Back to Journals » Infection and Drug Resistance » Volume 13
Ribaxamase, an Orally Administered β-Lactamase, Diminishes Changes to Acquired Antimicrobial Resistance of the Gut Resistome in Patients Treated with Ceftriaxone
Authors Kokai-Kun JF, Le C, Trout K, Cope JL ![]() , Ajami NJ
, Ajami NJ ![]() , Degar AJ, Connelly S
, Degar AJ, Connelly S ![]()
Received 28 April 2020
Accepted for publication 5 July 2020
Published 22 July 2020 Volume 2020:13 Pages 2521—2535
DOI https://doi.org/10.2147/IDR.S260258
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
John F Kokai-Kun,1 Chenxiong Le,1 Kenneth Trout,1 Julia L Cope,2 Nadim J Ajami,2 Andrew J Degar,2 Sheila Connelly1
1Synthetic Biologics, Inc., Rockville, MD, USA; 2Diversigen, Inc., Houston, TX, USA
Correspondence: Sheila Connelly Email [email protected]
Introduction: Intravenous (IV) β-lactam antibiotics, excreted through bile into the gastrointestinal (GI) tract, may disrupt the gut microbiome by eliminating the colonization resistance from beneficial bacteria. This increases the risk for Clostridium difficile infection (CDI) and can promote antimicrobial resistance by selecting resistant organisms and eliminating competition by non-resistant organisms. Ribaxamase is an orally administered β-lactamase for use with IV β-lactam antibiotics (penicillins and cephalosporins) and is intended to degrade excess antibiotics in the upper GI before they can disrupt the gut microbiome and alter the resistome.
Methods: Longitudinal fecal samples (349) were collected from patients who participated in a previous Phase 2b clinical study with ribaxamase for prevention of CDI. In that previous study, patients were treated with ceftriaxone for a lower respiratory tract infection and received concurrent ribaxamase or placebo. Extracted fecal DNA from the samples was subjected to whole-genome shotgun sequencing and analyzed for the presence of antimicrobial resistance (AMR) genes by alignment of sequences against the Comprehensive Antibiotic Resistance Database. A qPCR assay was also used to confirm some of the results.
Results: Database alignment identified ∼ 1300 acquired AMR genes and gene variants, including those encoding β-lactamases and vancomycin resistance which were significantly increased in placebo vs ribaxamase-treated patients following antibiotic exposure. qPCR corroborated the presence of these genes and supported both new acquisition and expansion of existing gene pools based on no detectable copy number or a low copy number in pre-antibiotic samples which increased post-antibiotics. Additional statistical analyses demonstrated significant correlations between changes in the gut resistome and clinical study parameters including study drug assignment and β-lactamase and vancomycin resistance gene frequency.
Discussion: These findings demonstrated that ribaxamase reduced changes to the gut resistome subsequent to ceftriaxone administration and may help limit the emergence of AMR.
Keywords: oral β-lactamase, ceftriaxone, gut resistome, whole-genome shotgun sequencing, clinical study, antimicrobial resistance
Introduction
Perturbation of the human gut microbiome has been associated with numerous diverse disease states including obesity, autoimmune diseases and Clostridium difficile infection (CDI).1–3 The human gut microbiome can be influenced by many factors including age, diet, geographic location and exposure to infectious diseases,1 but the most damaging are antibiotics.4 The broad spectrum β-lactam antibiotics, such as penicillins and cephalosporins, are particularly disruptive as their use significantly reduces certain families of bacteria, including obligate anaerobes, which are considered an essential component of a healthy gut ecosystem.5,6
Antibiotics are a major cause of gut microbiome disruption, and broad-spectrum antimicrobials are strongly associated with an increased risk for CDI4,7,8 including clindamycin, fluoroquinolones and β-lactam antibiotics (penicillins, cephalosporins and carbapenems).4,5,9-12 All intravenous (IV) β-lactam antibiotics are at least partially processed in the liver and excreted through the bile into the intestine as active antimicrobials.13 Biliary excretion of β-lactams can range from a small percentage of the input dose to up to 40% for antibiotics like ceftriaxone,13 resulting in high intestinal concentrations of up to 1 mg/mL.14 β-lactams are the most commonly used broad spectrum antibiotics and are especially harmful to the commensal organisms of the gut microbiome, particularly the beneficial anaerobes that play a role in colonization resistance.5
Exposure of the gut microbiome to antibiotics has the additional adverse consequence of propagating antimicrobial-resistant (AMR) organisms.15,16 As an antibiotic exerts its selective pressure on the gut microbiota, susceptible organisms are eliminated, allowing antimicrobial-resistant organisms to proliferate. Many AMR genes are found on mobile genetic elements facilitating their spread from one bacterial strain to another.16–18 For example, following antibiotic treatment for C. difficile, enterococci resistant to vancomycin commonly emerge.19 Vancomycin resistance can result from either transfer of resistance from commensal organisms to enterococci or from colonization by exogenous enterococci that are resistant to vancomycin.16,18 Vancomycin-resistant enterococci (VRE) are also observed following treatment with antibiotics other than vancomycin, due to antibiotic-mediated microbiome damage resulting in diminished competition within the gut leaving the host susceptible to colonization from the environment.19,20 This outcome is particularly devastating for immunocompromised patients, such as those receiving bone marrow transplants, where VRE is a leading cause of infection and mortality.21,22
Another consequence of antibiotic use, and in particular β-lactam use, is the emergence of extended spectrum, β-lactamase-producing-gram negative bacilli (ESBL-GNB). These bacteria, in the family Enterobacteriaceae, include Escherichia, Klebsiella and Salmonella organisms which are well adapted to the environment of the human gut and have become resistant to most antibiotics.23,24 These organisms can also carry β-lactamases such as Klebsiella pneumoniae carbapenemase25 and New Deli metallo-beta-lactamase26 that are active against all classes of β-lactam antibiotics including carbapenems. β-lactamase genes are often carried on plasmids with multiple resistance genes27 facilitating their spread to other organisms within the gut. Some of these bacteria are resistant to most classes of antibiotics therefore limiting treatment options to older antibiotics like colistin that have high toxicity profiles.25 Notably, recent reports of colistin-resistant strains of ESBL-GNB found in the United States have raised the specter of having no antibiotics available to treat these devastating infections.28
Strategies are needed to protect the gut microbiome from disruption by antibiotics and to prevent AMR phenotypes from emerging and propagating as a consequence of the selective pressure caused by antibiotic use. Ribaxamase is a recombinant β-lactamase designed to be orally administered with intravenous (IV) β-lactam antibiotics, specifically, most penicillins and cephalosporins, and functions in the proximal gastrointestinal (GI) tract to degrade excess antibiotics excreted into the intestine through the bile.29 Thus, antibiotic inactivation is expected to prevent disruption of the gut microbiome and reduce exposure of the gut bacteria to the selective pressure of the antibiotics. Using a pig model of β-lactam-induced dysbiosis, ribaxamase was demonstrated to protect the gut microbiome and prevent the emergence of AMR.30
Ribaxamase was evaluated in a Phase 2b proof-of-concept study performed in 412 patients who were admitted to the hospital for treatment of a lower respiratory tract infection (LRTI) with IV ceftriaxone.31 Patients were randomized one-to-one to receive co-treatment with either ribaxamase or placebo and could also receive macrolides as required by the attending physician. Patients were monitored for diarrhea and then tested for CDI during and for 6-weeks after the primary course of ceftriaxone. The study met its primary endpoint of significantly reducing the incidence of CDI in patients that received ribaxamase compared to placebo. A total of 3.4% of the placebo patients were diagnosed with CDI, while only 1.0% of ribaxamase patients developed CDI (risk reduction 2.4%, 95% CI –0.6 to 5.9; one-sided P=0.045), a 71% relative reduction in the incidence of CDI,31 including CDI caused by highly pathogenic 027 strains.32 Fecal samples, collected at three prescribed points, were analyzed to determine the composition of the fecal microbiome and compared to evaluate changes to the gut microbiome following antibiotic exposure.33 This analysis demonstrated that ribaxamase significantly diminished ceftriaxone-induced changes to the gut microbiome and allowed the microbiome to recover more quickly than placebo in patients treated with ceftriaxone.
In the present study, a set of the DNA samples, extracted from the fecal samples collected during the previous Phase 2b clinical study, were analyzed by whole-genome shotgun sequencing and alignment against the Comprehensive Antibiotic Resistance Database (CARD, https://card.mcmaster.ca/home). Some of the samples were also analyzed by qPCR and additional statistical analyses using clinical metadata and a linear model were performed to compare the change in the number of hits for AMR genes pre- to post-antibiotics. Here we report the findings of these analyses and demonstrate, consistent with its intended mechanism of action, ribaxamase appeared to reduce changes in the AMR gene profile (the resistome) in the GI tract of patients treated with ceftriaxone. Five-fold more AMR genes were significantly changed in terms of hits in placebo-treated patients as compared with ribaxamase-treated patients, including genes relevant to the clinical study such as β–lactamase, vancomycin resistance, and macrolide resistance genes. A qPCR assay confirmed these results for two representative genes.
Materials and Methods
Samples Sequenced and Analyzed
During the Phase 2b clinical study with ribaxamase, conducted in 84 hospitals in North America and Eastern Europe from Sept. 2016-Sept. 2017, 862 fecal samples for microbiome analysis were collected from patients who were receiving ceftriaxone for a LRTI.33 Of these samples, DNA was extracted with the MoBio PowerMag isolation kit on an automated KingFisher platform and 16S rRNA sequences were determined for 676 samples.31 Sample selection was based on the criteria that a patient had to have a fecal sample collected at screening so that they could serve as their own baseline prior to study-related treatments. Based on the funding contract with the Centers for Disease Control (CDC), a set of 349 of the extracted fecal DNA samples from the Phase 2b study were included in the present study (Supplemental Table S1). These samples represented 118 patients out of 412 from the study (66 ribaxamase and 52 placebo). Sample selection for analysis was based on the following criteria, 1) patients that had a full set of three longitudinal fecal samples (ie, screening [T0], 72 hours post-antibiotics [T1] and 4 weeks post-antibiotics [T2]),31,33 2) patients that had a screening sample and a T1 sample only, or 3) select samples from patients without a screening sample. For criteria 3, these samples were chosen from patients who met the Phase 2b clinical study endpoints of a) C. difficile infection (CDI), b) non-C. difficile antibiotic-associated diarrhea (AAD) or c) microbiologically confirmed colonization with one of two select pathogens, VRE or ESBL-GNB.
Whole-Genome Shotgun Sequencing and Change in Number of Hits per AMR Gene Variant
DNA extracted from fecal samples for microbiome analysis33 was shipped frozen to Diversigen, Inc. (Houston, TX) and libraries were prepared as per internal Diversigen standards. Briefly, fecal DNA (10 ng to 500 ng) was sheared into fragments of approximately 300–400 bp in a Covaris E210 system (96 well format, Covaris, Inc., Woburn, MA) followed by purification using AMPure XP beads. DNA end repair, 3ʹ-adenylation, ligation to Illumina (San Diego, CA) multiplexing PE adaptors, and ligation-mediated PCR (LM-PCR) were all completed using automated processes. Current standard library methods utilized KAPA HiFi polymerase (KAPA Biosystems Inc., Wilmington, MA) for PCR amplification (6–10 cycles). Prepared libraries were pooled according to the experimental plan and subjected to Illumina HiSeq 2000 sequencing.
Raw sequences with average raw reads per sample of 147,251,799 were trimmed of all primer sequences using BBDuK version 36.92, and filtered for human DNA and PhiX (Illumina control) with Bowtie2 version 2.3.0 against a prebuilt hg38-PhiX database (Illumina). Subsequently, reads were individually mapped against the CARD database (https://card.mcmaster.ca/home) using default parameters of 90% read coverage on a perfect match. Reads were aligned to CARD without prior assembly, therefore reads from complete genes were not distinguished from truncated or partial genes. A set of AMR hits was produced using the protein homolog model which detects proteins conferring antibiotic resistance based on similarity to the curated reference sequence. A “hit” was defined as each time the determined sequences of DNA within a sample matched the accessioned reference sequence for a specific entry within the CARD database. Further alignment of these protein homolog data were performed with linear discriminant analysis (LDA) effect size (LEfSe) analyses,34 an algorithm for high-dimensional biomarker discovery. Mann–Whitney (two-sample Wilcoxon) tests were used to compare between screening (T0) and post-antibiotics (T1) time points across treatment groups. Sequencing and resistome analyses data were submitted to the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra), accession PRJNA589866.
qPCR
Quantitative PCR of two representative genes (cfxA and vanRD), identified during sequence analysis as demonstrating a significant increase in number of hits in the placebo group vs the ribaxamase group from T0 to T1, was performed on a subset of 100 fecal DNA samples (Supplemental Table S2). The analyzed samples represented an equal number from each treatment group.
For each gene, primer sets (Table 1) were designed based on available sequences from the CARD database using Primer Express v3.0.1 and synthesized by Integrated DNA Technologies (IDT, Coralville, IA). Primers were purified by standard desalting, and for qPCR reactions, 10μM working stocks were prepared. Synthetic DNA gene fragments were created using IDT’s gBlocks Gene Fragments for positive controls and standard curves. A fragment was designed from each gene sequence, synthesized to 500ng, and concentrated to a stock of 10ng/μL.
 |
Table 1 Primer Sequences Used for qPCR Analysis |
For each qPCR experiment, a standardized thermocycling profile of initial denaturation of 95°C for 10 minutes, followed by 40 cycles of 10 seconds of denaturation at 95°C and 30 seconds of annealing/extension at 60°C, and a final dissociation stage to produce a melt curve were used. For each reaction, 5μL of PerfeCta SYBR Green FastMix, Low Rox 2x (QuantaBio, Beverly, MA), 1μL of primer set, and 5ng of fecal DNA template was added. All experiments and analyses were performed using an Applied Biosystems (Foster City, CA) QuantStudio 7 Real-Time PCR machine with v1.2 software. A no-template, negative control (NTC) was included in all sample runs using Applied Biosystems MicroAmp Fast Optical Reaction plates.
Samples for analysis were concentrated to 1.25ng/μL for a total of 5ng DNA per reaction. Each sample was tested in triplicate, against each of the two designed assays. Each assay was run individually, with a standard curve ranging from 104–108 copies using the gBlocks, with NTC included. The standard curve was determined by quantifying the concentration of the gBlock, and using the molar mass per base (650 g/mol/bp) and fragment length to calculate the proper copy number. Using linear regression analysis, copy numbers of 1 to 1010 were determined from the five point standard curve with R2 values consistently 0.99 in agreement with Diversigen internal standard operating procedures. Data are presented as copy number per 5ng of total DNA. If a sample did not produce a copy number above 1.0, as determined using linear regression, it was considered to be a false positive.
The copy numbers were also compared with the number of hits as determined by sequence alignment. Quartiles for number of hits and copy numbers were established using Microsoft Excel and the two parameters were compared for each sample to determine correlation between the two methods. Further, qPCR results across the longitudinal samples for each patient were examined to assess whether changes in the copy numbers of cfxA and vanRD over the course of the study were consistent with new acquisition of organisms carrying these antibiotic resistance genes or expansion of existing pools of organisms carrying the genes following selection by antibiotics.
Statistical Correlation Between Resistome Data and Clinical Study Data
Four classes of acquired antibiotic resistance genes, β-lactamase, vancomycin resistance, macrolide resistance, and multidrug efflux pump component genes, were chosen for analysis with respect to clinical metadata from the Phase 2b ribaxamase study. These classes were selected based on their medical importance as well as their relevance to the antibiotics administered during the study.31 A series of clinical parameters were also selected for statistical analysis (Supplemental Table S3) based on relevance to the clinical study design (20) and being known risk factors for gut microbiome disruption and CDI including, advanced age, use of macrolides, and use of proton pump inhibitors (PPI).35–39 Two demographic parameters, age and gender, three treatment-based parameters; study drug assignment (ribaxamase vs placebo), use of macrolides, and use of PPI, and one care-based parameter, whether at any time the patient was classified as no-food-by-mouth (NPO, as a surrogate for sicker patients), were investigated. CDI was not included as there were only seven post-antibiotic fecal samples available from CDI patients, an insufficient number for meaningful analysis.
A linear model was used to compare the change in the number of hits for each AMR gene in each class of genes from the T0, pre-ceftriaxone treatment, to the T1 collection point, 72 hrs after ceftriaxone was stopped. Analyses were conducted based on the log transformed values. For each gene, both the t-test and Wilcoxon test were used for the comparisons between the groups, and nominal P-values were provided from these tests without multiplicity adjustment. A linear regression was used to examine the impact of the identified clinical parameters on resistome changes from baseline (T0) to post-antibiotics (T1) for each gene in each subset. In the regression model, the baseline value and age were included as continuous variables, and the treatment group, gender, use of PPI, NPO and use of a ceftriaxone/macrolide combination were included as categorical variables.
Results
Whole-Genome Shotgun Sequencing and Identification of Acquired AMR Genes
Whole-genome shotgun sequences obtained from DNA extracted from the 349 fecal samples were aligned against CARD to identify the total number of hits for each acquired AMR gene present in each sample. This analysis identified DNA reads (hits) mapping to 1297 AMR genes or gene variants which ranged from one hit to five million hits per gene with a median of 64, and a range of 2001 to 277,184 hits per sample with a median of 49,378. The top ten AMR genes in terms of overall hits included five tetracycline resistance genes (tetQ, tetW, tetX, tetO and tet32), one erythromycin resistance gene (ermF) and four variants of the cfxA β-lactamase gene (cfxA2, cfxA3, cfxA4 and cfxA6).
Comparison of the Change in AMR Gene Hits from T0 to T1
Statistical analysis of the change in the number of hits from pre-ceftriaxone (screening [T0]) to post-ceftriaxone (72 hours post-antibiotics [T1]) for each of the AMR genes (or variants) and comparison of the change from T0 to T1 of placebo-treated versus the ribaxamase-treated patients were performed using LEfSe.34 This analysis identified 94 genes or gene variants in the placebo group and 19 genes or gene variants in the ribaxamase group that changed significantly (LDA score >2) from T0 to T1, an almost five-fold difference in the number of genes affected between treatment groups (Figure 1). Based on changes in hits, 68 genes in the placebo group and 11 genes in the ribaxamase group were significantly reduced and 26 genes in placebo and eight genes in ribaxamase were significantly increased in number of hits from pre- to post-antibiotics. The genes demonstrating significant decrease in both groups included several tet and erm genes as well as AMR genes representative of most other resistance classes. Interestingly, AMR genes demonstrating a significant increase in hits in the placebo group, but not in the ribaxamase group, included a family of cfxA β-lactamase gene variants and vancomycin resistance genes from the vanD operon.
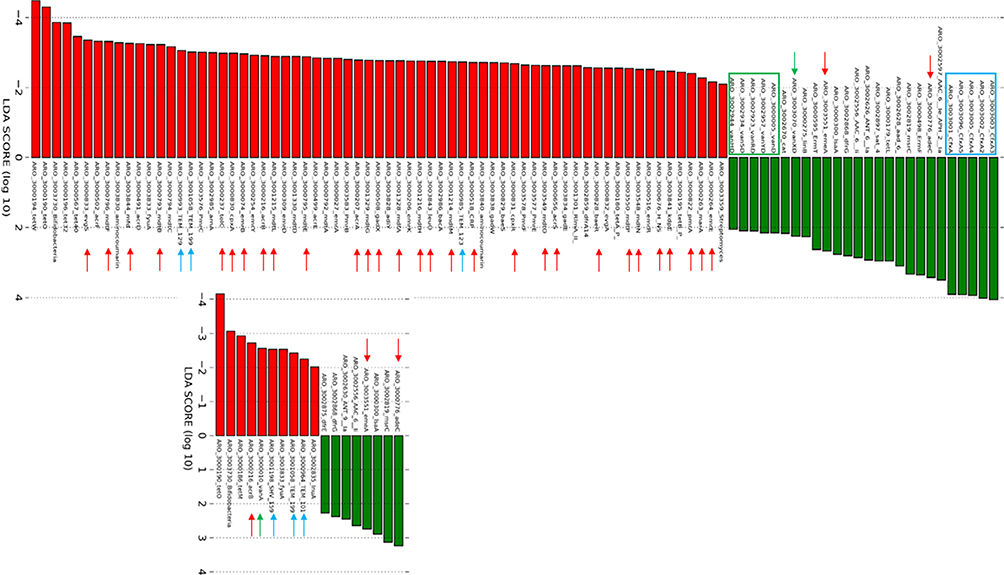 |
Figure 1 LEfSe Analysis of Change in Number of Hits from T0 to T1. Each column represents a different AMR gene that demonstrated a significant change (change in linear determinant analysis [LDA] of ≥2) in the number of hits from the T0 collection point (screening) to the T1 collection point (post-antibiotics). Top figure, placebo group and bottom figure, ribaxamase group. Red bars decreased significantly between these collection points while green bars increased significantly. Specific gene classes of interest are indicated by colors on the figure, blue β-lactamases, green vancomycin resistance genes and red efflux pump genes. Individual gene notations correspond to the Comprehensive Antimicrobial Resistance Database. |
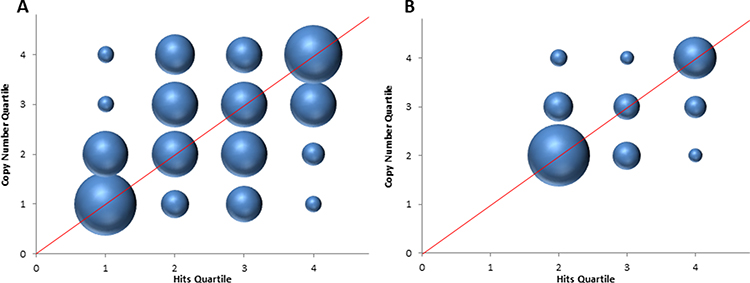 |
Figure 2 Comparing Number of Hits per Sample vs Copy Number per Sample. The graph compares data quartiles for the number of hits per sample with the copy number of the gene for each sample for, (A) cfxA and (B) vanRD. The data were separated into quartiles for the 100 samples that were subjected to qPCR analysis and compares the number of hits per sample, as determined by database alignment, vs the copy number per sample, as determined by qPCR analysis. The quartiles were as follows, cfxA Hits, Q1: 0–2658, Q2: 2659–52,776, Q3: 52,777–175,384, Q4: >175,384; Copy Number, Q1: 0–62, Q2: 63–410, Q3: 411–1244, Q4: >1244, vanRD Hits, Q1 & Q2 = 0, Q3: 1–33, Q4: >33, Copy Number, Q1 & Q2 = 0, Q3: 1–4, Q4: >4. Quartiles 1 and 2 were combined for both parameters on the figure for vanRD due to the abundance of samples with zero values in the sample set. The size of each sphere represents the number of samples, based on the copy number, which corresponded to the indicated quartile for hits. The red lines are added for emphasis. |
A subset of the identified AMR genes significantly increased in the placebo group vs the ribaxamase group was chosen for further statistical analysis based on AMR phenotype and overall number of hits (>500). This subset included 69 genes or variants (Supplemental Table S4), representing three classes of AMR chosen based on their relevance to the study including, β-lactamase, macrolide resistance and vancomycin resistance genes. An additional selection criterion included only genes that were represented in ≥25% of the samples. AMR genes found in <25% of the samples were excluded from further analysis because it was observed that those genes tended to be predominantly found in few samples with high hits thus skewing the results. In general, genes encoding β-lactamases had a greater number of hits and broader distribution across samples than vancomycin and macrolide resistance genes. The final analysis set included 17 AMR genes (or variants), eight β-lactamase, seven vancomycin resistance, and two macrolide resistance genes (Table 2). Statistical analysis of these genes of interest confirmed that seven genes demonstrated a significant increase in number of hits (P<0.05) from T0 to T1 in placebo compared to ribaxamase samples. The identified genes included, five cfxA β-lactamase variants (cfxA-A5) and two vanD operon accessory genes (vanRD and vanSD), while the vanD gene itself demonstrated a trend towards an increase in hits in the placebo group (Table 2).
 |
Table 2 Comparison of the Frequency of Antibiotic Resistance Genes at T0 vs T1 in the Ribaxamase vs Placebo-Treated Patients |
qPCR Analysis of Two Representative AMR Genes
To support the AMR sequencing analysis data demonstrating increased hit frequency of AMR genes in the placebo vs ribaxamase groups following antibiotic exposure, two representative genes, cfxA and vanRD, were selected for qPCR analysis. A subset of 100 extracted DNA samples from 35 patients (18 placebo and 17 ribaxamase) were analyzed by qPCR. For cfxA, 89 out of the 100 samples had at least one copy of the gene present by qPCR with a mean of 146,958, a median of 52,776 and a range of 0 to 2,325,389 copies. For vanRD, 46 out of the 100 samples had at least one copy, with a mean of 76, a median of 1 and a range of 0 to 1149 copies.
In general, there was a positive correlation between the number of hits for cfxA as determined by database alignment and copy number as determined by qPCR. Out of the 100 samples subjected to both analyses, 84 of the samples had a copy number in the same or an adjoining quartile of the data set as compared to the hit frequency data (Figure 2). Only 16 samples had either a lower hit frequency and higher copy number (eight samples) or vice versa (eight samples).
The correlation for the vanRD gene was also positive with 75 of the samples aligning in terms of quartiles of hit frequency and copy number (Figure 2) with those having at least one copy number of vanRD also displaying detectable hits for vanRD (second quartile for each). Of the 25 samples that did not align, 15 had no sequence reads mapping to vanRD but had at least one copy number of vanRD detected by qPCR, while 10 showed vanRD hits but were undetectable by qPCR.
Examination of the change in copy number of cfxA over the three collection points from the clinical study indicated that there was an overall increase in the mean copy number of cfxA in placebo-treated patients from T0 to T1 which remained elevated through T2 (4-weeks post-antibiotics), while there was an overall decrease in mean copy number in ribaxamase-treated patients from T0 to T1 which remained lower through the T2 collection point (Table 3). Similar results were observed with vanRD at the three collection points with the mean copy number increasing in the placebo group at T1 before returning to the starting level at T2, while the mean copy number trended lower at T1 for the ribaxamase samples before returning to the starting level at T2.
 |
Table 3 Change in Mean Copy Number Over Time for cfxA and vanRD in Placebo vs Ribaxamase-Treated Patients |
Fecal samples collected from 19 patients, who were determined to be colonized with VRE by microbiologic methods during the Phase 2b ribaxamase study,31 were included in the qPCR analysis set. Of these 19 patients, 15 were positive for the vanRD gene both by sequencing and qPCR at one or more of the collection points, while three were detectable by only one method (two by sequencing only and one by qPCR only). Only one VRE colonized patient was negative for vanRD by both detection methods. Of the 16 patients included in the qPCR analysis set who did not have microbiologically detectable VRE colonization during the clinical study, seven also did not have detectable vanRD by either method at any collection point. For the other nine patients, five were positive by both sequencing and qPCR, three were positive by sequencing only and one was positive by qPCR only.
Detection of the vanRD accessory gene by qPCR was correlated with detection of the vanD ligase gene by sequence analysis. Of the 35 patients represented by these qPCR samples in 32 cases, the presence or absence of vanRD by qPCR corresponded to the detection, or lack of detection, of vanD by sequence alignment. In only three cases, vanD was not detected by sequence alignment when vanRD was detected by qPCR, and in two of these cases vanRD was also detected by sequence alignment.
The change in copy number for cfxA and vanRD over the longitudinal collection points for individual patients appeared to follow one of three patterns. The first was an initially lower copy number followed by a higher copy number, consistent with expansion of existing AMR during ceftriaxone use (cfxA: 19 patients, vanRD: six patients). The second pattern was a higher copy number that decreased by T1 and then rebounded by T2 (cfxA: eight patients, vanRD: four patients). A variation was observed in two patients for whom the cfxA copy number started high and continued to decrease across the three collection points. For vanRD, three patients had a high copy number at screening and maintained this high copy number through T2. The final pattern was an undetectable copy number at screening with increasing copy numbers at T1 and T2 (cfxA: five patients, vanRD: seven patients), consistent with new acquisition of resistant organisms carrying these genes.
Correlation of AMR Data with Clinical and Demographic Data
Statistical analyses were conducted comparing the number of hits for a set of AMR genes and clinical data from the Phase 2b ribaxamase study. Six parameters were selected for this analysis (Supplemental Table S3). These parameters were demographic (age and gender), drug treatments (placebo vs ribaxamase, macrolide use and PPI use) and health status (no-food-by-mouth [NPO] as a surrogate for overall health). A linear regression model was used to determine correlation between these study data and changes in hit frequency of four selected classes of AMR genes, β-lactamase, vancomycin resistance, and macrolide resistance genes and multidrug efflux pumps. Significant correlations were observed with treatment group, macrolide or PPI use, and NPO.
β-Lactamases and β-Lactam Resistance Genes
A total of 781 genes or gene variants which confer β-lactam resistance were identified in the sample set, but ten genes accounted for the bulk of the hits in this class, the cfxA variant family (A-A6), cblA-1, PBP2a, OXA-347 and cepA. Linear regression analysis confirmed that five cfxA β-lactamase variants correlated with a statistically significant increase in the number of hits from T0 to T1 in the placebo group vs the ribaxamase group (Table 4). There was also a correlation between the use of PPI and changes in hit frequency from T0 to T1 for several β-lactamase genes (Table 5), with five of the cfxA family of variants and cblA-1 correlating with the use of PPI, while cfxA6 correlated with patients who did not receive PPI during the study.
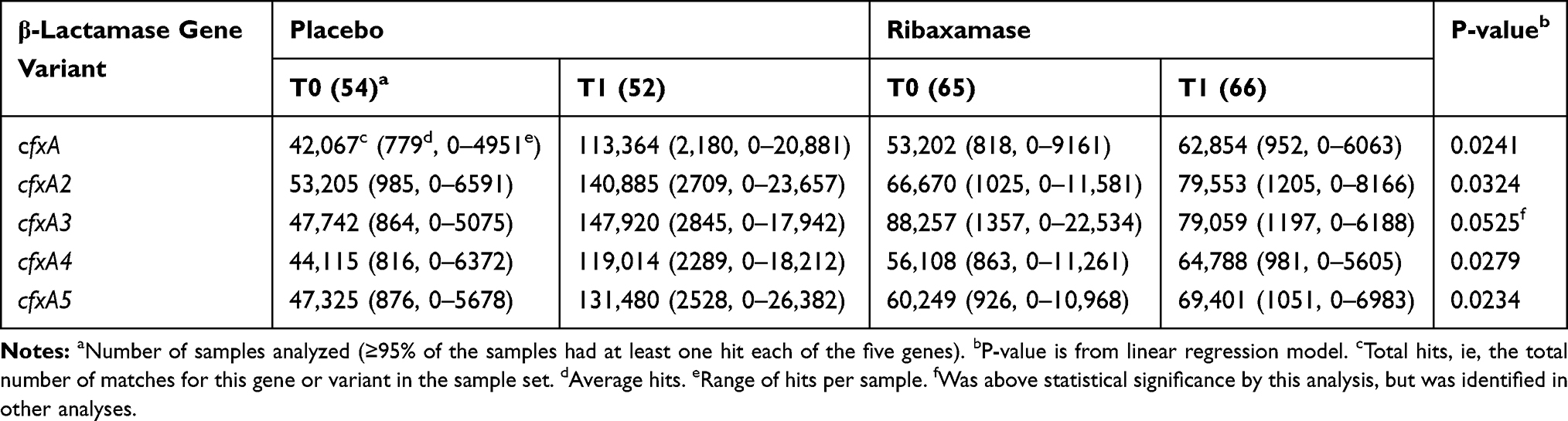 |
Table 4 β-Lactamase Gene Variants Demonstrating a Significant Increase in Placebo vs Ribaxamase-Treated Patients from T0 to T1 |
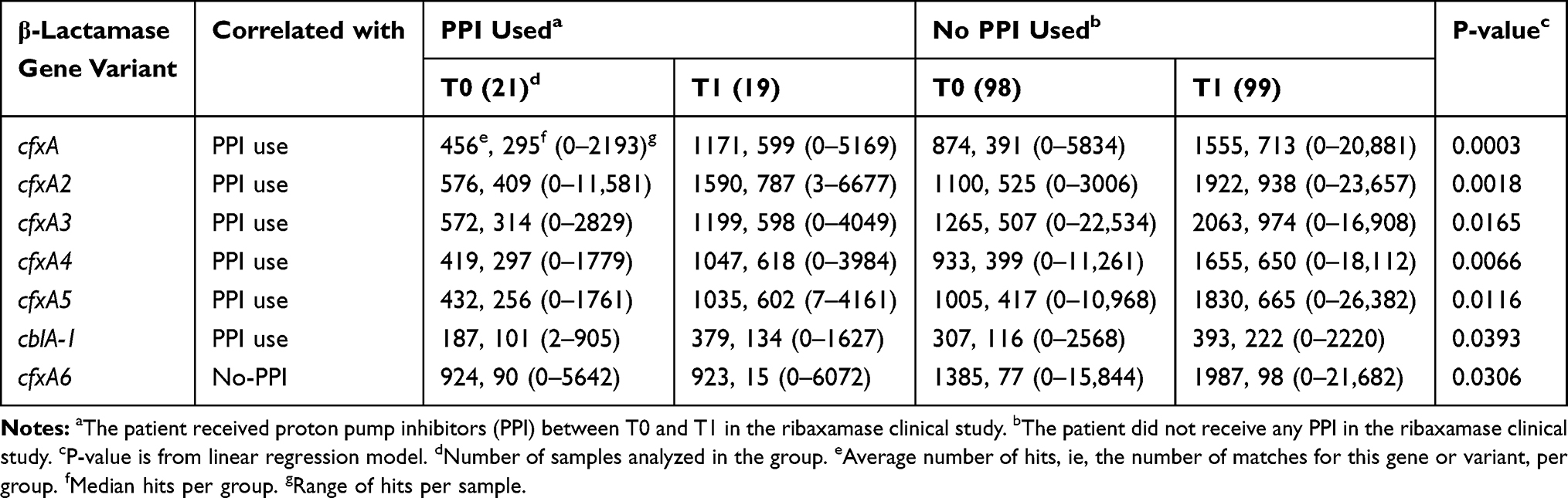 |
Table 5 β-Lactamase Gene Variants Demonstrating a Significant Increase in Hits with PPI-Use or Non-Use from T0 to T1 |
Vancomycin Resistance Genes
During the Phase 2b ribaxamase study, a significantly greater number of patients in the placebo group became newly colonized with VRE (P=0.0001) at the T1 collection point as compared with the ribaxamase group.31 A total of 34 vancomycin resistance genes were identified in the 349 fecal samples analyzed. These included genes from the vanA, vanB, vanC, vanD and vanG operons. Three van genes demonstrated a significant increase in total number of hits from pre-antibiotic (T0) to post-antibiotic exposure (T1) in placebo-treated patients vs ribaxamase-treated patients. These genes included vanRD and vanSD, previously identified by LEfSe analysis, and vanD (Table 6).
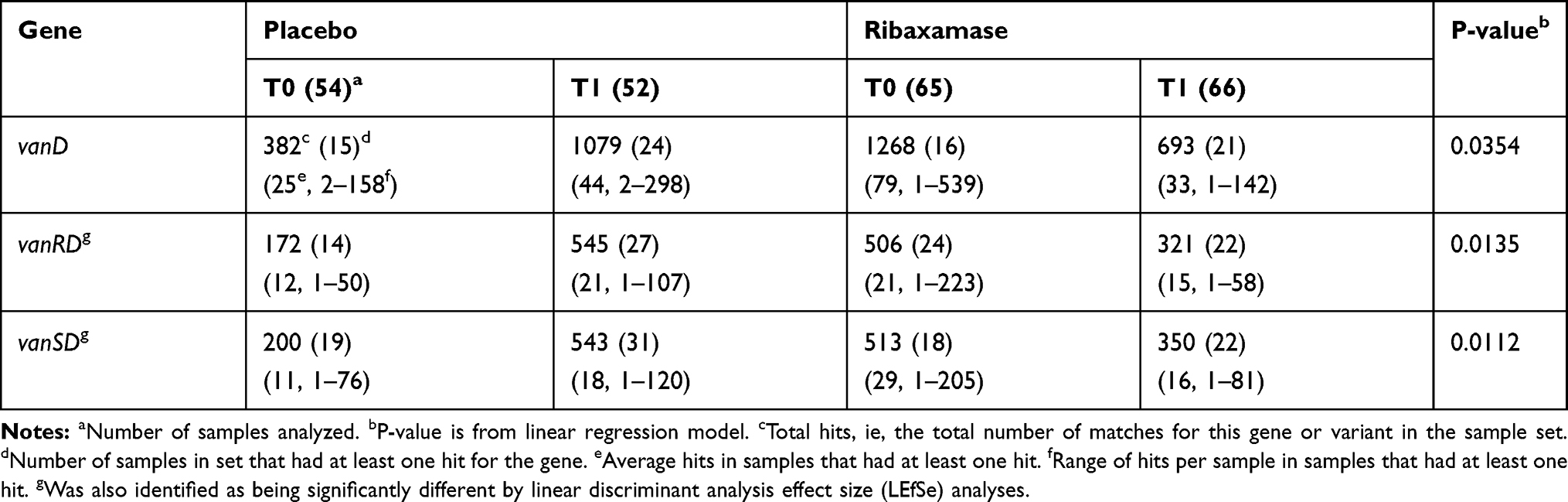 |
Table 6 Vancomycin Resistance Genes Demonstrating a Significant Increase in Placebo vs Ribaxamase-Treated Patients from T0 to T1 |
Macrolide Resistance Genes
During the Phase 2b study with ribaxamase, patients could also receive macrolides for their LRTI.31 Eight macrolide resistance genes were identified within the fecal sample set, and all but four of the 349 samples carried at least one sequence match for a macrolide resistance gene. Four genes accounted for the bulk of the macrolide resistance gene hits, mefA, mel, mrx and mphA, but only mefA demonstrated a significant correlation to any parameter examined in this analysis, a positive correlation with the use of PPI (P=0.0292). A trend toward correlation with macrolide treatment was also observed but did not reach significance (P=0.0785) for this gene.
Efflux Pumps
Efflux pumps were also selected for statistical analyses because of their involvement in AMR for many classes of antibiotics.40 Alignment of the sample sequences against CARD identified 39 efflux pump genes known to confer resistance to multiple drugs. All of the samples had at least one efflux pump gene present, but seven genes accounted for most of the hits, evgS, mdtF, acrF, acrD, mdtB, mdtC and tolC. Thirty-one efflux pump genes demonstrated a correlation with the treatment assignment (ribaxamase vs placebo), with 30 significantly increasing in hit frequency in ribaxamase-treated patients (Supplemental Table S5), while only one, adeC, demonstrated a significant increase in the number of hits correlating with placebo (P<0.0001). Notably, all 30 of these efflux pump genes demonstrated a positive correlation with macrolide exposure, while adeC did not. There was also a positive correlation in increased hit frequency following antibiotic exposure for 29 efflux pump genes in patients who were NPO at any time during the study, the exceptions being adeC, mdtO and CRP.
Discussion
In a Phase 2b clinical study, ribaxamase significantly reduced the incidence of CDI in patients treated with ceftriaxone for a LRTI.31 Consistent with this finding, ribaxamase protected the gut microbiome from damage caused by ceftriaxone as changes in microbiome diversity were significantly less in ribaxamase- vs placebo-treated patients by all measures examined (P<0.01), and appeared to allow the microbiome to recover more quickly.33 Therefore, reducing exposure of the gut microbiome to ceftriaxone excreted via bile into the GI tract was expected to attenuate changes to the gut resistome by eliminating antibiotic selective pressure. The results of the present study support this hypothesis.
Initial assessment of antibiotic resistance gene frequency in 349 fecal samples collected from the Phase 2b clinical study aligned against the CARD database, using the protein homolog model, yielded 21,070,360 hits for 1297 AMR genes or gene variants. This assessment was a semi-quantitative measurement of the DNA sequence matches to specific AMR genes or variants within a sample, and was based on the expectation that the greater the number of copies of an AMR gene present in a sample, the greater the number of short DNA sequences that were likely to be matched against the accessioned sequence data. A selection process, based on parameters including number of hits per gene, the evenness of distribution of hits across samples and the AMR gene phenotype, resulted in identification of 18 genes that had >500 total hits, ≥25% distribution, and were either β-lactam, vancomycin or macrolide resistance genes. These genes were highly relevant as the clinical study employed the β-lactam antibiotic, ceftriaxone, plus optional macrolide treatment, and demonstrated that the incidence of new colonization with VRE was significantly increased in placebo vs ribaxamase patients.31
Comparison of the number of hits for each resistance gene or variant in the screening samples (T0) vs the post-antibiotic samples (T1) identified seven genes in which the increase in hit frequency was significantly greater in placebo vs ribaxamase groups. These included, five β-lactamase variants, cfxA,41 cfxA2,42 cfxA3,43 cfxA444 and cfxA5,45 and two vancomycin resistance genes, vanRD and vanSD.46 The five identified cfxA family resistance genes encode Ambler class A β-lactamases associated with penicillin and cephalosporin resistance and are commonly found in multiple bacterial genera including Bacteroides.41,44,45,47 Notably, the relative abundance of Bacteroides species, including B. uniformis, was significantly increased in the gut microbiome of placebo patients but not in ribaxamase patients,33 suggesting that Bacteroides may have harbored the cfxA genes. In support of this assumption, the cfxA gene was correlated to cephalosporin resistance in clinically isolated B. fragilis strains,48 and B. uniformis is a species within the B. fragilis group of anaerobic pathogens linked to severe bacteremia and high mortality.49 Furthermore, the cfxA genes in Bacteroides are associated with the Tn4555 transposon which could facilitate gene transfer to other microbiota species in the gut.50 Indeed, Tn4555 has been identified in Shiga toxin producing E. coli51 and cfxA genes are associated with β-lactam resistance in oral pathogens Prevotella and Capnocytophaga.52,53
The two vancomycin resistance genes, vanSD and vanRD, that displayed increased frequency in placebo vs ribaxamase patients, are variants of the vanS/vanR two component regulator system that result in constitutive expression of vanD and confer high-level vancomycin resistance in E. faecium, E. faecalis and E. avium.54,55 Increased frequencies of vanSD and vanRD genes in the placebo group following antibiotic exposure are consistent with microbiologic culture data which demonstrated a significant increase in new colonization by VRE (P=0.0001) in placebo vs ribaxamase-treated patients from T0 to T1.31 Samples which were negative for VRE by microbiologic culture but positive for vanRD in this study may be due to colonization below the level of detection for culturing or that the detected vanRD gene was not part of an active vanD operon.
Since alignment of DNA sequences against a database is only semi-quantitative, yielding total hits based on DNA sequence matches within a sample, a qPCR strategy was pursued to corroborate these data. Two representative genes, one from each class of antibiotics discussed above, were selected for this work, cfxA and vanRD.41,55 The qPCR analysis yielded copy number data for cfxA and vanRD that was in concordance with the hit frequency for each gene. In general, samples with a low number of hits for cfxA or vanRD by sequence analysis also displayed low copy numbers, although there were some cases where these data did not correspond. This discordance could have been due to sequencing errors or other methodology shortcomings.56 Other possibilities include sequence variations affecting primer annealing efficiency or sequence divergence from the CARD database. It is also possible that samples that appeared to have high a high number of hits, but low copy number by qPCR, may be due to gene fragments or truncated genes that were detected by sequencing analyses but not amplifiable by qPCR. Regardless, these few discordances did not affect the overall conclusion that the qPCR data support the findings from sequencing analyses.
The change in copy number of cfxA and vanRD over the three sample collection points across treatment groups also reflected the overall conclusions from the ribaxamase clinical study. Placebo-treated patients had a general increase in copy number of both genes at T1 which dropped back to near baseline levels by T2 for vanRD but remained elevated for cfxA, while ribaxamase-treated patients displayed a decline in copy number of both genes at T1 and T2. These findings were also consistent with the significant increase in the hit frequency of cfxA and vanRD from T0 to T1 in placebo vs ribaxamase groups.
Examination of the copy number data from individual patients can also be helpful in determining whether changes in the hit frequency of certain AMR genes represented new gene acquisition or expansion of existing genes. For cfxA and vanRD, both scenarios may have occurred. Observations of low initial AMR gene frequency that increased following antibiotic exposure are consistent with propagation of existing AMR gene pools, while undetectable baseline levels of AMR genes that then became detectable post-antibiotics are consistent with new acquisition of organisms carrying with these genes. Another AMR gene pattern observed here was a high copy number at baseline which decreased at T1, and rose again by T2. This observation is consistent with an initial loss of organisms harboring these AMR genes during antibiotic exposure that then recolonized following cessation of antibiotic therapy.
A linear regression model was used to compare the change in the hit frequency of AMR genes from T0 to T1 for four antibiotic classes of interest, with specific clinical and demographic data from the study. This analysis confirmed the correlation between placebo treatment and increased hit frequency of the cfxA β-lactamase variants and the vanRD and vanSD vancomycin resistance genes that was also observed with LEfSe analysis. In contrast, increased abundance of efflux pump genes correlated with ribaxamase treatment, rather than placebo, suggesting that organisms carrying these genes were not resistant to ceftriaxone and could persist only when ceftriaxone was degraded by ribaxamase thus preserving these bacteria.
The linear regression analysis also revealed that the use of PPI correlated with changes in the gut resistome specifically leading to significant increases in several β-lactamase genes as well as the macrolide resistance gene, mefA. PPI are a known risk factor for CDI as they disrupt the balance of the gut microbiome.36,37,57 Notably, the cfxA family of β-lactamase genes is found in oral anaerobes,58 including Prevotella and Bacteroides species.50,52,53 Thus, it is interesting to speculate that the use of PPI may have allowed these oral anaerobes to survive stomach acidity and colonize the gut in the presence of β-lactams. The finding that the cfxA6 gene demonstrated a negative correlation with the use of PPI may be indicative of why this gene did not increase in hit frequency in placebo patients while the rest of the variant family did.
While all patients in the Phase 2b clinical study were to be treated with IV ceftriaxone for their LRTI, patients could also receive macrolides as deemed necessary by the treating physician. About a third of the patients in each group received macrolides.31 A previous study found that the use of macrolides significantly correlated with an increased relative abundance of numerous efflux pump genes, many of which confer macrolide resistance.59 Interestingly, in the present work, only one macrolide resistance gene, mefA, was associated with macrolide treatment, but did not reach significance (P=0.0785). Instead, mefA was significantly associated with the use of PPI (P=0.0292). The mefA gene is an erythromycin-inducible motive efflux pump found in S. pneumoniae.59 Since the patients in the Phase 2b study were admitted with a LRTI, most of which were pneumonias, it is possible that in the presence of PPI, lung bacteria carrying this gene survived passage through the stomach to at least transiently colonize the lower gut.
LEfSe is a tool developed to find biomarkers between two or more groups using relative abundances.34 LEfSe consistently provides lower false-positive rates and can effectively aid in explaining the biology underlying differences in microbial communities. This analysis indicated that genes encoding multidrug efflux pump components displayed a significantly decreased hit frequency in the placebo group and a significantly increased hit frequency with ribaxamase treatment. There was also a correlation of increased frequency of efflux pump genes with macrolide treatment. These observations are consistent with the hypothesis that bacteria harboring these genes were sensitive to ceftriaxone and had resistance to macrolides. An exception was the adeC gene, part of the AdeABC multidrug transporter from A. baumannii,60 that displayed a highly significant positive correlation with placebo-treatment but not with macrolide exposure. Acinetobacter relative abundance, detected with 16S rRNA sequencing, was extremely low in all fecal samples33 making the significance of this finding elusive.
There were shortcomings to the methodology used for this study. The total sample set was limited to 349 samples based on the contracted budget with the CDC, and it is possible that a larger set may have yielded stronger and/or additional correlations. For example, only seven post-antibiotic samples were available from patients with CDI. In addition, the fecal DNA sequencing analysis was performed by alignment with the CARD database. Limitations to this approach include 1) constraint by the accuracy and breadth of the CARD, 2) a sequence match indicates the presence of at least a fragment of an AMR gene but does not signify that the entire gene is present, 3) sequences with homology to several genes may read as multiple hits to one gene, 4) total number of hits is only semi-quantitative and thus the findings must be qualified, and 5) detection of an AMR gene does not necessarily mean that the gene is functional and the AMR phenotype expressed. However, in this study a good correlation between the sequencing and qPCR data was observed. Assembling the sequences prior to alignment may have eliminated some of these concerns, but would also eliminate the semi-quantitative nature of the analysis. As with any comparative study, innumerable additional analyses, such as metagenomics comparison of these AMR data to our previously reported microbiome composition findings,33 are warranted and thus additional discoveries are likely to be uncovered from such future investigations.
Conclusions
This study demonstrated that exposure of the gut microbiome to ceftriaxone resulted in significant changes in the gut resistome. AMR genes, such as those encoding β-lactamases, increased significantly and this finding is consistent with the mechanism of antibiotic-mediated selection of resistant organisms. Ceftriaxone treatment also significantly increased the hit frequency of certain vancomycin resistance genes which correlated with changes in VRE colonization that were seen in the Phase 2b ribaxamase clinical study.31,33 Additional factors including the use of PPI and macrolides also appeared to precipitate changes in the gut resistome. The Phase 2b clinical study with ribaxamase demonstrated a reduction in the relative risk of CDI by 71% and a significant reduction in new colonization by VRE.31 Here, we show ribaxamase also attenuated changes to the gut resistome suggesting that ribaxamase has the potential to reduce the emergence of AMR in patients treated with IV β-lactam antibiotics. Complementary to antibiotic stewardship, ribaxamase administration would allow the continued use of a highly effective class of broad-spectrum antibiotics, the β-lactams, with diminished concern over microbiome damage and the emergence of AMR.
Clinical Trial Data
The Phase 2b clinical with ribaxamase which was the source for the samples for this study was published previously31 (ClinicalTrials.gov, number NCT02563106). Individual, deidentified participant data and additional trial and participant information regarding the clinical study are available and will be shared through the Lancet Infectious Diseases portal. Fecal microbiome sequencing data and antimicrobial resistance gene sequencing data are available through the SRA, accession PRJNA589866. These data will be accessible for at least five years from the time of publication.
Ethics Approval and Patient Consent to Participate
The Phase 2b ribaxamase study protocol and informed consent forms were approved by the appropriate institutional review boards or ethics committees (Supplemental Table S6), and the clinical trial which was the original source of the samples for this analysis was conducted in accordance with the Declaration of Helsinki. All study patients provided written consent to participate in the study and have analysis of their fecal samples conducted.
Acknowledgments
We would like to thank the team at Diversigen for their excellent technical support and Emily Hollister for submitting the sequencing data to Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra), accession PRJNA589866. We thank the CDC for funding this work, and in particular, Cliff McDonald and Sarah Yi for their helpful discussions. We also thank the investigators and study coordinators from the Phase 2b ribaxamase study for their diligence in collecting the fecal samples used for this analysis and the patients for providing the samples.
Disclosure
John F. Kokai-Kun reports that the work detailed in the pending manuscript was supported by a contract from the US Centers for Disease Control and Prevention (CDC), during the conduct of the study; personal fees from Synthetic Biologics, Inc., outside the submitted work; is a former employee of Synthetic Biologics Inc. and was employed by them during the performance of the work detailed in the paper. Dr. Kokai-Kun's current affiliation is with the Unites States Pharmacopeia, Rockville, MD, USA. Chenxiong Le reports support through a contract with the CDC, during the conduct of the study; and is an employee of Synthetical Biologics, Inc. Kenneth Trout reports support through a contract with the CDC (contract # 200-2016-91935), during the conduct of the study; and is a former employee of Synthetic Biologics. Mr. Trout's current affiliation is with REGENXBIO, Inc., Rockville, MD USA. Julia L. Cope reports that SyntheticBiologics provided funding for the work with Diversigen, Inc., through a contract with the CDC, and that Diversigen, Inc. paid salary for Julia L. Cope and funded salary to Baylor College of Medicine for Andrew J. Degar, during the conduct of the study. Nadim J. Ajami's current affiliation is with the Department of Genomic Medicine, the University of Texas MD Anderson Cancer Center, Houston, TX, USA and has nothing to disclose. Andrew J. Degar reports their portion of work was performed in affiliation with Diversigen, Inc. at Baylor College of Medicine. There were no additional financial contributions to them personally, only the fee for service for their lab, during the conduct of the study. Sheila Connelly reports a contract from the CDC (contract # 200-2016-91935), during the conduct of the study; and is an employee of Synthetic Biologics, Inc. The authors report no other possible conflicts of interest in this work.
References
1. Kinross JM, Darzi AW, Nicholson JK. Gut microbiome-host interactions in health and disease. Genome Med. 2011;3(3):14. doi:10.1186/gm228
2. Britton RA, Young VB. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology. 2014;146(6):1547–1553. doi:10.1053/j.gastro.2014.01.059
3. Smits WK, Lyras D, Lacy DB, Wilcox MH, Kuijper EJ. Clostridium difficile infection. Nat Rev Dis Primers. 2016;2:16020. doi:10.1038/nrdp.2016.20
4. Crowther GS, Wilcox MH. Antibiotic therapy and Clostridium difficile infection – primum non nocere - first do no harm. Infect Drug Resist. 2015;8:333–337. doi:10.2147/IDR.S87224
5. Knecht H, Neulinger SC, Heinsen FA, et al. Effects of beta-lactam antibiotics and fluoroquinolones on human gut microbiota in relation to Clostridium difficile associated diarrhea. PLoS One. 2014;9(2):e89417. doi:10.1371/journal.pone.0089417
6. Panda S, El Khader I, Casellas F, et al. Short-term effect of antibiotics on human gut microbiota. PLoS One. 2014;9(4):e95476. doi:10.1371/journal.pone.0095476
7. Dubberke ER, Olsen MA, Stwalley D, et al. Identification of medicare recipients at highest risk for Clostridium difficile infection in the US by population attributable risk analysis. PLoS One. 2016;11(2):e0146822. doi:10.1371/journal.pone.0146822
8. Kuntz JL, Smith DH, Petrik AF, et al. Predicting the risk of Clostridium difficile Infection upon admission: a score to identify patients for antimicrobial stewardship efforts. Perm J. 2016;20(1):20–25. doi:10.7812/TPP/15-049
9. Slimings C, Riley TV. Antibiotics and hospital-acquired Clostridium difficile infection: update of systematic review and meta-analysis. J Antimicrob Chemother. 2015;69:881–891. doi:10.1093/jac/dkt477
10. Stevens V, Dumyati G, Fine LS, Fisher SG, van Wijngaarden E. Cumulative antibiotic exposures over time and the risk of Clostridium difficile infection. Clin Infect Dis. 2011;53(1):42–48. doi:10.1093/cid/cir301
11. Vincent C, Manges AR. Antimicrobial use, human gut microbiota and Clostridium difficile colonization and infection. Antibiotics (Basel). 2015;4(3):230–253. doi:10.3390/antibiotics4030230
12. Wieczorkiewicz JT, Lopansri BK, Cheknis A, et al. Fluoroquinolone and macrolide exposure predict Clostridium difficile infection with the highly fluoroquinolone- and macrolide-resistant epidemic C. difficile strain BI/NAP1/027. Antimicrob Agents Chemother. 2016;60(1):418–423. doi:10.1128/AAC.01820-15
13. Karachalios G, Charalabopoulos K. Biliary excretion of antimicrobial drugs. Chemotherapy. 2002;48(6):280–297. doi:10.1159/000069712
14. Kokai-Kun JF, Roberts T, Coughlin O, et al. The oral beta-lactamase SYN-004 (ribaxamase) degrades ceftriaxone excreted into the intestine in phase 2a clinical studies. Antimicrob Agents Chemother. 2017;61(1):3. doi:10.1128/AAC.02197-16
15. Johanesen PA, Mackin KE, Hutton ML, et al. Disruption of the gut microbiome: clostridium difficile infection and the threat of antibiotic resistance. Genes (Basel). 2015;6(4):1347–1360. doi:10.3390/genes6041347
16. van Schaik W. The human gut resistome. Philos Trans R Soc Lond B Biol Sci. 2015;370(1670):20140087. doi:10.1098/rstb.2014.0087
17. Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13(1):42–51. doi:10.1038/nrmicro3380
18. Miller WR, Munita JM, Arias CA. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther. 2014;12(10):1221–1236. doi:10.1586/14787210.2014.956092
19. Dumford DM, Nerandzic M, Chang S, Richmond MA, Donskey C. Epidemiology of Clostridium difficile and vancomycin-resistant Enterococcus colonization in patients on a spinal cord injury unit. J Spinal Cord Med. 2011;34(1):22–27. doi:10.1179/107902610x12883422813822
20. Fujitani S, George WL, Morgan MA, Nichols S, Murthy AR. Implications for vancomycin-resistant Enterococcus colonization associated with Clostridium difficile infections. Am J Infect Control. 2011;39(3):188–193. doi:10.1016/j.ajic.2010.10.024
21. Taur Y, Jenq RR, Perales MA, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood. 2014;124(7):1174–1182. doi:10.1182/blood-2014-02-554725
22. Kamboj M, Chung D, Seo SK, et al. The changing epidemiology of vancomycin-resistant Enterococcus (VRE) bacteremia in allogeneic hematopoietic stem cell transplant (HSCT) recipients. Biol Blood Marrow Transplant. 2010;16(11):1576–1581. doi:10.1016/j.bbmt.2010.05.008
23. Boyle DP, Zembower TR. Epidemiology and management of emerging drug-resistant gram-negative bacteria: extended-spectrum beta-lactamases and beyond. Urol Clin North Am. 2015;42(4):493–505. doi:10.1016/j.ucl.2015.05.005
24. Cornaglia G, Giamarellou H, Rossolini GM. Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect Dis. 2011;11(5):381–393. doi:10.1016/S1473-3099(11)70056-1
25. Nordmann P, Cuzon G, Naas T. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis. 2009;9(4):228–236. doi:10.1016/S1473-3099(09)70054-4
26. Zmarlicka MT, Nailor MD, Nicolau DP. Impact of the New Delhi metallo-beta-lactamase on beta-lactam antibiotics. Infect Drug Resist. 2015;8:297–309. doi:10.2147/IDR.S39186
27. Carattoli A. Plasmids and the spread of resistance. Int J Med Microbiol. 2013;303(6–7):298–304. doi:10.1016/j.ijmm.2013.02.001
28. McGann P, Snesrud E, Maybank R, et al. Escherichia coli harboring mcr-1 and blaCTX-M on a novel IncF plasmid: first report of mcr-1 in the USA. Antimicrob Agents Chemother. 2016;60(7):4420–4421.
29. Kaleko M, Bristol JA, Hubert S, et al. Development of SYN-004, an oral beta-lactamase treatment to protect the gut microbiome from antibiotic-mediated damage and prevent Clostridium difficile infection. Anaerobe. 2016;41:58–67. doi:10.1016/j.anaerobe.2016.05.015
30. Connelly S, Bristol JA, Hubert S, et al. SYN-004 (ribaxamase), an oral beta-lactamase, mitigates antibiotic-mediated dysbiosis in a porcine gut microbiome model. J Appl Microbiol. 2017;123(1):66–79. doi:10.1111/jam.13432
31. Kokai-Kun JF, Roberts T, Coughlin O, et al. Use of ribaxamase (SYN-004), a β-lactamase, to prevent Clostridium difficile infection in β-lactam-treated patients: a double-blind, phase 2b, randomised placebo-controlled trial. Lancet Infect Dis. 2019;19(5):487–496. doi:10.1016/S1473-3099(18)30731-X
32. Kokai-Kun JF, Sarver JL, Carman RJ. Characterization of Clostridium difficile isolates collected during a phase 2b clinical study with SYN-004 (ribaxamase) for the prevention of C. difficile infection. Anaerobe. 2018;53:30–33. doi:10.1016/j.anaerobe.2018.07.002
33. Kokai-Kun JF, Connelly S. Ribaxamase, an orally administered β-lactamase, protects the gut microbiome in patients treated with ceftriaxone. J Trans Sci. 2019;6:1–9.
34. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60
35. Chalmers JD, Akram AR, Singanayagam A, Wilcox MH, Hill AT. Risk factors for Clostridium difficile infection in hospitalized patients with community-acquired pneumonia. J Infect. 2016;73(1):45–53. doi:10.1016/j.jinf.2016.04.008
36. Imhann F, Bonder MJ, Vich Vila A, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016;65(5):740–748. doi:10.1136/gutjnl-2015-310376
37. Jackson MA, Goodrich JK, Maxan ME, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut. 2016;65(5):749–756. doi:10.1136/gutjnl-2015-310861
38. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–834. doi:10.1056/NEJMoa1408913
39. Zapata HJ, Quagliarello VJ. The microbiota and microbiome in aging: potential implications in health and age-related diseases. J Am Geriatr Soc. 2015;63(4):776–781. doi:10.1111/jgs.13310
40. Fernandez L, Hancock RE. Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin Microbiol Rev. 2012;25(4):661–681. doi:10.1128/CMR.00043-12
41. Parker AC, Smith CJ. Genetic and biochemical analysis of a novel Ambler class A beta-lactamase responsible for cefoxitin resistance in Bacteroides species. Antimicrob Agents Chemother. 1993;37(5):1028–1036. doi:10.1128/AAC.37.5.1028
42. Madinier I, Fosse T, Giudicelli J, Labia R. Cloning and biochemical characterization of a class A beta-lactamase from Prevotella intermedia. Antimicrob Agents Chemother. 2001;45(8):2386–2389. doi:10.1128/AAC.45.8.2386-2389.2001
43. Jolivet-Gougeon A, Tamanai-Shacoori Z, Desbordes L, Burggraeve N, Cormier M, Bonnaure-Mallet M. Genetic analysis of an ambler class A extended-spectrum beta-lactamase from Capnocytophaga ochracea. J Clin Microbiol. 2004;42(2):888–890. doi:10.1128/JCM.42.2.888-890.2004
44. Meggersee R, Abratt V. The occurrence of antibiotic resistance genes in drug resistant Bacteroides fragilis isolates from Groote Schuur Hospital, South Africa. Anaerobe. 2015;32:1–6. doi:10.1016/j.anaerobe.2014.11.003
45. Molina J, Barrantes G, Quesada-Gomez C, Rodriguez C, Rodriguez-Cavallini E. Phenotypic and genotypic characterization of multidrug-resistant Bacteroides, Parabacteroides spp., and Pseudoflavonifractor from a Costa Rican hospital. Microb Drug Resist. 2014;20(5):478–484. doi:10.1089/mdr.2013.0180
46. Courvalin P. Vancomycin resistance in gram-positive cocci. Clin Infect Dis. 2006;42 Suppl 1(SUPPL1):S25–S34. doi:10.1086/491711
47. Soki J, Gonzalez SM, Urban E, Nagy E, Ayala JA. Molecular analysis of the effector mechanisms of cefoxitin resistance among Bacteroides strains. J Antimicrob Chemother. 2011;66(11):2492–2500. doi:10.1093/jac/dkr339
48. Kierzkowska M, Majewska A, Szymanek-Majchrzak K, Sawicka-Grzelak A, Mlynarczyk A, Mlynarczyk G. The presence of antibiotic resistance genes and bft genes as well as antibiotic susceptibility testing of Bacteroides fragilis strains isolated from inpatients of the Infant Jesus Teaching Hospital, Warsaw during 2007–2012. Anaerobe. 2019;56:109–115. doi:10.1016/j.anaerobe.2019.03.003
49. Aldridge KE, Ashcraft D, O’Brien M, Sanders CV. Bacteremia due to Bacteroides fragilis group: distribution of species, beta-lactamase production, and antimicrobial susceptibility patterns. Antimicrob Agents Chemother. 2003;47(1):148–153. doi:10.1128/AAC.47.1.148-153.2003
50. Ferreira LQ, Avelar KE, Vieira JM, et al. Association between the cfxA gene and transposon Tn4555 in Bacteroides distasonis strains and other Bacteroides species. Curr Microbiol. 2007;54(5):348–353. doi:10.1007/s00284-006-0411-0
51. Novais RC, Chaves MC, Gonzalez AGM, Andrade JRC. Molecular investigation of tRNA genes integrity and its relation to pathogenicity islands in Shiga toxin-producing Escherichia coli (STEC) strains. Genet Mol Biol. 2004;27:589–593. doi:10.1590/S1415-47572004000400020
52. Garcia N, Gutierrez G, Lorenzo M, Garcia JE, Piriz S, Quesada A. Genetic determinants for cfxA expression in Bacteroides strains isolated from human infections. J Antimicrob Chemother. 2008;62(5):942–947. doi:10.1093/jac/dkn347
53. Iwahara K, Kuriyama T, Shimura S, et al. Detection of cfxA and cfxA2, the beta-lactamase genes of Prevotella spp., in clinical samples from dentoalveolar infection by real-time PCR. J Clin Microbiol. 2006;44(1):172–176. doi:10.1128/JCM.44.1.172-176.2006
54. Casadewall B, Courvalin P. Characterization of the vanD glycopeptide resistance gene cluster from Enterococcus faecium BM4339. J Bacteriol. 1999;181(12):3644–3648. doi:10.1128/JB.181.12.3644-3648.1999
55. Depardieu F, Foucault ML, Bell J, et al. New combinations of mutations in VanD-Type vancomycin-resistant Enterococcus faecium, Enterococcus faecalis, and Enterococcus avium strains. Antimicrob Agents Chemother. 2009;53(5):1952–1963. doi:10.1128/AAC.01348-08
56. Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi:10.1038/nature08821
57. Tariq R, Singh S, Gupta A, Pardi DS, Khanna S. Association of gastric acid suppression with recurrent Clostridium difficile infection: a systematic review and meta-analysis. JAMA Intern Med. 2017;177(6):784–791. doi:10.1001/jamainternmed.2017.0212
58. Binta B, Patel M. Detection of cfxA2, cfxA3, and cfxA6 genes in beta-lactamase producing oral anaerobes. J Appl Oral Sci. 2016;24(2):142–147. doi:10.1590/1678-775720150469
59. Ambrose KD, Nisbet R, Stephens DS. Macrolide efflux in Streptococcus pneumoniae is mediated by a dual efflux pump (mel and mef) and is erythromycin inducible. Antimicrob Agents Chemother. 2005;49(10):4203–4209. doi:10.1128/AAC.49.10.4203-4209.2005
60. Wieczorek P, Sacha P, Hauschild T, Zórawski M, Krawczyk M, Tryniszewska E. Multidrug resistant Acinetobacter baumannii– the role of AdeABC (RND family) efflux pump in resistance to antibiotics. Folia Histochem Cytobiol. 2008;46(3):257–267. doi:10.2478/v10042-008-0056-x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.