")
Back to Journals » International Journal of Nanomedicine » Volume 16
Rhamnolipid-Based Liposomes as Promising Nano-Carriers for Enhancing the Antibacterial Activity of Peptides Derived from Bacterial Toxin-Antitoxin Systems
Authors Sanches BCP, Rocha CA, Martin Bedoya JG, Silva VL, Silva PB, Fusco-Almeida AM, Chorilli M, Contiero J, Crusca E, Marchetto R
Received 24 September 2020
Accepted for publication 25 December 2020
Published 10 February 2021 Volume 2021:16 Pages 925—939
DOI https://doi.org/10.2147/IJN.S283400
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Beatriz Cristina Pecoraro Sanches,1 Camila Aguiar Rocha,1 Jose Gregorio Martin Bedoya,1 Vinicius Luiz da Silva,2 Patrícia Bento da Silva,3 Ana Marisa Fusco-Almeida,4 Marlus Chorilli,3 Jonas Contiero,2 Edson Crusca,1 Reinaldo Marchetto1
1São Paulo State University (UNESP), Institute of Chemistry, Department of Biochemistry and Organic Chemistry, Araraquara, SP, Brazil; 2São Paulo State University (UNESP), Institute of Biosciences, Department of General and Applied Biology, Rio Claro, SP, Brazil; 3São Paulo State University (UNESP), School of Pharmaceutical Sciences, Department of Drugs and Medicines, Araraquara, SP, Brazil; 4São Paulo State University (UNESP), School of Pharmaceutical Sciences, Department of Clinical Analysis, Araraquara, SP, Brazil
Correspondence: Reinaldo Marchetto
UNESP Institute of Chemistry, Rua Prof. Francisco Degni, 55, Araraquara, 14.800-060, SP, Brazil
Tel +55 16 3301 9670
Fax +55 16 3322 2308
Email [email protected]
Background: Antimicrobial resistance poses substantial risks to human health. Thus, there is an urgent need for novel antimicrobial agents, including alternative compounds, such as peptides derived from bacterial toxin-antitoxin (TA) systems. ParELC3 is a synthetic peptide derived from the ParE toxin reported to be a good inhibitor of bacterial topoisomerases and is therefore a potential antibacterial agent. However, ParELC3 is inactive against bacteria due to its inability to cross the bacterial membranes. To circumvent this limitation we prepared and used rhamnolipid-based liposomes to carry and facilitate the passage of ParELC3 through the bacterial membrane to reach its intracellular target - the topoisomerases.
Methods and Results: Small unilamellar liposome vesicles were prepared by sonication from three formulations that included 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and cholesterol. ParELC3 was loaded with high efficiency into the liposomes. Characterization by DLS and TEM revealed the appropriate size, zeta potential, polydispersity index, and morphology. In vitro microbiological experiments showed that ParELC3 loaded-liposomes are more efficient (29 to 11 μmol·L− 1) compared to the free peptide (> 100 μmol·L− 1) at inhibiting the growth of standard E. coli and S. aureus strains. RL liposomes showed high hemolytic activity but when prepared with POPC and Chol this activity had a significant reduction. Independently of the formulation, the vesicles had no detectable cytotoxicity to HepG2 cells, even at the highest concentrations tested (1.3 mmol·L− 1 and 50 μmol·L− 1 for rhamnolipid and ParELC3, respectively).
Conclusion: The present findings suggest the potential use of rhamnolipid-based liposomes as nanocarrier systems to enhance the bioactivity of peptides.
Keywords: rhamnolipid liposomes, biosurfactants, bioactive peptides, antimicrobial activity
Introduction
Resistant bacterial infections have aroused interest due to the considerable threat to human health.1 If bacterial resistance continues on its current trajectory, there will be 10 million deaths worldwide by 2050 (overcoming cancer deaths) at a cost of up to 100 trillion dollars.2 To eradicate bacterial resistance researchers are developing novel synthetic antimicrobial peptides derived from bacterial toxin-antitoxin systems and new drug-release systems, such as rhamnolipid-based liposomes. Toxin-antitoxin (TA) systems are genetic modules found on bacterial plasmids and chromosomes. These modules are commonly two-component systems, composed of a toxin and an antitoxin.3,4 The toxin represses the metabolism and arrests growth by interfering with essential cellular process. The antitoxin is responsible for neutralizing the activity of the toxin. Under unfavorable conditions, especially stress, the antitoxin is depleted, enabling the toxin to become free and exert its toxic effect.3,4 These systems have been widely studied in recent years due to their vital functions such as programmed cellular death, the formation of biofilms, protection against bacteriophages, repairing chromosomal DNA, and responses to stress conditions, which are related to the control and regulation of bacterial growth.5–7 TA systems are grouped into seven types, based on how the antitoxin neutralizes the toxin.8 The type II system, in which both the toxin and antitoxin are proteins, has been the most widely studied.5,9 An example of a type II TA system is the ParE/ParD system identified in the RK2 plasmid of a range of prokaryotes, in which ParE is the toxin (± 100 amino acids) and ParD is the antitoxin (± 80 amino acids).10,11 ParE blocks DNA replication by inhibiting DNA gyrase activity.12–14 Studies involving synthetic peptides derived from the ParE toxin have revealed novel inhibitors of bacterial topoisomerases.15 However, the low or null capacity to permeate the bacterial plasma membrane has hindered the use of these peptides as antibacterial agents. In an attempt to circumvent this limitation and exploit the biotechnological potential of these topoisomerase inhibitors, ParELC3 - a 21-amino acid peptide from ParE toxin-15 was synthesized, purified and entrapped in liposomes in order to enhance its bioavailability and stability in a solution.
Liposomes are small, spherical, artificial vesicles generally composed of one or more phospholipid bilayers that surround an aqueous compartment.16 Liposomes are among the various delivery system for drugs or biomolecules and are effective at transporting molecules into cells. The liposome lipid bilayer can fuse with other bilayers (eg, the cell membrane), thereby delivering its contents where the free form would not reach.17 Like other microstructures, liposomes may also be formed by the self-assembly of surfactant molecules in an aqueous medium, which are classified as synthetic surfactants and biosurfactants.18 Synthetic surfactants are produced by organic chemical reactions from non-renewable resources, whereas biosurfactants, also known as surface-active agents, are produced through biological processes involving bacteria, fungi or yeast.19 There are five major categories of biosurfactants: glycolipids; phospholipids and fatty acids; lipopeptides and lipoproteins; polymeric biosurfactants; and particulate biosurfactants. These natural compounds have applications in the agricultural, pharmaceutical, food, cosmetic, and detergent industries.20 Biosurfactants offer a number of advantages, such as high biodegradability, high selectivity, and specific activity at extreme temperatures, pH, and salinity.21,22 Rhamnolipids (RLs) are among the most important biosurfactants and are predominantly produced by Pseudomonas aeruginosa. RLs are part of the glycolipid family and are composed of one or two β-hydroxy fatty acid chains connected by the hydroxyl group to one or two rhamnose molecules.23,24 In recent years, research conducted on RLs has revealed many of the astonishing applications of these natural compounds, which has led to an increase in their popularity among all categories of biosurfactants in the global market.20
The current global interest in RL production owes to their physicochemical and surfactant properties, which give these glycolipids a wide spectrum of applications in bioremediation processes, the tertiary recovery of petroleum, food additives, drugs (antifungal, antibiotic and antibiofilm agents), cleaning products, and cosmetics, as well their many “eco-friendly” properties.20,23,25–27 The antimicrobial activity of RLs has been the subject of several studies, but most authors relate this activity to their ability to interact with polysaccharides of bacterial plasma membranes, creating domains of permeability in the cells, producing pores disorganizing the cellular balance and causing cell death.26,28 RLs are well-characterized biosurfactants but are little studied and underused for nanotechnology purposes. The literature reports a patent related to the use of rhamnolipids and their application as liposomes, studies involving the production and characterization of vesicles, emulsion, microemulsion and more recently nanoemulsion for applications in the food, pharmaceutical and agricultural industries.29–33 However, very little is known regarding the formation of rhamnolipid liposomes containing encapsulated peptides.
The aim of the present study was to use RLs produced by Pseudomonas aeruginosa for the production of liposomes containing encapsulated ParELC3 in order to enable the peptide to pass through the bacterial membrane and reach its intracellular target - the topoisomerases. We also evaluated the recognized activity of both (the rhamnolipid and peptide), as well as the biotechnological potential of these molecules for combating bacterial infections.
Materials and Methods
Materials
Fmoc-Rink amide MBHA resin (Rink amide resin; 0.52 mmol/g), N-hydroxybenzotriazole (HOBt), N,N′-diisopropylcarbodiimide (DIC), and all 9-fluorenylmethyloxycarbonyl-amino acids (Fmoc-amino acids) in their appropriately protected form were purchased from AAPPTec (Louisville, KY, USA). 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, >99% purity) and cholesterol (Chol, >99% purity) were purchased from the Avanti Polar Lipids, Inc (Alabaster, AL, USA). Resazurin (AlamarBlue®) and fetal bovine serum (FBS) were purchased from ThermoFisher (Waltham, MA, USA). Roswell Park Memorial Institute (RPMI 1640) medium was acquired from Sigma (St. Louis, MO, USA). The solvents and all other reagents were purchased from Merck. All chemicals were used as received without further purification.
Methods
Peptide Synthesis
The peptides ParELC3, WParELC3 and CFParELC3 (Figure 1) were synthesized manually through to solid-phase synthesis method using standard Fmoc (9-fluorenylmethoxycarbonyl) chemistry with Rink amide resin and DIC-HOBt activation, as previously reported.15 Deprotection and cleavage were achieved by treatment with trifluoroacetic acid (TFA) solution (TFA/water/1.2-ethanedithiol/triisopropylsilane, 94:2.5:2.5:1, v/v), 10 mL/g peptidyl-resin for 2 h at room temperature. Crude peptides were precipitated in cold diethyl ether, centrifuged (four times), dissolved in 10% aqueous acetic acid and lyophilized. Peptide purification and analysis were carried out by semi-preparative and analytical reverse phase high-performance liquid chromatography (RP-HPLC), respectively, using the Prominence HPLC System (Shimadzu, Japan). Peptide identities were checked by electrospray–ion trap mass spectrometry (ESI-MS) using an Amazon mass spectrometer (Bruker Daltonics, Billerica, MA, USA). Pure WParELC3 and CFParELC3 concentrations were determined by UV spectroscopy, using an extinction coefficient of 5600 M−1·cm−1 at 280 nm and 83,000 M−1·cm−1 at 495 nm, respectively. Peptide purity was estimated to be higher than 98% by HPLC analysis.
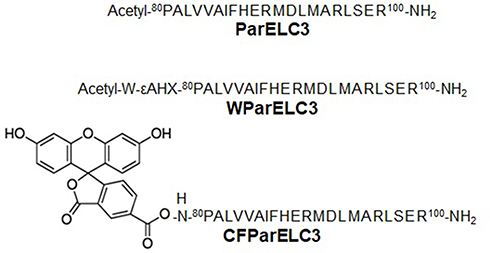 |
Figure 1 Primary structure of synthetized peptides (εAHX = ε-aminocaproic acid). |
Rhamnolipid Production
A mutant strain of Pseudomonas aeruginosa LBI characterized by high productivity (P. aeruginosa LBI 2A1) was used. RL production was conducted as previously described, in a 1-L Erlenmeyer flask containing 300 mL of a Ca-free mineral salt medium consisting of 15.0 g.L−1 NaNO3, 0.5 g.L−1 MgSO4×7 H2O, 1.0 g.L−1 KCl and 0.3 g.L−1 K2HPO4.34 Soybean oil was added (2%, v/v) as the carbon source. We also added 1 mL·L−1 of a trace element solution containing 2.0 g.L−1 sodium citrate×2 H2O, 0.28 g.L−1 FeCl3×6 H2O, 1.4 g.L−1 ZnSO4×7 H2O, 1.2 g.L−1 CoCl2×6 H2O, 1.2 g.L−1 CuSO4×5 H2O, and 0.8 g.L−1 MnSO4 x H2O. The flasks were incubated at 30°C for 120 h and 200 rpm on a rotary shaker. The initial pH of the broth was adjusted to 6.8. RLs were extracted from the cell-free culture medium through centrifugation at 12,000 rpm for 20 min. The supernatant pH was adjusted to 2.0 with 3.0 mol·L−1 H2SO4 and 300 mL of ethyl acetate was added. The mixture was vigorously shaken for 10 min and allowed to settle until phase separation. The organic phase was removed and the operation was repeated. The RL product was concentrated from the pooled organic phases in a rotary evaporator. The product was dissolved in methanol and concentrated again for solvent evaporation at 45°C.
Preparation and Characterization of Liposomes
Lipid films were prepared using different formulations (Table 1). All films were prepared by adding the appropriate amounts of lipid in chloroform to a test tube and drying with a nitrogen flow. Residual traces of organic solvent were further removed under vacuum in a SpeedVac system overnight. The lipid film was hydrated with 1 mL of PBS buffer (pH 7.4) or 1 mL of a 100 µmol·L−1 peptide (ParELC3, WParELC3 or CFParELC3) solution in PBS (pH 7.4) at a temperature above the phase transition temperature of the lipids. The suspension of large multilamellar vesicles was submitted to sonication to produce large unilamellar vesicles using a Q700 sonicator (QSonica, Newtown, CT, USA) at 21% amplitude for 6 minutes with a 20-second pause every 1 minute.
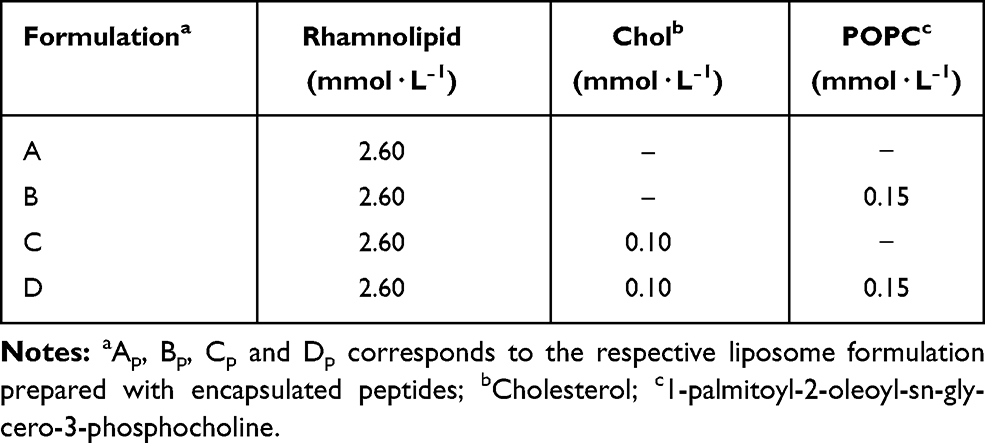 |
Table 1 Liposome Compositions |
The mean hydrodynamic diameter (HD), polydispersity index (PDI), and zeta potential (ZP) were determined using a combined dynamic light scattering/particle electrophoresis instrument (Zetasizer Nano ZS system, Malvern Instruments Ltd., UK). The samples consisted of 1 mL of a suspension of empty and peptide-loaded liposomes in PBS buffer, pH 7.4. All experiments were run in triplicate at 25°C. Transmission electron microscopy (TEM) was used to analyze the structure and morphology of the liposomes. A 20-µL aliquot of each sample (empty and peptide-loaded liposomes) was loaded on a carbon film-coated copper grid and allowed to stand for 10 min, followed by the removal of excess sample with filter paper. Sample-loaded grids were stained using a drop of solution containing 1% uranyl acetate (w/v) in water. After 2 min, the excess liquid was removed and the sample was dried at room temperature. Images were acquired using a JEOL 100CX transmission electron microscope (Jeol USA, Inc, Peabody, MA, USA) operating at an accelerating voltage of 100 kV.
Peptide Encapsulation Efficiency (EE)
For the encapsulation efficiency (EE) analysis, the liposomes were prepared using the ParELC3 peptide derivative containing an N-terminal tryptophan residue (WParELC3). EE was determined by centrifugation. With this method, the encapsulated peptides (Penc) within the liposomes are separated from the unencapsulated peptides (Punenc) by passing the prepared samples through AMICON Ultra-0.5 (50 KDa cutoff) device (Merck, Germany) at 14,000 x g, for 14 minutes at 25°C. The unencapsulated peptides crossed through the membrane and were then collected and quantified in triplicate at 280 nm on a UV-1601PC spectrophotometer (Shimadzu, Japan), using a previously designed standard curve (y = 0.0055x + 0.00002, R2 = 0.9999, in which y and x correspond to absorbance and peptide concentration (μmol·L−1), respectively). The EE was calculated by Equation 1, in which Ptotal is the initial peptide quantity (in 100 µL solution) used in the vesicle preparation.
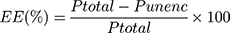
Storage Stability
Liposome formulations Ap, Bp, Cp and Dp (Table 1) were stored at 4°C in PBS buffer (pH 7.4) for the evaluation of storage stability. The HD and PDI were analyzed for 30 days after preparation.
Peptide Release
Centrifugation using an AMICON Ultra-0.5 (50 KDa cutoff) device (Merck, Germany) was employed for the in vitro peptide release assay. A suspension of liposomes containing encapsulated WParELC3, in PBS buffer (12 mL) was prepared and kept under agitation at 150 rpm for 48 hours at 37°C. Hourly, 0.5-mL aliquots were removed, immediately placed in the device and centrifuged at 14,000 x g for 14 minutes at 37°C. The released peptides were collected and quantified in triplicate at 280 nm on a UV-1601PC spectrophotometer (Shimadzu, Japan), using a previously designed standard curve, as in the EE assay. For the controls, the same procedure was performed using empty liposomes or free peptide rather than peptide-loaded liposomes.
Microorganisms and Growth Conditions
Escherichia coli O157:H17 (ATCC 43895) and Staphylococcus aureus (ATCC 14458) were used in the membrane permeability and antimicrobial susceptibility experiments. Briefly, bacteria from aliquots stored at −80°C were sub-cultured in Muller Hinton Broth (MHB) for 24 h at 37°C. A single pure colony of each bacteria was inoculated into 10 mL of MHB and incubated for 24 h at 37°C and 150 rpm in an orbital shaker incubator (Tecnal, Brazil). Cell growth was standardized spectrophotometrically (λ = 600 nm) to give 1 x 108 CFU.mL−1 (0.5 McFarland scale) and then diluted to obtain the final concentration of 1 x 107 CFU.mL−1, used in the experiments.
Membrane Permeability Assay
Liposomes prepared with the peptide derivative containing an N-terminal 5-carboxyfluorescein residue (CFParECL3) were used in this study. Aliquots of 100 μL of liposomes with encapsulated CFParELC3 (100 µmol·L−1) were transferred to the wells of a sterile polypropylene 96-well microplate containing 80 µL of MHB. Next, 20 µL of the standardized bacterial cell suspension was added, followed by incubation for 24 h at 37°C. The bacterial cells were then suspended in filtered PBS buffer, pH 7.4, and centrifuged at 2,000 x g for 3 min. This process was repeated three times to ensure the complete removal of the medium. The samples were examined under a Zeiss LSM 780 Confocal Microscope (Zeiss, Jena, Germany) with a 63x oil objective and an excitation wavelength of 488 nm to determine the presence of peptide in the cells. Images obtained at 522 nm (emission wavelength) were treated using the ZEN Black software. Free CFParELC3 peptide (100 µmol·L−1) was also used for comparative purposes.
Antimicrobial Susceptibility
The antimicrobial activity of peptide-loaded liposomes was determined in comparison to that of the free peptide using the broth microdilution method described in the M7-A6 reference guide of the Clinical and Laboratory Standards Institute, with modifications.35 Eighty µL of standardized bacterial inoculum in MHB were added to each well of a sterile 96-well microplate. Aliquots of 20 µL of each serially diluted (100 to 3.125 µmol·L−1) ParELC3-loaded liposome preparation were added to each well of the microplate for incubation. Ciprofloxacin (100 µmol·L−1) was used as the positive control and MHB was used as the negative control. Empty liposomes from each formulation and free ParELC3 were also tested. After incubation for 24 h at 37°C, 30 μL of resazurin solution (100 µg.mL−1) were added followed for future incubation at 37°C for 2 hr. The color change was assessed visually. Pink indicates bacteria growth, whereas blue indicates growth inhibition. The minimum inhibitory concentration (MIC) was determined as the lowest peptide concentration in the liposome (considering EE values) at which no bacterial growth was observed.36 This assay was performed in triplicate.
Hemolytic Activity
Human erythrocytes were prepared right before the experiments from red blood cell concentrates supplied by Hemonúcleo Regional de Araraquara - UNESP. Cells were washed three times with PBS, pH 7.4, followed by three centrifugations for 5 min at 2,000 x g. After washing, the erythrocytes were resuspended to 1% (v/v) in PBS. The erythrocyte suspension (100 µL) was incubated with 100 µL of free RL and each empty or peptide-loaded liposome preparation serially diluted in PBS (1.3 to 0.041 mmol·L−1) at 37°C for 1 h. After incubation, the microtubes were centrifuged for 5 min at 3,000 x g. The supernatants (100 µL) were transferred to a 96-microwell plate and the absorbance read at 540 nm with a Biotek Epoch microplate reader. A solution containing 0.1% Triton X-100 was used as a positive control of hemolysis and PBS was used as a negative control. Three independent experiments were performed for each condition.
Cytotoxicity Evaluation
The cytotoxic effect of the free peptide and respective loaded-liposomes was investigated by the Resazurin assay on HepG2 cells (human liver carcinoma, ATCC HB-8065) obtained from the cell bank of Rio de Janeiro (BCRJ).37 The cells were cultured in RPMI-1640 medium supplemented with 10% (v/v) FBS, 2 mmol·L−1 glutamine, 100 μg·mL−1 streptomycin and 100 U.mL−1 penicillin, under a humidified atmosphere of 5% CO2 at 37°C in the incubator (New Brunswick Galaxy 48R, Eppendorf, Germany). Growing cells were seeded in the sterile 96-wells of flat bottom plates at a density of the 5 × 104 cells/well and allowed to attach for 24 h at 37°C in 5% CO2. The medium was then removed and the cells were washed with PBS and SDS, pH 7.4. Aliquots of 100 µL of free ParELC3 and of each peptide-loaded liposome preparation serially diluted in the culture medium (50 to 6.25 µmol·L−1) were added to each well of the microplate, followed by incubation for 24 h at 37°C. Subsequently, 10 µL of Resazurin reagent (10% of incubation volume) were added, followed by incubation for 4 h at 37°C. Absorbance was then read at 570 nm using an Epoch microplate reader (BioTek, USA). Negative and positive control treatments were carried out with culture medium and DMSO, respectively. All samples were analyzed three times. Cell viability was obtained by the ratio between the optical density (OD) of the treated cells and that of the negative control. Empty liposomes from each formulation were tested and used for the OD corrections (when incubated in the absence of cells).
Statistical Analysis
Hemolysis and cytotoxic effect assays data were analyzed by one-way analysis of variance ANOVA using R version 3.6.1 software with Tukey’s multiple comparison test. The data are presented as the means ± standard deviation. The statistical significance was defined as a P-value of less than 0.05.
Results and Discussion
Peptides from toxin proteins of TA systems have been described as a new class of molecules with inhibitory activity toward bacterial topoisomerases.15,38,39 However, the low capacity to permeate the bacterial plasma membrane prevents the use of these peptides as antibacterial agents. RL liposome production and use as a peptide delivery constitutes an attempt to circumvent this limitation. In this context, ParELC3 (MW = 2495.97), a 21-amino acid peptide recognized for inhibiting bacterial DNA gyrase and topoisomerase IV activity, and its fluorescent derivatives - WParELC3 and CFParELC3 - were chemically synthesized with good yield (48 to 55%) and high purity (98 to 99%). ParELC3 is a water-soluble peptide. ParELC3 is a water-soluble peptide. However, it cannot pass through by diffusion channels of bacterial outer membranes, formed by Porins. Porins are proteins found in the membranes of Gram-negative bacteria, intended for passive diffusion of hydrophilic molecules with the molecular weight of no more than 600 Da.40,41 They form transmembrane water-filled channels (or pores) that provide the exchange of low-molecular substances between cell and environment. ParELC3 is too large to fit and pass by these channels.41 . Thus, ParELC3 and its fluorescent derivatives were encapsulated in RL liposomes prepared by sonication to generate the respective nanosized unilamellar vesicles. RL extracted from the liquid culture of P.aeruginosa LBI 2A1 was used as the primary lipid at a constant concentration of 2.60 mmol·L−1 or 6.5 times the critical micelle concentration for all formulations (Table 1).31 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, 16:0–18:1Δ9c), which is an excellent synthetic substitute for Egg PC, was chosen as the co-surfactant (formulations B and D) to influence the fluidity and confer greater stability to the vesicles. Moreover, the low transition temperature of this surfactant (−2°C) ensures that the resulting lipid bilayers remain fluid at room temperature.28,42,43 The ideal POPC concentration employed in the preparation of the vesicles (0.15 mmol·L−1) was determined after a set of stability tests in the range of 0.05 to 1.0 mmol·L−1. To provide stability and regulate the fluidity of the proposed lipid bilayers, we also used cholesterol (Chol) in two liposome formulations (C and D - Table 1). The Chol concentration was 0.10 mmol·L−1, as a higher concentration would significantly reduce the vesicle size, and consequently, encapsulation efficiency besides, causing the incomplete incorporation of the Chol in the RL vesicles, leading to the formation of Chol precipitates.31 The synthesis procedure of ParELC3 as well as rhamnolipid production and liposome formation is illustrated in Figure 2.
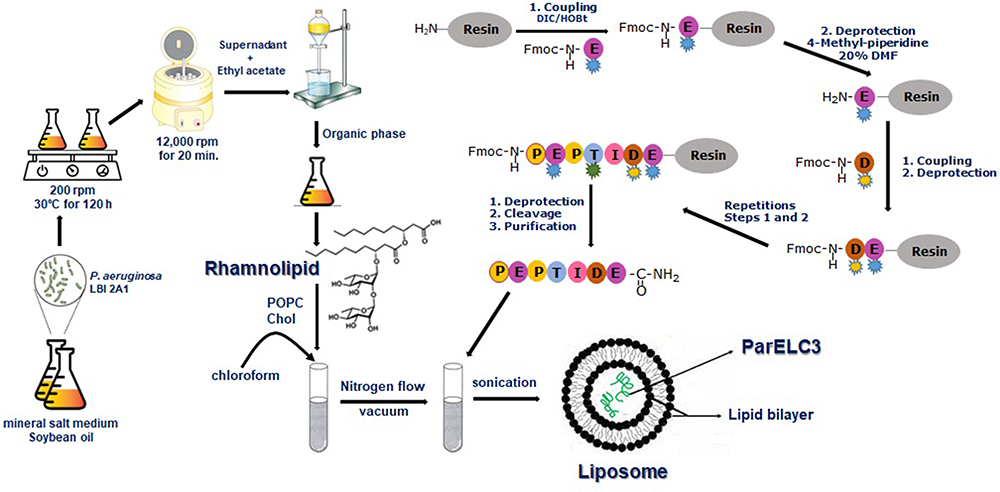 |
Figure 2 Schematic illustration of ParELC3 synthesis, rhamnolipid (RL) production and liposomes preparation. The ParELC3 (identified as PEPTIDE) was prepared by solid-phase method using standard Fmoc chemistry. The RL was produced by P. aeruginosa LBI 2A1 in a mineral salt medium and by organic extraction. Empty and peptide-loaded RL liposomes were prepared by sonication method in the POPC and Cholesterol presence. |
Vesicle Characterization
The average size and size distribution of liposomes are important aspects to consider, especially when intended for use as hydrophilic or hydrophobic drug carriers. TEM and DLS were used to evaluate the mean diameter and size distribution of liposomes formed. The TEM images of the empty liposomes show a unilamellar structure for all formulations (Figure 3), which is an important feature for the peptide encapsulation process, as the internal aqueous volume of a unilamellar vesicle is greater than that of a multilamellar one. However, these images offer little utility in evaluating absolute sizes, as the liposomes appear in the form of non-spherical structures due to the vacuum and, in principle, the high-energy electron beam used for the imaging.44 Therefore, DLS was used to evaluate the mean diameter and size distribution of liposomes, which measures the hydrodynamic diameter of particles suspended in a solution. The HD, ZP and PDI for empty and peptide-loaded liposomes are displayed in Table 2. For the different formulations, the HD of empty liposomes ranged from 93.3 to 118.6 nm and that of the peptide-loaded liposomes ranged from 128.3 to 212.3 nm independently of the peptide used (ParELC3 or WParELC3). This increase in size indicates that the peptides were incorporated into the liposomes, probably in the inner aqueous compartment because it is a water-soluble molecule.45 ZP is a measure related to the electrical charge and stability of the vesicles. As a rule, a system is considered electrostatically stable if the absolute ZP is higher than above 20 mV.28,46 The ZP of the empty liposomes was slightly negative (close to −20 mV) for all prepared formulations due to their rhamnolipid-based composition and became more negative after peptide incorporation. ParELC3 and WParELC3 have no net charge at pH 7.4 and, therefore, do not affect the final charge of liposomes. However, the insertion of the peptide into the liposome is accompanied by a reduction in external ionic strength, which is an effect that may have led to the slight increase in the negative ZP found in the Ap, Bp, Cp and Dp formulations.47 POPC is a zwitterionic phospholipid that can have a slightly negative charge in neutral pH and under the ionic strength conditions used in this study.48 This could explain the slight increase in the negative ZP values obtained for formulations B and D compared to A and C, respectively, as well as their respective encapsulated forms. POPC should also establish a molecular interaction between the acyl chain and the RL molecule as a consequence of the intercalation of the POPC molecule between the rhamnolipids, characterizing the observed reduction in the HD of formulations B and D, which has previously been reported for phospholipid vesicles as well as other lipid compositions.42,49,50 Chol had no apparent influence on the size or ZP of the RL liposomes. Chol is one of the most widely used co-solutes in vesicle systems.31 The incorporation of Chol in the lipid bilayer provides stability and alters the fluidity of hydrocarbon chains in the bilayer assisting in the prevention of peptide leakage.31,51 PDI values determine particle size distribution. Values of approximately 0.20 indicate monodispersed particles, whereas values >0.4 indicate polydisperse distribution or a high standard deviation.52,53 The PDI values for the empty liposomes were close to 0.20, which characterizes monodispersion. The insertion of the peptide increased the PDI values to 0.37, on average, for all lipid preparations, indicating homogeneity among the samples analyzed. However, the liposomes with encapsulated peptides were less homogeneous compared to the respective empty samples. Despite the differences, both the HD and PDI values were within the acceptable range for lipid nanoparticles (HD < 300 nm and PDI < 0.4) characterized as a monodisperse system, even after insertion of the peptides.52
 |
Figure 3 TEM micrographs of the rhamnolipid vesicles produced by sonication method: (A) formulation A; (B) formulation B; (C) formulation C; (D) formulation D. |
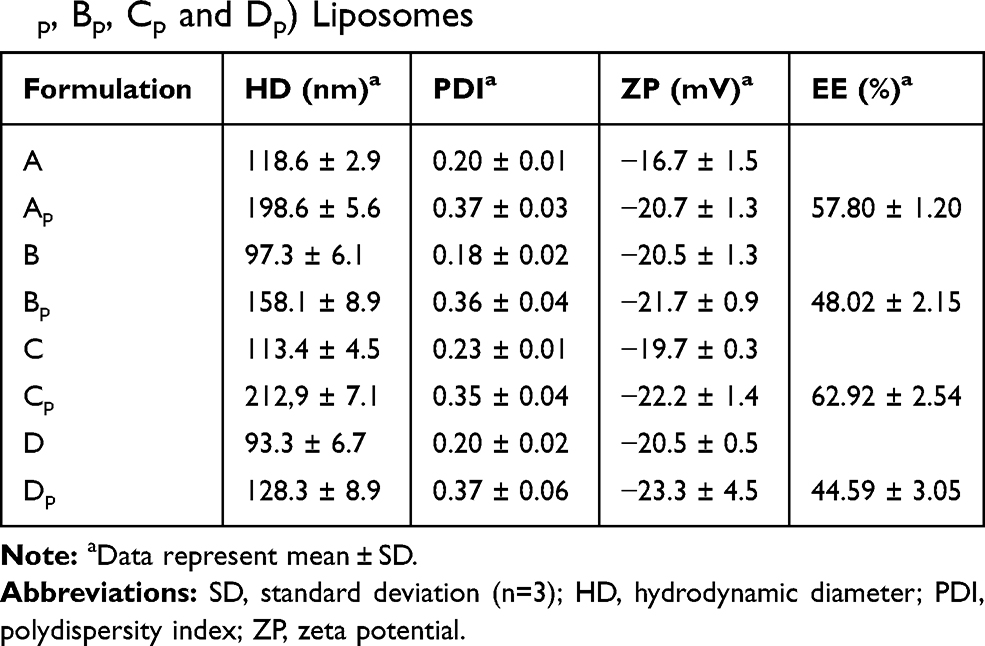 |
Table 2 Dynamic Light Scattering (DLS) and Encapsulation Efficiency (EE) Results for Empty (A, B, C and D) and Loaded (Ap, Bp, Cp and Dp) Liposomes |
Encapsulation Efficiency (EE)
EE is the expression of the amount of peptides loaded into the liposomes. A high EE is desirable because it increases the amount of peptide to be transported and made available to reach its target within bacterial cells. EE was highest in formulation C (62.92%) among all liposome formulations. This could be attributed to the presence of Chol. RL liposomes without Chol had a good EE that improved when Chol was in the membrane (Table 2). This increase in EE value may be due to the increase in rigidity and stability of the bilayer membrane due to the Chol.54 Indeed, the improvement in the EE of the RL vesicles observed at Chol concentrations of up to 100 μmol·L−1 has been attributed to the increase in the rigidity, stability and hydrophobicity of the bilayer membrane as well as the reduction in membrane permeability.31,54 EE decreased significantly with the incorporation of POPC to liposome formulation, which was an expected result, given the smaller particle size of the empty forms. Moreover, POPC and RLs constitute a mixture of unsaturated/saturated lipid characterized by both high stability and fluidity.55 These should make the membrane more permeable, which possibly explains the lower observed EE found for formulations Bp and Dp.
Stability Evaluation
To evaluate the storage stability of the samples, the liposomes were stored at 4°C and HD and PDI were analyzed as stability variables. The temperature of 4°C was used because, according to some studies, it favors the storage of phospholipid liposomes obtained through both exclusion and sonication techniques.56 All samples exhibited stability after 10 days, as evidenced by the constant HD and PDI values. After 30 days, formulations A and C had an increase in HD, whereas this variable remained practically constant for B and D (Figure 4). The presence of POPC conferred greater stability to formulations B and D, as evidenced by the lack of a significant change in PDI for 30 days. POPC confers greater fluidity to the lipid bilayer, which can result in a better adjustment of the bilayer components throughout the storage period, as reported for other lipid structures.50,56 The addition of POPC to the RL liposomes made peptide encapsulation more difficult, as seen previously, but may improve liposome stability. Comparing the characteristics between formulations B and D, formulation D was even more stable, probably due to the Chol used in its composition. In this case, Chol must prevent the packing of POPC, inducing its orientation, which decreases its interaction with the RLs, conferring greater rigidity to the liposomes and preventing their aggregation, as evidenced by the insignificant change in PDI. No differences were found in the ZP among the samples throughout the storage period.
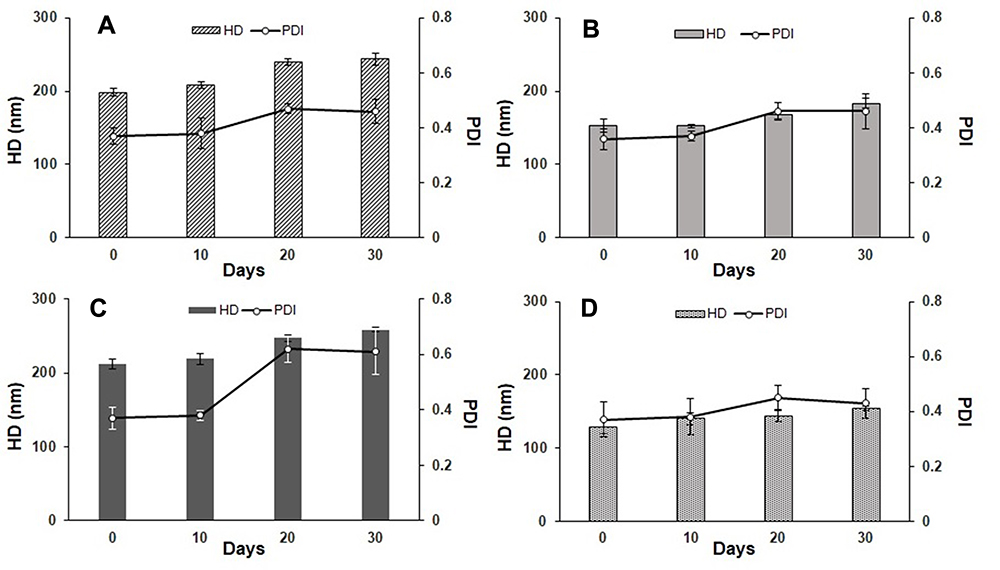 |
Figure 4 Hydrodynamic diameter (HD) and polydispersity index (PDI) for (A) Ap, (B) Bp, (C), Cp and (D) Dp formulations stored at 4°C for 30 days. |
In vitro Peptide Release
The release profiles of WParELC3 from liposome preparations demonstrated a gradual release for up to 15 hours, indicating that the peptide is effectively incorporated within the liposome and not on its surface.50 The release behavior of the liposomal samples is summarized in the cumulative percentage release up to the maximum value predicted by the EE studies (Figure 5). The use of POPC in the liposome preparations resulted in a reduction in particle size and the fastest peptide release for the Bp sample (6 h for complete release). Comparatively to the Ap sample (8 h for 100% release), this result shows that smaller liposomes release drugs more readily than larger liposomes. The curvature of the vesicle is greater in smaller liposomes and, consequently, the packing between the lipids in the membrane is looser, which facilitates peptide release.57 However, this effect was not found for the Dp sample, which also had POPC in its formulation and a smaller particle size than the corresponding formulation without this phospholipid (Cp). Complete release was found after 15 h for formulation Dp and 10 h for formulation Cp. This longer release of formulation Dp may be attributed to the presence of Chol in its composition. The incorporation of Chol in the vesicle membrane leads to a compact well-organized bilayer, which slows the release of the encapsulated peptide from the vesicles.51 These results together with the stability results clearly indicate that the RL liposome prepared with POPC and Chol (formulation Dp) can be used effectively as a ParELC3 delivery system. Variables such as composition, peptide loading, peptide release from lipid vesicles, particle size, and stability, which should be considered when liposomes are used for peptide delivery systems, were positively evaluated in the present study.
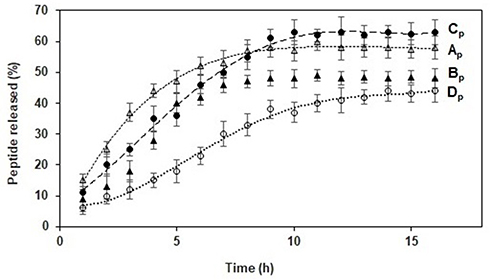 |
Figure 5 In vitro release profile of WParELC3 from Ap, Bp, Cp and Dp formulations. |
Membrane Permeability
The ability of the RL vesicles to entrap and release peptides suggests their potential use as a nanocarrier system. However, to evaluate the ability of RLs liposomes to deliver the peptide content directly into the cell cytoplasm, confocal laser scanning microscopic analysis was performed with E. coli and S. aureus cultures incubated with free or encapsulated CFParELC3 in the RL liposomes. The green fluorescence visible in Figure 6A–D clearly shows that the fluorescent peptide is accumulated in the cytoplasm of both bacteria. In contrast, no labeling was observed when the cells were incubated under the same conditions with the free fluorescent peptide (Figure 6E). This demonstrates that encapsulation in the RL liposomes enabled the peptide to pass through the plasma membrane and accumulate inside the bacteria. Liposomes can interact with cells by four different mechanisms and it often is difficult to determine which mechanism is occurring.58 However, fusion of the lipid bilayer of the liposome with the bacterial plasma membrane is likely. In this case, the fusion of the vesicle bilayers could be causing destabilization of the cell membrane, thereby, facilitating the observed release of the peptide in the cytoplasm. Bacteria incubated with liposomes of formulations C and D exhibited greater fluorescence than those incubated with formulations A and B. Chol was present in both C and D. As mentioned above, Chol reduces membrane fluidity and confers resistance to membrane deformation, increasing its stability.31,51,54 However, contrary to what might be expected, Chol seems to facilitate the merger process. In this case, a factor to consider is the increase in the average curvature of the liposome lipid bilayer induced by Chol. Indeed, Chol has been shown to increase the average membrane curvature and cause an uneven distribution of this curvature.59 The high curvature induces defects in such a way that the membrane obtains the required energy for the fusion of lipids, which corroborates our results.60
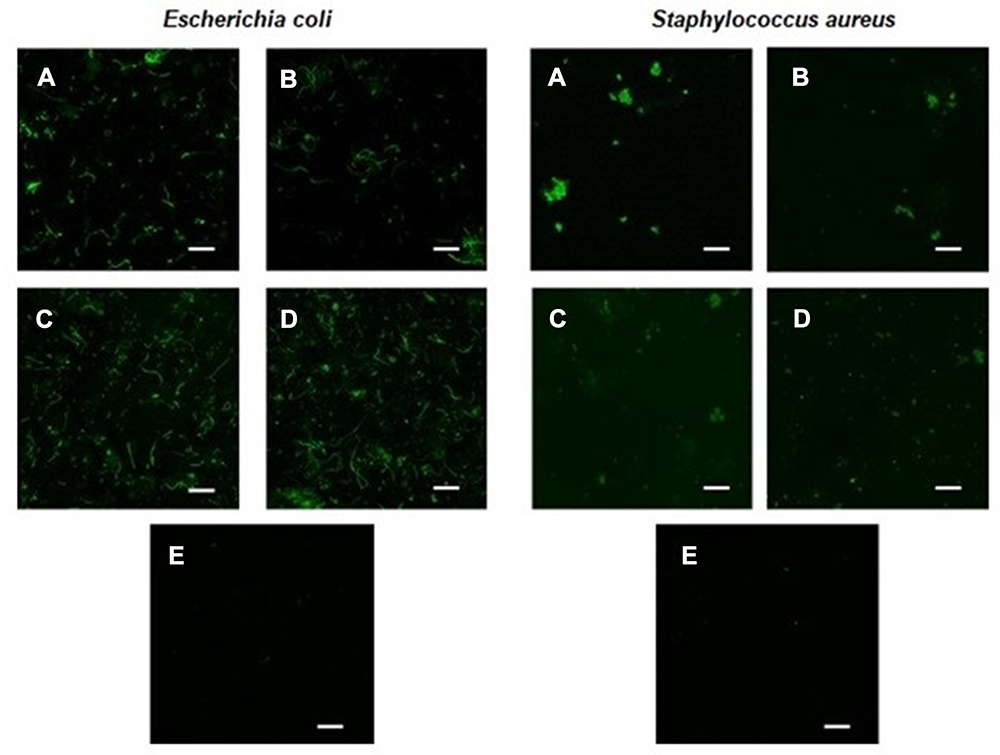 |
Figure 6 Fluorescence confocal microscopy images of Escherichia coli and Staphylococcus aureus treated with encapsulated CFParELC3 in RL liposomes (A–D formulations) and CFParELC3 free (E). Scale bar: 10 µm. |
Antibacterial Susceptibility
In the microbiological assay, free ParELC3 was inactive against both strains tested (MIC > 100 µmol·L−1). In contrast, when the peptide was loaded into liposomes of different formulations, good MIC values were found (Table 3), such as 11 µmol·L−1 for S. aureus. As control experiments, the respective empty liposomes were unable to affect bacterial growth. It is clear that encapsulation in the RL liposomes enabled the peptide to pass through the plasma membrane and accumulated in the cytoplasm, as showed by membrane permeability assay. Otherwise, if attached to the bacterial surface, the peptide would be unable to affect bacterial growth since its target is intracellular. Peptide-loaded liposomes C and D were the most effective against the microorganisms tested, which is in agreement with the results of the permeability tests. Comparatively to A and B formulations, the Chol, in C and D must increase the average curvature of the liposome lipid bilayer, inducing defects in the bilayer that generate the required energy for the lipid fusion process. Thus, the released peptide accumulated in the bacterial cytoplasm, enabling it able to reach its target - the DNA topoisomerases. Differences in inhibitory patterns against the two bacterial strains were also observed for the peptide-loaded liposomes C and D. The growth of E. coli was inhibited more efficiently by formulation C, possibly due to its higher peptide content (EE = 62.92%) and, consequently, the greater amount of peptide released in the bacterial cytoplasm. For S. aureus, which is a Gram-positive strain, greater inhibition was achieved with formulation D, even with the lower peptide content (EE = 44.59%). In this type of bacteria, the thick external peptidoglycan layer acts as a barrier that hinders direct contact with the liposomes. In this case, the fluidity of the liposomal bilayer is particularly important for the release process.61 A more fluid liposomal bilayer is able to release its compounds after interaction with the external peptidoglycan barrier, whereas liposomes with more rigid structure (such as formulation C) release compounds slowly and are therefore less effective against Gram-positive bacteria.61,62 POPC (in formulation D) increased the fluidity of the liposomal bilayer, promoted the reduction in the mean diameter of the vesicles, and enabled stronger interaction between the liposomes and cells, resulting in greater peptide release. Moreover, the affinity of the peptide with the topoisomerases of the different strains is another factor that influences bacterial susceptibility. Previous enzymatic inhibition studies have shown that ParELC3 achieved the complete inhibition of the activity of Escherichia coli DNA gyrase and topoisomerase IV at 20 and 10 µmol·L−1, respectively.15 These values are in agreement with the MIC values obtained for liposomes C and D loaded with ParELC3, demonstrating that the observed antibacterial activity can be attributed exclusively to the peptide and not to a possible synergism of the activity of both the rhamnolipid and peptide.
 |
Table 3 Antimicrobial Activity of Empty (A, B, C and D) and Peptide-Loaded Liposomes (Ap, Bp, Cp and Dp) in Compare to Free ParELC3 |
Hemolytic Activity
In addition to the antibacterial assays, the effects of RL vesicles on human healthy cells were also investigated since pure RL has been described as a hemolytic lipid.63 The hemolytic activity assays confirmed this effect of RL on the red blood cells (Figure 7). High hemolytic activity was obtained both at concentrations above (1.3 mmol·L−1) and below (0.163 mmol·L−1) of CMC. (>75% of hemolysis), a result previously reported for pure dirhamnolipid.64 From this point, hemolysis was not detected for RL. Hemolysis decrease was observed when RL is structured in liposomes (sample A). Indeed, hemolysis induced by RL liposomes remained between 60 and 40%; on the contrary, the free RL hemolytic activity has always been greater than 75%. This significant difference can be attributed to the higher adsorption of the free RL onto red cell membranes. In this case, in a similar manner to antimicrobial peptides, RL intercalates into the phospholipid bilayers and produces a structural perturbation that affects the membrane function.64 The membrane is subsequently ruptured and the hemoglobin released. In its turn, the adsorption of RL liposomes must be comparatively smaller or slower, which may explain the reduction in hemolytic activity obtained for the RL in this structured form. At 1.3 mmol·L−1 concentration, the B, C and D liposomes prepared with POPC and Chol showed hemolytic activity significantly lowers (p < 0.05) than A liposome. POPC, as a consequence of its intercalation between the RL molecules, should establish molecular interactions between acyl chain and the RL molecule resulting in a bilayer more stable. On the other hand, Chol provides stability and alters the fluidity of hydrocarbon chains in the bilayer conferring greater rigidity to the liposomes and preventing their aggregation, which may be reflected in the hemolytic activity reduction of these liposomes. At lower concentrations, these factors appear to have no significant influence on hemolytic activity due to the dilution effect. The hemolysis (%) induced by the different peptide-loaded liposome types, presented in Figure 7B, was significantly lower than the respective empty liposomes, but with the maintenance of the effects caused by POPC and cholesterol as commented above. The peptide incorporation into the liposomes is accompanied by a relative increase of the liposomes size (Table 2). In terms of hemocompatibility, the surface of the liposome plays an unquestionable role so that hemolysis rates are closely related to the vesicle size.65,66 The vesicle size increase with consequent surface area reduction produced by peptide encapsulation may explain the observed lower hemolytic activity of peptide-loaded liposomes. For higher RL concentrations and therefore of encapsulated peptide, the lower hemolysis values were 22.75 ± 5.37% and 15.81 ± 6.29% for Bp and Dp liposomes at 0.325 mmol·L−1 (12.5 µmol·L−1 peptide). Based on its physical and chemical properties, the Dp liposome had been previously highlighted as an effective delivery system for the ParELC3 peptide. Since formulations with a hemolysis value of <10% to be nonhemolytic while values >25% to be at risk for hemolysis, the Dp liposome use as delivery system becomes even more attractive.67 No statistically significant hemolytic activity was observed for free ParELC3 in the concentration range of the assay (50 to 1.75 µmol·L−1).
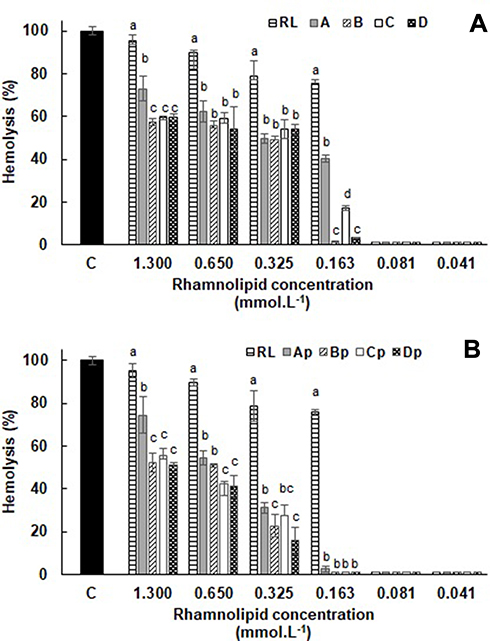 |
Figure 7 Hemolytic activity of (A) empty and (B) peptide-loaded liposomes in human erythrocytes. Cells were treated with RL, empty liposomes (A, B, C and D formulations) and peptide-loaded liposomes (Ap, Bp, Cp and Dp formulations) at RL concentrations ranging from 0.041 to 1.3 mmol·L−1. All concentration groups were compared with each other and different letters indicate statistical differences. ANOVA with Tukey’s post-test (p < 0.05). Abbreviations: C, control; RL, rhamnolipid. |
Cytotoxicity Effect
The peptide ParELC3 has been demonstrated to be a simultaneous inhibitor of DNA gyrase and topoisomerase IV activity.15 RLs, in turn, have a wide spectrum of applications in food products (additives), drugs (antifungal, antibiotic and antibiofilm agents), and cosmetics.25–27 Hence, the evaluation of the toxicity of these compounds is crucial prior to the development of products for pharmaceutical applications. To examine the biocompatibility effect of the free and encapsulated peptide, HepG2 cells were treated with different concentrations of the samples for 24 h and the Resazurin assay was used to investigate the effect of the encapsulated peptide formulation on the viability of living cells. The human hepatocellular carcinoma cell line HepG2 was used because of its availability, unlimited cell growth, and the high reproducibility of the results, as well as its many liver-specific functions and its previous use as a screening model for cytotoxic substances.68 Moreover, studies have demonstrated that RLs elicit the same level of cytotoxicity between normal and cancer cells.69 No statistically significant differences were observed between the control and the ParELC3 or the peptide-loaded liposomes. As shown in Figure 8, cellular proliferation was not inhibited either by free peptide or the peptide-loaded liposomes indicating the maintenance of cell viability even at the highest concentrations tested (1.3 mmol·L−1 and 50 µmol·L−1 for RL and ParELC3, respectively). No previous toxicity evaluations of free ParELC3 or RL liposomes have been performed, but similar results have been obtained with RLs on HepG2 cells.70 RLs produced by three different strains of P. aeruginosa had no cytotoxic effect on L6 and HepG2 except at high concentration.70 This cytotoxic effect has been attributed to the enhancement of the cell membrane permeability when the surface tension of culture medium drops below a certain threshold.71 The surface tension of the culture medium can be affected by the RL structure, which influences cell viability.69 Therefore, when in a liposome composition, the RL structure should not affect the surface tension of the medium and, consequently, not exert an influence on cell viability, as observed.
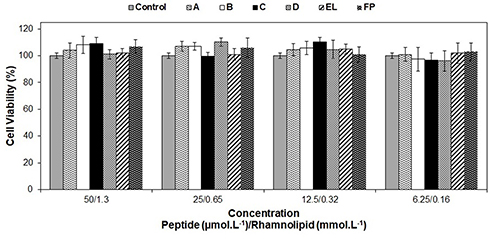 |
Figure 8 Cell Viability diagram of different amount of free peptide and peptide-loaded in A, B, C and D liposomes on HepG2 cell after 24 h of treatment. Control = culture medium; FP = free ParELC3; EL = empty liposome of respective A, B, C or D formulation. The differences between the means were not statistically significant (P<0.05). |
Conclusion
In this study, for the first time, a peptide derivative from a bacterial TA system was successfully encapsulated in rhamnolipid-based liposomes prepared by sonication. Different lipid compositions were used to alter the fluidity and rigidity of the RL liposomes. The DLS and TEM results demonstrated the appropriate size, zeta potential, polydispersity index, and morphology. The studies showed that the RLs together with POPC and Chol produce liposomes with high peptide encapsulation capacity, good colloidal stability, and the sustained release of loaded peptides. The in vitro release assay showed the peptide ParELC3 was released for up to 15 h. Formulation A had the fastest release profile and formulation D had the slowest. Moreover, confocal microscopy revealed that the liposomes facilitated the passage of the peptide through the plasma membrane and, consequently, its accumulation within the bacteria. Thus, ParELC3, which had no activity in its free form, showed a good antimicrobial activity, especially for Gram-positive bacteria, when encapsulated in RL liposome. The cytotoxicity assays indicated that cellular proliferation was not inhibited by either ParELC3 or the peptide-loaded liposomes, indicating the maintenance of cell viability. The results support the initial hypothesis that RL liposomes are able to carry a bioactive compound through the bacterial membrane and into the cytoplasm, where their antibacterial activity can be exerted.
Acknowledgments
We would like to thank the CAPES for the student´s scholarships awarded to B.C.P. Sanches, C.A. Rocha and J.G.M. Bedoya. We also thank to FAPESP for financial support (2015/17183-6) and CNPq for the research fellowship awarded to R. Marchetto.
Disclosure
All authors declare that there are no conflicts of interest.
References
1. Khameneh B, Diab R, Ghazvini K, et al. Breakthroughs in bacterial resistance mechanisms and the potential ways to combat them. Microb Pathog. 2016;95:32–42. doi:10.1016/j.micpath.2016.02.009
2. Luepke KH, Suda KJ, Boucher H, et al. Past, present, and future of antibacterial economics: increasing bacterial resistance, limited antibiotic pipeline, and societal implications. Pharmacotherapy. 2017;37(1):71–84. doi:10.1002/phar.1868
3. Harms A, Brodersen DE, Mitarai N, et al. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol Cell. 2018;70(5):768–784. doi:10.1016/j.molcel.2018.01.003
4. Page R, Peti W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol. 2016;12(4):208–214. doi:10.1038/nchembio.2044
5. Van Melderen L, de Bast MS. Bacterial toxin antitoxin systems: more than selfish entities? PLoS Genet. 2009;5(3):e1000437. doi:10.1371/journal.pgen.1000437
6. Pandey DP, Gerdes K. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33(3):966–976. doi:10.1093/nar/gki201
7. Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microb. 2005;3(5):371–382. doi:10.1038/nrmicro1147
8. Kim JS, Schantz AB, Song S, et al. GhoT of the GhoT/GhoS toxin/antitoxin system damages lipid membranes by forming transient pores. Biochem Biophys Res Commun. 2018;497(2):467–472. doi:10.1016/j.bbrc.2018.01.067
9. Leplae R, Geeraerts D, Hallez R, et al. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011;39(13):5513–5525. doi:10.1093/nar/gkr131
10. Jiang Y, Pogliano J, Helinski DR, et al. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol. 2002;44(4):971–979. doi:10.1046/j.1365-2958.2002.02921.x
11. Barbosa LCB, Garrido SS, Garcia A, et al. Function inferences from a molecular structural model of bacterial ParE toxin. Bioinformation. 2010;4(10):438–440. doi:10.6026/97320630004438
12. Hayes F. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science. 2003;301(5639):1496–1499. doi:10.1126/science.1088157
13. Roberts RC, Helinski DR. Definition of a minimal plasmid stabilization system from the broad-host-range plasmid RK2. J Bacteriol. 1992;174(24):8119–8132. doi:10.1128/jb.174.24.8119-8132.1992
14. Fiebig A, Castro Rojas CM, Siegal-Gaskins D, et al. Interaction specificity, toxicity and regulation of a paralogous set of ParE/RelE‐family toxin–antitoxin systems. Mol Microbiol. 2010;77(1):236–251. doi:10.1111/j.1365-2958.2010.07207.x
15. Barbosa LCB, Garrido SS, Garcia A, et al. Design and synthesis of peptides from bacterial ParE toxin as inhibitors of topoisomerases. Eur J Med Chem. 2012;54:591–596. doi:10.1016/j.ejmech.2012.06.008
16. Sharma D, Ali AAE, Trivedi LR. An updated review on: liposomes as drug delivery system. PharmaTutor. 2018;6(2):50–52. doi:10.29161/PT.v6.i2.2018.50
17. Shashi K, Satinder K, Bharat P. A complete review on: liposomes. Int Res J Pharm. 2012;3:10–16.
18. Marques EF, Silva BFB. Surfactant self-assembly. In: Tadros T, editor. Encyclopedia of Colloid and Interface Science. Berlin, Heidelberg: Springer; 2013:1202–1241. doi:10.1007/978-3-642-20665-8_169
19. Liu H, Shao B, Long X, et al. Foliar penetration enhanced by biosurfactant rhamnolipid. Colloid Surface B. 2016;145:548–554. doi:10.1016/j.colsurfb.2016.05.058
20. Sekhon Randhawa KK, Rahman PKSM. Rhamnolipid biosurfactants-past, present, and future scenario of global market. Front Microbiol. 2014;5:454. doi:10.3389/fmicb.2014.00454
21. Banat IM, Makkar RS, Cameotra SS. Potential commercial applications of microbial surfactants. Appl Microbiol Biotechnol. 2000;53(5):495–508. doi:10.1007/s002530051648
22. Mukherjee S, Das P, Sen R. Towards commercial production of microbial surfactants. Trends Biotechnol. 2006;24(11):509–515. doi:10.1016/j.tibtech.2006.09.005
23. Dwivedi S, Saquib Q, Al-Khedhairy AA, et al. Rhamnolipids functionalized AgNPs-induced oxidative stress and modulation of toxicity pathway genes in cultured MCF-7 cells. Colloid Surface B. 2015;132:290–298. doi:10.1016/j.colsurfb.2015.05.034
24. Moussa Z, Chebl M, Patra D. Interaction of curcumin with 1, 2-dioctadecanoyl-sn-glycero-3-phosphocholine liposomes: intercalation of rhamnolipids enhances membrane fluidity, permeability and stability of drug molecule. Colloid Surface B. 2017;149:30–37. doi:10.1016/j.colsurfb.2016.10.002
25. Henkel M, Müller MM, Kügler JH, et al. Rhamnolipids as biosurfactants from renewable resources: concepts for next-generation rhamnolipid production. Process Biochem. 2012;47(8):1207–1219. doi:10.1016/j.procbio.2012.04.018
26. Cai J, Huang H, Song W, et al. Preparation and evaluation of lipid polymer nanoparticles for eradicating H. pylori biofilm and impairing antibacterial resistance in vitro. Int J Pharm. 2015;495(2):728–737. doi:10.1016/j.ijpharm.2015.09.055
27. Ramos MAS, Da Silva PB, Spósito L, et al. Nanotechnology-based drug delivery systems for control of microbial biofilms: a review. Int J Nanomedicine. 2018;13:1179–1213. doi:10.2147/IJN.S146195
28. Haba E, Pinazo A, Pons R, et al. Complex rhamnolipid mixture characterization and its influence on DPPC bilayer organization. BBA Biomembranes. 2014;1838(3):776–783. doi:10.1016/j.bbamem.2013.11.004
29. Ishigami Y, Gama Y, Nagahora H, et al., inventors; Agency of Industrial Science and Technology and Shin-Etsu Chemical Co., Ltd., assignee. Rhamnolipid liposome. United States patent US 4902512. 1990 Feb 20.
30. Pornsunthorntawee O, Chavadej S, Rujiravanit R. Solution properties and vesicle formation of rhamnolipid biosurfactants produced by Pseudomonas aeruginosa SP4. Colloid Surface B. 2009;72(1):6–15. doi:10.1016/j.colsurfb.2009.03.006
31. Pornsunthorntawee O, Chavadej S, Rujiravanit R. Characterization and encapsulation efficiency of rhamnolipid vesicles with cholesterol addition. J Biosci Bioeng. 2011;112(1):102–106. doi:10.1016/j.jbiosc.2011.03.009
32. Bai L, McClements DJ. Formation and stabilization of nanoemulsions using biosurfactants: rhamnolipids. J Colloid Interf Sci. 2016;479:71–79. doi:10.1016/j.jcis.2016.06.047
33. Nitschke M, Silva SS. Recent food applications of microbial surfactants. Crit Rev Food Sci Nutr. 2018;58(4):631–638. doi:10.1080/10408398.2016.1208635
34. Lovaglio RB, Silva VL, Capelini TL, et al. Rhamnolipids production by a Pseudomonas aeruginosa LBI mutant: solutions and homologs characterization. Tenside Surfact Det. 2014;51(5):397–405. doi:10.3139/113.110321
35. CLSI. Manual Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standards- 6th Ed. Document M7-A6 Performance Standards for Antimicrobial Susceptibility Testing. Wayne, PA.: Clinical and Laboratory Standards Institute; 2006.
36. Balouiri M, Sadiki M, Ibnsouda SK. Methods for in vitro evaluating antimicrobial activity: a review. J Pharm Anal. 2016;6(2):71–79. doi:10.1016/j.jpha.2015.11.005
37. Ahmed SA, Gogal RM, Walsh JEA. New rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes an alternative to [3H] thymidine incorporation assay. J Immunol Methods. 1994;170(2):211–224. doi:10.1016/0022-1759(94)90396-4
38. Trovatti E, Cotrim CC, Garrido SS, et al. Peptides based on CcdB protein as novel inhibitors of bacterial topoisomerases. Bioorg Med Chem Lett. 2008;18(23):6161–6164. doi:10.1016/j.bmcl.2008.10.008
39. Barbosa LCB, Dos Santos Carrijo R, da Conceição MB. Characterization of an OrtT-like toxin of Salmonella enterica serovar Houten. Braz J Microbiol. 2019;50(3):839–848. doi:10.1007/s42770-019-00085-3
40. Vergalli J, Bodrenko IV, Mais M, et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat Rev Microbiol. 2020;18(3):164–176. doi:10.1038/s41579-019-0294-2
41. Novikova OD, Solovyeva TF. Nonspecific porins of the outer membrane of gram-negative bacteria: structure and functions. Biochem Moscow Suppl Ser A. 2009;3(1):3–15. doi:10.1134/S1990747809010024
42. Ortiz A, Teruel JA, Espuny MJ, et al. Effects of dirhamnolipid on the structural properties of phosphatidylcholine membranes. Int J Pharm. 2006;325(1–2):99–107. doi:10.1016/j.ijpharm.2006.06.028
43. Kovnova R, Caffrey M. Phases and phase transitions of the phosphatidylcholines. Biochem Biophys Acta. 1998;1376(1):91–145. doi:10.1016/s0304-4157(98)00006-9
44. Lapinski MM, Castro-Forero A, Greiner AJ, et al. Comparison of liposomes formed by sonication and extrusion: rotational and translational diffusion of an embedded chromophore. Langmuir. 2007;23(23):11677–11683. doi:10.1021/la7020963
45. Nii T, Ishii F. Encapsulation efficiency of water-soluble and insoluble drugs in liposomes prepared by the microencapsulation vesicle method. Int J Pharm. 2005;298(1):198–205. doi:10.1016/j.ijpharm.2005.04.029
46. Lozano N, Pinazo A, La Mesa C, et al. Catanionic vesicles formed with arginine-based surfactants and 1,2-dipalmitoyl-sn-glycero-3-phosphate monosodium salt. J Phys Chem B. 2009;113(18):6321–6327. doi:10.1021/jp810671p
47. Carrión FJ, De La Maza A, Parra JL. The influence of ionic strength and lipid bilayer charge on the stability of liposomes. J Colloid Interf Sci. 1994;164:78–87. doi:10.1006/jcis.1994.1145
48. Makino K, Yamada T, Kimura M, et al. Temperature- and ionic strength-induced conformational changes in the lipid head group region of liposomes as suggested by zeta potential data. Biophys Chem. 1991;41:175–183. doi:10.1016/0301-4622(91)80017-l
49. Schwarz C, Mehnert W, Lucks JS, et al. Solid lipid nanoparticles (SLN) for controlled drug delivery. I. Production, characterization and sterilization. J Control Release. 1994;30(1):83–96. doi:10.1016/0168-3659(94)90047-7
50. Do Prado AH, Araújo VHS, Eloy JO, et al. Synthesis and characterization of nanostructured lipid nanocarriers for enhanced sun protection factor of octyl p-methoxycinnamate. AAPS PharmSciTech. 2020;21(4):125. doi:10.1208/s12249-019-1547-0
51. Hao Y, Zhao F, Li N, et al. Studies on a high encapsulation of colchicine by a noisome system. Int J Pharm. 2002;244(1–2):73–80. doi:10.1016/s0378-5173(02)00301-0
52. Sato MR, Oshiro Junior JA, Machado RTA, et al. Nanostructured lipid carriers for incorporation of copper(II) complexes to be used against Mycobacterium tuberculosis. Drug Des Devel Ther. 2017;11:909–920. doi:10.2147/DDDT.S127048
53. Krause B, Mende M, Pötschke P, et al. Dispersability and particle size distribution of CNTs in an aqueous surfactant dispersion as a function of ultrasonic treatment time. Carbon. 2010;48(10):2746–2754. doi:10.1016/j.carbon.2010.04.002
54. Hudiyanti D, Aminah S, Hikmahwati Y, et al. Cholesterol implications on coconut liposomes encapsulation of beta-carotene and vitamin C. IOP Conf Ser Mater Sci Eng. 2019;509:012037. doi:10.1088/1757-899X/509/1/012037
55. Liu W, Wang Z, Fu L, et al. Lipid compositions modulate fluidity and stability of bilayers: characterization by surface pressure and sum frequency generation spectroscopy. Langmuir. 2013;29(48):15022–15031. doi:10.1021/la4036453
56. Zambom CR, Fonseca FH, Crusca E, et al. A novel antifungal system with potential for prolonged delivery of Histatin 5 to limit growth of Candida albicans. Front Microbiol. 2019;10:1667. doi:10.3389/fmicb.2019.01667
57. Yamauchi M, Tsutsumi K, Abe M, et al. Release of drugs from liposomes varies with particle size. Biol Pharm Bull. 2007;30(5):963–966. doi:10.1248/bpb.30.963
58. Shareen SM, Shakil Ahmed FR, Hossen MN, et al. Liposome as a carrier for advanced drug delivery. Pak J Biol Sci. 2006;9(6):1181–1191. doi:10.3923/pjbs.2006.1181.1191
59. Yesylevskyy SO, Demchenko AP, Kraszewski S, et al. Cholesterol induces uneven curvature of asymmetric lipid bilayers. Sci World J. 2013;2013:965230. doi:10.1155/2013/965230
60. Roy SM, Sarkar M. Membrane fusion induced by small molecules and ions. J Lipids. 2011;2011:528784. doi:10.1155/2011/528784
61. Furneri PM, Fresta M, Puglisi G, et al. Ofloxacin-loaded liposomes: in vitro activity and drug accumulation in bacteria. Ant Agents Chem. 2000;44(9):2458–2464. doi:10.1128/AAC.44.9.2458-2464.2000
62. Ma Y, Wang Z, Zhao W, et al. Enhanced bactericidal potency of nanoliposomes by modification of the fusion activity between liposomes and bacterium. Int J Nanomedicine. 2013;8:2351–2360. doi:10.2147/IJN.S42617
63. Zaragoza A, Aranda FJ, Espuny MJ, et al. Hemolytic activity of a bacterial trehalose lipid biosurfactant produced by rhodococcus sp.: evidence for a colloid-osmotic mechanism. Langmuir. 2010;26(11):8567–8572. doi:10.1021/la904637k
64. Sánchez M, Aranda FJ, Teruel JÁ, et al. Permeabilization of biological and artificial membranes by a bacterial dirhamnolipid produced by Pseudomonas aeruginosa. J Colloid Interf Sci. 2010;341(2):240–247. doi:10.1016/j.jcis.2009.09.042
65. de la Harpe KM, Kondiah PPD, Choonara YE, et al. The hemocompatibility of nanoparticles: a review of cell–nanoparticle interactions and hemostasis. Cells. 2019;8(10):1209. doi:10.3390/cells8101209
66. Mourtas S, Michanetzis GPA, Missirlis YF, Antimisiaris SG. Haemolytic activity of liposomes: effect of vesicle size, lipid concentration and polyethylene glycol-lipid or arsonolipid incorporation. J Biomed Nanotechnol. 2009;5:1–7. doi:10.1166/jbn.2009.1050
67. Amin K, Dannenfelser RM. In vitro hemolysis: guidance for the pharmaceutical scientist. J Pharm Sci. 2006;95(6):1173–1176. doi:10.1002/jps.20627
68. Elje E, Hesler M, Rundén-Pran E, et al. The comet assay applied to HepG2 liver spheroids. Mutat Res Gen Tox En. 2019;845:403033. doi:10.1016/j.mrgentox.2019.03.006
69. Jiang L, Shen C, Long X, et al. Rhamnolipids elicit the same cytotoxic sensitivity between cancer cell and normal cell by reducing surface tension of culture medium. Appl Microb Biotechnol. 2014;98(24):10187–10196. doi:10.1007/s00253-014-6065-0
70. Tamothran AM, Sevakumaran V, Bhubalan K. Production and toxicity evaluation of rhamnolipids produced by Pseudomonas strains on L6 and HepG2 cells. Malays Appl Biol. 2019;48:149–156.
71. Xia WJ, Onyuksel H. Mechanistic studies on surfactant-induced membrane permeability enhancement. Pharm Res. 2000;17(5):612–618. doi:10.1023/A:1007581202873
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.