms in non-small cell lung cancer. J Thorac Oncol. 2009;4(6):761–767. | ||
MA K. NF-κB mediated adaptive resistance to ionizing radiation. FREE Radic Biol Med. 2008;44(1):1–13. | ||
Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59(4):928–942. | ||
Bernier I, Jolles P. Purification and characterization of a basic 23 kDa cytosolic protein from bovine brain. Biochim Biophys Acta. 1984;790(2):174–181. | ||
Lamiman K, Keller JM, Mizokami A, Zhang J, Keller ET. Survey of Raf kinase inhibitor protein (RKIP) in multiple cancer types. Crit Rev Oncog. 2014;19(6):455–468. | ||
Yeung KC, Rose DW, Dhillon AS, et al. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol. 2001;21(21):7207–7217. | ||
Sara HYNK, Angeles HC. Expression of phosphorylated raf kinase inhibitor protein (pRKIP) is a predictor of lung cancer survival. BMC Cancer. 2011;11(1):259. | ||
Gao C, Pang L, Ren C, Ma T. Prognostic value of raf kinase inhibitor protein in esophageal squamous cell carcinoma. Pathol Oncol Res. 2012;18(2):471–477. | ||
Ruan L, Wang GL, Yi H, et al. Raf kinase inhibitor protein correlates with sensitivity of nasopharyngeal carcinoma to radiotherapy. J Cell Biochem. 2010;110(4):975–981. | ||
Baritaki S, Bonavida B. Viral infection and cancer: the NF-kappaB/Snail/RKIP loop regulates target cell sensitivity to apoptosis by cytotoxic lymphocytes. Crit Rev Immunol. 2010;30(1):31–46. | ||
Baritaki S, Katsman A, Chatterjee D, Yeung KC, Spandidos DA, Bonavida B. Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J Immunol. 2007;179(8):5441–5453. | ||
Baritaki S, Yeung K, Palladino M, Berenson J, Bonavida B. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res. 2009;69(21):8376–8385. | ||
Chatterjee D, Bai Y, Wang Z, et al. RKIP sensitizes prostate and breast cancer cells to drug-induced apoptosis. J Biol Chem. 2004;279(17):17515–17523. | ||
Woods Ignatoski KM, Grewal NK, Markwart SM, et al. Loss of Raf kinase inhibitory protein induces radioresistance in prostate cancer. Int J Radiat Oncol Biol Phys. 2008;72(1):153–160. | ||
He QY, Yi HM, Yi H, et al. Reduction of RKIP expression promotes nasopharyngeal carcinoma invasion and metastasis by activating Stat3 signaling. Oncotarget. 2015;6(18):16422–16436. | ||
Li Y, Zhang H, Zhu X, et al. Oncolytic adenovirus-mediated short hairpin RNA targeting MYCN gene induces apoptosis by upregulating RKIP in neuroblastoma. Tumour Biol. 2015;36(8):6037–6043. | ||
Bible KC, Suman VJ, Molina JR, et al. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 2010;11(10):962–972. | ||
Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22(18):2454–2472. | ||
Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet. 2000;67(5):1047–1054. | ||
Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30(6):303–312. | ||
Cohen MM Jr. The hedgehog signaling network. Am J Med Genet A. 2003;123A(1):5–28. | ||
Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–3087. | ||
Kump E, Ji J, Wernli M, Hausermann P, Erb P. Gli2 upregulates cFlip and renders basal cell carcinoma cells resistant to death ligand-mediated apoptosis. Oncogene. 2008;27(27):3856–3864. | ||
ten Haaf A, Bektas N, von Serenyi S, et al. Expression of the glioma-associated oncogene homolog (GLI) 1 in human breast cancer is associated with unfavourable overall survival. BMC Cancer. 2009;9(1):298. | ||
Yoshikawa K, Shimada M, Miyamoto H, et al. Sonic hedgehog relates to colorectal carcinogenesis. J Gastroenterol. 2009;44(11):1113–1117. | ||
He B, Barg RN, You L, et al. Wnt signaling in stem cells and non-small-cell lung cancer. Clin Lung Cancer. 2005;7(1):54–60. | ||
Hong Z, Bi A, Chen D, Gao L, Yin Z, Luo L. Activation of hedgehog signaling pathway in human non-small cell lung cancers. Pathol Oncol Res. 2014;20(4):917–922. | ||
Bora-Singhal N, Perumal D, Nguyen J, Chellappan S. Gli1-mediated regulation of sox2 facilitates self-renewal of stem-like cells and confers resistance to EGFR inhibitors in non-small cell lung cancer. Neoplasia. 2015;17(7):538–551. | ||
Mizugaki H, Sakakibara-Konishi J, Kikuchi J, et al. CD133 expression: a potential prognostic marker for non-small cell lung cancers. Int J Clin Oncol. 2014;19(2):254–259. | ||
Merchant AA, Matsui W. Targeting Hedgehog – a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130–3140. | ||
Alison MR, Lin WR, Lim SM, Nicholson LJ. Cancer stem cells: in the line of fire. Cancer Treat Rev. 2012;38(6):589–598. | ||
Biswas S, Guix M, Rinehart C, et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117(5):1305–1313. | ||
Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3(2):117–129. | ||
Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel). 2016;8(2):22. | ||
Bai XY, Zhang XC, Yang SQ, et al. Blockade of hedgehog signaling synergistically increases sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer cell lines. PLoS One. 2016;11(3):e0149370. | ||
Fu Z, Kitagawa Y, Shen R, et al. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate. 2006;66(3):248–256. | ||
Wang Q, Wu X, Wu T, Li GM, Shi Y. Clinical significance of RKIP mRNA expression in non-small cell lung cancer. Tumour Biol. 2014;35(5):4377–4380. | ||
Granovsky AE, Rosner MR. Raf kinase inhibitory protein: a signal transduction modulator and metastasis suppressor. Cell Res. 2008;18(4):452–457. | ||
Yuan L, Yi HM, Yi H, et al. Reduced RKIP enhances nasopharyngeal carcinoma radioresistance by increasing ERK and AKT activity. Oncotarget. 2016;7(10):11463–11477. | ||
Wu X, Yang Y, Xu Z, et al. Raf kinase inhibitor protein mediated signaling inhibits invasion and metastasis of hepatocellular carcinoma. Biochim Biophys Acta. 2016;1860(2):384–391. | ||
Huangfu D, Anderson KV. Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development. 2006;133(1):3–14. | ||
Murone M, Rosenthal A, de Sauvage FJ. Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr Biol. 1999;9(2):76–84. | ||
Walterhouse DO, Yoon JW, Iannaccone PM. Developmental pathways: Sonic hedgehog-Patched-GLI. Environ Health Perspect. 1999;107(3):167–171. | ||
Yoshida GJ, Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016;107(1):5–11. | ||
O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–110. | ||
Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–699. | ||
Wang J, Li ZH, White J, Zhang LB. Lung cancer stem cells and implications for future therapeutics. Cell Biochem Biophys. 2014;69(3):389–398. | ||
Song W, Li H, Tao K, et al. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int J Clin Pract. 2008;62(8):1212–1218. | ||
Varnat F, Duquet A, Malerba M, et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1(6–7):338–351. | ||
Zhang S, Wang Y, Mao JH, et al. Inhibition of CK2alpha down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells. PLoS One. 2012;7(6):e38996. | ||
Naka K, Hoshii T, Hirao A. Novel therapeutic approach to eradicate tyrosine kinase inhibitor resistant chronic myeloid leukemia stem cells. Cancer Sci. 2010;101(7):1577–1581. | ||
Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8(2):97–106. | ||
Ieta K, Tanaka F, Haraguchi N, et al. Biological and genetic characteristics of tumor-initiating cells in colon cancer. Ann Surg Oncol. 2008;15(2):638–648. | ||
Todaro M, Alea MP, Di Stefano AB, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1(4):389–402. | ||
Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. | ||
Martinho O, Pinto F, Granja S, et al. RKIP inhibition in cervical cancer is associated with higher tumor aggressive behavior and resistance to cisplatin therapy. PLoS One. 2013;8(3):e59104. | ||
Wang A, Duan G, Zhao C, et al. Reduced RKIP expression levels are associated with frequent non-small cell lung cancer metastasis and STAT3 phosphorylation and activation. Oncol Lett. 2017;13(5):3039–3045. | ||
Wang Y, Wang LY, Feng F, et al. Effect of Raf kinase inhibitor protein expression on malignant biological behavior and progression of colorectal cancer. Oncol Rep. 2015;34(4):2106–2114. | ||
Dai H, Chen H, Liu W, et al. Effects of Raf kinase inhibitor protein expression on pancreatic cancer cell growth and motility: an in vivo and in vitro study. J Cancer Res Clin Oncol. 2016;142(10):2107–2117. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.