Back to Journals » OncoTargets and Therapy » Volume 12
Reversing platinum resistance in ovarian cancer multicellular spheroids by targeting Bcl-2
Authors Yang Y, Li S, Sun Y, Zhang D, Zhao Z, Liu L
Received 24 September 2018
Accepted for publication 1 January 2019
Published 29 January 2019 Volume 2019:12 Pages 897—906
DOI https://doi.org/10.2147/OTT.S187015
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Ya’nan Yang,1–3,* Song Li,1,* Yiting Sun,1 Di Zhang,1 Zeyi Zhao,1 Lian Liu1
1Department of Chemotherapy, Cancer Center, Qilu Hospital of Shandong University, Jinan, Shandong 250012, China; 2Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai 200032, China; 3Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China
*These authors contributed equally to this work
Purpose: Peritoneal metastasis is the most common pathway for the spread of ovarian cancer. Ovarian cancer cells in ascites prefer to aggregate into the more chemoresistant multicellular spheroids (MCSs), leading to treatment failure and disease recurrence. We previously established a suspension MCS model of ovarian cancer cells in vitro and found that the MCS cells acquired drug resistance to cisplatin. In the present study, we aimed to uncover the underlying mechanism of the platinum resistance of MCS and the potential targets to reverse the drug resistance.
Materials and methods: MCS models were established for the phenotypic studies, including proliferation, invasion, migration, drug resistance, apoptosis assays, and signaling pathway analysis. The key molecule, Bcl-2, was screened by profile analysis and validated by Western blotting. siRNA was used to verify the anti-cisplatin-induced apoptosis effect of Bcl-2. The Bcl-2 inhibitor, ABT-737, was used for improving the sensitivity of MCS to cisplatin. The 50% inhibitory concentrations (IC50) were measured by viability assays treated with different concentrations of cisplatin. Flow cytometry and Western blotting were used for quantification of drug-induced apoptosis.
Results: The ovarian cancer MCS showed a proliferation-stagnant but invasive phenotype when resuspended. When treated with cisplatin, MCS cells showed much higher viability, with significantly fewer apoptotic cells than the adherent cells. Levels of Bcl-2 were upregulated in ovarian cancer ascitic cells and MCS cells. Bcl-2 knockdown by siRNA or blockage by ABT-737 enhanced the cisplatin-induced apoptosis and reduced the 50% inhibitory concentrations of cisplatin for MCS by 58.5% and 88.2%, respectively.
Conclusion: The upregulated Bcl-2 contributes to cisplatin resistance in our MCS model and targeting it sensitizes the MCS to cisplatin treatment. This provides us a preliminary treatment method for ovarian cancer peritoneal metastasis.
Keywords: Bcl-2, drug resistance, molecular targeted therapy, multicellular spheroids, ovarian neoplasms
Introduction
Ovarian cancer is one of the most common and lethal gynecologic malignancies in the world.1 Despite increasing surgical approaches and treatment regimens, prognosis for women with ovarian cancer has remained largely unchanged in the past 30 years.2,3 This fact is primarily due to the inability to detect the disease at early stage; about 75% of them are diagnosed at late stage, with widely metastatic lesions beyond the ovaries.3 Unlike other malignancies, ovarian cancer predominately disseminates within the peritoneal cavity, closely associated with ascites formation.4–6 The peritoneal carcinomatosis as well as ascites production is associated with poor prognosis and deterioration of the quality of life of the patients.3,7 Incomplete understanding of the underlying cellular and molecular mechanisms of peritoneal metastasis hinders the development of effective therapies to improve the prognosis of ovarian cancer.
Peritoneal metastasis of ovarian cancer involves the shedding of cells from the primary tumor, dissemination into the abdominal cavity, attachment and invasion into the mesothelial lining of the peritoneum, followed by formation of metastatic outgrowth.8,9 During these processes, disseminated tumor cells may aggregate into multicellular spheroids (MCSs) in ascites with several advantages for further metastasis and chemotherapy resistance.5,10 First, the suspended MCS cells live much longer than the floating single cells that undergo anoikis, representing an important intermediate survival mechanism that can facilitate ovarian cancer dissemination.11 Second, the unvascularized spheroid generates a metabolite density gradient that inhibits access of chemotherapy agents to the internal cells.12 Third, the MCS enters slowly cycling and/or quiescent states that are resistant to therapies that specifically kill proliferating cells, such as taxanes.13,14 Therefore, it is necessary to eradicate MCS completely in peritoneal cavity during the treatment of ovarian cancer to prevent tumor recurrence and metastasis.
It has been previously revealed that formation of MCS leads to development of taxane resistance in ovarian peritoneum metastasis.15,16 Targeting inhibition of the molecules necessary for MCS formation, such as p27 or P-glycoprotein, reversed this resistance.15,16 Platinum is another most effective first-line reagent against ovarian cancer as well as the most frequently used reagent for intraperitoneal chemotherapy clinically.17 Resistance to platinum, whether primary or secondary, relates to a poor prognosis.18 In previous studies, we established a suspension MCS model of ovarian cancer cells in vitro and revealed that E-cadherin promoted the formation of MCS as well as drug resistance to cisplatin.19 The underlying mechanism of cisplatin resistance in ovarian cancer MCS was further investigated in the present study.
Materials and methods
Cell culture
SKOV-3 and OVCAR-3 cell lines were obtained from the Cell Bank, Chinese Academy of Science (Shanghai, China). The cells were cultured in RPMI-1640 (Gibco, Suzhou, China) with 10% FBS (ScienCell, Carlsbad, CA, USA). All cell incubations were maintained in a 37°C incubator with a relative humidity of 90% and 5% CO2. Cells were passaged when the confluence reached 80%–90%.
Establishment of the MCS models
The method to establish MCS was the same as in our previous publications.19 Briefly, poly 2-hydroxylethyl methacrylate (Poly-HEMA) gel (Sigma, St Louis, MO, USA) was diluted into 12 mg/mL and coated on the bottoms of 24-well plates by 500 μL per well. Then, the plate was air-dried in a laminar flow cabinet and washed with PBS three times before use. A total of 5×104 cells were cultured in each Poly-HEMA-treated and untreated well. Cells were morphologically monitored daily and used for subsequent experiments.
Migration and invasion assays
In vitro analysis of invasion and migration was performed using a Transwell system (8 μm pore size; Corning, NY, USA), according to the manufacturers’ instructions. Matrigel (BD Bioscience, Bedford, MA, USA) was diluted in serum-free DMEM (Hyclone, Logan, UT, USA) with the volume ratio 1:8 and then coated into the upper chamber for invasion assay. Cells were trypsinized and resuspended in the medium without serum or growth factors. Basal mediums with 20% FBS were added to the bottom of the transwells. The above-described cell suspensions (400 μL containing 50,000 and 100,000 cells for migration and invasion assays, respectively) were added to the insert and incubated for 24 hours at 37°C. The cells were fixed with methanol, and the cells inside the inserts were removed with cotton swabs. The invasive and migratory cells were stained with 0.1% crystal violet for 20 minutes and then were photographed through microscope.
Cell viability assay
Cell viability was measured utilizing Cell Counting Kit-8 (CCK-8) cell assay kits (BestBio, Shanghai, China) according to the manufacturer’s protocol. For cell proliferation assay, 5×104 cells were seeded in the Poly-HEMA treated or untreated wells of a 24-well plate in triplicate. The proliferation curves were made based on OD values of each well at four time points (24, 48, 72, and 96 hours) from three independent experiments. To measure the concentration-dependent cytotoxicity of cisplatin in ovarian cancers MCS cells and adherent cells (ADCs), cells after 3 days of growth in suspension or adhesion were exposed to 1, 5, 10, 30, 50, 70, or 100 μM of cisplatin for 36 hours before CCK-8 assay. Cell viability was calculated according to the manufacturer’s formula and 50% inhibitory concentrations (IC50) value was calculated from the manufacturer’s software. To measure the effect of targeting Bcl-2 in platinum resistance, cells were pretreated with 0.1 μM ABT-737 (Selleckchem, Houston, TX, USA) or siRNA before exposure to cisplatin.
Gene expression profile mining
The Gene Expression Omnibus (GEO) dataset, including the expression data, metadata, and probe set annotations, were downloaded from the publicly available GEO databases (http://www.ncbi.nlm.nih.gov/geo). Differentially expressed genes between ascitic and metastatic tumor cells were identified by R limma packages. The cutoff values for differentially expressed genes were set at greater than or equal to two-fold difference and the P-value <0.05. Gene ontology (GO) analysis was performed by The Database for Annotation, Visualization and Integrated Discovery v6.8 (https://david.ncifcrf.gov/tools.jsp), and the differentially expressed genes in the GO term “intrinsic apoptotic signaling pathway in response to DNA damage” were further analyzed.
Apoptosis analysis
After treatment with cisplatin for 24 hours, the cells, including ADC, MCS, ABT-737-treated, and siRNA-treated, were harvested with 0.25% trypsin and washed twice with cold PBS. Then, the cells were resuspended and incubated with propidium iodide and fluorescein isothiocyanate-Annexin V (BD Bioscience) for 15 minutes in the dark. Analysis of apoptotic cells was performed on a FACScan flow cytometer, and the data were analyzed using the software Flowjo.
siRNA transfection
Apoptosis analysis Bcl-2 siRNA and scramble siRNA were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Cells were transfected with siRNA after 2 days of growth in suspension or adhesion, using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions.
Western blotting
The protein extraction and SDS-PAGE gel electrophoresis were described as earlier.19 Total protein concentrations were measured with a Bradford protein assay kit (Beyotime, Shanghai, China). The anti-Bcl-2 antibody was purchased from ImmunoWay Biotechnology Company (Suzhou, China). The anti-β-actin antibody was purchased from Abcam (Cambridge, UK). The others were purchased from Cell Signaling Technology (Danvers, MA, USA). The immunoreactive proteins were visualized using the MiniBIS Pro gel imaging system (DNR, Jerusalem, Israel). All the experiments were independently repeated at least three times. One representative picture of each experiment is shown in the figures. The results were quantified by ImageJ software and normalized to b-actin.
Graphs and statistical analyses
The heatmap was graphed by Heml 1.0.3.7. All the other data were graphed and analyzed using software Graphpad Prism 6. Migration rates, invasion rates, frequencies of dead cells, cell viability, and IC50 were compared by two-tailed, unpaired Student’s t-test. A P-value of 0.05 was considered to indicate statistical significance.
Results
Ovarian cancer MCS is proliferation arrested but invasive
In our previous study, we established suspended MCS models of several ovarian cancer cells and observed that the MCS cells had stagnant proliferation, prolonged survival time, and cisplatin resistance in comparison with ADCs.19 In this study, we chose SKOV-3 cells due to their satisfying suspension effect. From the second day of suspension, the floating cells aggregated into compact oval or round masses with tight intercellular connections, which is morphologically different from ADCs (Figure 1A). Compared with ADCs, the MCS cells remained in a proliferation quiescent state, with stable numbers of cells during the cultivation periods (P<0.05 from days 2 to 4; Figure 1B). However, when the cell migration and invasion were examined, the trypsinized MCS cells showed a significant increase in the number of tumor cells that invaded through Matrigel and migrated through filters compared with ADCs (P=0.0126 and P<0.0001; Figure 1C–F). These results indicate that MCS has its own advantages not only in cell survival but also in tumor metastasis.
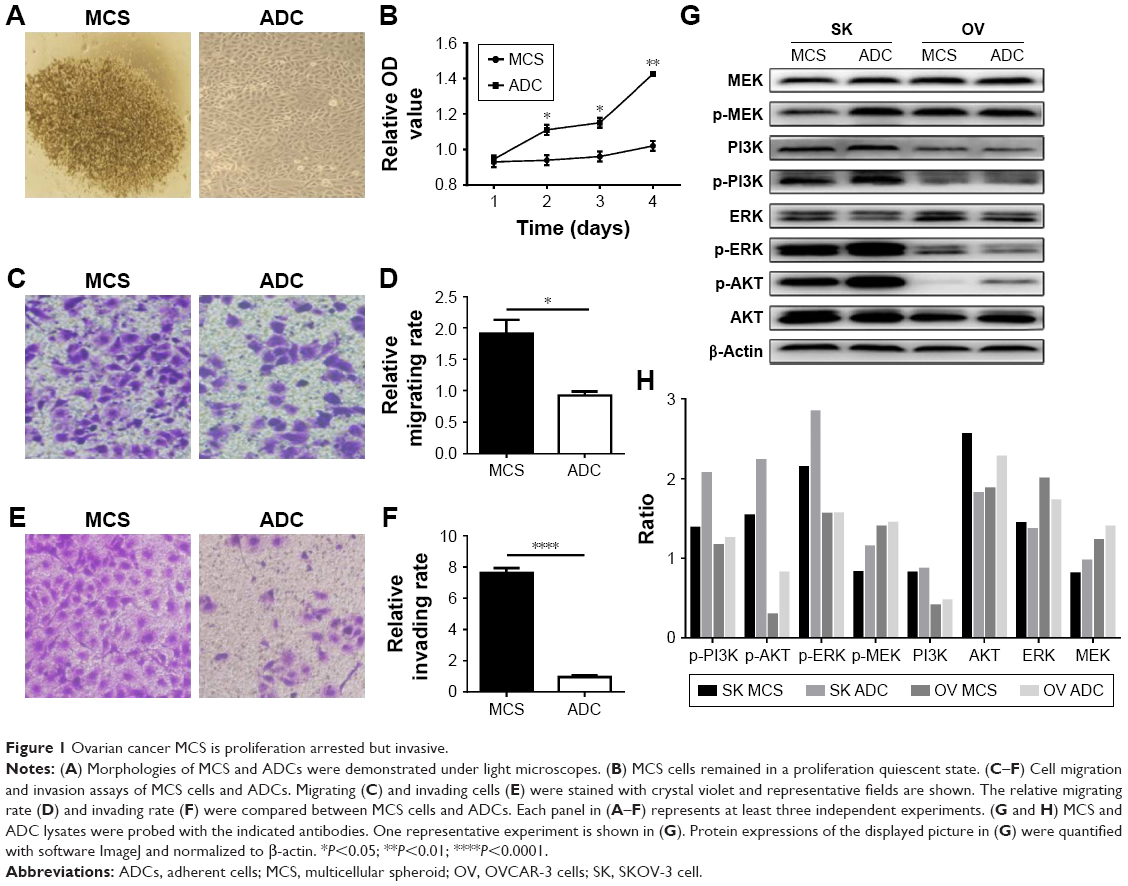 | Figure 1 Ovarian cancer MCS is proliferation arrested but invasive. |
Next, we accessed several proteins playing key roles in ovarian cancers to reveal the possible signaling pathways behind the unique behaviors of MCS. In SKOV-3 cells, phosphorylation of AKT, ERK, and MEK was downregulated in MCS (Figure 1G and H). In MCS of OVCAR-3 cells which has inferior ability to form MCS, phosphorylation of AKT was also decreased, but that of MEK/ERK changed little (Figure 1G and H).
Ovarian cancer MCS is highly resistant to platinum-induced apoptosis
To investigate the drug resistance of MCS cells, cisplatin with different concentrations was used to treat the suspended and ADCs. After exposure to increasing concentrations of cisplatin, MCS cells showed a lower percentage of dead cells than that of ADCs (Figure 2A). MCS demonstrated significant superiority over ADCs in cell viability when cisplatin ranged between 5 and 70 μM (P<0.05). Accordingly, cisplatin achieved a much higher IC50 in MCS cells than that in ADC (34.53 vs 76.49 μM, P=0.002; Figure 2B). These indicated that the MCS cells have a greater drug resistance to cisplatin than ADCs. To investigate whether apoptosis was responsible for this drug resistance, we assessed the frequencies of cisplatin-induced apoptosis by flow cytometry and Western blot. With the same concentration (30 μg/mL) of cisplatin, the frequencies of apoptotic ADCs were significantly higher than those of MCS cells (15.95% vs 9.45%, P=0.0467; Figure 2C and D). In accordance, less caspase-3 and caspase-9 were activated in MCS cells than those in ADCs in the presence of cisplatin (Figure 2E and F). These data revealed that MCS cells developed cisplatin resistance, at least partially, through avoiding platinum-induced apoptosis in this process.
 | Figure 2 Ovarian cancer MCS is highly resistant to platinum-induced apoptosis. |
Bcl-2 levels are augmented in ovarian cancer MCS with cisplatin
To uncover the molecules that were responsible for the impaired drug-induced apoptosis in MCS, we searched GEO databases and found one dataset (GSE73064) that compared expression profiles of magnetic-sorted ovarian cancer cells from matched primary tumors, ascites, and metastases of five high-grade serous ovarian cancer patients. GO analyses were performed in genes with differential expressions between metastatic and ascitic cells (P<0.05 and fold change >2 or <0.5). Thirteen genes were identified within the GO term “intrinsic apoptotic signaling pathway in response to DNA damage”, with Bcl-2, CDIP1, RPS27L, PYCARD, CIDERB, and PIK3R1 upregulated in ascitic cells and EPHA2, SFN, ST20, TP73, HIPK2, PML, and TP63 downregulated (Figure 3A). Among the six upregulated genes, we first excluded CDIP1, RPS27L, PYCARD, and CIDEB, due to their proapoptotic roles by literature. Between the rest two genes, we chose Bcl-2 for further investigation because it is a potent antiapoptotic gene as well as a potential target for ovarian cancer therapy. In the same dataset, the expression level of Bcl-2 was elevated in ascitic cells compared with that in metastatic cells and primary cells (Figure 3A).
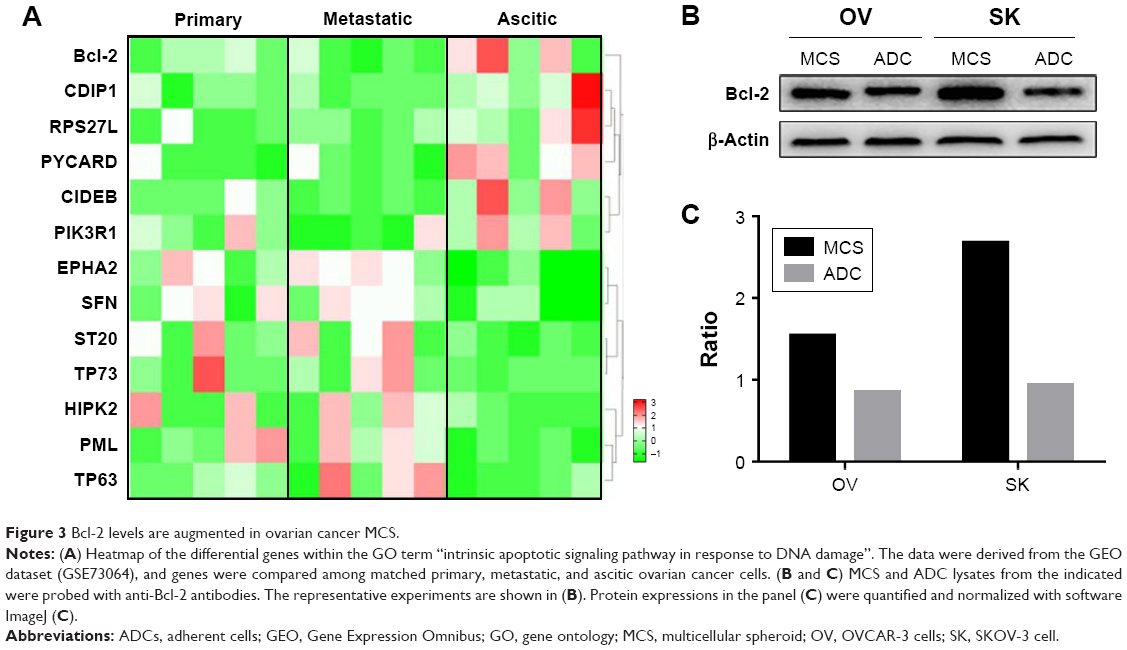 | Figure 3 Bcl-2 levels are augmented in ovarian cancer MCS. |
We then examined the expression level of Bcl-2 in MCS cells in the presence of cisplatin. It revealed that Bcl-2 was indeed upregulated in MCS cells in comparison to ADCs in both SKOV-3 cells and OVCAR-3 cell lines (Figure 3B and C). In addition, the levels of Bcl-2 in MCS cells and ADCs were not significantly altered when treated with cisplatin (Figure 2E and F). Therefore, we speculated that the antiapoptotic property of Bcl-2 might contribute to the platinum resistance of MCS.
Downregulation of Bcl-2 reverses cisplatin resistance in ovarian cancer MCS
To examine whether Bcl-2 contributes to the anti-cisplatin-induced apoptosis in ovarian cancer MCS, we used a commercial siRNA to block the effect of Bcl-2. Downregulation of Bcl-2 indeed enhanced the sensitivity of MCS cells to cisplatin (Figure 4A) and thus lowered the IC50 of cisplatin by 58.5% (P=0.0018; Figure 4B). The siRNA did not significantly enhance the antitumor ability of cisplatin in ovarian ADCs (P=0.0947; Figure 4B). Importantly, after treatment with the siRNA, the IC50 value of cisplatin in MCS cells became no longer significantly different from that in ADCs (P=0.2754; Figure 4B). These results indicated that interference with Bcl-2 enhanced the antitumor activity of cisplatin, especially in ovarian cancer MCS cells.
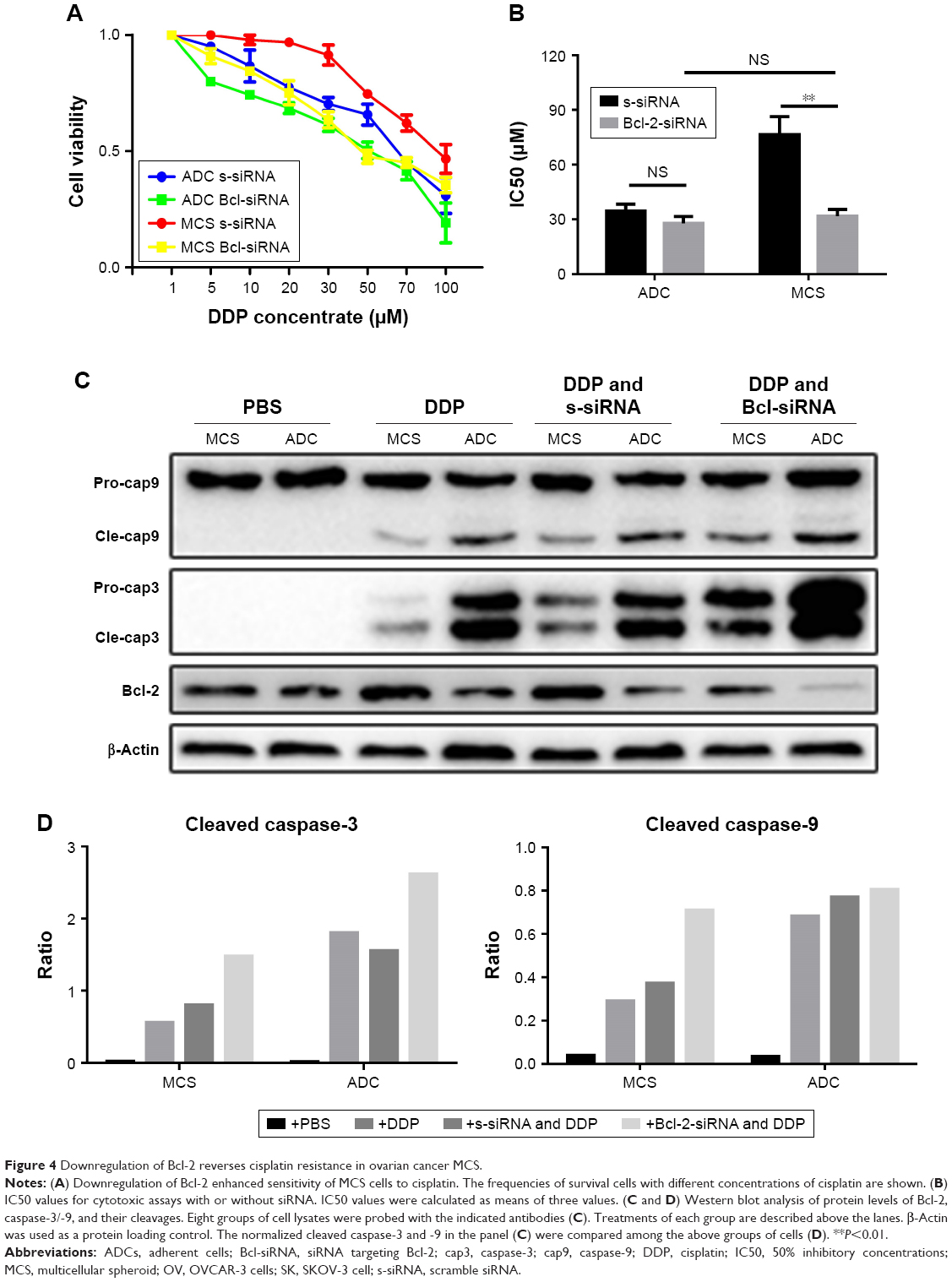 | Figure 4 Downregulation of Bcl-2 reverses cisplatin resistance in ovarian cancer MCS. |
Western blot revealed that downregulation of Bcl-2 raised expression of caspase-3 in both MCS cells and ADCs in the presence of cisplatin (Figure 4C). Importantly, the cleavages of caspase-3 and -9 were obviously elevated by the siRNA in the drug-exposed ovarian cancer MCS (Figure 4C and D). We also observed an increased cleaved caspase-3, but not cleaved caspase-9, in ADCs with cisplatin when Bcl-2 was knocked down (Figure 4C and D). The activation of caspase-3 and -9 indicates that downregulation of Bcl-2 could reverse the cisplatin resistance in ovarian cancer MCS by inhibition of its antiapoptotic effect.
Targeted inhibitor of Bcl-2 sensitizes ovarian cancer MCS to cisplatin treatment
Based on the above results, we speculated that the Bcl-2 inhibitors could be utilized as a chemosensitizer for treatment of ovarian cancer peritoneal metastasis with cisplatin. Therefore, we utilized ABT-737, a specific targeting inhibitor of Bcl-2, to block the effect of Bcl-2 in MCS. Treatment with ABT-737 significantly enhanced MCS cells’ sensitivity to cisplatin (Figure 5A), and thus lowered the IC50 to cisplatin by 88.2% (P=0.0027; Figure 5B). Besides, when treated with ABT-737, the IC50 of cisplatin in MCS cells became similar to that in ADCs with no significant difference (P=0.1929; Figure 5B). We also found that ABT-737 sensitized ovarian ADCs to cisplatin and reduced the IC50 by 61.8% in ADCs (P=0.0261; Figure 5B). In the ABT-737-treated cells, caspase-3 and -9 were both further activated in MCS cells and ADCs (Figure 5C and D). Based on these, we believe that targeting Bcl-2 sensitizes ovarian cancer MCS to cisplatin treatment in our MCS model.
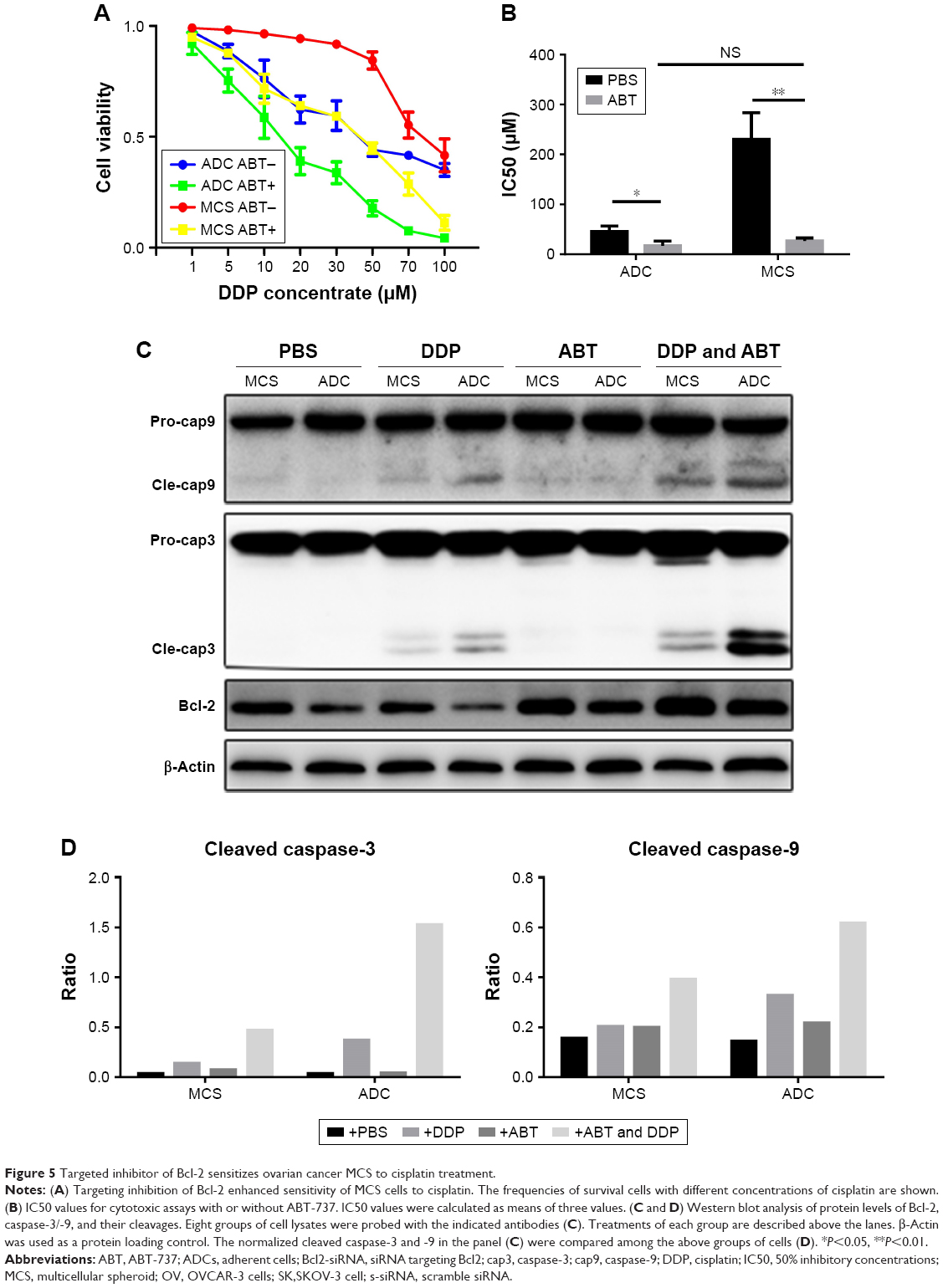 | Figure 5 Targeted inhibitor of Bcl-2 sensitizes ovarian cancer MCS to cisplatin treatment. |
Discussion
Instead of the classic patterns of hematogenous or lymphatic metastasis, peritoneal seeding is the most common pathway for the spread of ovarian cancer. At the first diagnosis of ovarian cancer, approximately two-thirds of the patients developed peritoneal carcinomatosis and one-third of the patients developed ascites, which are closely associated with poor survival.20 Furthermore, 30%–50% of patients responded to primary treatments and had relapses mainly intraperitoneally.21–23 The spreading cells that shed from the local ovarian cancer formed MCS with multiple advantages in survival and invasion. In our previous and present studies, MCS maintained its structure as long as 30 days in vitro and invaded and migrated exceedingly, behaving likes cancer stem cells.19 The quiescent MCS that survived the treatments may wake up in a later phase and cause tumor relapse as well as chemoresistance. Especially, despite quiescent growth, the MCS cells showed more aggressive phenotypes in migration and invasion ability when resuspended. Therefore, it is critical to overcome the MCS-related chemoresistance during the treatment of advanced ovarian cancer with peritoneal metastasis. When comparing the signaling pathways in the two MCS models, the MEK/ERK pathway was depressed in SKOV-3 MCS, but changed little in the OVCAR-3 cells which had inferior suspension ability. Thus, we speculate that the MEK/ERK signaling pathway may be involved in the formation of MCS. The depressed AKT in MCS of both cell lines was probably a result of proliferation quiescent or nonadherence state in MCS.
Nonetheless, present treatment methods can hardly solve peritoneal metastasis, which is closely associated with the effect of MCS for several reasons. First of all, intravenous or oral chemotherapy has to pass the peritoneal–plasma barrier, which creates a concentration differential favorable to tumors in peritoneal cavities.24 Secondly, intraperitoneal chemotherapy achieves high local concentration, but does not pass the barrier of drug resistance of MCS, as indicated above. In the present study, the IC50 for SKOV-3 cell MCS was as high as 76.5 μg/mL. Thus, overcoming the chemoresistance of MCS is one of the most important issues for treatment of ovarian cancer peritoneal carcinomatosis. Xing et al reported that target inhibiting p27 or P-glycoprotein reversed resistance to taxanes in MCS.15,16 However, platinum is the most important chemotherapeutic agent both intravenously and intraperitoneally for ovarian cancer. For this reason, we aimed to uncover therapeutic targets for platinum resistance in ovarian cancer MCS.
Dysregulation of apoptosis, especially the intrinsic apoptotic pathway, is considered a hallmark of cancer.25 Apoptosis induced by platinum is generally considered a response to the subsequent DNA damage.26 We studied the differentially expressed genes in the ascitic cells and their primary/metastatic compartments in patients with ovarian cancer (GSE73064) and found 13 such genes, including Bcl-2, within the GO term “intrinsic apoptotic signaling pathway in response to DNA damage”. High level of Bcl-2 expression was detected in primary ovarian cancers.27 Bcl-2 is an antiapoptotic molecular that modulates the intrinsic apoptotic pathway by neutralizing the mitochondrial permeabilizers such as Bax and Bak.28 The downstream caspase cascade includes caspase-3 and caspase-9.28 In the online dataset analysis, we found an even higher level of Bcl-2 expression in the ascitic cells. This augmentation was also validated in our following study, with inactivation of caspase-3/-9 in MCS derived from two ovarian cancer cell lines. It is plausible that the increased Bcl-2 contributed to the inactivation of caspase-9/-3 as well as the attenuated cisplatin-induced apoptosis. In consideration of its potential role in antiapoptosis and drug resistance, we attempted to examine whether targeting this molecule could reverse the chemoresistance in ovarian cancer MCS. We found that knockdown of Bcl-2 by siRNA or blockage of Bcl-2 by a proteinase inhibitor, ABT-737, resulted in enhancement of cisplatin-induced apoptosis of MCS. Downregulation of Bcl-2 expression reduced the IC50 of cisplatin for MCS by 58.5%, and targeting inhibition of Bcl-2 reduced the IC50 by 88.2%. These results indicate that the elevated Bcl-2 accounts for cisplatin resistance in the SKOV-3 MCS model, and targeting it may reverse this chemoresistance. This resistance-reversing effect in MCS is of potentially great value because peritoneal metastasis and ascites formation in refractory ovarian cancers remain a rough issue with inferior treatment outcome and survival, especially when platinum resistance occurs. Intractable ascites also impairs the patients’ quality of life and even leads to significant morbidity.7 Our in vitro data indicate that the Bcl-2 inhibition strategy may overcome the problem of platinum resistance of MCS, and may help improve the chemotherapy effect on ovarian cancer peritoneal metastasis, and therefore, potentially benefit the patients’ survival and quality of life clinically. Yet, our data were based on limited types of cell models, and this presumption needs further investigations by other cell lines or primary cells.
ABT-737 and its derivatives ABT-263 (navitoclax) and ABT-199 (venetoclax) are Bcl-2 family protein inhibitors that have an anticancer potential as single agents or in combination with conventional chemotherapy.29 Many clinical trials with them are currently undergoing for various solid cancers and hematological cancers.29 Particularly, the trial NCT02591095 is a Phase II clinical study on the efficacy of ABT-263 in platinum-resistant/refractory ovarian cancer. However, there are no such clinical studies on ovarian cancer peritoneal metastasis. Thrombocytopenia is the main adverse reaction and this limits the usage of Bcl-2 inhibitors.30 Intraperitoneal infusion of chemotherapeutic agents has been widely accepted; thus, we considered that the Bcl-2 inhibitors could also be administrated intraperitoneally for the treatment of MCS as well as for avoidance of their severe side effects.
Conclusion
In summary, upregulation of Bcl-2 modulated platinum-induced apoptosis in the SKOV-3 MCS model, and targeting this molecule reversed this effect. This suggests that the Bcl-2 may be a key gene in chemoresistance of MCS and provides us a preliminary strategy to help overcome chemoresistance of advanced ovarian cancer with peritoneal metastasis and/or refractory ascites. These conclusions are yet to be studied and confirmed in other cell lines and primary cells.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (NSFC 81172487 to Lian Liu and 81500092 to Song Li) and the Natural Science Foundation of Shandong Province, China (ZR201702180008 to Lian Liu).
Author contributions
LL designed the experiment. YY, YS, DZ, and ZZ carried out the experiments. SL performed the GEO dataset analysis. YY, SL, and LL interpreted the result, analyzed the data, prepared the figures and tables, and wrote and revised the manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384(9951):1376–1388. | ||
Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177(3):1053–1064. | ||
Vaughan S, Coward JI, Bast RC, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11(10):719–725. | ||
Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer. 2005;5(5):355–366. | ||
Tan DS, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006;7(11):925–934. | ||
Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Front Oncol. 2013;3:256. | ||
Weidle UH, Birzele F, Kollmorgen G, Rueger R. Mechanisms and targets involved in dissemination of ovarian cancer. Cancer Genomics Proteomics. 2016;13(6):407–424. | ||
Yeung TL, Leung CS, Yip KP, Au Yeung CL, Wong ST, Mok SC. Cellular and molecular processes in ovarian cancer metastasis. A review in the theme: cell and molecular processes in cancer metastasis. Am J Physiol Cell Physiol. 2015;309(7):C444–C456. | ||
Desoize B, Gimonet D, Jardiller JC. Cell culture as spheroids: an approach to multicellular resistance. Anticancer Res. 1998;18(6A):4147–4158. | ||
Shield K, Riley C, Quinn MA, Rice GE, Ackland ML, Ahmed N. Alpha2beta1 integrin affects metastatic potential of ovarian carcinoma spheroids by supporting disaggregation and proteolysis. J Carcinog. 2007;6:11. | ||
Shield K, Ackland ML, Ahmed N, Rice GE. Multicellular spheroids in ovarian cancer metastases: biology and pathology. Gynecol Oncol. 2009;113(1):143–148. | ||
Dong Y, Stephens C, Walpole C, et al. Paclitaxel resistance and multicellular spheroid formation are induced by kallikrein-related peptidase 4 in serous ovarian cancer cells in an ascites mimicking microenvironment. PLoS One. 2013;8(2):e57056. | ||
Peart T, Ramos Valdes Y, Correa RJ, et al. Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget. 2015;6(26):22424–22438. | ||
Xing H, Wang S, Hu K, et al. Effect of the cyclin-dependent kinases inhibitor p27 on resistance of ovarian cancer multicellular spheroids to anticancer chemotherapy. J Cancer Res Clin Oncol. 2005;131(8):511–519. | ||
Xing H, Wang S, Weng D, et al. Knock-down of P-glycoprotein reverses taxol resistance in ovarian cancer multicellular spheroids. Oncol Rep. 2007;17(1):117–122. | ||
Muñoz-Casares FC, Rufián S, Rubio MJ, et al. Treatment of peritoneal carcinomatosis from ovarian cancer. Present, future directions and proposals. Clin Transl Oncol. 2007;9(10):652–662. | ||
Jelovac D, Armstrong DK. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J Clin. 2011;61(3):183–203. | ||
Xu S, Yang Y, Dong L, et al. Construction and characteristics of an E-cadherin-related three-dimensional suspension growth model of ovarian cancer. Sci Rep. 2014;4:5646. | ||
Ayantunde AA, Parsons SL. Pattern and prognostic factors in patients with malignant ascites: a retrospective study. Ann Oncol. 2007;18(5):945–949. | ||
Ghaemmaghami F, Hassanzadeh M, Karimi-Zarchi M, Modari-Gilani M, Behtash A, Mousavi N. Centralization of ovarian cancer surgery: do patients benefit? Eur J Gynaecol Oncol. 2010;31(4):429–433. | ||
Behtash N, Karimi Zarchi M, Ashraf-Ganjoei T. Uterine involvement in advanced epithelial ovarian cancer. Eur J Gynaecol Oncol. 2010;31(1):99–101. | ||
Ghaemmaghami F, Karimi-Zarchi M, Modares-Gilani M, Mousavi A, Behtash N. Clinical outcome of Iranian patients with advanced ovarian cancer with neoadjuvant chemotherapy versus primary debulking surgery. Asian Pac J Cancer Prev. 2008;9(4):719–724. | ||
Hasovits C, Clarke S. Pharmacokinetics and pharmacodynamics of intraperitoneal cancer chemotherapeutics. Clin Pharmacokinet. 2012;51(4):203–224. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Evans DL, Dive C. Effects of cisplatin on the induction of apoptosis in proliferating hepatoma cells and nonproliferating immature thymocytes. Cancer Res. 1993;53(9):2133–2139. | ||
Fauvet R, Dufournet C, Poncelet C, Uzan C, Hugol D, Daraï E. Expression of pro-apoptotic (p53, p21, Bax, Bak and Fas) and anti-apoptotic (Bcl-2 and Bcl-X) proteins in serous versus mucinous borderline ovarian tumours. J Surg Oncol. 2005;92(4):337–343. | ||
Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. | ||
Opydo-Chanek M, Gonzalo O, Marzo I. Multifaceted anticancer activity of BH3 mimetics: current evidence and future prospects. Biochem Pharmacol. 2017;136:12–23. | ||
Scarfò L, Ghia P. Reprogramming cell death: Bcl2 family inhibition in hematological malignancies. Immunol Lett. 2013;155(1–2):36–39. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.