")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Reversal of corticosteroid insensitivity by p38 MAPK inhibition in peripheral blood mononuclear cells from COPD
Authors Khorasani N, Baker J, Johnson M, Chung KF, Bhavsar P
Received 7 August 2014
Accepted for publication 10 October 2014
Published 4 February 2015 Volume 2015:10(1) Pages 283—291
DOI https://doi.org/10.2147/COPD.S72403
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Richard Russell
Nadia Khorasani,1 Josephine Baker,1 Malcolm Johnson,2 Kian Fan Chung,1 Pankaj K Bhavsar1
1Experimental Studies, Airway Disease Section, National Heart and Lung Institute, Imperial College and Biomedical Research Unit, Royal Brompton and Harefield NHS Trust Hospital, London, UK; 2GlaxoSmithKline, Uxbridge, UK
Background: Corticosteroids (CS) have limited efficacy in the treatment of chronic obstructive pulmonary disease (COPD). p38 mitogen-activated protein kinase (MAPK) activation is increased in lung macrophages of COPD. We investigated whether p38 MAPK inhibition can modulate CS insensitivity of peripheral blood mononuclear cells (PBMCs) from patients with COPD.
Methods: PBMCs from patients with COPD (n=8) or healthy smokers (n=8) were exposed to lipopolysaccharide (LPS) with a selective p38 MAPK inhibitor (GW856553; 10–10–10–6 M), with dexamethasone (10–10–10–6 M), or with both. Phosphorylated glucocorticoid receptor (GR) was measured by Western blot.
Results: Baseline (P<0.01) and LPS-induced (P<0.05) CXCL8 release was greater in PBMCs from COPD compared to healthy smokers. Inhibition of LPS-induced CXCL8 release by dexamethasone (10–6 M) was reduced, and baseline and LPS-induced p38 MAPK activation increased in PBMCs of COPD. GW856553 (10–9 and 10–10 M) synergistically increased the inhibitory effect of dexamethasone (10–8 and 10–6 M) on LPS-induced CXCL8 release in COPD. Similar results were obtained for IL-6 release. GW856553 inhibited dexamethasone- and LPS-activated phosphorylation of serine 211 on GR. CS insensitivity in COPD PBMCs is reversed by inhibition of p38 MAPK activity, partly by preventing phosphorylation of GR at serine 211.
Conclusion: p38 MAPK inhibition may be beneficial in COPD by restoring CS sensitivity.
Keywords: glucocorticoid receptor, p38 mitogen-activated protein kinase
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive disease affecting both the airways and lungs, largely caused by cigarette smoking. It is a major cause of global morbidity and mortality, predicted to become the third leading cause of death by 2020.1 COPD is characterized by chronic airflow obstruction of the small airways that is poorly responsive to β2-agonist bronchodilators and corticosteroids (CS). The chronic airway inflammatory response in COPD is characterized by a predominance of neutrophils and macrophages in the lung.2 Increased activation of the proinflammatory transcription factor, nuclear factor-κB, has been reported in bronchial biopsies of stable COPD patients and in sputum macrophages of patients with COPD exacerbation.3 There is also evidence of systemic inflammation with raised serum levels of inflammatory biomarkers such as interleukin (IL)-6 and C-reactive protein (CRP).4
Inhaled CS (ICS) are recommended anti-inflammatory treatment for COPD patients with a forced expiratory volume in 1 second (FEV1) of less than 60% predicted and who are prone to frequent exacerbations. ICS are combined with long-acting β-agonists (LABA) when symptoms persist or worsen. CS diffuse across the plasma membrane, and bind to and activate the glucocorticoid receptor (GR) in the cytoplasm, which then translocates into the nucleus, where it can either activate the transcription of anti-inflammatory genes or suppress proinflammatory gene expression. GR phosphorylation at serine (Ser) 203, Ser211, and Ser226 has been reported to be enhanced upon binding of CS to GR, linking GR phosphorylation with transcriptional activity.5 However, hyperphosphorylation of GR can have a detrimental effect on ligand binding6 as well as on nuclear DNA and protein interactions.7
The p38 MAPK family of serine/threonine protein kinases consist of four isoforms (p38α, p38β, p38γ, and p38δ) that are activated by inflammatory stimuli that include Toll-like receptor agonists.8 Activated p38 MAPK phosphorylates a number of intracellular proteins, including transcription factors such as GR,9 and regulates the translation and the stability of inflammatory mRNAs.10 The p38α isoform is expressed in endothelial, immune, and inflammatory cells and regulates the expression of the proinflammatory cytokines TNF-α, IL-1β, CXCL8, and IL-6.11 Alveolar macrophages from patients with COPD express a greater degree of activation of p38 MAPK, as compared to cells from healthy smokers,12 and are less sensitive to inhibition of CXCL8 and GM-CSF release13 by dexamethasone. Cross talk between the p38 MAPK signaling pathways and GR has been reported, such as Ser211 on GR being a potential substrate for p38 MAPK.14
p38 MAPK inhibitors suppress inflammatory mediator release from alveolar macrophages of patients with COPD.15 We now extend these in vitro studies to peripheral blood mononuclear cells (PBMCs) of patients with COPD. Because exacerbations of COPD are frequently caused by bacterial infections,16 we used lipopolysaccharide (LPS) to activate PBMCs. Although CS are used in treating exacerbations of COPD, their anti-inflammatory response is limited.
Anti-inflammatory therapies for COPD, such as ICS, may provide partial benefit, although there is a degree of CS insensitivity in these patients.17 There is an unmet need to develop novel anti-inflammatory therapies that could slow or stop disease progression, but one other approach would be to reverse CS insensitivity. To explore a potential role for p38 MAPK activation in CS insensitivity, we examined whether an inhibitor of p38 MAPK could improve the anti-inflammatory ability of dexamethasone to suppress cytokine release in PBMCs from patients with COPD in response to LPS stimulation. We studied the effect of p38 MAPK activation on the phosphorylation status of GR at Ser211.
Methods
Study participants
Patients with COPD were recruited from the clinics of the Royal Brompton Hospital (London, UK), and smokers were recruited by local advertisement (Table 1). Patients with COPD were diagnosed on the basis of a ratio of FEV1/forced vital capacity <0.7 with a cigarette-smoking history of more than 10 pack-years, and classified according to the Global initiative of Chronic Obstructive Lung Disease (GOLD) criteria based on the predicted FEV1. Healthy smokers had a cigarette smoking history of >10 pack-years but had an FEV1/forced vital capacity ratio >70% and FEV1 > 80% predicted. The study protocol was approved by the Ethics Committee of Royal Brompton and Harefield NHS Trust/National Heart and Lung Institute, London, UK (09/H0708/19). All volunteers gave their written informed consent.
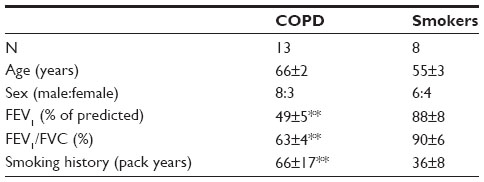 | Table 1 Clinical characteristics of subjects |
Isolation and stimulation of PBMCs
PBMCs were isolated as previously described.18 PBMCs (7.5×105 cells/well) were stimulated with LPS (10 ng/mL), and with or without dexamethasone (10−10–10−6 M) or with or without GW856553 (10−10–10−6 M). Supernatants were removed 18 hours later and analyzed for CXCL8 or IL-6 by enzyme-linked immunosorbent assay (R&D Systems, Inc., Minneapolis, MN, USA).
p38 MAPK inhibitor
GW856553 (GlaxoSmithKline plc, London, UK) is a p38 MAPK inhibitor of the nicotinamide class, with a p38α pKi =8.1 and a p38β pKi =7.6.19 It has a pIC50 =7.6 in blocking LPS-induced TNFα production in human whole blood and is 100-fold selective for p38 MAPK when tested against a panel of 67 other kinases.19
Western blotting
Protein membranes were incubated with rabbit antibody for anti-phospho-p38 followed by anti-rabbit-horseradish peroxidase antibody. Antibody-bound proteins were visualized by ECL or ECL plus (GE Healthcare UK Ltd, Little Chalfont, UK). The membranes were then reprobed with rabbit anti-total p38 (Cell Signaling Technology, Inc., Danvers, MA, USA) or with mouse anti-β-actin monoclonal antibody (Santa Cruz Biotechnology Inc., Dallas, TX, USA) to control for protein loading. Relevant band intensities were quantified by scanning densitometric analysis (VisionWorks V7; UVP Corp, Upland, CA, USA).
Data analysis
Cytokine release induced by LPS with or without dexamethasone or p38 MAPK inhibitor was calculated by subtraction of baseline release. The release of each cytokine following dexamethasone plus LPS was calculated as a percentage of cytokine release following LPS stimulation alone. We performed Spearman’s rank correlations to determine the relationships between baseline or LPS-induced CXCL8 release and FEV1. CS responsiveness between COPD patients and healthy smokers was compared using the Mann–Whitney U test. Concentration-dependent responses were examined using one-way analysis of variance (Kruskal–Wallis test) followed by a Dunn’s Multiple Comparison test. Results are expressed as mean ± standard error. P<0.05 was taken as a significant difference.
Results
Baseline data of patients with COPD and healthy smokers are presented in Table 1. Patients with COPD had severe airflow obstruction (P<0.01).
Regulation of CXCL8 by dexamethasone
Baseline and LPS-induced CXCL8 release were higher in patients with COPD compared to healthy smokers (P<0.01 and P<0.05, respectively; Figure 1A). Baseline and induced CXCL8 release correlated inversely with FEV1 (r=−0.55, P=0.04; and r=−0.71, P=0.008, respectively; Figure 1B and C). Dexamethasone inhibited CXCL8 release in a concentration-dependent manner (10−10–10−6 M) in the COPD (P<0.001, Kruskal–Wallis) and healthy-smoker (P<0.001, Kruskal–Wallis) groups, with a significantly reduced suppression in PBMCs of patients with COPD compared to healthy smokers (Figure 2A). Dexamethasone (10−6 M) led to a maximal suppression of LPS-induced CXCL8 release in PBMCs of patients with COPD of 41%±3.5% compared to 58.9%±4.5% in healthy smokers (P<0.01).
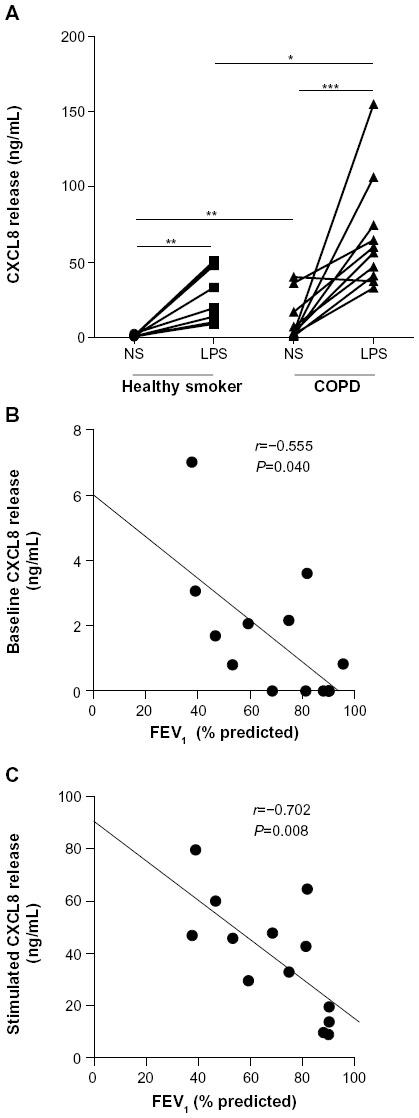 | Figure 1 Baseline and LPS-induced CXCL8 release from PBMCs of patients with COPD and healthy smokers. |
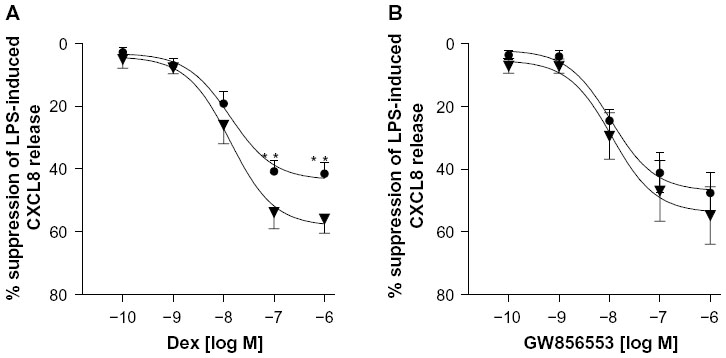 | Figure 2 Relative corticosteroid insensitivity in PBMCs of COPD patients. |
Effect of p38 MAPK inhibitor on LPS-induced CXCL8 release
GW856553 inhibited LPS-induced CXCL8 release in a concentration-dependent manner (10−10–10−6 M) in both the COPD (P<0.0001, Kruskal–Wallis) and healthy-smoker (P<0.0001, Kruskal–Wallis) groups. A maximal suppression of 47.2%±6.7% (half maximal inhibitory concentration [IC50] 9×10−6 M) and 53.8%±9.2% (Figure 2B) from PBMCs of patients with COPD and healthy subjects, respectively, was achieved at a concentration of 10−6 M. There was no difference in suppression in PBMCs from patients with COPD compared to that in healthy smokers.
p38 MAPK activity
We compared induced-p38 MAPK activity at 30 minutes poststimulation in PBMCs of patients with COPD and healthy smokers. p38 phosphorylation in COPD was higher than in smokers both at baseline (P<0.05; Figure 3A and B) and after LPS stimulation (P<0.05; Figure 3C).
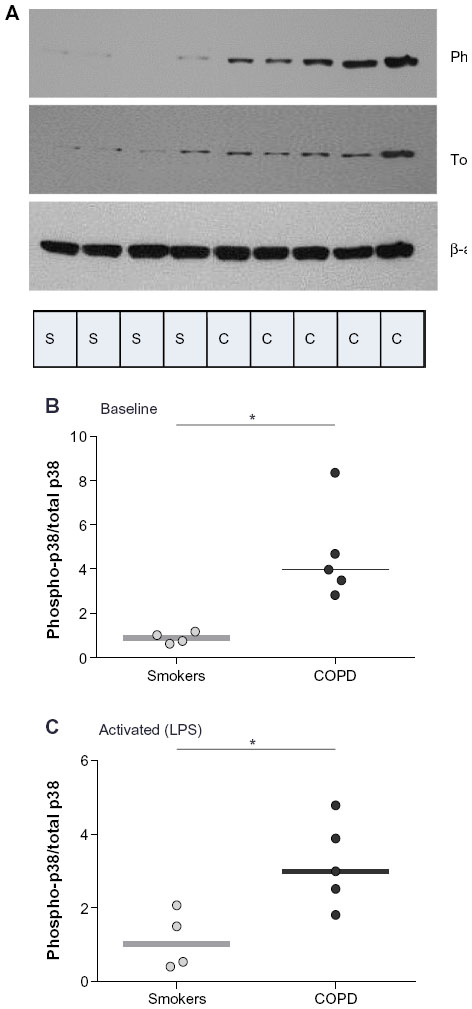 | Figure 3 Comparison of baseline and induced p38 MAPK activation in PBMCs of COPD patients and healthy smokers. |
Effect of dexamethasone and GW856553 on CXCL8 and IL-6 release
For all concentrations of dexamethasone, the addition of GW856553 resulted in a concentration-dependent improvement in the suppressive effect of the glucocorticoid. The effect of dexamethasone at 10−6 M (Figure 4A) in suppressing CXCL8 release was increased in the presence of GW856553 (10−10–10−6 M) (P<0.0001), with a maximal suppression of 80.1%±4% (IC50 2.6×10−11 M) with GW856553 at 10−6 M compared to 47.2%±6.7% suppression with dexamethasone alone (P<0.001). With GW856553 at 10−9 M and 10−10 M, which alone has a suppressive effect of less than 3%, CXCL8 suppression by dexamethasone (10−6 M) was greater at 65.2%±4.1% (P<0.001) and 57.4%±5.5% (P<0.01), respectively (Figure 4B). At dexamethasone 10−8 M, the suppressive effect was also increased by GW856553 (P<0.001), with maximal suppression with GW856553 (10−6 M) of 67.5%±5.6% (IC50 1.4×10−9 M) as compared to 20.2%±4.1% with dexamethasone alone. Similar effects were observed with dexamethasone at a concentration of 10−9 M in combination with GW856553 (IC50 3.7×10−7 M).
 | Figure 4 Effect of p38 MAPK inhibition on dexamethasone-mediated suppression of LPS-induced CXCL8 release from PBMCs of COPD patients. |
Improvements in the suppressive effects of dexamethasone by GW856553 were also observed for IL-6 release (Figure 5A). Compared with the effect of GW856553 alone, the combination of dexamethasone at 10−6 M and GW856553, at 10−9 M and 10−10 M (Figure 5B), increased maximal suppression of IL-6 (62%±7% versus 9.1%±2.0% and 51%±7% versus 10.3%±2.8%, respectively; P<0.001).
 | Figure 5 Effect of p38 MAPK inhibition on dexamethasone-mediated suppression of LPS-induced IL-6 release from PBMCs of COPD patients. |
GW856553 was a potent steroid-sparing agent: the maximal suppression of CXCL8 release obtained with the highest concentration of dexamethasone (10−6 M) can also be achieved by a 100-fold lower concentration of dexamethasone (10−8 M) in the presence of GW856553 (10−9 M).
p38 MAPK inhibition reverses CS insensitivity in COPD
The maximal CS suppression achieved in combination with GW856553 (10−6 M) was 87.1%±3.1% in PBMCs from healthy smokers, which is not different from PBMCs from COPD patients (80.1%±4%; Figure 6). For all concentrations of GW856553, there was an equivalent improvement in the suppressive effect of dexamethasone (10−6 M) in PBMCs from both patients with COPD and healthy smokers. There was no difference in the suppressive effect of dexamethasone between COPD and healthy smokers in the presence of the p38 MAPK inhibitor.
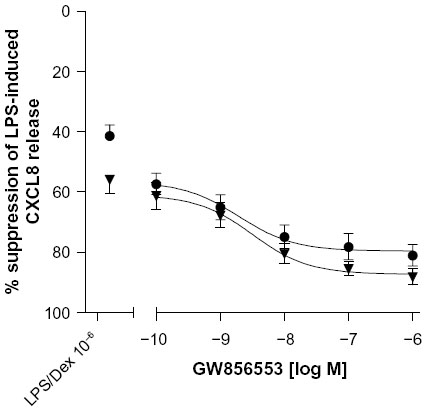 | Figure 6 Reversal of corticosteroid insensitivity in PBMCs of COPD patients. |
p38 MAPK phosphorylation of Ser211 on GR
To explore a mechanism by which GW856553 improves the suppressive effects of dexamethasone, we examined the phosphorylation of Ser211 on GR in response to dexamethasone and/or LPS. PBMCs of patients with COPD were pretreated with dexamethasone and/or GW856553 (both at 10−6 M) for 1 hour and then stimulated with LPS (10 ng/mL) for 30 minutes. Dexamethasone induced a fourfold increase in Ser211 phosphorylation over baseline (P<0.05; Figure 7A), which was prevented by GW856553 (P<0.05; Figure 7B). LPS with dexamethasone induced a sixfold increase in phosphorylation of Ser211 over baseline (P<0.05) while GW856553 suppressed this induction to the level observed for dexamethasone alone (P<0.05).
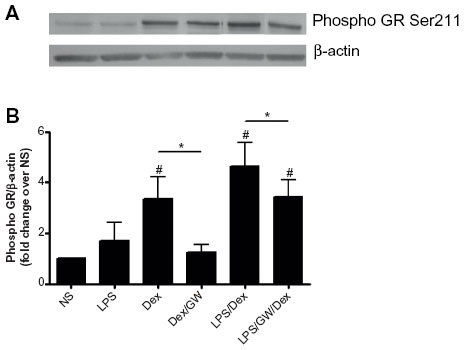 | Figure 7 Effect of GW865553 on dexamethasone-induced phosphorylation of Ser211 on the glucocorticoid receptor in PBMCs of COPD patients. |
Discussion
PBMCs of patients with COPD have higher baseline and induced CXCL8 release compared with healthy smokers, which correlates with impaired lung function. We showed relative CS insensitivity in PBMCs of patients with COPD when compared to PBMCs from healthy smokers, together with heightened p38 MAPK activation at both baseline and in response to LPS stimulation. To investigate the interaction between p38 MAPK activation and CS, we studied the effect of the selective p38 MAPK inhibitor, GW856553, and showed an enhancement of the suppressive effects of dexamethasone in PBMCs from patients with COPD at very low concentrations of GW856553, which had minimal effects alone, on CXCL8 release. This enhancement in the inhibitory effects of dexamethasone was synergistic. p38 MAPK inhibition also reversed CS insensitivity because, over the complete range of GW856553 concentrations used, the improvement in the suppressive effects of dexamethasone (10−6 M) in COPD was identical to that achieved in healthy smokers. Our data is supported by a recent study that showed that a p38 MAPK inhibitor, in combination with dexamethasone, caused a greater suppression of gene expression induced by LPS in alveolar macrophages.15
An increase in the number of macrophages expressing phosphorylated p38 MAPK in lung tissues from patients with COPD has been reported.12 In a study of oral p38 MAPK inhibitor GW856553 (Losmapimod) in patients with COPD, there was a reduction in plasma fibrinogen by 11%, with a trend towards lower CXCL8, IL-6, and CRP plasma levels.20 This effect could result from an inhibitory effect of GW856553 on the release of CXCL8 and IL-6 from circulating PBMCs, as p38 MAPK activation is reported to enhance CXCL8 mRNA stability and reduce protein turnover,20 leading to increased CXCL8 release.21 Cigarette-smoke-induced CXCL8 release from airway epithelial cells in vitro is also attenuated by a p38 MAPK inhibitor (SB203580) at both transcriptional and translational levels.22 Using another p38 MAPK inhibitor (PH-797804), a decrease in serum C-reactive protein in moderate to severe COPD patients was observed, despite these patients already being treated with ICS. This would indicate an improvement in CS insensitivity, as illustrated by an improvement in anti-inflammatory effect by p38 MAPK inhibition above that provided by ICS/LABA alone.23 In rodent models of tobacco-smoke-induced inflammation, a p38 inhibitor given therapeutically or prophylactically was able to inhibit tobacco-smoke-induced inflammatory responses and increases in CXCL8 production,24 whereas dexamethasone was ineffective.25
Our data show the potential downstream effects of heightened p38 MAPK activation, either at baseline or in response to LPS stimulation, can lead to a reduction in the effectiveness of CS. Heightened p38 MAPK activity may result in the failure of CS to suppress inflammation in severe asthma,18 and we now report similar findings in COPD. We also report an increase in baseline p38 MAPK activity which has been shown to have an effect on GR function. For example, in airway smooth muscle cells, baseline levels of p38 MAPK activation maintains unliganded GR in an inactive state.26 Thus, an increase in baseline p38 MAPK activation, as reported in our study, may induce CS insensitivity in COPD by decreasing the ability of GR to switch from an inactive to active state in response to CS.
We also showed that CS-dependent phosphorylation of Ser211, on the human GR, is a p38 MAPK-dependent process. However, we did not determine whether the effect is a result of direct or indirect action of p38 MAPK on GR Ser211. GR phosphorylation is enhanced upon glucocorticoid ligand binding, suggesting a link between GR hormone-dependent phosphorylation and transcriptional activity.5 The human GR phosphorylation sites, Ser203, Ser211, and Ser226, lie within the N-terminal AF1 region of the receptor. GR Ser203 and Ser226 are phosphorylated in both the absence and presence of agonists, whereas phosphorylation of Ser211 is only observed in a ligand-dependent manner. Recent studies have suggested that Ser211 is not a primary target for p38 MAPK activity27 and that p38 MAPK primarily phosphorylates Ser134 in a ligand-independent manner.28 In airway smooth muscle cells, Bouazza et al showed that Ser203 but not Ser211 is a direct substrate for p38 MAPK29 and suggested that blockade of p38 MAPK, or mutation of Ser211 to alanine, results in a decrease in Ser203 phosphorylation with a concomitant increase in Ser211 phosphorylation. A possible explanation for this could be the result of a conformational change in GR upon Ser203 phosphorylation, which hinders kinase access to Ser211. Our findings confirm Ser211 phosphorylation in PBMCs requires p38 MAPK activity, while findings from other studies would indicate that phosphorylation of Ser211 may be a cell-type specific process.
In support of our findings, other studies have shown that GR phosphorylation on Ser211 enhances GR-mediated transcription.7,29,30 For example, in transformed cells it has been suggested that phosphorylation of Ser211 enhances GR-mediated transcription by having an effect on the conformation of GR, rendering it more accessible to interaction with cofactors such as GR-interacting protein-1.31 Paradoxically, sustained, dysregulated phosphorylation of GR can impair GR function.32 Potential mechanisms of CS insensitivity resulting from hyper-phosphorylation of GR include reduction in: 1) GR–glucocorticoid response element interaction; 2) GR ligand binding affinity; and 3) GR recycling following nuclear export.7 Hyperphosphorylation has been shown to inhibit GR-mediated transcriptional activation, promote GR export from the nucleus upon hormone withdrawal,33 and alter cofactor recruitment to modulate the transcriptional response of GR to attenuate CS signaling.32 Direct involvement of Ser211 in these mechanisms requires further investigation.
Conclusion
Our study points to a novel approach to the treatment of COPD. A p38 MAPK inhibitor may not only be used as an anti-inflammatory agent, but it also induces reversal of CS insensitivity, which would lead to improved therapeutic effects of CS.
Acknowledgments
We thank Florence Chow and Sally Meah for the recruitment of patients. This study was supported by the National Institute of Health Research Respiratory Disease Biomedical Research Unit at the Royal Brompton Hospital and Harefield Foundation NHS Trust and Imperial College London.
Author contributions
NK participated in the design of the study, performed the majority of experiments and statistical analyses, and drafted the manuscript. JB contributed to the acquisition and interpretation of data and to the initial draft of the manuscript. MJ participated in the design and critically revised the manuscript. PKB and KFC conceived the study, participated in the design and coordination of the study, and critically revised the manuscript. All authors read and approved the final manuscript.
Disclosure
PKB has received research support from GlaxoSmithKline, UK. KFC has been on Advisory Boards for GlaxoSmithKline, Merck, and Gilead, and has received research support from National Institute of Health, Medical Research Council UK, Asthma UK, and the Wellcome Trust. MJ is an employee of GSK. NK and JB report no conflicts of interest in this work.
References
Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555. | |
Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–1356. | |
Caramori G, Romagnoli M, Casolari P, et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax. 2003;58:348–351. | |
Hansel TT, Barnes PJ. New drugs for exacerbations of chronic obstructive pulmonary disease. Lancet. 2009;374:744–755. | |
Weigel NL, Moore NL. Steroid receptor phosphorylation: a key modulator of multiple receptor functions. Mol Endocrinol. 2007;21:2311–2319. | |
Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol. 2002;109:649–657. | |
Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 2009;61:979–986. | |
Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. | |
Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3:69–75. | |
Dean JL, Sully G, Clark AR, Saklatvala J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 2004;16:1113–1121. | |
Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis. 2008;67:909–916. | |
Renda T, Baraldo S, Pelaia G, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31:62–69. | |
Culpitt SV, Rogers DF, Shah P, et al. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167:24–31. | |
Miller AL, Webb MS, Copik AJ, et al. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 2005;19:1569–1583. | |
Armstrong J, Harbron C, Lea S, et al. Synergistic Effects of p38 mitogen-activated protein kinase inhibition with a corticosteroid in alveolar macrophages from patients with chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2011;338:732–740. | |
Mackay AJ, Hurst JR. COPD exacerbations: causes, prevention, and treatment. Immunol Allergy Clin North Am. 2013;33:95–115. | |
Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131:636–645. | |
Bhavsar P, Khorasani N, Hew M, Johnson M, Chung KF. Effect of p38 MAPK inhibition on corticosteroid suppression of cytokine release in severe asthma. Eur Respir J. 2010;35:750–756. | |
Goldstein DM, Kuglstatter A, Lou Y, Soth MJ. Selective p38alpha inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J Med Chem. 2010;53:2345–2353. | |
Tebo J, Der S, Frevel M, Khabar KS, Williams BR, Hamilton TA. Heterogeneity in control of mRNA stability by AU-rich elements. J Biol Chem. 2003;278:12085–12093. | |
Bhattacharyya S, Gutti U, Mercado J, Moore C, Pollard HB, Biswas R. MAPK signaling pathways regulate IL-8 mRNA stability and IL-8 protein expression in cystic fibrosis lung epithelial cell lines. Am J Physiol Lung Cell Mol Physiol. 2011;300:L81–L87. | |
Lau WK, Chan SC, Law AC, Ip MS, Mak JC. The role of MAPK and Nrf2 pathways in ketanserin-elicited attenuation of cigarette smoke-induced IL-8 production in human bronchial epithelial cells. Toxicol Sci. 2012;125:569–577. | |
Fogel R, Shields K, Christensen J, et al. A phase II, randomised, placebo controlled trial of 12 weeks treatment with an oral p38 inhibitor in patients with COPD on a background of ICS/LABA. Eur Respir J. 2013;42(Suppl 57):186. | |
Birrell MA, Wong S, Catley MC, Belvisi MG. Impact of tobacco-smoke on key signaling pathways in the innate immune response in lung macrophages. J Cell Physiol. 2008;214:27–37. | |
Medicherla S, Fitzgerald MF, Spicer D, et al. p38alpha-selective mitogen-activated protein kinase inhibitor SD-282 reduces inflammation in a subchronic model of tobacco smoke-induced airway inflammation. J Pharmacol Exp Ther. 2008;324:921–929. | |
Bouazza B, Debba-Pavard M, Amrani Y, et al. Basal p38 mitogen-activated protein kinase regulates unliganded glucocorticoid receptor function in airway smooth muscle cells. Am J Respir Cell Mol Biol. 2014;50(2):301–315. | |
Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766. | |
Galliher-Beckley AJ, Williams JG, Cidlowski JA. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol. 2011;31:4663–4675. | |
Bouazza B, Krytska K, Debba-Pavard M, et al. Cytokines alter glucocorticoid receptor phosphorylation in airway cells: role of phosphatases. Am J Respir Cell Mol Biol. 2012;47:464–473. | |
Wang B, Palomares K, Parobchak N, et al. Glucocorticoid receptor signaling contributes to constitutive activation of the noncanonical NF-κB pathway in term human placenta. Mol Endocrinol. 2013;27:203–211. | |
Avenant C, Kotitschke A, Hapgood JP. Glucocorticoid receptor phosphorylation modulates transcription efficacy through GRIP-1 recruitment. Biochemistry. 2010;49:972–985. | |
Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3beta-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 2008;28:7309–7322. | |
Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol. 2002;16:2382–2392. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.