Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Reversible Neuropsychiatric Disturbances Caused by Nitrous Oxide Toxicity: Clinical, Imaging and Electrophysiological Profiles of 21 Patients with 6–12 Months Follow-up
Authors Zheng R, Wang Q, Li M, Liu F, Zhang Y, Zhao B, Sun Y, Zhang D, Yan C, Zhao Y, Li W ![]()
Received 7 July 2020
Accepted for publication 18 October 2020
Published 23 November 2020 Volume 2020:16 Pages 2817—2825
DOI https://doi.org/10.2147/NDT.S270179
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jun Chen
Rui Zheng,1,* Qinzhou Wang,2,* Mingyuan Li,3 Fuchen Liu,4 Yongqing Zhang,1 Bing Zhao,1 Yuan Sun,1 Dong Zhang,2 Chuanzhu Yan,1,2 Yuying Zhao,2 Wei Li2
1Department of Neurology, Qilu Hospital (Qingdao), Cheeloo College of Medicine, Shandong University, Qingdao City, People’s Republic of China; 2Department of Neurology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan City, People’s Republic of China; 3EMG-Evoked Potential Room, Binzhou Medical University Hospital, Binzhou City, People’s Republic of China; 4Department of Neuroscience, Yale School of Medicine, New Haven, CT, USA
*These authors contributed equally to this work
Correspondence: Wei Li
Department of Neurology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, No. 107 Wenhua West Road, Jinan City, Shandong Province 250012, People’s Republic of China
Tel +86-185 60085517
Email [email protected]
Purpose: Nitrous oxide (N2O) abuse has become an increasingly severe problem in China. The aim of the study was to summarize the features of N2O-induced neurology and enhance the awareness of this disease among physicians.
Patients and Methods: We retrospectively reviewed the clinical, imaging, electrophysiological characteristics and the prognosis of patients with N2O neurotoxicity in our hospital from January 2016 to August 2019.
Results: Twenty-one patients (average age: 22.6± 4.6 years) were collected. Eighty-six percent (18/21) patients presented with acute or subacute neurological disorders as their initial symptoms. The remaining fourteen percent (3/21) had psychiatric symptoms as the earliest symptoms. With progression, movement dysfunction appeared in ninety percent (19/21) of the patients with fifty-three percent (10/19) presented with weakness limited to both lower extremities. Sixty-two percent (13/21) of the patients presented with subjective sensory deficit. Seventy-one percent (15/21) had vibration sense impairment and positive Romberg’s sign. Sixty-seven percent of the patients had hyporeflexia or areflexia. Fourteen percent (3/21) showed positive Babinski’s sign. Seventy-eight percent (14/18) showed significantly increased homocysteine (HCY) level and only seventeen percent (3/18) showed decreased serum vitamin B12 level. T2 hyperintensity involving the posterior columns and lateral columns with inverted V sign in cervical spinal MRI had been observed in forty-seven percent (8/17) of the patients. Axonal peripheral neuropathy occurred in eighty-five percent (17/20) of the patients. The level of serum vitamin B12 and HCY, as well as imaging findings, were rapidly recovered after supplementation of Vitamin B12.
Conclusion: The N2O-induced neuropsychiatric disturbances mainly occurred in the young groups and should be recognized by clinicians. The prognosis of N2O intoxication is relatively good.
Keywords: nitrous oxide toxicity, peripheral neuropathy, psychiatric disturbance, subacute combined degeneration, vitamin B12 deficiency
Introduction
Nitrous oxide (N2O) was first discovered by British scientist Joseph Priest in 1799.1 Due to its effect of analgesia, it was widely applied in surgery and dental operations as inhaled anesthetic at the end of nineteeth century.2 In addition to its clinical application, N2O was widely used in the food industry as an aerosol spray propellant to produce whipped cream due to its bacteriostatic effect. It is also used in racecar and rocket engines to increase thrust owing to its property of combustion-supporting.3 In addition, due to its euphoric effect, N2O is becoming increasingly popular among youngsters as a recreational drug. However, Layzer et al revealed the neurological toxicity of N2O initially in 1978.4 And in the following 20 years, hundreds of cases with neurologic symptoms after prolonged exposure of N2O were reported.5–8 The main neurologic syndromes include myelopathy, peripheral neuropathy, subacute combined degeneration (SCD). Notably, some patients may also present with psychological, emotional, or mental features.3
The first case of N2O induced myelopathy in China was reported in 2016.9 Until now, only around 60 cases had been reported.9–14 Due to the lack of epidemiological data and the unawareness by the clinical practitioners, most N2O abuse cases have been misdiagnosed for other diseases such as copper deficiency, hypothyroidism, hepatic or uremic encephalopathy, infection or medication intoxication. In this study, we summarized the clinical characteristics and prognosis of N2O-related neuropathy in order to enhance the awareness of this disease among physicians.
Patients and Methods
Twenty-one patients who were diagnosed with N2O-induced neuropathy from January 2016 to August 2019 were collected. Clinical severity was assessed by the modified Rankin Scale (mRS) scored as 0–5. All the cases had undergone a series of laboratory examinations including blood cell analysis, biochemistry analysis, vitamin B12, folate, HCY, creatine kinase (CK), thyroid function, rheumatism-related antibody detection, tumor markers and HIV antibody tests. Several patients had an MRI scan of spinal cord and brain, electrophysiological examination included nerve conduction velocity (NCV) and electromyogram (EMG), cerebrospinal fluid (CSF) analysis and urine methylmalonic acid (MMA) detection. All patients were evaluated when they were discharged and five of them were followed-up by telephone six months later. One case was also followed-up in clinic at one month, five months and one year after discharge. Follow-up blood cell analysis, serum vitamin B12 level, HCY, MRI and EMG were performed in this case.
Results
Basic Demographic Data and Clinical Manifestations
Table 1 contains the basic demographic data, clinical characteristics, physical examinations and mRS of these twenty-one patients (fourteen males and seven females). None of them had medical or family history of neurological or psychiatric disorders. The mean age of this group was 22.6±4.6 years. All patients had histories of recreational exposure at pubs, nightclubs or karaoke bars. The duration of N2O exposure ranged from two to twenty-four months (7.2±5.1 months). The time from the initial symptom onset to admission varied from five to 180 days (35.6±40.4 days). The mean mRS was 2.9±0.9, which indicated a moderate level of severity. Eighty-six percent (18/21) of the patients presented with acute or subacute neurological disorders, including motor (17/19) or sensory (8/19) abnormalities as their initial symptoms. The typical neurologic manifestations include muscle weakness, numbness, or stumbling. The remaining fourteen percent (3/21) of them had psychiatric symptoms such as depression, hallucination, or delusion as the earliest symptoms. One case presented with cognitive decline (Mini-Mental State Examination Score is 20). With progression, movement dysfunction appeared in ninety percent (19/21) of the patients with fifty-three percent (10/19) of them presenting with weakness limited to both lower extremities. Muscle strength of the feet dorsiflexion was obviously declined with sixty-three percent (12/19) of them was less than or equal to grade 3 (Medical Research Council Scale). Sixty-two percent (13/21) of the patients presented with subjective sensory deficit such as numbness or algesia. Sixty-nine percent (9/13) showed peripheral sensory deficit while thirty-one percent% (4/13) of them had sensory plane related to spinal lesion, which mainly manifested as pain and temperature sense dysfunction. Seventy-one percent (15/21) presented with vibration sense impairment and positive Romberg’s sign. Sixty-seven percent (14/21) of the patients had hyporeflexia or areflexia. Ten percent (2/21) had hyperreflexia. Fourteen percent (3/21) showed positive Babinski’s sign. Two patients showed autonomic dysfunction with urination disorder and constipation.
 |
Table 1 Basic Demographic Data and Clinical Manifestations |
Laboratory, MRI, and Electrophysiological Characteristics
We summarized the laboratory, MRI, and electrophysiological characteristics in Table 2. We checked serum Vitamin B12, folate and HCY levels in eighteen patients and only seventeen percent (3/18) showed lower level of vitamin B12 and one patient had mildly decreased folic levels. HCY levels were elevated in seventy-eight (14/18) patients. Methylmalonate urinary levels were checked in seven patients and only two presented with methylmalonateuria. Complete blood count was checked in twenty patients and only ten percent (2/20) had anemia. The other laboratory data included biochemistry analysis, CK, thyroid function, rheumatism-related antibody detection, ANCA, tumor markers and HIV testing are normal or nonspecific. We also performed lumbar puncture in nine patients. All of them had normal opening pressure. Forty-four percent (4/9) of them showed slightly elevated CSF protein without increase of cell count, which is similar to finding in Guillain–Barré syndrome (GBS), reflecting the widespread inflammation of the nerve roots. MMA gene detections in three patients were negative, ruling out the hereditary methylmalonic acidemia.
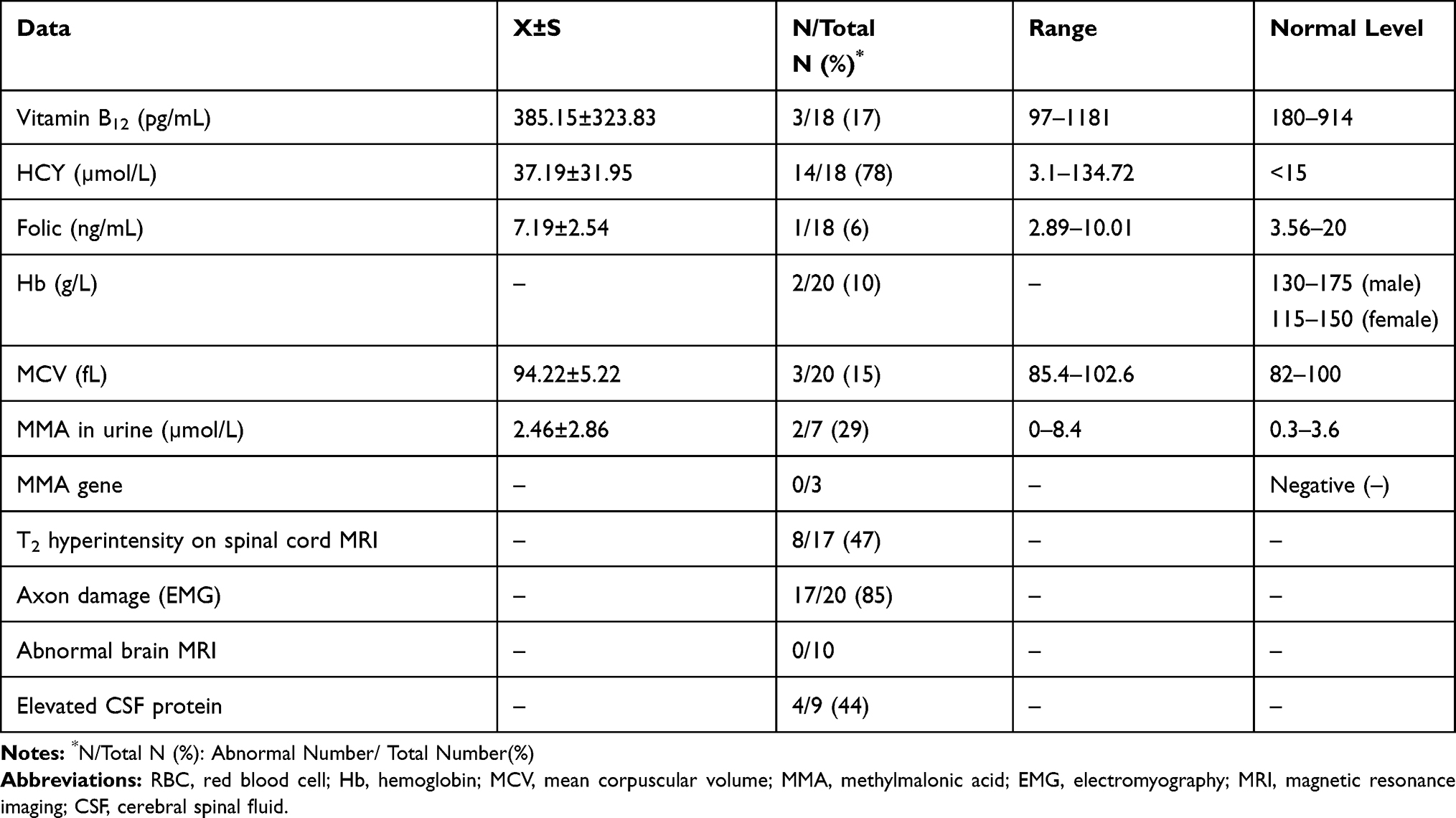 |
Table 2 Laboratory, MRI and Electrophysiological Data |
Seventeen patients had spinal cord MRI examinations. Forty-seven percent (8/17) patients showed abnormally high signal involving the posterior columns and/or lateral column of cervical cord on T2-weighted sagittal images (T2WI). Seven of them had inverted “‘V’” sign. One case also demonstrated severe surrounding edema (Figure 1A and D). Ten patients had brain MRI scans and no abnormalities were found.
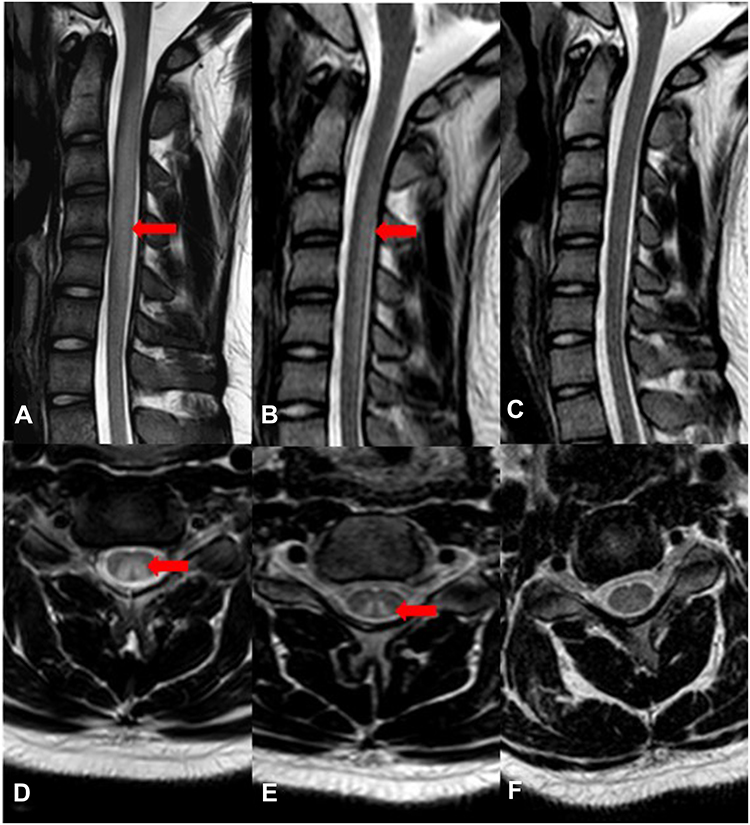 |
Figure 1 Serial cervical spinal cord MRI in patient with N2O intoxication. (A and D) MRI was performed at the time of first admission. T2-weighted sagittal images showed increased signal intensity and obviously swelling in the cervical spinal cord extending from C2 to C5. T2-weighted axial image showed abnormal signal involving the posterior columns and lateral columns of cervical cord with inverted V sign. (B and E) MRI performed one month later showed the spinal swelling mitigated. T2-weighted axial image still showed inverted V sign. (C and F) MRI performed one year later showed the disappearance of T2WI abnormal signal. |
Ninety-five percent (20/21) of the patients had electrophysiologic testing of the peripheral nerves. We evaluated the damage types of peripheral nerves by summarizing the amplitude of complex muscle action potential (CMAP) and sensory nerve action potential (SNAP), as well as NCV of the motor and sensory nerves (MNCV and SNCV). The decrease of CMAP and SNAP referred to axonal injuries while the slow-down of the NCV referred to the demyelinating lesions. In our patients, the most common type of N2O-induced neuropathy was axonal neuropathy (85%, 17/20), followed by demyelinating neuropathy (5%, 1/20). The remaining two patients only exhibited mildly abnormal findings and cannot be classified as axonal or demyelinating lesions. In the 17 cases of axonal injury, seven of them showed axonal damages involving both the sensory and motor nerves. Eight patients showed only motor axonal injuries and the other two had only sensory axonal injuries.
Follow-Up Evaluation
All of our patients were treated with vitamin B12 supplements by injection or oral administration. The average duration of treatment was more than half a year.
Assessment of the patients’ overall physical and mental status was performed before discharge. All patients demonstrated improvement to some degree in the aspect of sensation and motor domain. The subjective paresthesia improved quickly, while the motor dysfunction recovered slowly, especially the strength of distal extremities of both lower extremities. Five patients who had been followed-up by telephone six months after discharge recovered satisfactorily. They reported that their neurological symptoms were gradually mitigated or disappeared and finally returned to normal daily activities of life. Only one patient was left with sequela of mild foot drop which is correlated to longer N2O exposure duration.
We performed MRI, laboratory, and electrophysiologic studies in one patient during a 12-month period after discharge (Figures 1–3). On the initial admission, serum vitamin B12 level of this patient was extremely low and the level HCY was very high. When vitamin B12 supplementation therapy was started, the level of serum vitamin B12 was increased and HCY was returned to the normal range quickly (one month later). Unfortunately, the patient was addicted and exposed to N2O again 12 months later and the abnormal vitamin B12 and HCY level reappeared, indicating that N2O deactivates vitamin B12 very rapidly. Such laboratory abnormalities also happen in patients with vitamin B12 deficiency from malabsorption or malnutrition. However, the process of depletion usually takes three to four years, which is much longer than that due to N2O deactivation. By observing serial cervical spinal MRI, we found that abnormal T2WI signals and spinal swelling in the cervical spinal cord gradually and completely disappeared with the supplement of vitamin B12. Similarly, electrophysiologic studies demonstrated that the amplitude of CMAP improved along with the vitamin B12 supplementation, especially that in both lower extremities.
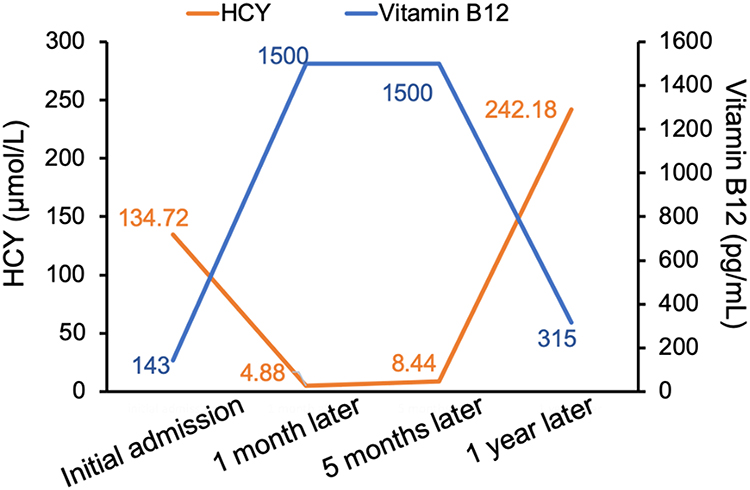 |
Figure 2 The serial serum vitamin B12 and HCY level during 12 months follow-up. |
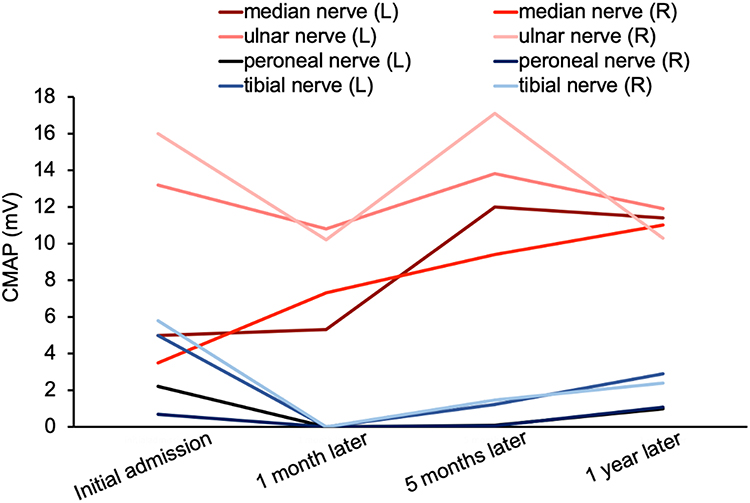 |
Figure 3 The changes of the nerves’ complex muscle action potential (CMAP) in patient with N2O intoxication. On initial admission, the patient developed severe symptoms and the CMAP of the peripheral nerve deteriorated rapidly. After stopping the exposure of N2O and high-dose vitamin B12 supplementation therapy, the CMAP of these nerves recovered gradually. |
Discussion
N2O was once widely used in surgical auxiliary anesthesia.2 In 1978, Layzer et al reported neurological damage due to N2O exposure for the first time.4 Since then, more cases about N2O toxicities such as neurotoxicity, reproductive and bone marrow toxicity of N2O were reported.15 Recently, several studies found that N2O might precipitate complications after major surgery and was associated with long-term risk of myocardial infarction.16,17 As the toxicities of N2O had been discovered gradually, there has been an obvious decrease in the use of N2O as an analgesic drug in modern clinical practice. However, since the 1970s, inhalation of N2O became a popular public entertainment due to its pleasurable properties. A study in 1979 showed that more than 20% of dental students in the US had tried inhaling N2O to achieve a “high” condition.18 Another study in 2003 indicated that more than 12% of freshmen at the University of Auckland had inhaled N2O as a recreational activity.19 Taiwanese scholars even realized that N2O-induced neuropathy has become a common disease in the outpatient department of neurology.20 In the recent years, the recreational use of N2O in China has become more and more prevalent as well, especially in the adolescent group.
Consistent with previous studies,11,21 we found that the main group of N2O abuse is the young whose age ranges from 14 to 34-year-olds. All patients were exposed to N2O at some entertainment venues. Theoretically, using a high dose of N2O in a short period is more likely to induce neurological symptoms and the severity of clinical manifestations is mostly correlated with the amount of N2O inhaled.22,23
Most researchers referred that N2O-induced neuropathy is related to vitamin B12 deficiency.6,24–26 Vitamin B12, also known as cobalamin (Cbl), is composed of cobalt ions (Co+) in the center and the surrounding corrin ring.27 Combined with different modified groups, there are diverse Cbl including cyanocobalamin (CNCbl), hydroxycobalamin (HOCbl), methycobalamine (MeCbl), and adenosine cobalamin (AdoCbl). MeCbl is the cofactor of methionine synthetase which participants in the reaction of N5-methyltetrahydrofolate (N5-methyl-THF) with HCY to produce tetrahydrofolate (THF) and methionine (Met). Met is then converted to s-adenosylmethionine (SAM), a donor of methyl groups participating in the methylation of myelin. AdoCbl is a cofactor of methylmalonyl CoA mutase (MMCoAM) which takes part in the oxidation of long-chain fatty acid and converts methylmalonyl CoA to succinyl CoA, which then enters tricarboxylic acid cycle.28 N2O, however, is cleaved into free nitrogen and oxygen after inhalation and, subsequently, the oxygen then rapidly oxidizes Co+ to Co3+ and thus irreversibly inactivates vitamin B12, leading to the inhibition of both pathways. SAM synthesis reduced when methionine cycle was inhibited by N2O, leading to demyelination of peripheral nerves by decreasing the methylation of myelin. In addition, when the AdoCbl-MMCoAM pathway was inhibited, the normal fatty acid synthesis was blocked while the abnormal fatty acid synthesis increased, leading to demyelination. In addition, the accumulation of HCY,24 the oxidative stress injury,29 and the change of cytokines and growth factors30 were also recognized to be associated with N2O-induced neuropathy. N2O can also affect the activity of MMCoAM to interfere with the cell energy metabolism, which is accompanied by accumulation of metabolites such as MMA and methylcitrate et al, and finally leads to dysfunction of multiple organs such as bone marrow, nerve, liver, and kidney.25
SCD and peripheral neuropathy are the two main neuropathies caused by N2O toxicity.
In our study, the main neurological symptoms and signs were weakness, numbness and hyporeflexia of distal extremities with acute or subacute onset, which were easily misdiagnosed as GBS. Moreover, the CSF albuminocytologic dissociation in part of our patients further complicates the diagnosis. In fact, two of them were diagnosed as GBS at the beginning and treated with intravenous immunoglobulin (IVIG). Hence, when we encountered young patients who complained of acute weakness and/or numbness, we should be aware of the possibility of N2O abuse. The history of prodromal infection, the serum HCY level and the cervical spinal MRI sign maybe more helpful for the differential diagnosis at the early stage of the course. In addition, N2O-induced neuropathy should be differentiated from other acute motor axonal neuropathy, diabetic peripheral neuropathy, alcoholic peripheral neuropathy, multiple peripheral neuropathy after HIV infection, HIV-related vacuolar myelopathy, vitamin E deficiency myelopathy and copper deficiency myelopathy.31
It is worth noting that some of our patients suffered from psychiatric symptoms such as hallucination, delusion, or depression. Two of our patients initially experienced transient hallucination before limb symptoms appeared. Similarly, the other patient started off with depression and was misdiagnosed as tristimania until he developed cognition impairment and weakness of his both lower limbs. Therefore, when we engage a patient with acute mental symptoms, we should not ignore the probability of N2O abuse, especially when the patient has no history and family history of mental disorders. Moreover, autoimmune encephalitis and some rarely genetic metabolic encephalopathies also need to be excluded.
Elevated HCY level is common in our patients. However, serum vitamin B12 is usually in the normal range. There are several mechanisms responsible for such a finding. Firstly, vitamin B12 is unevenly distributed in our body which means the level of serum vitamin B12 does not completely represent the level of intracellular vitamin B12.32 Furthermore, N2O affects not only the amount of vitamin B12' but also the activity of it. In addition, some patients took vitamin B12 supplement on their own before they came to us. For these reasons above, most previous studies regarded the elevated HCY level as one of the more valuable diagnostic indicators.3,20 The other specific characteristics of N2O intoxication is hyperintensity of T2-weighted image mainly involving the posterior columns with inverted “V” sign over the spinal MRI especially the cervical spinal cord.
Electrophysiological study is also characteristic. In our research, almost all patients had abnormal results and most of them showed bilaterally symmetrical, length-dependent, axonal damage of nerve, and motor axonal dysfunction was more prominent. Only very few patients presented with demyelinating damage. This result is completely consistent with previous studies.11,20,33 Li et al, reported 33 cases in Taiwan and revealed that the most common peripheral neuropathy of N2O-induced neuropathy was mixed axonal and demyelinating neuropathy (36%), followed by axonal neuropathy (30%) and demyelinating neuropathy (6%).20 Sural nerve biopsy further confirmed the axonal damage as well as secondary demyelinating damage.34 Thus, axonal neuropathy appeared to be more common than the demyelinating in N2O-induced neuropathy.
However, this result cannot be fully explained by the mechanism of hypomyelination in peripheral nerves caused by vitamin B12 metabolic dysfunction alone. The exact pathophysiology of axonal injury had not been clearly explained in current studies but several possibilities had been raised. Studies had shown that the accumulation of HCY was considered to be related to the decrease of CMAP and SNAP20 suggesting that high HCY may be an independent factor for axonal injury. As mentioned above, N2O irreversibly inactivated vitamin B12 leading to HCY accumulation.25 High HCY level contributes to oxidative stress injury and finally results in mitochondrial dysfunction and neuronal necrosis.24 In addition, high HCY can induce nerve ischemia by damaging microvascular endothelial cells and lead to axonal injury.24,29
Previous reports had also referred to the fact that N2O might exert its neurotoxicity by other mechanisms such as antagonism of NMDA receptors.2,3,35,36 Morris et al, reported a delayed phenomenon which referred to the delayed deterioration of motor nerves in patient with N2O-induced neuropathy after vitamin B12 supplementation treatment and suggested that N2O toxicity has an unknown mechanism different from the induction of functional vitamin B12 deficiency.26 Tani et al, observed a unique nerve excitability pattern in N2O abuse patients showing prominent motor superexcitability changes and less prominent sensory superexcitability changes, revealing that N2O abuse might exert its toxicity via affecting the paranodal region, possibly affecting fast K+ channel.37
Avoiding exposure to N2O and high-dose vitamin B12 supplementation are the main treatment of N2O-induced neuropathy. The symptoms generally improved rapidly in a short time, and the injury is reversible. However, some patients can develop sequelae, as well. The standard treatment regimen38 is injection of 1000 µg vitamin B12 per day for one week, then weekly injection for four weeks, followed by monthly injections or oral supplementations (1000–2000 µg per day) until the clinical symptoms are completely relieved. Met might be an assistant treatment which can accelerate the regeneration of myelin sheath.39
There are several limitations in our study. Firstly, it is a retrospective research, some patients’ information is not complete. Most of the patients refused to offer their detailed history in N2O abuse. Secondly, it is difficult to estimate how much N2O the patients inhaled. Thus, the dose–response relationship is difficult to calculate. In addition, vitamin B included mecobalamin tablets are widely used in a variety of neuropathies with clinical manifestations of numbness and weakness. Therefore, the vitamin B12 level and HCY level of a few patients might not be reliable indicators in our research.
Conclusion
In conclusion, the N2O-induced neuropathy mainly occurred in adolescents and the young groups. In patients presenting with acute or subacute onset of myelopathy, psychosis as well as axonal polyneuropathy, N2O intoxication should be at the top of the differential diagnosis.
Ethics Approval and Informed Consent
This study was approved by the Ethics Committee of Qilu Hospital, Shandong University and informed consent was obtained from all subjects in this study. The protocols were in accordance with the Declaration of Helsinki. Written informed consent was acquired from all participants or their legal guardians.
Acknowledgments
This research was supported by the key Research and Development Plan of Shandong Province (2017GSF18139 to QZ. Wang) and Innovative Research Project of Undergraduate Clinical Medicine Teaching of Qilu Hospital, Shandong University (No. 2019QLJY13). We thank the physicians who provided clinical support. We also thank our patients and their families. Rui Zheng and Qinzhou Wang contributed equally to the article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Weimann J. Toxicity of nitrous oxide. Best Pract Res Clin Anaesthesiol. 2003;17(1):47–61. doi:10.1053/bean.2002.0264
2. Jevtović-Todorović V, Todorović SM, Mennerick S, et al. Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin. Nat Med. 1998;4:460–463. doi:10.1038/nm0498-460
3. Garakani A, Jaffe RJ, Savla D, et al. Neurologic, psychiatric, and other medical manifestations of nitrous oxide abuse: a systematic review of the case literature. Am J Addict. 2016;25:358–369. doi:10.1111/ajad.12372
4. Layzer RB, Fishman RA, Schafer JA. Neuropathy following abuse of nitrous oxide. Neurology. 1978;28(5):504–506. doi:10.1212/WNL.28.5.504
5. Blanco G, Peters HA. Myeloneuropathy and macrocytosis associated with nitrous oxide abuse. Arch Neurol. 1983;40(7):416–418.
6. Pema PJ, Horak HA, Wyatt RH. Myelopathy caused by nitrous oxide toxicity. AJNR Am J Neuroradiol. 1998;19:894–896.
7. Mancke F, Kaklauskaitė G, Kollmer J, Weiler M. Psychiatric comorbidities in a young man with subacute myelopathy induced by abusive nitrous oxide consumption: a case report. Subst Abuse Rehabil. 2016;7:155–159. doi:10.2147/SAR.S114404
8. Sahenk Z, Mendell JR, Couri D, Nachtman J. Polyneuropathy from inhalation of N2O cartridges through a whipped cream dispenser. Neurology. 1978;28:485–487. doi:10.1212/WNL.28.5.485
9. Wang L, Fan QJ, Dong MR, et al. Nervous system disorder caused by nitrous oxide intoxication: one case report. Chin J Contemp Neurol Neurosurg. 2016;16:533–537.
10. Shen Q, Lu H, Wang H, Xu Y. Acute cognitive disorder as the initial manifestation of nitrous oxide abusing: a case report. Neurol Sci. 2019. doi:10.1007/s10072-019-04183-w
11. Zheng D, Ba F, Bi G, Guo Y, Gao Y, Li W. The sharp rise of neurological disorders associated with recreational nitrous oxide use in China: a single-center experience and a brief review of Chinese literature. J Neurol. 2020;267(2):422–429. doi:10.1007/s00415-019-09600-w
12. Yuan JL, Wang SK, Jiang T, Hu WL. Nitrous oxide induced subacute combined degeneration with longitudinally extensive myelopathy with inverted V-sign on spinal MRI: a case report and literature review. BMC Neurol. 2017;17(1):222. doi:10.1186/s12883-017-0990-3
13. Dong X, Ba F, Wang R, Zheng D. Imaging appearance of myelopathy secondary to nitrous oxide abuse: a case report and review of the literature. Int J Neurosci. 2019;129(3):225–229. doi:10.1080/00207454.2018.1526801
14. Li XY, Li YY, Jia J, Du P, Feng GD, Jin LR. Nervous system damage caused by laughing gas abuse (with a case report and literature review). Stroke Neurol Dis. 2018;25:59–64.
15. Louis-Ferdinand RT. Myelotoxic, neurotoxic and reproductive adverse effects of nitrous oxide. Adverse Drug React Toxicol Rev. 1994;13:193–206.
16. Myles PS, Leslie K, Chan MT, et al. Avoidance of nitrous oxide for patients undergoing major surgery: a randomized controlled trial. Anesthesiology. 2007;107:221–231. doi:10.1097/01.anes.0000270723.30772.da
17. Leslie K, Myles PS, Chan MT, et al. Nitrous oxide and long-term morbidity and mortality in the ENIGMA trial. Anesth Analg. 2011;112:387–393. doi:10.1213/ANE.0b013e3181f7e2c4
18. Rosenberg H, Orkin FK, Springstead J. Abuse of nitrous oxide. Anesth Analg. 1979;58(2):104–106. doi:10.1213/00000539-197903000-00009
19. Ng J, O’Grady G, Pettit T, Frith R. Nitrous oxide use in first-year students at Auckland University. Lancet. 2003;361(9366):1349–1350. doi:10.1016/S0140-6736(03)13045-0
20. Li HT, Chu CC, Chang KH, et al. Clinical and electrodiagnostic characteristics of nitrous oxide-induced neuropathy in Taiwan. Clin Neurophysiol. 2016;127:3288–3293. doi:10.1016/j.clinph.2016.08.005
21. Lin RJ, Chen HF, Chang YC, Su JJ. Subacute combined degeneration caused by nitrous oxide intoxication: case reports. Acta Neurol Taiwan. 2011;20:129–137.
22. Cheng HM, Park JH, Hernstadt D. Subacute combined degeneration of the spinal cord following recreational nitrous oxide use. BMJ Case Rep. 2013;2013(mar08 1):bcr2012008509–bcr2012008509. doi:10.1136/bcr-2012-008509
23. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21:1063–1068. doi:10.1111/j.1525-1497.2006.00525.x
24. Savage S, Ma D. The neurotoxicity of nitrous oxide: the facts and “putative” mechanisms. Brain Sci. 2014;4(1):73–90. doi:10.3390/brainsci4010073
25. Hathout L, El-Saden S. Nitrous oxide-induced B₁₂ deficiency myelopathy: perspectives on the clinical biochemistry of vitamin B₁₂. J Neurol Sci. 2011;301:1–8. doi:10.1016/j.jns.2010.10.033
26. Morris N, Lynch K, Greenberg SA. Severe motor neuropathy or neuronopathy due to nitrous oxide toxicity after correction of vitamin B12 deficiency. Muscle Nerve. 2015;51:614–616.
27. Hodgkin DC, Karnper J, Mackay M, Pickworth J. Structure of Vitamin B12. Nature. 1956;178:64–66. doi:10.1038/178064a0
28. Kräutler B. Vitamin B12: chemistry and biochemistry. Biochem Soc Trans. 2005;33:806–810. doi:10.1042/BST0330806
29. Singh SK, Misra UK, Kalita J, Bora HK, Murthy RC. Nitrous oxide related behavioral and histopathological changes may be related to oxidative stress. Neurotoxicology. 2015;48:44–49. doi:10.1016/j.neuro.2015.03.003
30. Veber D, Mutti E, Galmozzi E, et al. Increased levels of the CD40: CD40ligand dyad in the cerebrospinal fluid of rats with vitamin B12 (cobalamin)-deficient central neuropathy. J Neuroimmunol. 2006;176:24–33. doi:10.1016/j.jneuroim.2006.04.002
31. Sotirchos ES, Saidha S, Becker D. Neurological picture. Nitrous oxide-induced myelopathy with inverted V-sign on spinal MRI. J Neurol Neurosurg Psychiatry. 2012;83:915–916. doi:10.1136/jnnp-2012-303105
32. Scalabrino G, Nicolini G, Buccellato FR, et al. Epidermal growth factor as a local mediator of the neurotrophic action of vitamin B12 (cobalamin) in the rat central nervous system. FASEB J. 1999;13:2083–2090. doi:10.1096/fasebj.13.14.2083
33. Blanco G, Peters HA. Myeloneuropathy and macrocytosis associated with nitrous oxide abuse. Arch Neurol. 1983;40:416–418.
34. Kalita J, Chandra S, Bhoi SK, et al. Clinical, nerve conduction and nerve biopsy study in vitamin B12 deficiency neurological syndrome with a short-term follow-up. Nutr Neurosci. 2014;17:156–163. doi:10.1179/1476830513Y.0000000073
35. Maze M, Fujinaga M. Recent advances in understanding the actions and toxicity of nitrous oxide. Anaesthesia. 2000;55:311–314. doi:10.1046/j.1365-2044.2000.01463.x
36. Richardson KJ, Shelton KL. N-methyl-D-aspartate receptor channel blocker-like discriminative stimulus effects of nitrous oxide gas. J Pharmacol Exp Ther. 2015;352:156–165. doi:10.1124/jpet.114.218057
37. Tani J, Weng HY, Chen HJ, Chang TS, Sung JY, Lin CS. Elucidating unique axonal dysfunction between nitrous oxide abuse and Vitamin B12 deficiency. Front Neurol. 2019;10:704. doi:10.3389/fneur.2019.00704
38. Stabler SP. Vitamin B12 Deficiency. N Engl J Med. 2013;368(2):149–160. doi:10.1056/NEJMcp1113996
39. Scott JM, Dinn JJ, Wilson P, Weir DG. Pathogenesis of subacute combined degeneration: a result of methyl group deficiency. Lancet. 1981;318(8242):334–337. doi:10.1016/S0140-6736(81)90649-8
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.