")
Back to Journals » International Journal of General Medicine » Volume 14
Risk of Colorectal Carcinoma May Predispose to the Genetic Variants of the GST, CYP450, and TP53 Genes Among Nonsmokers in the Saudi Community
Authors Sindi IA, Babalghith AO , Tayeb MT , Mufti AH, Naffadi H, Ekram SN , Elhawary EN , Alenezi M, Elhawary NA
Received 7 January 2021
Accepted for publication 25 March 2021
Published 15 April 2021 Volume 2021:14 Pages 1311—1323
DOI https://doi.org/10.2147/IJGM.S294802
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Ikhlas A Sindi,1 Ahmed O Babalghith,2 Mohammed T Tayeb,2 Ahmad H Mufti,2 Hind Naffadi,3 Samar N Ekram,2,4 Ezzeldin N Elhawary,5,6 Munaifah Alenezi,2 Nasser A Elhawary2,7
1Department of Biotechnology, Faculty of Science, King Abdulaziz University, Jeddah, 21589, Saudi Arabia; 2Department of Medical Genetics, Faculty of Medicine, Umm Al-Qura University, Mecca, 21955, Saudi Arabia; 3Common Science, First Year Deanship, Umm Al-Qura University, Mecca, Saudi Arabia; 4Department of Medical Oncology, King Abdullah City Hospital, Mecca, Saudi Arabia; 5MS Genomic Medicine Program, Faculty of Medicine, University of Southampton, Southampton General Hospital, Southampton, UK; 6Faculty of Biotechnology, October Modern Sciences and Arts University, Giza, Egypt; 7Department of Genetics, Faculty of Medicine, Ain Shams University, Cairo, Egypt
Correspondence: Nasser A Elhawary
Department of Medical Genetics, Faculty of Medicine, Umm Al-Qura University, P.O. Box 57543, Mecca, 21955, Saudi Arabia
Tel +966 55 369 2180
Email [email protected]
Ikhlas A Sindi
Department of Biotechnology, Faculty of Science, King Abdulaziz University, Jeddah, 21589, Saudi Arabia
Email [email protected]
Purpose: Colorectal carcinoma (CRC) represents a considerable public health burden in Saudi Arabia. Several candidate genes and genetic variants have been associated with morbidity and mortality among patients with CRC. We explored whether allelic variants of the GSTM1, GSTT1, CYP450 (rs4646903 and rs1048943), and TP53 (rs1042522) genes predisposed nonsmoking Saudi individuals to increased risk for CRC.
Patients and Methods: DNA from buccal cells of 158 participants (80 with CRC and 78 healthy controls) were analyzed for five SNPs using conventional PCR and TaqMan genotyping assays. The SNPStats software was utilized to choose the best interactive inheritance mode for selected SNPs (https://www.snpstats.net).
Results: The mean age of diagnosis was 62.4± 13.5 years (range, 40– 83 years), with those aged 71– 80 years and those aged 40– 50 years accounting for the most diagnoses (35.7% and 28.6% of diagnosis, respectively). The GSTM1 and TP53 rs1042522 SNPs were associated with CRC (OR= 3.7; P< 0.0001, and OR= 1.6; P= 0.033, respectively). A plausible contribution to CRC was observed for the GSTM1 and TP53 rs1042522 SNPs (x2Yates= 14.7; P= 0.00013, and x2Yates= 11.2; P= 0.0008, respectively), while the GSTT1 null variant did not affect risk. Heterozygosity in the CYP450 (rs4646903 and rs1048943 SNPs) was associated with a significant risk for CRC. The GSTM1/GSTT1 and CYP450 rs4646903/rs1048943 SNP pairs were in linkage disequilibrium, and the associations were statistically significant (P= 0.01 and P= 4.6x10‒7, respectively).
Conclusion: The GSTM1 and TP53 rs1042522 variants can increase the development of CRC in Saudi nonsmokers. Even the presence of one copy of a variant allele in the CYP1A1 gene can predispose CRC risk. Additional studies should also examine other SNP combinations with lifestyle factors that may help prevent, rather than facilitate, colorectal tumorigenesis.
Keywords: colorectal carcinoma, single nucleotide polymorphism, TaqMan genotyping, linkage disequilibrium, age at diagnosis, nonsmokers
Plain Language Summary
Colorectal cancer (CRC) is the second most common cancer in men and the third most common in women in Saudi Arabia. The current study focuses on specific genes predisposed to the CRC risk among nonsmokers. Multiple genes involved in CRC development with strong genetic impact have been identified in different cultures and ethnic peoples. The heterogeneity and the interaction effects of genetic variants could play a considerable role in complex multifactorial disorders. Advanced methods such as whole-exome analysis may unveil further predictive factors and help explain a susceptibility to the disease risk. The increasing trend of CRC among young adults suggests that significant lifestyle modifications are necessary. Unfortunately, malnutrition or insufficient food will also inhibit cell apoptosis and thus enhance tumor cells’ growth. Several popular foods and other dietary and lifestyle products and smoking habits are considered risk factors for CRC in young adults. Under the environmental pressure of malnutrition or insufficient nutrition, cancer cells grow better than normal cells.
Introduction
Genetic factors are known to cause the development of many common cancers. In some countries, genetic susceptibility accounts for 35% of colorectal carcinoma (CRC) cases,1 most of which remain unexplained. In Saudi Arabia, CRC is the second most common cancer among men and the third most common among women.2 In the past two decades, the CRC prevalence in Saudi Arabia has nearly doubled from 4.8% to 10.1%.3,4 The differences in CRC prevalence among distinct countries, with marked regional variation, indicate that environmental factors, such as diet and exposure to carcinogens, could have an important role in cancer risk.5
The selection of candidate genes is always laborious, especially in multifactorial disorders and cancer in which exposure to endogenous and exogenous toxins is problematic. Thus, various members of the Phase I cytochrome P450 (CYP) and Phase II glutathione S-transferase (GST) gene families are of interest in various cancers. GSTs play significant roles in detoxifying environmental pollutants, carcinogenic compounds, reactive oxidative species to protect DNA from oxidative damage.5–7 Loss or reduction of the enzyme activity inhibits toxin neutralization and may indirectly affect the risk of cancer development.7 Several tumor studies focus on the most common GST classes, namely mu (μ) and theta (θ). GSTM1 (MIM #138350) and GSTT1 (MIM #600436) genes are commonly focused due to their high frequency of polymorphisms and broad expression in gastrointestinal tissue.8–10
CYPs are heme proteins with an important function in detoxifying, activating, and metabolizing several endogenous and exogenous toxins by adding an oxygen atom to their substrate (Stavrinou et al, 2015).11–13 CYP1A1, an extra enzyme responsible for the aryl-hydrocarbon hydroxylase activity, was already seen in the context of CRC 28 years ago.14,15 This enzyme is involved in the metabolic activation of several carcinogenic substances.16 Moreover, CYP1A1 play a crucial role in the metabolic activation of polyaromatic hydrocarbons and heterocyclic amines, both known to cause CRC.7,17 Although, there is potential support for several studies on smoking-related cancers and CYP1A1 polymorphisms,18–20 few studies found no significant associations between CYP1A1 and enzyme inducibility.16,21 Two common gene polymorphisms (CYP1A1*2A rs4646903 and CYP1A1*2C rs1048943) are correlated with a predisposition to different cancer types,22 and a significant association has been observed between these CYP1A1 variants and in situ CRC.23
Reports from various types of carcinomas also reported the importance of genes involved in cell cycle control.24,25 p53 protein, a tumor suppressor, is considered one of the most significant cancer development drivers in various organs, including the colon.26 TP53 contributes to cell monitoring, including cell cycle control, DNA repair, genomic plasticity, differentiation, and cell apoptosis.27 Once the cell is damaged, p53 protein changes the cell cycle or induces apoptosis by repairing DNA.28 Otherwise, the genomic instability caused by the deletion of p53 may make tumor cells accumulate more cancer drivers, thus accelerating carcinogenesis, tumor metastasis, and drug resistance.29 The TP53 gene (TP53, MIM #191170) mutation rate in non-hypermutated CRC is about 60%, making TP53 mutations the second most frequent mutations seen in CRC. The mutation rate of TP53 is lower (almost 20%) in hypermutated CRC30,31 but is significantly increased in advanced CRC patients (higher than 60%).32
Previous studies addressing the impact of polymorphisms in GSTs, CYP450, and TP53 have found different effects on cancer types among different ethnic populations.19,33–35 This study investigated associations between the common GSTM1, GSTT1, CYP450 (rs4646903 and rs1048943), and TP53 (rs1042522) polymorphisms and the risk of CRC in the non-smoking Saudi community.
Patients and Methods
Ethics Statement
This study was conducted in accordance with the Declaration of Helsinki. Ethical approval was obtained for the study from the Institutional Biomedical Ethics Committee at Medicine college-Umm Al-Qura University (reference #HAPO-02K-012), licensed from the National Committee of Medical and Bioethics-Riyadh (http://bioethics.kacst.edu.sa/About.aspx?lang=en-US). All individuals provided informed consent before enrollment in the study.
Study Population
Our study included eighty nonsmoking patients from the Oncology Department at King Abdullah City Hospital (Western region of Saudi Arabia, Mecca) who had CRC confirmed by a histopathological diagnosis of specimens collected during colonoscopy surgery (unpublished data). Individuals who currently or previously smoked were excluded from this study due to correlations between tobacco smoke and the examined polymorphic variants. Epidemiological data for eligible individuals, including sex, age, and history of histopathology other than CRC, were obtained. Patients who had undergone radiotherapy or chemotherapy before surgery were excluded. Nonsmoking individuals (n = 78; age 51–88 years) were selected as healthy controls if they had no clinical evidence of malignancies or ulcerative colitis.
DNA Isolation
Within the Molecular Diagnostic Laboratory in the Medicine College of Umm Al-Qura University, DNA was extracted from buccal cells (Oragene OGR-575 kit, DNA Genotek Inc., Ottawa, ON, Canada). Briefly, buccal cells were collected in the Oragene tube within 30 s and capped immediately. OGR-lysis buffer was added, and the cells were incubated in a 53°C water bath for an hour. DNA samples were precipitated by ethanol and dissolved in an aqueous elution buffer.36
Diplex Amplifications of GSTM1/GSTT1 Loci
We investigated the GSTM1 and GSTT1 gene deletions using a diplex PCR strategy, as described by Hezova et al37. The amplicon fragments (215 bp and 480 bp) were separated on a 2% agarose/ethidium bromide gel and visualized. These fragments were aligned with internal control and a blank test to help confirm the successful PCR amplification.
TaqMan Genotyping Analysis
We implemented TaqMan Real-Time PCR assays (Fast Dx Real-Time PCR System, Model 7500, Thermo Fisher Inc., USA) to genotype the individuals for the CYP450 (rs4646903, and rs1048943) and TP53 rs1042522 SNPs. One hundred fifty-eight cases and controls and eight negative controls were loaded in a 96-well plate to validate the genotype results. Assays were repeated for 10% of the genotypes for confirmation.
Bioinformatics Analysis
We utilized Sorting Intolerant from Tolerant (SIFT) (https://sift.bii.a-star.edu.sg), Polymorphism Phenotyping v2 (PolyPhen-2) (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (https://www.mutationtaster.org/ChrPos.html), Functional Analysis through Hidden Markov Models (FATHMM), Mutation Assessor, and the in-silico LoFtool to predict the effects of the SNPs on the functional proteins (Ensemble Variant Effect Predictor; https://www.ensembl.org/vep).
Statistical Analysis
Hardy-Weinberg equilibrium (HWE) was evaluated to detect any significant differences between the observed and expected genotypic distributions for CRC cases using the chi-square (χ2) test (https://www.genecalculators.net/pq-chwe-genotypes.html). The SNPStats software (https://www.snpstats.net) was used to choose the best interactive model of inheritance for all the examined markers among cases and controls (adjusted by gender) and to examine linkage disequilibrium (LD) between the polymorphic markers. Moreover, the statistical significance of the LD was calculated based on the coefficient of LD (D’) and the correlation coefficient between pairs of loci (r). All statistical parameters utilized in this study, including the odds ratio (OR), 95% confidence interval (CI), z-test, and χ2-test in terms of P-value, were calculated for genotype distributions and allele frequencies with Social Science Statistics (https://www.socscistatistics.com/tests/) and MedCalc Statistical Software (https://www.medcalc.org).
Results
Characteristics of the Study Population
Eighty eligible Saudi nonsmokers with CRC (55 men: 25 women; a ratio of 2.2: 1) and 78 controls (54 men: 24 women; a ratio of 2.25: 1) were enrolled in the study. Thirty-nine individuals who had undergone radiotherapy or chemotherapy (n = 12), had ulcerative colitis (n= 21) or had other cancer types (n= 6) were excluded from the study (Figure 1).
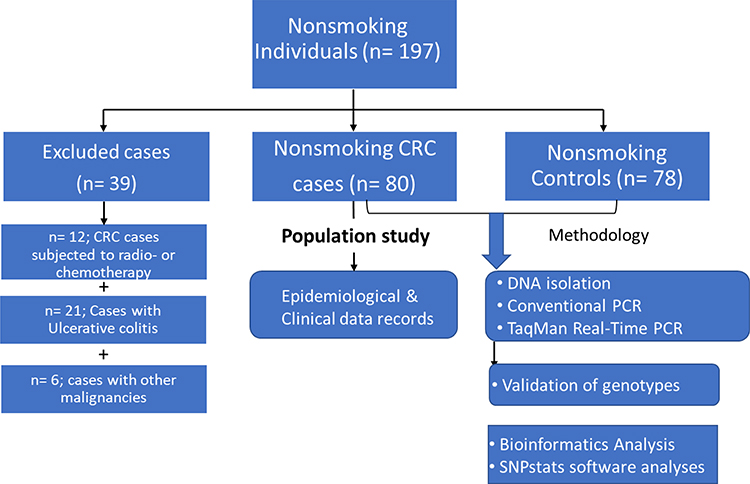 |
Figure 1 Flow chart of the eligible nonsmoking individuals and applied methodology in the study population. |
The mean age of diagnosis was 62.4 ± 13.5 years (range, in the 40–83 years), with no significant difference (t = 0.84; P = 0.4) when compared with controls (64.0 ± 10.1 years). The mean age of diagnosis was lower among men than women (51.3 ± 18.23 years versus 54.8 ± 21.91 years), but not significantly (t = 0.97; P = 0.335). The frequency of CRC was highest in those aged 71–80 years (35.7%). The second highest frequency was in adults aged 40–50 years (28.6%), followed by those aged 51–60 (Figure 2). Several popular foods and other dietary and lifestyle products and habits are considered risk factors for CRC in young Saudi adults. Thus, the high frequency in younger adults may highlight the importance of lifestyle modifications and physical activity for prevention.
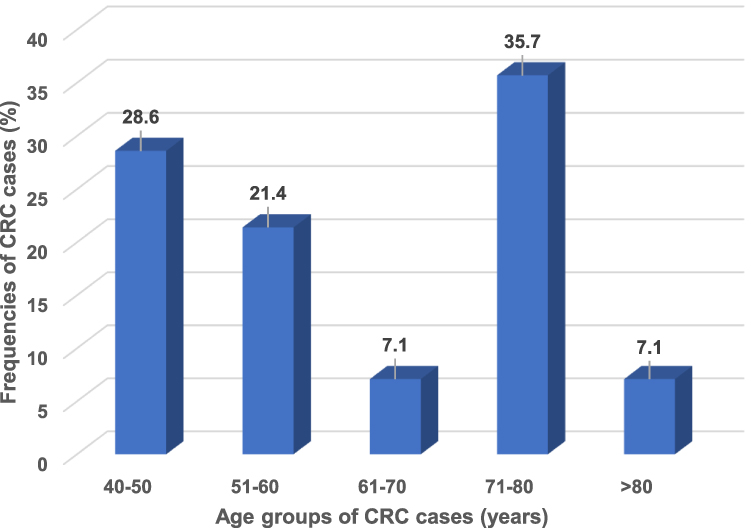 |
Figure 2 A schematic histogram showing the distribution of CRC cases (n = 80) according to their age groups. A maximum frequency of 35.7% is found at the 71–80 years age group. The young age group (40–50 years) showed a frequency of 28.6%. |
Hardy-Weinberg Equilibrium
The genotypic distribution of the CYP450 rs1048943 A>G SNP was consistent with HWE in both cases and controls (χ2 = 0.86; P = 0.35, and χ2 = 0.29; P = 0.56, respectively), but the CYP450 rs4646903 T>C and TP53 rs1042522 G>C SNPs deviated from HWE in cases and controls (P > 0.05) (Table 2). The disagreement of CYP450 rs4646903 T>C with HWE might be due to the absence of the homozygous C/C genotype. For the TP53 rs1042522 G>C SNP, the small number of cases with the normal G/G genotype compared with the G/C genotype might explain the deviation from the HWE. We could not examine HWE for the GSTM1 or GSTT1 genotypes, as the heterozygotes could not be tested using the conventional PCR protocol.
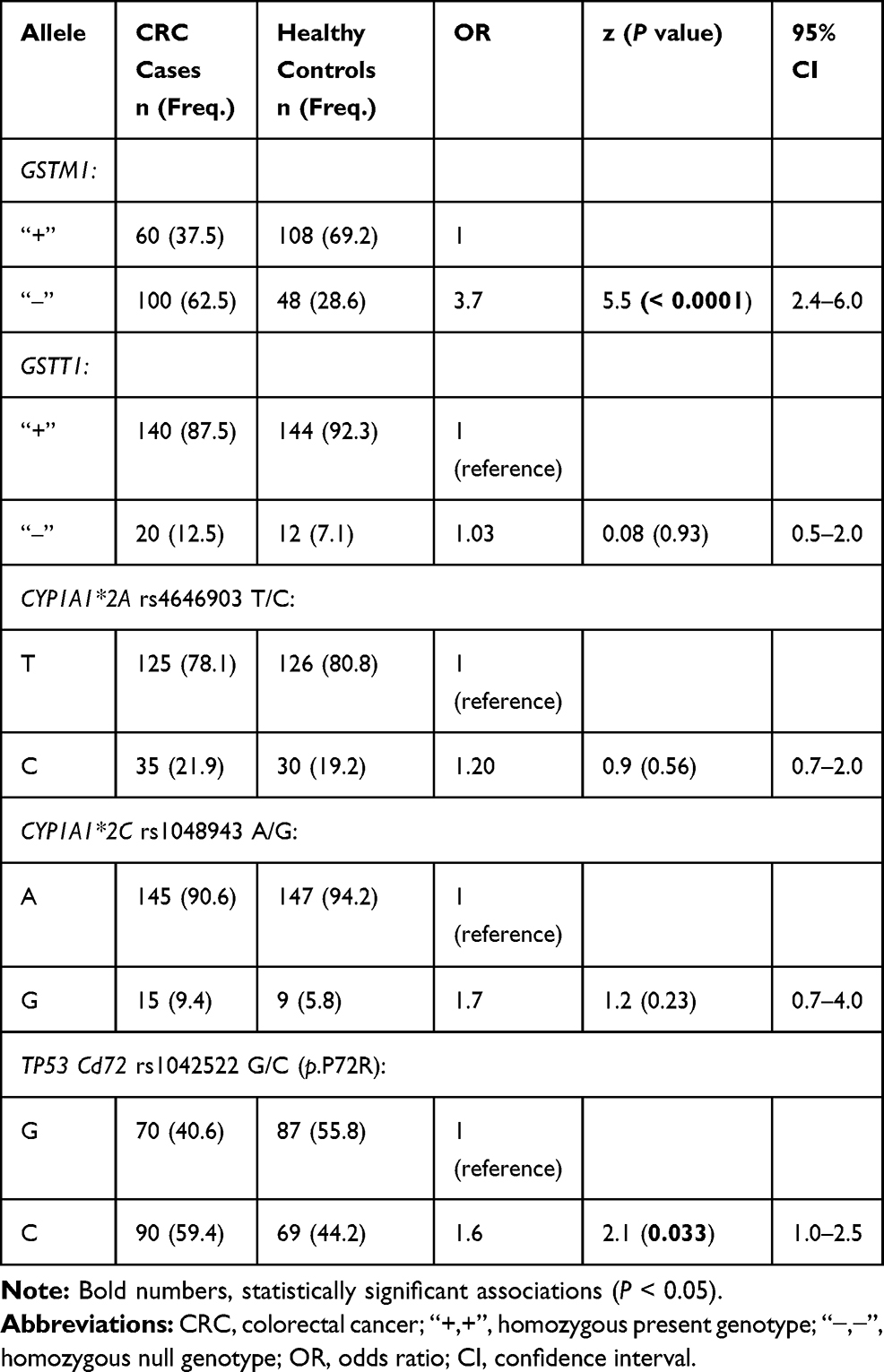 |
Table 1 Allele Frequencies of the Examined SNPs in CRC Cases and Controls |
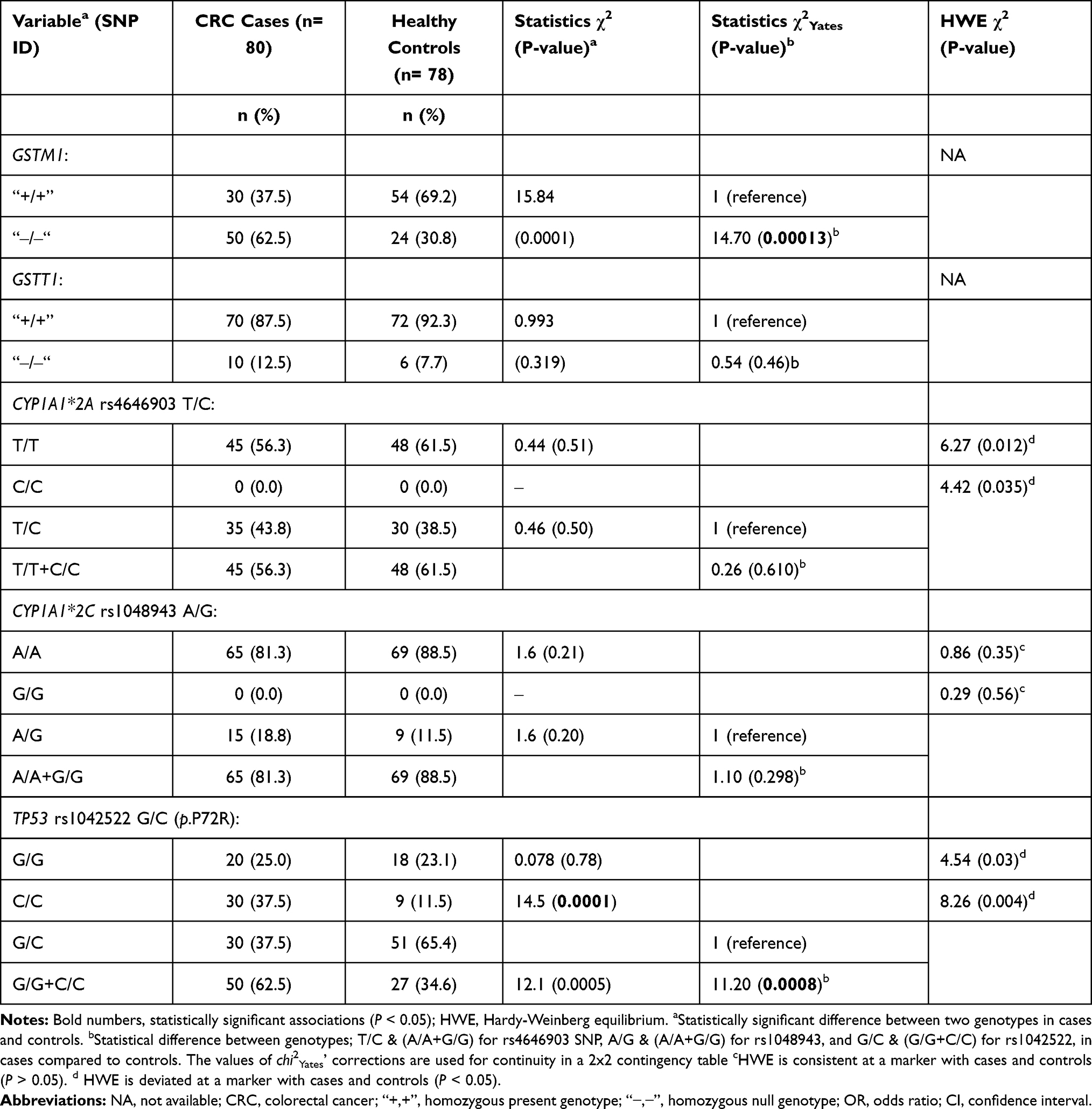 |
Table 2 Genotype Distributions of the Examined SNPs in CRC Cases and Healthy Controls (Adjusted by Gender) |
GSTM1/GSTT1 Variants in CRC
The frequencies of the GSTM1 null allele were significantly higher in CRC cases than in controls (62.5% versus 28.6%; OR = 3.7 95% CI, 2.4–6.0, z = 5.5 and p = 0.0001. Results were similar for the GSTT1 null allele (12.5% for cases versus 7.1% for controls; OR = 1.03, 95% CI, 0.5–2.0, z = 0.08, and z = 0.93 (Table 1). The genotypic distribution of the GSTM1 null genotype showed a significantly higher frequency in CRC cases than in controls (62.5% versus 30.8%; x2Yates= 14.70, p = 0.00013) (Table 2). The GSTM1 null allele was strongly associated with CRC (OR = 3.7; 95% Cl, 2.4–6.0, z = 5.5; p = 0.0001). Neither the homozygous null GSTT1 genotype “‒/‒” nor its allelic frequency “‒” were significantly different in cases than in controls (x2Yates = 0.54, p = 0.46, and OR = 1.0; 95% Cl, 0.5–2.0, z = 0.08; p = 0.93). However, the frequency of GSTT1 null homozygotes was increased in cases when compared with controls (12.5% versus 7.7%) (Table 2).
CYP450 rs4646903 and rs1048943 Variants in CRC
Frequency differences between variant alleles in cases and controls were not statistically significant for the CYP450 rs4646903 T/C (OR =1.20, 95% CI, 0.7–2.0, z = 0.90; p = 0.56) and rs1048943 A/G (OR = 1.7, 95% CI, 0.7–4.0, z = 1.2; z = 0.23) SNPs. Although these two SNPs were not associated with CRC, heterozygosity at each of the two loci was more common in cases than in controls (73.8% versus 38.5% and 18.8% versus 11.5%, respectively) (Table 2). The normal genotypes were less frequent in cases than in controls (56.3% versus 61.5% for rs4646903 T/T and 81.3% versus 88.5% for rs1048943 A/A).
TP53 rs1042522 G>C Variant in CRC
A statistically significant difference was found between allele frequencies of the TP53 rs1042522 G/C polymorphism in cases versus controls (OR = 1.6, 95% CI, 1.0–2.5, p = 2.1, z = 0.033) (Table 1). Interacting the G/C versus G/G+C/C genotypes (overdominant model of inheritance) in cases and controls revealed a strongly significant difference within the rs1042522 SNP (x2Yates = 11.20, z = 0.0008). C/C homozygosity was associated with a three-fold increase in risk of CRC in cases when compared with controls (37.5% versus 11.5%; x2= 14.5; z = 0.0001), and G/G homozygosity was not associated with any difference in risk between cases and controls (25.0% versus 23.1%; x2= 0.08; z = 0.78) (Table 2). At the TP53 rs1042522 loci, G/C heterozygosity was more common in controls than in cases, suggesting a significant protective effect (65.4% versus 37.5%; x2= 12.2; P = 0.0005).
Gene-Gene Interactions
Figure 3 presented the gene-network interaction of the examined genes was created with STRING software. The CYP1A1 gene was exhibited to interact with the GSTM1 gene strongly and, to a lesser extent, with the TP53 gene. A weak gene-gene interaction was found between the GSTM1-TP53 network. The scores of the gene-gene interactions among CYP1A1-GSTM1, CYP1A1-TP53, and GSTM1-TP53 were 0.962, 0.754 and 0.543, respectively (https://string-db.org/).
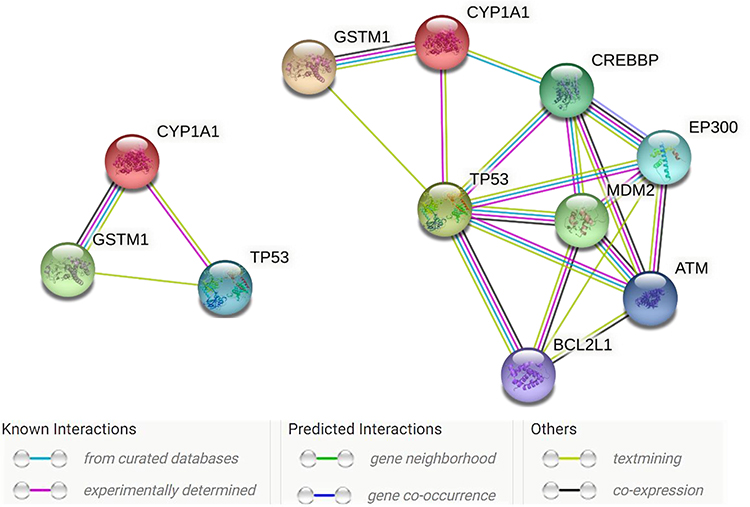 |
Figure 3 Gene-network interactions contained the CYP1A1, GSTM1, and TP53 genes examined in this study (left side) created with STRING (https://string-db.org/). On the Right side: More extended genes, namely, CREBBP, EP300, MDM2, ATM, and BCL2L1 genes, strongly interacted with the TP53 genes. Each node represents all the proteins produced by a single, protein-coding gene locus. Colored nodes describe proteins and the first shell of interactors. Edges represent protein-protein associations that are meant to be specific and meaningful, ie, proteins jointly contribute to a shared function; this does not necessarily mean they are physically binding each other. |
Linkage Disequilibrium
Table 3 shows the correlation coefficients of linkage disequilibrium among the examined SNPs. The CYP450 rs4646903 and rs1048943 polymorphisms were in LD, and their association was statistically significant (D’ = 0.999, r = 0.550; P = 4.6x10−7) (Table 3). The GSTM1 and GSTT1 genetic loci were also found to be in LD with a statistically significant association (D’ = 0.998, r = 0.280; P = 0.010).
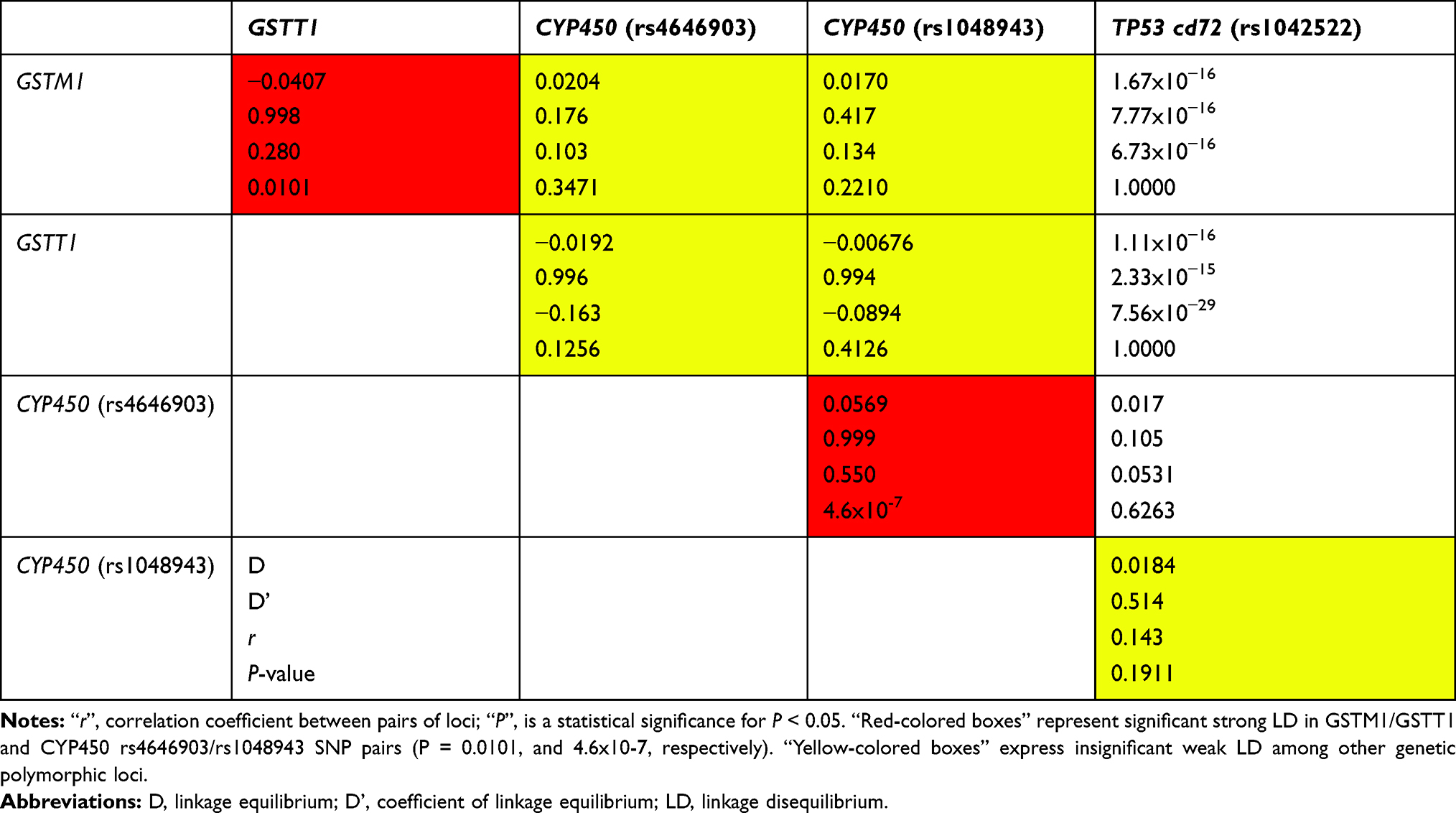 |
Table 3 Coefficients of Linkage Disequilibrium (LD) Among the GSTM1, GSTT1, CYP450 (rs4646903 and rs1048943), and TP53 rs1042522 SNPs |
Bioinformatics and Functional Data
Table 4 shows the predicted functional consequences of the examined SNPs. The missense CYP450 rs1048943 SNP (c.1506A/G; p.I462V) was found to have “possibly damaging” effects on the functional protein according to the MutationaTaster and Mutation Assessor tools and a loss-of-function effect (score, 0.627; possibly damaging) according to the LoFtool. The missense TP53 rs1042522 SNP (c.348G/C; p.P72R) was also predicted to have negative effects according to PolyPhen-2 (possibly damaging), FATHMM (score, ‒5.23; damaging), and the LoFtool (score, 0.00096; probably damaging). The CYP450 rs4646903 SNP, supposed to form a complex with a lncRNA in the 3’untranslated region, was predicted to have a possibly damaging effect (score, 0.627, LoFtool) on the functional protein (https://www.ensembl.org/vep).
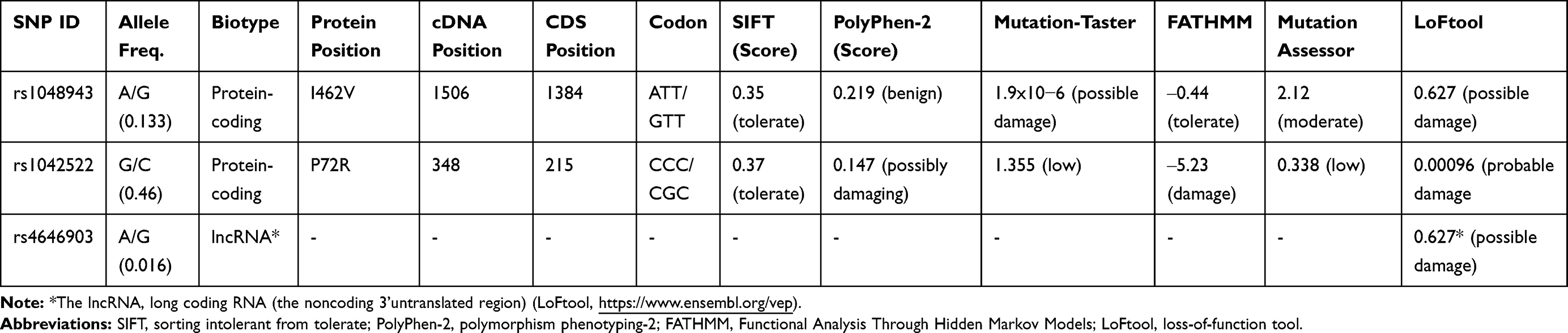 |
Table 4 In-silico Functional Predictions of Non-Synonymous SNPs |
Discussion
Our hospital-based case-control study is the first investigation of GSTM1/GSTT1, CYP450 (rs4646903 and rs1048943), and TP53 (rs1042522) polymorphisms and CRC in Saudi non-smokers. Overall, our results provide strong evidence of an association between the GSTM1 SNP and CRC risk and the TP53 rs1042522 SNP and CRC risk. Moreover, we found strong LD between the polymorphic GSTM1/GSTT1 and CYP450 rs4646903/rs1048943 pairs. Although the CYP450 rs4646903 and rs1048943 SNPs were not shown to be associated with CRC risk, heterozygosity at these two loci was more common in CRC cases than in controls.
The absence of an association between the null GSTT1 allele and CRC in this study is aligned with the several other studies’ results.38,39 The null genotype of the GSTM1 allele indicates a loss of the entire gene and is considered the most common polymorphism in CRC. Enzyme activities of GSTM1 are absent in individuals with a homozygous null allele.40 The ability to detoxify carcinogens decreases in individuals with the null GSTM1 homozygous genotype.7 This genotype appears to be linked to a low detoxifying capacity of some xenobiotics and a reduced regulating oxidative stress caused by free radical activity.41
Moreover, the null GSTM1 genotype has been associated with lung, hepatocellular, breast, and prostate cancers.41–46 Studies conducted in different populations have reported a remarkable association between the GSTM1 null genotype and CRC.46–48 However, other studies could not find a significant relationship between the null GSTM1 genotype and cancers.49,50 Khabaz51 revealed the GSTM1 null genotype’s effect on increasing the risk of CRC in the Saudi population, while Saeed et al52 demonstrated that the GSTM1 null genotype was found in 2% of CRC patients in the Saudi population. These contradictory results suggest the need to conduct genotyping studies of GSTM1 in larger samples.
Despite much less is known about the properties, functions, and biological significance, long noncoding RNAs (lncRNAs) were later found to be associated with many human diseases, including cancers (http://bioinfo.life.hust.edu.cn/lncRNASNP2).10 The mechanistic interactions between CYPs and ncRNAs are associated with environmental chemicals’ toxicity and carcinogenicity.53 A lncRNA RP11-108K3.2, mapped to a 15q21 chromosome locus, overlaps with a CYP19A1 gene, changing the CRC risk inflammation-related mechanism.34,54,55 In patients with CRC, the lncRNA CCAT1 and CCAT2 are highly expressed with a low survival rate and recurrence rate of the disease.56
Neither the noncoding rs4646903 nor the missense rs1048943 variant in CYP450 was associated with the risk of CRC in the present study; hence, the relatively small sample size among the Saudi population might have made it difficult to detect associations with CRC. Previous studies have shown contradictory data regarding associations between CYP1A1 SNPs and CRC susceptibility.38,57 A meta-analysis study investigating the CYP450 (rs1048943 A/G and rs4646903 T/C SNPs) has shown an increased risk of CRC with rs1048943, but not with the rs4646903.58 Although our study did not examine gene-environmental correlations due to insufficient data, additional studies should evaluate potential gene-environmental interactions involving smoking, CYP1A1 rs4646903, CYP1A1*2C rs1048943 SNPs, and CRC.57,58
Given the importance of p53 in multiple cellular functions, including gene transcription, DNA repair, and apoptosis, it is biologically reasonable that TP53 polymorphisms could be associated with CRC risk.59 Several studies have provided evidence that TP53 rs1042522 (c.348G>C; p.P72R) could be associated with CRC, but results are still conflicting. Consistent with our study results, 72Arg is associated with CRC risk in populations in Greece,60 Argentina,61 Germany,62 and Iran.63 On the other hand, 72Pro is associated with a higher risk of CRC in Turkey64 and Malaysia.65 At the same time, other studies in different populations have failed to link TP53 rs1042522 to CRC.66,67
The distribution of the TP53 rs1042522 G/C polymorphism differs based on geographic regions and ethnicity. General populations from Latin America, the United States, and Europe show higher C-allele frequencies than the G-allele. On the other hand, the C-allele is less prevalent in African and Asian populations.68,69 The present study showed that the TP53 rs1042522 SNP was significantly associated with susceptibility to CRC. C/C homozygosity conferred a triple risk of CRC in cases compared with controls. Since the G/C genotype was more common in controls, the C-allele may be a protective moiety in the G/C heterozygous genotype. Thus, only one copy of the rs1042522 variant C-allele may not be sufficient to impact CRC development.
Our results revealed that CRC is more frequent in men than women (a ratio of 2.2: 1), with the most common diagnosis among 71–80 years. These outcomes are consistent with a 7849-case cohort study that reported the highest CRC diagnosis frequency in those ages 60–75 and older.70 However, the mean age at diagnosis is still conflicting among different ethnic populations; our mean age at diagnosis was older than that reported in South-Eastern Asians (62.4 ± 13.5 years versus 59.3 ± 14.6 years).71–73 Our finding of a relatively high frequency of CRC (28.6%) in those ages 40–50 years agrees with results from the United States that also showed a high percentage in those ages 35–49 years,74 but disagrees with research from Japan.75
Esophageal, stomach, colorectal, hepatic, and pancreatic cancers are the major gastrointestinal cancers. Worldwide, CRC is the most common cancer with high age-standardized incidence rates (ASIRs) in both sexes,76,77 but it is not among the top ten carcinomas in the Saudi population.78 Recent studies have reported an increasing trend in ASIRs of CRC in Riyadh, Mecca, and the Eastern region in both sexes.70 The ASIR and age-standardized mortality rate (ASMR) of CRC in the Saudi community are estimated to be 13.1 and 6.3 per 100,000 people, respectively. The highest ASIRs and ASMRs of CRC among both sexes were recorded in 2018 in Austria, New Zealand, Mongolia, and Hungary.79
The increasing CRC trend among young Saudi adults is alarming, signaling the need for significant lifestyle modifications. Siegel et al80 have reported that CRC incidence in adults younger than 50 years old rose by 1.6% from 2000 to 2013–2014, and that mortality rose by 13% in the same period. In addition to tobacco-smoking habits being implicated in the development of various cancers, several popular foods and other dietary and lifestyle products and habits are well known as serious risk factors for CRC.81 Lifestyle modifications could prevent about 50–60% of incident CRC cases in the United States.82,83 Smoking, high levels of body fat, and consumption of red and processed meat have been established to increase CRC risk,71 whereas physical activity and consumption of dietary fiber, whole grains, dairy products, calcium supplements, vitamin D, and marine omega-3 fatty acid may lower disease risk (https://www.wcrf.org/sites/default/files/Colorectal-Cancer-2017-Report.pdf). Under the environmental pressure of malnutrition or insufficient nutrition, cancer cells grow better than normal cells.84,85
Study Limitations
Conflicting results in establishing genetic associations with CRC are common. Poor replication of results could be due to several factors. First, previous studies that did not separate smokers and nonsmokers suggested positive associations between CRC and GST, CYP, and TP53 genes. Excluding Saudi smokers from the present study resulted in a much smaller sample size, making it more difficult to detect associations. However, to improve accuracy, we used nonsmoker conditions to exclude 39 individuals (those who have ulcerative colitis, having other malignancies, or cases subjected to radio- or chemotherapy) with CRC (approximately 50%) originally recruited. Second, some studies have been conducted in populations with admixed ethnicities, while we confined our criteria to only individuals from the Western region. Third, some studies used multiple sources for CRC cases, which would decrease the overall results’ power. Fourth, environmental and lifestyle factors (eg, fatty/red meat consumption, vegetable consumption, physical exercise) should be addressed in CRC risk studies.
Conclusion
The present study represents unreeled investigation of GST, CYP450, and TP53 with colorectal carcinoma (CRC) among the nonsmoking Saudi community. The null GSTM1 allele and the TP53 rs1042522 G/C polymorphism were targeting risk factors in CRC nonsmokers. Even though the GSTT1 and CYP450 (rs4646903 and rs1048943) SNPs may not be individual risk factors for CRC, the LD of the polymorphic GSTM1/GSTT1 and rs4646903/rs1048943 pairs showed potential significant effects on CRC risk. However, heterozygosity of CYP450 rs4646903T/C, CYP450 rs1048943A/G, and TP53 rs1042522 G/C cannot be considered risk factors for CRC in this cohort. So far, these outcomes should be taken with caution, as the examined SNPs do not act alone to explain such complex multifactorial malignancies.
Moreover, mutations in non-coding regions may play an important role in the development and progression of tumors. Thus, an in-depth prospective study of the molecular mechanism and clinical application of lncRNAs helps explain the mechanistic binding with the CYP and p53 for CRC development and provides new prognosis and management targets. More linkage outcomes based on a next-generation sequencing approach instead of a single gene or a few candidate genes may help discover new genes associated with susceptibility to CRC. An ongoing large-scale whole-exome analysis may identify further predictive factors and help explain a predisposition to the disease. The increasing trend of CRC among young Saudi adults suggests that significant lifestyle modifications are necessary. Unfortunately, malnutrition or insufficient food will also inhibit cell apoptosis and thus enhance tumor cells’ growth.
Data Sharing Statement
Data sets analyzed during this study are provided by the corresponding authors.
Ethics Approval and Consent to Participate
Written informed consent was obtained from all study participants enrolled in this project, approved by the Institutional Biomedical Ethics Committee of Umm Al-Qura University (reference #HAPO-02-K-012) licensed from the National Committee Medical and Bioethics, KACST (http://bioethics.kacst.edu.sa/About.aspx?lang=en-US).
Consent for Publication
Written informed consent was taken from all study participants to publish the results.
Acknowledgments
The authors wish to thank the clinical oncologists in the Department of Medical Oncology, King Abdullah City Hospital, and the Molecular Diagnostic Laboratory researchers, Umm Al-Qura University, for collecting buccal cells and performing the laboratory work.
Author Contributions
NAE, and IAS designed the research (Corresponding authors); SNE, NAE, MTT, and MA made the clinical investigations, surgery, and managements; IAS, AOB, AHM, HMN, ENE, MA, and NAE performed the practical work; IAS, NAE, AHM, and ENE work for in-silico predictions and statistical analysis. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85. doi:10.1056/NEJM200007133430201
2. Alsanea N, Abduljabbar AS, Alhomoud S, Ashari LH, Hibbert D, Bazarbashi S. Colorectal cancer in Saudi Arabia: incidence, survival, demographics and implications for national policies. Ann Saudi Med. 2015;35(3):196–202. doi:10.5144/0256-4947.2015.196
3. Tomlinson IP, Webb E, Carvajal-Carmona L, et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008;40(5):623–630. doi:10.1038/ng.111
4. Bazarbashi S, Al Eid H, Minguet J. Cancer Incidence in Saudi Arabia: 2012 Data from the Saudi Cancer Registry. Asian Pac J Cancer Prev. 2017;18(9):2437–2444. doi:10.22034/APJCP.2017.18.9.2437
5. Sugimura T. Nutrition and dietary carcinogens. Carcinogenesis. 2000;21(3):387–395. doi:10.1093/carcin/21.3.387
6. Nagao M, Sugimura T. Carcinogenic factors in food with relevance to colon cancer development. Mutat Res. 1993;290(1):43–51. doi:10.1016/0027-5107(93)90031-a
7. Hayes JD, Strange RC. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology. 2000;61(3):154–166. doi:10.1159/000028396
8. Ryberg D, Skaug V, Hewer A, et al. Genotypes of glutathione transferase M1 and P1 and their significance for lung DNA adduct levels and cancer risk. Carcinogenesis. 1997;18(7):1285–1289. doi:10.1093/carcin/18.7.1285
9. Luo W, Kinsey M, Schiffman JD, Lessnick SL. Glutathione s-transferases in pediatric cancer. Front Oncol. 2011;1:39. doi:10.3389/fonc.2011.00039
10. Gong M, Dong W, Shi Z, Xu Y, Ni W, An R. Genetic polymorphisms of GSTM1, GSTT1, and GSTP1 with prostate cancer risk: a meta-analysis of 57 studies. PLoS One. 2012;7(11):e50587. doi:10.1371/journal.pone.0050587
11. Estabrook RW. A passion for P450s (rememberances of the early history of research on cytochrome P450). Drug Metab Dispos. 2003;31(12):1461–1473. doi:10.1124/dmd.31.12.1461
12. Nebert DW, Dalton TP. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer. 2006;6(12):947–960. doi:10.1038/nrc2015
13. Stavrinou P, Mavrogiorgou MC, Polyzoidis K, et al. Expression profile of genes related to drug metabolism in human brain tumors. PLoS One. 2015;10(11):e0143285. doi:10.1371/journal.pone.0143285
14. Rosenberg DW, Leff T. Regulation of cytochrome P450 in cultured human colonic cells. Arch Biochem Biophys. 1993;300(1):186–192. doi:10.1006/abbi.1993.1026
15. Sivaraman L, Leatham MP, Yee J, Wilkens LR, Lau AF, Le Marchand L. CYP1A1 genetic polymorphisms and in situ colorectal cancer. Cancer Res. 1994;54(14):3692–3695.
16. Androutsopoulos VP, Tsatsakis AM, Spandidos DA. Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC Cancer. 2009;9(1):187. doi:10.1186/1471-2407-9-187
17. Shimada T, Fujii-Kuriyama Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004;95(1):1–6. doi:10.1111/j.1349-7006.2004.tb03162.x
18. Pande M, Amos CI, Eng C, Frazier ML. Interactions between cigarette smoking and selected polymorphisms in xenobiotic metabolizing enzymes in risk for colorectal cancer: a case-only analysis. Mol Carcinog. 2010;49(11):974–980. doi:10.1002/mc.20682
19. Elhawary NA, Nassir A, Saada H, et al. Combined genetic biomarkers confer susceptibility to risk of urothelial bladder carcinoma in a Saudi Population. Dis Markers. 2017;2017:1474560. doi:10.1155/2017/1474560
20. Vidigal VM, Silva TD, de Oliveira J, Pimenta CAM, Felipe AV, Forones NM. Genetic polymorphisms of vitamin D receptor (VDR), CYP27B1 and CYP24A1 genes and the risk of colorectal cancer. Int J Biol Markers. 2017;32(2):e224–e230. doi:10.5301/jbm.5000248
21. Hanna IH, Dawling S, Roodi N, Guengerich FP, Parl FF. Cytochrome P450 1B1 (CYP1B1) pharmacogenetics: association of polymorphisms with functional differences in estrogen hydroxylation activity. Cancer Res. 2000;60(13):3440–3444.
22. Kawajiri K, Nakachi K, Imai K, Yoshii A, Shinoda N, Watanabe J. Identification of genetically high risk individuals to lung cancer by DNA polymorphisms of the cytochrome P450IA1 gene. FEBS Lett. 1990;263(1):131–133. doi:10.1016/0014-5793(90)80721-t
23. Shimada T, Gillam EM, Sandhu P, Guo Z, Tukey RH, Guengerich FP. Activation of procarcinogens by human cytochrome P450 enzymes expressed in Escherichia coli. Simplified bacterial systems for genotoxicity assays. Carcinogenesis. 1994;15(11):2523–2529. doi:10.1093/carcin/15.11.2523
24. Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015;21(1):84–93. doi:10.3748/wjg.v21.i1.84
25. Hou Z, Guo K, Sun X, et al. TRIB2 functions as novel oncogene in colorectal cancer by blocking cellular senescence through AP4/p21 signaling. Mol Cancer. 2018;17(1):172. doi:10.1186/s12943-018-0922-x
26. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54(18):4855–4878.
27. Chang YT, Lin TP, Tang JT, et al. HOTAIR is a REST-regulated lncRNA that promotes neuroendocrine differentiation in castration resistant prostate cancer. Cancer Lett. 2018;433:43–52. doi:10.1016/j.canlet.2018.06.029
28. Huang Y, Zhang J, Hou L, et al. LncRNA AK023391 promotes tumorigenesis and invasion of gastric cancer through activation of the PI3K/Akt signaling pathway. J Exp Clin Cancer Res. 2017;36(1):194. doi:10.1186/s13046-017-0666-2
29. Kanwal R, Plaga AR, Liu X, Shukla GC, Gupta S. MicroRNAs in prostate cancer: functional role as biomarkers. Cancer Lett. 2017;407:9–20. doi:10.1016/j.canlet.2017.08.011
30. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi:10.1038/nature12634
31. Giannakis M, Mu XJ, Shukla SA, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15(4):857–865. doi:10.1016/j.celrep.2016.03.075
32. Brannon AR, Vakiani E, Sylvester BE, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014;15(8):454. doi:10.1186/s13059-014-0454-7
33. Reis AA, Silva DM, Curado MP, da Cruz AD. Involvement of CYP1A1, GST, 72TP53 polymorphisms in the pathogenesis of thyroid nodules. Genet Mol Res. 2010;9(4):2222–2229. doi:10.4238/vol9-4gmr807
34. Jiang D, Jin M, Ye D, et al. Polymorphisms of a novel long non-coding RNA RP11-108K3.2 with colorectal cancer susceptibility and their effects on its expression. Int J Biol Markers. 2020;35(1):3–9. doi:10.1177/1724600819888512
35. Li J, Xu W, Liu F, Huang S, He M. GSTM1 polymorphism contribute to colorectal cancer in Asian populations: a prospective meta-analysis. Sci Rep. 2015;5:12514. doi:10.1038/srep12514
36. Elhawary NA, Jiffri EH, Jambi S, et al. Molecular characterization of exonic rearrangements and frame shifts in the dystrophin gene in Duchenne muscular dystrophy patients in a Saudi community. Hum Genomics. 2018;12(1):18. doi:10.1186/s40246-018-0152-8
37. Hezova R, Bienertova-Vasku J, Sachlova M, et al. Common polymorphisms in GSTM1, GSTT1, GSTP1, GSTA1 and susceptibility to colorectal cancer in the Central European population. Eur J Med Res. 2012;17:17. doi:10.1186/2047-783X-17-17
38. Sachse C, Smith G, Wilkie MJ, et al. A pharmacogenetic study to investigate the role of dietary carcinogens in the etiology of colorectal cancer. Carcinogenesis. 2002;23(11):1839–1849. doi:10.1093/carcin/23.11.1839
39. Huang K, Sandler RS, Millikan RC, Schroeder JC, North KE, Hu J. GSTM1 and GSTT1 polymorphisms, cigarette smoking, and risk of colon cancer: a population-based case-control study in North Carolina (United States). Cancer Causes Control. 2006;17(4):385–394. doi:10.1007/s10552-005-0424-1
40. Tetlow N, Robinson A, Mantle T, Board P. Polymorphism of human mu class glutathione transferases. Pharmacogenetics. 2004;14(6):359–368. doi:10.1097/00008571-200406000-00005
41. Wei B, Xu Z, Zhou Y, et al. Association of GSTM1 null allele with prostate cancer risk: evidence from 36 case-control studies. PLoS One. 2012;7(10):e46982. doi:10.1371/journal.pone.0046982
42. Seidegård J, Pero RW, Miller DG, Beattie EJ. A glutathione transferase in human leukocytes as a marker for the susceptibility to lung cancer. Carcinogenesis. 1986;7(5):751–753. doi:10.1093/carcin/7.5.751
43. Maugard CM, Charrier J, Bignon YJ. Allelic deletion at glutathione S-transferase M1 locus and its association with breast cancer susceptibility. Chem Biol Interact. 1998;111-112:365–375. doi:10.1016/s0009-2797(97)00173-7
44. Murata M, Shiraishi T, Fukutome K, et al. Cytochrome P4501A1 and glutathione S-transferase M1 genotypes as risk factors for prostate cancer in Japan. Jpn J Clin Oncol. 1998;28(11):657–660. doi:10.1093/jjco/28.11.657
45. Wang B, Huang G, Wang D, et al. Null genotypes of GSTM1 and GSTT1 contribute to hepatocellular carcinoma risk: evidence from an updated meta-analysis. J Hepatol. 2010;53(3):508–518. doi:10.1016/j.jhep.2010.03.026
46. Cai X, Yang L, Chen H, Wang C. An updated meta-analysis of the association between GSTM1 polymorphism and colorectal cancer in Asians. Tumour Biol. 2014;35(2):949–953. doi:10.1007/s13277-013-1125-0
47. Teng Z, Wang L, Zhang J, Cai S, Liu Y. Glutathione S-transferase M1 polymorphism and colorectal cancer risk in Chinese population. Tumour Biol. 2014;35(3):2117–2121. doi:10.1007/s13277-013-1281-2
48. Djansugurova L, Zhunussova G, Khussainova E, et al. Association of DCC, MLH1, GSTT1, GSTM1, and TP53 gene polymorphisms with colorectal cancer in Kazakhstan. Tumour Biol. 2015;36(1):279–289. doi:10.1007/s13277-014-2641-2
49. Lin D, Tang Y, Peng Q. [Genetic polymorphisms of cytochrome P450 2E1 and glutathione S-transferase P1 and susceptibility to esophageal cancer]. Zhonghua Zhong Liu Za Zhi [Chinese J Oncol]. 1998;20(2):94–97. Chinese.
50. Liu G, Ghadirian P, Vesprini D, et al. Polymorphisms in GSTM1, GSTT1 and CYP1A1 and risk of pancreatic adenocarcinoma. Br J Cancer. 2000;82(10):1646–1649. doi:10.1054/bjoc.2000.1221
51. Khabaz MN. The GSTP1 Ile105Val polymorphism is not associated with susceptibility to colorectal cancer. Asian Pac J Cancer Prev. 2012;13(6):2949–2953. doi:10.7314/apjcp.2012.13.6.2949
52. Saeed HM, Alanazi MS, Nounou HA, et al. Cytochrome P450 1A1, 2E1 and GSTM1 gene polymorphisms and susceptibility to colorectal cancer in the Saudi population. Asian Pac J Cancer Prev. 2013;14(6):3761–3768. doi:10.7314/apjcp.2013.14.6.3761
53. Li D, Tolleson WH, Yu D, et al. Regulation of cytochrome P450 expression by microRNAs and long noncoding RNAs: epigenetic mechanisms in environmental toxicology and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2019;37(3):180–214. doi:10.1080/10590501.2019.1639481
54. Lin JH, Manson JE, Kraft P, et al. Estrogen and progesterone-related gene variants and colorectal cancer risk in women. BMC Med Genet. 2011;12:78. doi:10.1186/1471-2350-12-78
55. Slattery ML, Lundgreen A, Herrick JS, et al. Variation in the CYP19A1 gene and risk of colon and rectal cancer. Cancer Causes Control. 2011;22(7):955–963. doi:10.1007/s10552-011-9768-x
56. Selvaraj S, Dhoke NR, Kiley J, et al. Gene Correction of LGMD2A Patient-Specific iPSCs for the development of targeted autologous cell therapy. Mol Ther. 2019;27(12):2147–2157. doi:10.1016/j.ymthe.2019.08.011
57. Slattery ML, Samowtiz W, Ma K, et al. CYP1A1, cigarette smoking, and colon and rectal cancer. Am J Epidemiol. 2004;160(9):842–852. doi:10.1093/aje/kwh298
58. Zhu X, Wang Z, He J, et al. Associations between CYP1A1 rs1048943 A > G and rs4646903 T > C genetic variations and colorectal cancer risk: proof from 26 case-control studies. Oncotarget. 2016;7(32):51365–51374. doi:10.18632/oncotarget.10331
59. Russo A, Bazan V, Iacopetta B, et al. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol. 2005;23(30):7518–7528. doi:10.1200/JCO.2005.00.471
60. Dakouras A, Nikiteas N, Papadakis E, et al. P53Arg72 homozygosity and its increased incidence in left-sided sporadic colorectal adenocarcinomas, in a Greek-Caucasian population. Anticancer Res. 2008;28(2A):1039–1043.
61. Perez LO, Abba MC, Dulout FN, Golijow CD. Evaluation of p53 codon 72 polymorphism in adenocarcinomas of the colon and rectum in La Plata, Argentina. World J Gastroenterol. 2006;12(9):1426–1429. doi:10.3748/wjg.v12.i9.1426
62. Schneider-Stock R, Boltze C, Peters B, et al. Selective loss of codon 72 proline p53 and frequent mutational inactivation of the retained arginine allele in colorectal cancer. Neoplasia. 2004;6(5):529–535. doi:10.1593/neo.04178
63. Dastjerdi MN. TP53 codon 72 polymorphism and P53 protein expression in colorectal cancer specimens in Isfahan. Acta Med Iran. 2011;49(2):71–77.
64. Eren F, Akkiprik M, Atug O, et al. R72P polymorphism of TP53 in ulcerative colitis patients is associated with the incidence of colectomy, use of steroids and the presence of a positive family history. Pathol Oncol Res. 2010;16(4):563–568. doi:10.1007/s12253-010-9255-9
65. Aizat AA, Shahpudin SN, Mustapha MA, et al. Association of Arg72Pro of P53 polymorphism with colorectal cancer susceptibility risk in Malaysian population. Asian Pac J Cancer Prev. 2011;12(11):2909–2913.
66. Koushik A, Tranah GJ, Ma J, et al. p53 Arg72Pro polymorphism and risk of colorectal adenoma and cancer. Int J Cancer. 2006;119(8):1863–1868. doi:10.1002/ijc.22057
67. Song HR, Kweon SS, Kim HN, et al. p53 codon 72 polymorphism in patients with gastric and colorectal cancer in a Korean population. Gastric Cancer. 2011;14(3):242–248. doi:10.1007/s10120-011-0034-4
68. Beckman G, Birgander R, Sjalander A, et al. Is p53 polymorphism maintained by natural selection? Hum Hered. 1994;44(5):266–270. doi:10.1159/000154228
69. Lu XM, Zhang YM, Lin RY, et al. p53 polymorphism in human papillomavirus-associated Kazakh’s esophageal cancer in Xinjiang, China. World J Gastroenterol. 2004;10(19):2775–2778. doi:10.3748/wjg.v10.i19.2775
70. Almatroudi A. The incidence rate of colorectal cancer in saudi arabia: an observational descriptive epidemiological analysis. Int J Gen Med. 2020;13:977–990. doi:10.2147/IJGM.S277272
71. Lim KG. A review of colorectal cancer research in malaysia. Med J Malaysia. 2014;69(Suppl A):23–32.
72. Chong VH, Telisinghe PU, Bickle I, Abdullah MS, Lim E, Chong CF. Increasing incidence of colorectal cancer, starting at a younger age for rectal compared to colon cancer in brunei darussalam. Asian Pac J Cancer Prev. 2015;16(12):5063–5067. doi:10.7314/apjcp.2015.16.12.5063
73. Koh KS, Telisinghe PU, Bickle I, Abdullah MS, Chong CF, Chong VH. Characteristics of young colorectal cancer in Brunei Darussalam: an epidemiologic study of 29 years (1986-2014). Asian Pac J Cancer Prev. 2015;16(8):3279–3283. doi:10.7314/apjcp.2015.16.8.3279
74. Bailey CE, Hu CY, You YN, et al. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975-2010. JAMA Surg. 2015;150(1):17–22. doi:10.1001/jamasurg.2014.1756
75. Minami Y, Nishino Y, Tsubono Y, Tsuji I, Hisamichi S. Increase of colon and rectal cancer incidence rates in Japan: trends in incidence rates in Miyagi Prefecture, 1959-1997. J Epidemiol. 2006;16(6):240–248. doi:10.2188/jea.16.240
76. Arnold M, Abnet CC, Neale RE, et al. Global burden of 5 major types of gastrointestinal cancer. Gastroenterology. 2020;159(1):335–349e15. doi:10.1053/j.gastro.2020.02.068
77. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–1953. doi:10.1002/ijc.31937
78. Chaudhri E, Fathi W, Hussain F, Hashmi SK. The increasing trends in cases of the most common cancers in Saudi Arabia. J Epidemiol Glob Health. 2020. doi:10.2991/jegh.k.200515.001
79. Ferlay J, Colombet M, Soerjomataram I, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries and 25 major cancers in 2018. Eur J Cancer. 2018;103:356–387. doi:10.1016/j.ejca.2018.07.005
80. Siegel RL, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974-2013. J Natl Cancer Inst. 2017;109(8). doi:10.1093/jnci/djw322
81. Song M, Chan AT. Environmental factors, gut microbiota, and colorectal cancer prevention. Clin Gastroenterol Hepatol. 2019;17(2):275–289. doi:10.1016/j.cgh.2018.07.012
82. Song M. Giovannucci E. preventable incidence and mortality of carcinoma associated with lifestyle factors among white adults in the United States. JAMA Oncol. 2016;2(9):1154–1161. doi:10.1001/jamaoncol.2016.0843
83. Islami F, Goding Sauer A, Miller KD, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68(1):31–54. doi:10.3322/caac.21440
84. Huo X, Han S, Wu G, et al. Dysregulated long noncoding RNAs (lncRNAs) in hepatocellular carcinoma: implications for tumorigenesis, disease progression, and liver cancer stem cells. Mol Cancer. 2017;16(1):165. doi:10.1186/s12943-017-0734-4
85. Huang J, Li J, Li Y, et al. Interferon-inducible lncRNA IRF1-AS represses esophageal squamous cell carcinoma by promoting interferon response. Cancer Lett. 2019;459:86–99. doi:10.1016/j.canlet.2019.05.038
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.